

Unpredicted selectivity in the aggregation of partially labeled peptides is revealed by AFM-STED nanoscopy.
Abstract
It is known that the presence of fluorophores can influence the dynamics of molecular processes. Despite this, an affordable technique to control the fluorophore distribution within the sample, as well as the rise of unpredictable anomalous processes induced by the fluorophore itself, is missing. We coupled a stimulated emission depletion (STED) microscope with an atomic force microscope to investigate the formation of amyloid aggregates. In particular, we studied the in vitro aggregation of insulin and two alloforms of β amyloid peptides. We followed standard methods to induce the aggregation and to label the molecules at different dye-to-protein ratios. Only a fraction of the fibrillar aggregates was displayed in STED images, indicating that the labeled molecules did not participate indistinctly to the aggregation process. This finding demonstrates that labeled molecules follow only selected pathways of aggregation, among the multiple that are present in the aggregation reaction.
INTRODUCTION
Far-field optical microscopy is an essential tool not only in scientific research but also in clinical analysis and industrial quality control. The limit of the classical optical observations, fixed by Abbe’s diffraction limit (1) that imposes a maximum resolution of ~250 nm, is a severe limitation for analysis at the nanometer scale. In the past two decades, a series of original approaches open the way to a new class of far-field microscopy, defined as super-resolution (SR) optical microscopy, that shattered the diffraction barrier, improving spatial resolution by an order of magnitude or more over the diffraction limit. These techniques provide new insights into biological processes at the, hitherto inaccessible, molecular scale (2).
SR techniques are fluorescence microscopy modes (3–6) based on the use of fluorescent molecules. Ideal molecular dyes must provide high specificity, brightness, and stability. While the two latter properties are mostly related to the molecular structure of the fluorochrome itself, the specificity depends on the method used to recognize and bind a particular molecular target (7). Further, the influence of the fluorescent dye on the biological processes under investigation should be minimal.
In particular, covalent fluorescent labeling is commonly applied in vitro (8). The fluorophores used for covalent labeling are small molecules (<1 kDa) and are widely used in the study of dynamic processes of molecular interaction. Despite this, previous reports highlighted that the covalent linkage of fluorophores could affect the visualization, dynamics, or biological properties of the molecular system (9, 10). However, it is not trivial to have a direct and unambiguous assessment of the efficiency and homogeneity of the labeling, as well as on the influence that the fluorescent tag has on the molecular activity.
We propose a new approach in the study of misfolded protein aggregates based on the use of a combined system that couples the capability of an atomic force microscope (AFM) (11), a primary technique in the study of amyloid aggregation in vitro (12–14), with that of a stimulated emission depletion (STED) microscope (15). In particular, we investigated the aggregation process of a nonpathological protein, insulin from the bovine pancreas, covalently labeled with N-hydroxysuccinimide (NHS)–dye fluorophore, testing the aggregation at different dye-to-protein ratios (1:19, 1:99, and 1:499). In the second set of experiments, we used the same approach on widely used and commercially available fluorescently labeled Aβ1–42 and Aβ1–40 (16).
The formation of misfolded protein aggregates is associated with several pathological conditions (17), such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, characterized by the conversion of specific peptides or proteins from their soluble functional states into highly organized and insoluble fibrillar aggregates. In the past decade, SR microscopies were applied in the study of the molecular mechanism associated with misfolded protein diseases.
Direct stochastic optical reconstruction microscopy (dSTORM) was used to investigate the aggregation of β amyloid (Aβ) both in vitro and in situ. Morphological differences between the species obtained in vitro and in vivo were detected (18). With two-color dSTORM, the polarized elongation of Aβ1–42 fibrils was also demonstrated (19). Immunolabeled tau filaments from brain sections were imaged with a resolution of 77 nm using STED microscopy (20). In a recent study, the preferential localization of γ-secretase in the presynaptic and postsynaptic compartment has been characterized by STORM and STED microscopy (21).
In the field of Parkinson’s disease, amyloid fibrils from α-synuclein were imaged with an unprecedented resolution using binding-activated localization microscopy (22). Two-color dSTORM was used to study the elongation of preformed α-synuclein fibrils, providing new insights into the role of seeding in the fibrillogenesis (23). Last, the fibrillation of human lysozyme, involved in a fatal form of hereditary systemic amyloidosis, was characterized in detail (24).
Studying aggregates from both insulin and Aβ, we had direct and incontestable proof of the influence that the molecular dyes have on the aggregation process. In particular, we demonstrated that a fraction of labeled peptides follows selected aggregation pathways. The slowing of the kinetics of aggregation—detectable with several techniques such as thioflavin T (ThT) binding, Fourier transform infrared spectroscopy, and time-lapse AFM—is just an aspect of the influence of the labeled monomers on the intermolecular processes. The results obtained by correlative AFM-STED microscopy highlight a new and essential aspect and generate a warning: Fluorescence techniques are not able to characterize all the products derived from the in vitro aggregation of misfolded proteins. This statement reveals the importance of correlating fluorescence data with others derived from label-free techniques. This loss of information that affects not only the imaging but also the spectroscopic techniques based on the fluorescence of a dye molecule suggests the primary role that can be played by label-free techniques. This phenomenon is demonstrated here for three particular peptides (insulin, Aβ1–42, and Aβ1–40) and uses well-established and largely diffused labeling methods, but it can likely be common to other molecular systems and extended to other molecular mechanisms.
RESULTS
Analysis of insulin fibrillar aggregates
ThT fluorescence assay and AFM were used as consolidated techniques to follow the aggregation of insulin and to characterize the morphology of the fibrils. Insulin fibrils were obtained at three different dye-to-protein ratios, called 1:19, 1:99, and 1:499 (see Materials and Methods). The kinetics of aggregation was slightly delayed by the presence of a fraction of labeled monomers (fig. S1). We apply correlative AFM-STED microscopy in the study of mature fibrils, i.e., aggregates after 14 days of incubation in denaturing conditions. Fibrils have a variable length, and the thickness is slightly affected by the presence of the dye. In particular, the mean thickness calculated from the analysis of images acquired in liquid is 6.3 ± 2.7 nm for 1:19 samples, 7.5 ± 2.5 nm for 1:99 samples, and 7.6 ± 2.5 nm for 1:499 samples, while the thickness of unlabeled insulin fibrils is 5.6 ± 1.7 nm. These results indicate that the fibril thickness at different dye-to-protein ratios is compatible within the experimental errors, although the unlabeled fibrils are slightly thinner, likely due to steric effect or a different organization of the protofilaments within the fibrillary aggregates.
Considering the tip broadening effects on the AFM images, under the approximation of cylindrical symmetry, the expected apparent width derived from the relation
where R is the radius of curvature of the tip and h is the measured thickness (25). The reported heights and the nominal R value of the tip used allow us to conclude that insulin fibrils are notably flattened, likely due to the interaction and binding at the glass substrate. In particular, the mean value of W/W', where W is the measured fibril width, is 1.7 ± 0.5 in 1:19, 2.6 ± 0.6 in 1:99, 2.0 ± 1.1 in 1:499, and 1.5 ± 0.4 in the absence of labeled peptides.
The fibrillar aggregates at the three different dye-to-protein ratios were imaged using correlative AFM-STED microscopy (Fig. 1), following the procedure described in Materials and Methods. Images acquired on the 1:19 sample shows a significant presence of unlabeled fibrils (Fig. 2). The analysis of 10 images derived from three different preparations indicates a value of correlation of 0.42 ± 0.11 (see Materials and Methods). The same experiment was repeated at 1:99 (Fig. 3, A to C) and 1:499 (Fig. 3, D to F) samples (three preparations per sample). So far, the correlation between AFM and STED data drops down at lower dye-to-protein ratios, and the ratios of colocalization are 0.29 ± 0.07 (1:99, n = 10) and 0.19 ± 0.09 (1:499, n = 10). The single labeled/unlabeled fibrils are displayed in correlative images on a smaller area (fig. S2). The pixel-by-pixel correlation is shown in the scatter plots in fig. S3. All the confocal images acquired on the same area of the STED images shown here are presented in fig. S4.
Fig. 1. Correlative imaging.
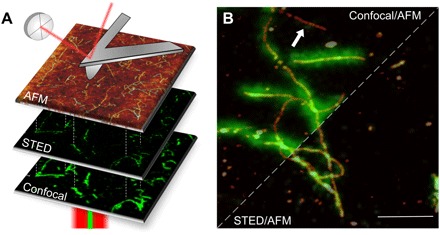
(A) Images of the same area are acquired in different modes. The confocal and STED images are acquired in reflection, while the AFM probe reaches the sample from above, providing a three-dimensional topographical view at high resolution. (B) Amyloid fibrils from bovine insulin labeled with fluorescence dye ATTO 488 NHS ester with a 1:19 dye-to-protein ratio. The STED and confocal images are overlaid with AFM topography. The resolution of the STED microscopy image is significantly enhanced with respect to the confocal microscopy image. At the same time, the AFM provides the topographical image on the same sample area. Some fibrillar aggregates are not displayed in fluorescence microscopy (e.g., white arrow). Scale bar, 1 μm.
Fig. 2. Insulin fibrils at a dye-to-protein ratio of 1:19.
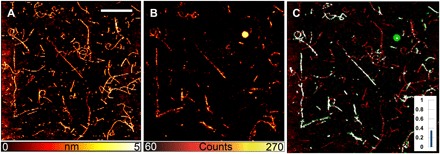
Insulin fibril (dye-to-protein ratio, 1:19) images with AFM (A), STED microscopy (B), and the correlative images obtained by (A) and (B) (C). Only a fraction of the fibrillary aggregates is displayed in the optical image. In particular, some fibrils are completely missing, indicating that the labeled monomers were not taking part in the formation of these aggregates. On the contrary, a continuous distribution of fluorophores is present in other aggregates. The ratio of colocalization for this particular field of view is indicated by the histogram. Scale bar, 5 μm. Inset (C), overlay graph.
Fig. 3. Insulin fibrils at dye-to-protein ratios of 1:99 and 1:499.
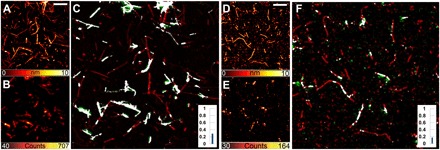
Insulin fibrils obtained from 1:99 (A to C) and 1:499 (D to F) dye-to-protein ratios. AFM images (A and D), STED microscopy images (B and E), and correlative images (C and F). Several fibrils are totally unlabeled, while a homogeneous fluorescence is emitted by some other aggregates. The ratio of colocalization is shown by the histograms (C and F, insets). Scale bars, 2 μm.
Analysis of Aβ1–42 and Aβ1–40 fibrillar aggregates
The aggregation of Aβ1–42 was induced using standard protocol proposed in the literature (26) and described in Materials and Methods. We characterize the initial state of the aggregation to exclude the presence of preexistent fibrillar aggregates that could induce important artifact in the analysis. AFM images acquired just after the resuspension of the lyophilized peptide in the aggregation medium are shown in fig. S5 and indicate the presence of a monodisperse distribution of globular aggregates. The dynamic light scattering (DLS) experiments confirm the data with a mean size of 2.0 ± 0.4 nm. The absence of fibrillary aggregates at the initial stage of the aggregation process is confirmed. Aβ1–42 aggregates faster than insulin, and after 4 hours of aggregation, fibrillar aggregates, often assembled in clusters, are present (fig. S6, A to D). ThT assay demonstrated that the kinetics of aggregation of the labeled peptides is slower (fig. S6, G and H). In addition, in Aβ1–42 samples, the correlative images indicated the presence of two different types of aggregates (Fig. 4), with a significant unlabeled fraction of fibrils already present at a dye-to-protein ratio of 1:19 (Fig. 4, A to C). The correlation between AFM and STED images, i.e., the population of fibrils that are visible in fluorescence microscopy, decreases at lower dye-to-protein ratios: 0.45 ± 0.13 (1:19), 0.26 ± 0.11 (1:99), and 0.16 ± 0.04 (1:499). The results derived from two different preparations (15 images per sample).
Fig. 4. Correlative images of fibrils from Aβ.
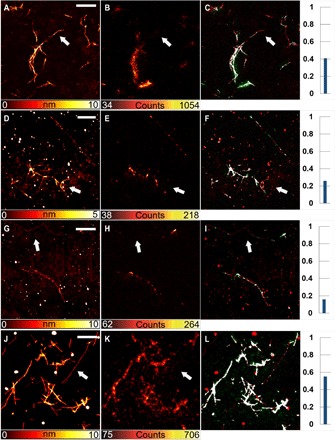
Amyloid aggregates from Aβ1–42 (A to I). Different concentrations of fluorescent peptides were present. The dye-to-protein ratios were 1:19 (A to C), 1:99 (D to F), and 1:499 (G to I). In addition, at the higher dye-to-protein ratio (A to C), some fibrils were not displayed by fluorescence images (e.g., see the white arrows). For amyloid aggregates from Aβ1–40, the dye-to-protein ratio is 1:19 (J to L). Scale bars, 2 μm.
Further, these results are confirmed on Aβ1–40 (two preparations at a 1:19 dye-to-protein ratio), analyzing 22 different images. The presence of a significant population of unlabeled fibrils is evident in Fig. 4 (J to L). In addition, in this case, we use AFM (fig. S5, E and F) and DLS techniques to exclude the presence of preformed fibrils at the initial stage of the aggregation process. The average size of the peptides just after the resuspension in the aggregation buffer is 2.4 ± 0.3 nm. Fibrillar aggregates after 1 day of aggregation time are shown in fig. S6. The scatter plot analysis is shown in fig. S3. All the confocal images acquired on the same area of the STED images shown here are presented in fig. S4.
Characterization of immunolabeled Aβ1–42 aggregates
Amyloid fibrils from Aβ1–42 were labeled via indirect immunolabeling, following a well-established procedure (see Materials and Methods). After the conjugation with the fluorescent secondary antibody, we found that all the fibrillar aggregates displayed by AFM were also visible in the STED image. As a consequence of this, the correlation between the AFM and STED images (Fig. 5) is very high for all the images acquired (n = 12) and for all the preparations (n = 2). The level of colocalization between AFM and STED images has been quantified to 0.92 ± 0.05. The typical cross section of the fibrils, measured by AFM, is significantly increased as a consequence of a massive binding of antibody molecules at the fibril surface, with a mean height value of 20.4 ± 7 nm. This indirect proof of the presence of the molecular dyes is a final example of the potentialities that are opened by the use of a correlative technique in the study of molecular interaction.
Fig. 5. Correlative images from immunolabeled samples.
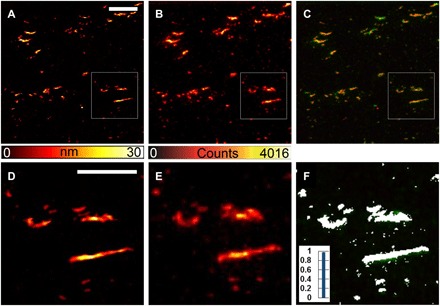
(A to F) Amyloid aggregates formed from Aβ1–42 in the absence of fluorescent monomers and subsequently labeled via indirect immunolabeling. All the features displayed in the AFM images are also visible in the STED images and are colocalized. The fibril size, measured by AFM, is larger with respect to the unlabeled fibrils, confirming the presence of a large amount of antibody on the fibrillar aggregates. Scale bars, 2 μm (A to C) and 1 μm (D to F).
DISCUSSION
Amyloid aggregates derived from insulin and two different alloforms of Aβ peptides, Aβ1–42 and Aβ1–40, were imaged by correlative AFM-STED microscopy. Our research is mainly focused on mature fibrils. The aggregation started from solutions of mixed unlabeled and labeled proteins/peptides at different dye-to-protein ratios (1:19, 1:99, and 1:499). The dye-to-protein ratio of 1:19 was chosen following well-established methods previously presented and largely used (19, 23, 27). We used 1:99 and 1:499 as controls to evaluate how the presence of fluorescent aggregates rescales with the concentration of the dye. Insulin was labeled by covalently binding a fluorophore at the free amine groups, while fluorescent Aβ1–42 presented a single fluorophore at a terminal group and was purchased by an external company. In both cases, we found that, for all three dye-to-protein ratios under investigation, fluorescent fibrils coexist with a significant fraction of aggregates that are mainly or entirely unlabeled.
Insulin from the bovine pancreas is not an amyloidogenic protein in vivo. On the other hand, it has been demonstrated that peptides and proteins that are unrelated to disease have a generic ability to form amyloid fibrils upon destabilization of their native structure induced by particular environmental conditions (28–32). In particular, we chose insulin because the structure of insulin fibrils, as well as the number of monomers that are present in a unit length of an insulin fibril, is well known (33). From small-angle scattering experiments, considering the radius of gyration and the molecular weight of a fibril nucleus before the elongation phase, it was calculated that an insulin fibril with a circular cross section of 6 nm in diameter is formed by seven protofilaments, and the number of monomer per unit length was calculated. Comparing this model with the morphological features of our insulin fibrils (average thickness and aspect ratio are reported in Results) and with the assumption of an indistinct aggregation, in which fibrils are the result of the stochastic assembly of labeled and unlabeled monomers, we deduce that at least one labeled molecule every 6 nm along the fibril axis must be expected in 1:19 samples, one every 30 nm in 1:99 samples, and one every 150 nm in 1:499 samples. This means that, considering the resolution achieved in our experimental conditions, the fibrils must appear entirely fluorescent upon STED microscopy visualization, at least in the case of 1:19 and 1:99 samples. The presence of a large population of unlabeled fibrils indicates the coexistence of distinct aggregation processes; some of them allow the contribution of the labeled peptide in the formation of aggregates, and the others preclude this contribution (Fig. 6A).
Fig. 6. Summary and ratios of colocalization.
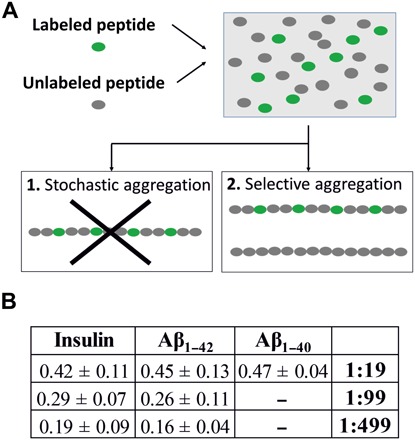
(A) A stochastic aggregation of the labeled/unlabeled peptides should bring to the formation of uniformly labeled fibrils. Our experiments indicate a different scenario where only a fraction of the fibrils has a labeled component. Other fibrils, dark in the STED analysis, are unlabeled. These two products are the results of different aggregation pathways. (B) Table showing the ratio of colocalization calculated for the different peptide species and dye-to-protein ratios.
The presence of directly labeled monomers not only slows the kinetics of the aggregation (see fig. S1) of insulin but also favors the coexistence of different aggregation pathways, bringing two distinct final products: labeled and unlabeled fibrils. We pointed out that the presence of these two species of aggregates can be evinced only by applying AFM fluorescence. However, we also noted that super-resolved fluorescence microscopy is unique in providing the sufficient capability of discerning between different tightly packed filaments arranged in a confined space.
The same procedure has been applied to the Alzheimer’s disease–associated peptides Aβ1–42 and Aβ1–40, demonstrating that the phenomenon described in detail for insulin is general in the process of aggregation of misfolded proteins. Further, unlabeled aggregates are also present when the peptides are labeled with a single dye at the N terminus, a part of the polypeptide chain that has been classically considered as scarcely relevant in fibrillation processes (19, 27). The ratio of colocalization obtained as a function of different conditions is summarized in Fig. 6B. Using a standard indirect immunolabeling method, we were able to display the entire population of fibrils within the sample, demonstrating that the loss of information evinced from directly labeled samples is not related to an instrumental limit but is inherently related to the sample properties.
These findings define a new and not expected scenario and suggest the need for new quantitative investigations aimed to characterize the physicochemical properties of the two species of aggregates, i.e., fluorescent and nonfluorescent aggregates. Further, the analysis should be extended to the bioactivity of the same species with a particular focus on their toxicity.
As a whole, these results indicate that the integration between AFM and SR fluorescence microscopy represents not only a complement but also a fundamental tool to validate the results deriving from fluorescence microscopy and that it can drive the experiments toward more accurate and less invasive methods of labeling. AFM-STED microscopy could be a fundamental test in the validation and in the quality control of the new dyes used for the in vitro study of molecular processes.
MATERIALS AND METHODS
Insulin labeling and fibrillation
Bovine insulin was purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. The protein was labeled with fluorescent dyes, ATTO 488 NHS ester (Sigma-Aldrich, St. Louis, MO, USA). NHS esters readily react with amino groups of proteins, i.e., the ε-amino groups of lysine or the N terminus, forming a chemically stable amide bond. Labeling was done in a solution of 100 mM sodium bicarbonate at pH 8.3 and by mixing monomeric insulin at a final concentration of 2 mg/ml (0.35 mM) with 1 mM ATTO 488, previously diluted in dimethyl sulfoxide (Thermo Fisher Scientific, Waltham, MA, USA). After 1 hour under stirring, the proteins were separated from the excess of free dye via filtration with Amicon Ultra centrifugal filters (molecular weight cutoff, 3000; Merck Millipore, Milan, Italy). Immediately after filtration, the concentration of the labeled proteins was determined spectrophotometrically (Cary spectrophotometer, Agilent Technologies, Santa Clara, CA, USA). The unlabeled insulin monomers were added in HCl (pH 2) solution to a final dye-to-protein ratio (i.e., labeled:unlabeled insulin) of 1:19, 1:99, and 1:499. The protein concentration was 1 mg/ml. Fibrillation took place at 60°C in glass vials, without agitation (34). AFM was used to verify the fibrillation process in the absence and presence of fluorescent molecules (fig. S1).
Aβ1–42 and Aβ1–40 fibrillation
Lyophilized synthetic human Aβ1–42/Aβ1–40 (Bachem, Bubendorf, Switzerland) was dissolved in 100% hexafluoroisopropanol solution (1 mg/ml), aliquoted, and then stored at −20°C. The solvent was evaporated, and Aβ was resuspended in 50 mM sodium hydroxide. Synthetic Aβ1–42/Aβ1–40 HiLyte Fluor 488 (HF488) (AnaSpec, San Jose, CA, USA) was dissolved in 1% ammonium hydroxide solution (1 mg/ml), aliquoted, and then stored at −20°C. Labeled peptides were used without further purification. Fluorescently labeled amyloid fibrils were prepared by dissolving unlabeled Aβ in buffer [phosphate-buffered saline (PBS)] and adding HF488-labeled Aβ to give a final dye-to-protein ratio (i.e., labeled:unlabeled Aβ) of 1:19, 1:99, and 1:499 at the 25 μM final protein concentration. The solutions were sonicated and centrifuged for 10 min at 15,000g to separate possible aggregates, and the pelleted fractions were discarded. The supernatant was incubated at 25°C for 3 days without agitation. AFM (fig. S5) and DLS were used to exclude the presence of preaggregated peptides at the beginning of the fibrillation process.
Immunolabeling analysis
Labeling of the fibrils was done by standard immunofluorescence staining. Aβ1–42 was prepared as described above, and a drop of solution after 4 days of aggregation in vitro was deposited on the microscope coverslip previously cleaned and treated with 1 M MgCl2. The fibrils were then slightly rinsed and incubated overnight with mouse monoclonal Aβ1–42 antibody (Abcam, Cambridge, UK) (which recognizes N-terminal residues 1 to 17 of human Aβ) diluted in 1% bovine serum albumin (BSA). The samples were rinsed three times with 1% BSA, then incubated with Alexa Fluor 488 secondary antibody (Sigma-Aldrich, St. Louis, MO, USA) diluted in 1% BSA for 2 hours, and rinsed again with 1% BSA and PBS.
Light scattering
DLS measurements were performed with a Zetasizer Nano ZS system (Malvern Instruments Ltd., Worcestershire, UK) at a protein concentration of 0.05 mg/ml. Aβ1–42 and Aβ1–40 were dissolved in water after pretreatment in 100% hexafluoroisopropanol and centrifugation for seed removal. The measurements were done immediately after centrifugation.
ThT assay
ThT solutions were purchased from Abcam (Cambridge, UK). The stock solutions were prepared by dissolving 30 mg of dry powder in 1 ml of water. The solution was filtered through 0.22-μm syringe filters and stored protected from light at 4°C for less than 2 weeks. The ThT solution was diluted at different concentrations for each protein. The final concentration of 100 μM with 50 μM insulin protein was used, while ThT at 50 μM with 25 μM Aβ was used. A twofold molar excess of ThT was used to ensure saturation of the binding sites. The fluorescence was measured using a FluoroMax-4 spectrofluorometer (HORIBA Jobin Yvon Inc.) using an excitation of 450 nm. The highest emission maximum observed was in the insulin-bound ThT at 490 nm, while that in Aβ-ThT at 488 nm.
AFM control measurements
AFM control measurements were performed with NanoWizard 3 (JPK Instruments, Berlin, Germany) mounted on an Axio Observer D1 (Carl Zeiss, Oberkochen, Germany) inverted optical microscope. Following a well-established methodology (13, 31, 35), a drop of the sample solution (20 μl) was deposited on cut muscovite mica mounted on glass slides. The samples were adsorbed, rinsed with deionized water to remove salts, and dried under a mild nitrogen flow before AFM imaging. All imaging was performed in tapping mode both in air and in liquid. Triangular silicon nitride cantilevers (DNP, Bruker, MA, USA) with a tip, with a typical curvature radius of 20 to 60 nm, were used for imaging in liquid. All images were acquired as 512 pixel–by–512 pixel images with a scan rate between 0.6 and 1.0 Hz.
Correlative AFM/STED
A NanoWizard II AFM head (JPK, JPK Instruments, Berlin, Germany) was mounted on a STED continuous wave gated microscope (Leica TCS SP5 Gated STED CW, Leica Instrument). STED images were collected with a Hybrid detector in a spectral range of 470 to 520 nm. We used an HCX PL APO CS 100× 1.4 numerical aperture oil objective (Leica Microsystems, Mannheim, Germany) and a scan speed of 1000 Hz for 1024 pixels per line with a 32-line average and a time-gated detection of 1.2 ns. The optical microscope stage was exchanged with an AFM compatible stage (JPK Instruments, Berlin, Germany), which features minimum mechanical coupling of noise to the AFM cantilever. The direct overlay was available in the AFM software. A short AFM scan helps fix a common point of origin between the two image coordinates by shifting the STED image (36). Direct overlay requires an image calibration file created for an objective and field of view from an image sequence of AFM movements.
All the images of insulin samples were acquired in liquid (PBS, 1×) working in tapping mode using triangular silicon nitride cantilevers (DNP, Bruker, MA, USA) with a tip, with a typical curvature radius of 20 to 60 nm and a nominal elastic constant of 0.24 N/m. The images on Aβ samples were performed in air, using single-beam silicon cantilever probes (TESPA; Bruker, USA) with a typical radius of curvature of 7 nm and a nominal spring constant of 37 N/m.
The correlative images and the level of colocalization [Pearson’s correlation coefficient (37, 38)] between AFM and optical images were calculated using the colocalization function of ImageJ (Bethesda, USA). An example is shown in fig. S7. In particular, the Colocalization Threshold plugin was used. The threshold was automatically determined by the software using the Costes autothreshold method (39). The scatter plots of the pixel by pixel correlation were also calculated using the same ImageJ function and are shown in fig. S3.
Supplementary Material
Acknowledgments
Funding: No funding was received. Author contributions: C.C. and A.D. designed the study; M.C. and C.C. performed the experiments; M.C. and P.B. carried out the data analysis; C.C. wrote the paper with help from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav8062/DC1
Fig. S1. AFM and ThT study of insulin aggregation process.
Fig. S2. Correlative images of insulin fibrils on a smaller area.
Fig. S3. Colocalization analysis between AFM and STED images.
Fig. S4. Confocal microscopy images.
Fig. S5. Aβ after resuspension of the aggregation medium.
Fig. S6. Amyloid aggregates from Aβ peptides.
Fig. S7. Example of correlation between AFM-confocal and AFM-STED images.
REFERENCES AND NOTES
- 1.Abbe E., Beiträge zur theorie des mikroskops und der mikroskopischen wahrnehmung. Arch. Mikrosk. Anat. 9, 413–418 (1873). [Google Scholar]
- 2.A. Diaspro, Nanoscopy and Multidimensional Optical Fluorescence Microscopy (Chapman and Hall/CRC, 2010).
- 3.Hell S. W., Wichmann J., Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Rust M. J., Bates M., Zhuang X., Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–796 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Vicidomini G., Bianchini P., Diaspro A., STED super-resolved microscopy. Nat. Methods 15, 173–182 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Fornasiero E. F., Opazo F., Super-resolution imaging for cell biologists. Bioessays 37, 436–451 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Apetri M. M., Harkes R., Subramaniam V., Canters G. W., Schmidt T., Aartsma T. J., Direct observation of α-synuclein amyloid aggregates in endocytic vesicles of neuroblastoma cells. PLOS ONE 11, e0153020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luitz M. P., Barth A., Crevenna A. H., Bomblies R., Lamb D. C., Zacharias M., Covalent dye attachment influences the dynamics and conformational properties of flexible peptides. PLOS ONE 12, e0177139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Ham T. J., Esposito A., Kumita J. R., Hsu S.-T. D., Kaminski Schierle G. S., Kaminski C. F., Dobson C. M., Nollen E. A. A., Bertoncini C. W., Towards multiparametric fluorescent imaging of amyloid formation: Studies of a YFP model of α-synuclein aggregation. J. Mol. Biol. 395, 627–642 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Binnig G., Quate C. F., Gerber C., Atomic force microscope. Phys. Rev. Lett. 56, 930–933 (1986). [DOI] [PubMed] [Google Scholar]
- 12.Mangione M. R., Vilasi S., Marino C., Librizzi F., Canale C., Spigolon D., Bucchieri F., Fucarino A., Passantino R., Cappello F., Bulone D., San Biagio P. L., Hsp60, amateur chaperone in amyloid-beta fibrillogenesis. Biochim. Biophys. Acta Gen. Subj. 1860, 2474–2483 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Di Gaetano S., Guglielmi F., Arciello A., Mangione P., Monti M., Pagnozzi D., Raimondi S., Giorgetti S., Orrù S., Canale C., Pucci P., Dobson C. M., Bellotti V., Piccoli R., Recombinant amyloidogenic domain of ApoA-I: Analysis of its fibrillogenic potential. Biochem. Biophys. Res. Commun. 351, 223–228 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Calamai M., Canale C., Relini A., Stefani M., Chiti F., Dobson C. M., Reversal of protein aggregation provides evidence for multiple aggregated states. J. Mol. Biol. 346, 603–616 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Harke B., Chacko J. V., Haschke H., Canale C., Diaspro A., A novel nanoscopic tool by combining AFM with STED microscopy. Opt. Nanoscopy 1, 3 (2012). [Google Scholar]
- 16.Esbjörner E. K., Chan F., Rees E., Erdelyi M., Luheshi L. M., Bertoncini C. W., Kaminski C. F., Dobson C. M., Kaminski Schierle G. S., Direct observations of amyloid β self-assembly in live cells provide insights into differences in the kinetics of Aβ(1–40) and Aβ(1–42) aggregation. Chem. Biol. 21, 732–742 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiti F., Dobson C. M., Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Kaminski Schierle G. S., van de Linde S., Erdelyi M., Esbjörner E. K., Klein T., Rees E., Bertoncini C. W., Dobson C. M., Sauer M., Kaminski C. F., In situ measurements of the formation and morphology of intracellular β-amyloid fibrils by super-resolution fluorescence imaging. J. Am. Chem. Soc. 133, 12902–12905 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Young L. J., Kaminski Schierle G. S., Kaminski C. F., Imaging Aβ(1–42) fibril elongation reveals strongly polarised growth and growth incompetent states. Phys. Chem. Chem. Phys. 19, 27987–27996 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benda A., Aitken H., Davies D. S., Whan R., Goldsbury C., STED imaging of tau filaments in Alzheimer’s disease cortical grey matter. J. Struct. Biol. 195, 345–352 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Schedin-Weiss S., Caesar I., Winblad B., Blom H., Tjernberg L. O., Super-resolution microscopy reveals γ-secretase at both sides of the neuronal synapse. Acta Neuropathol. Commun. 4, 29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ries J., Udayar V., Soragni A., Hornemann S., Nilsson K. P. R., Riek R., Hock C., Ewers H., Aguzzi A. A., Rajendran L., Superresolution imaging of amyloid fibrils with binding-activated probes. ACS Chem. Neurosci. 4, 1057–1061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinotsi D., Buell A. K., Galvagnion C., Dobson C. M., Kaminski Schierle G. S., Kaminski C. F., Direct observation of heterogeneous amyloid fibril growth kinetics via two-color super-resolution microscopy. Nano Lett. 14, 339–345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn M., Ghaemmaghami S., Huang Y., Phuan P.-W., May B. C. H., Giles K., DeArmond S. J., Prusiner S. B., Pharmacokinetics of quinacrine efflux from mouse brain via the P-glycoprotein efflux transporter. PLOS ONE 7, e39112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.C. Canale, B. Torre, D. Ricci, P. C. Braga, in Atomic Force Microscopy in Biomedical Research: Methods and Protocols (Humana Press, 2011), pp. 31–43. [Google Scholar]
- 26.Ladiwala A. R. A., Litt J., Kane R. S., Aucoin D. S., Smith S. O., Ranjan S., Davis J., Van Nostrandand W. E., Tessier P. M., Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 287, 24765–24773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vestergaard M., Hamada T., Saito M., Yajima Y., Kudou M., Tamiya E., Takagi M., Detection of Alzheimer’s amyloid beta aggregation by capturing molecular trails of individual assemblies. Biochem. Biophys. Res. Commun. 377, 725–728 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Guijarro J. I., Sunde M., Jones J. A., Campbell I. D., Dobson C. M., Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. U.S.A. 95, 4224–4228 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Litvinovich S. V., Brew S. A., Aota S., Akiyama S. K., Haudenschild C., Ingham K. C., Formation of amyloid-like fibrils by self-association of a partially unfolded fibronectin type III module. J. Mol. Biol. 280, 245–258 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Chiti F., Webster P., Taddei N., Clark A., Stefani M., Ramponi G., Dobson C. M., Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. U.S.A. 96, 3590–3594 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Relini A., Torrassa S., Rolandi R., Gliozzi A., Rosano C., Canale C., Bolognesi M., Plakoutsi G., Bucciantini M., Chiti F., Stefani M., Monitoring the process of HypF fibrillization and liposome permeabilization by protofibrils. J. Mol. Biol. 338, 943–957 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Vetri V., Canale C., Relini A., Librizzi F., Militello V., Gliozzi A., Leone M., Amyloid fibrils formation and amorphous aggregation in concanavalin A. Biophys. Chem. 125, 184–190 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Nayak A., Sorci M., Krueger S., Belfort G., A universal pathway for amyloid nucleus and precursor formation for insulin. Proteins 74, 556–565 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Jansen R., Dzwolak W., Winter R., Amyloidogenic self-assembly of insulin aggregates probed by high resolution atomic force microscopy. Biophys. J. 88, 1344–1353 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrotta R., Canale C., Diaspro A., Trapani A., Biagio P. L. S., Bulone D., Inhibiting effect of αs1-casein on Aβ1–40 fibrillogenesis. Biochim. Biophys. Acta Gen. Subj. 1820, 124–132 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Chacko J. V., Canale C., Harke B., Diaspro A., Sub-diffraction nano manipulation using STED AFM. PLOS ONE 8, e66608 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolte S., Cordelières F. P., A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Manders E. M. M., Verbeek F. J., Aten J. A., Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169, 375–382 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., Lockett S., Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86, 3993–4003 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav8062/DC1
Fig. S1. AFM and ThT study of insulin aggregation process.
Fig. S2. Correlative images of insulin fibrils on a smaller area.
Fig. S3. Colocalization analysis between AFM and STED images.
Fig. S4. Confocal microscopy images.
Fig. S5. Aβ after resuspension of the aggregation medium.
Fig. S6. Amyloid aggregates from Aβ peptides.
Fig. S7. Example of correlation between AFM-confocal and AFM-STED images.