

Conspectus
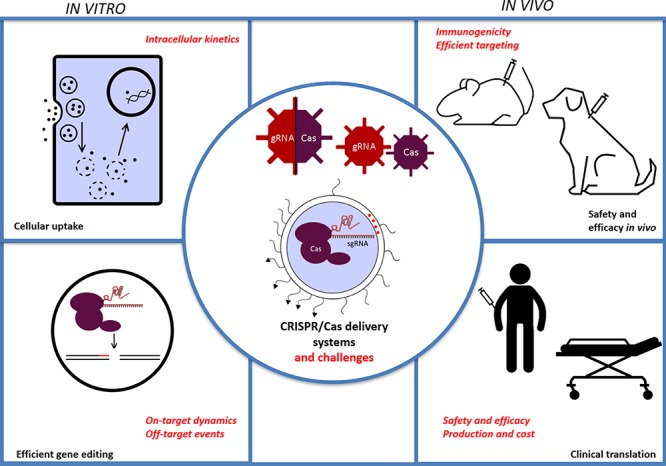
The discovery of CRISPR/Cas has revolutionized the field of genome editing. CRIPSR/Cas components are part of the bacterial immune system and are able to induce double-strand DNA breaks in the genome, which are resolved by endogenous DNA repair mechanisms. The most relevant of these are the error-prone nonhomologous end joining and homology directed repair pathways. The former can lead to gene knockout by introduction of insertions and deletions at the cut site, while the latter can be used for gene correction based on a provided repair template. In this Account, we focus on the delivery aspects of CRISPR/Cas for therapeutic applications in vivo. Safe and effective delivery of the CRISPR/Cas components into the nucleus of affected cells is essential for therapeutic gene editing. These components can be delivered in several formats, such as pDNA, viral vectors, or ribonuclear complexes. In the ideal case, the delivery system should address the current limitations of CRISPR gene editing, which are (1) lack of targeting specific tissues or cells, (2) the inability to enter cells, (3) activation of the immune system, and (4) off-target events.
To circumvent most of these problems, initial therapeutic applications of CRISPR/Cas were performed on cells ex vivo via classical methods (e.g., microinjection or electroporation) and novel methods (e.g., TRIAMF and iTOP). Ideal candidates for such methods are, for example, hematopoietic cells, but not all tissue types are suited for ex vivo manipulation. For direct in vivo application, however, delivery systems are needed that can target the CRISPR/Cas components to specific tissues or cells in the human body, without causing immune activation or causing high frequencies of off-target effects.
Viral systems have been used as a first resort to transduce cells in vivo. These systems suffer from problems related to packaging constraints, immunogenicity, and longevity of Cas expression, which favors off-target events. Viral vectors are as such not the best choice for direct in vivo delivery of CRISPR/Cas. Synthetic vectors can deliver nucleic acids as well, without the innate disadvantages of viral vectors. They can be classed into lipid, polymeric, and inorganic particles, all of which have been reported in the literature. The advantage of synthetic systems is that they can deliver the CRISPR/Cas system also as a preformed ribonucleoprotein complex. The transient nature of this approach favors low frequencies of off-target events and minimizes the window of immune activation. Moreover, from a pharmaceutical perspective, synthetic delivery systems are much easier to scale up for clinical use compared to viral vectors and can be chemically functionalized with ligands to obtain target cell specificity. The first preclinical results with lipid nanoparticles delivering CRISPR/Cas either as mRNA or ribonucleoproteins are very promising. The goal is translating these CRISPR/Cas therapeutics to a clinical setting as well. Taken together, these current trends seem to favor the use of sgRNA/Cas ribonucleoprotein complexes delivered in vivo by synthetic particles.
Introduction
RNA-guided endonucleases derived from the bacterial CRISPR/Cas system have gained tremendous popularity over the use of protein-guided nucleases for genome editing during the past years. This is owed to the ease at which target gene specificity can be changed, enabling precise genome surgery on-targeted diseased cells. This gene surgery method has widespread applications, including crop manipulation, cancer diagnostics, and gene therapy. Preclinical data demonstrate the power of this technology in correcting genetic diseases, and we start to better understand the CRISPR/Cas machinery from a molecular perspective. However, despite CRIPSR/Cas technology slowly moving into the clinic, there remain some critical questions unanswered. One of these questions is whether CRISPR/Cas can be administered safely and effectively to humans via direct intravenous administration. For this, the delivery method being used is critically important and should ideally restrict genome editing to affected target cells only, and thereby avoid gene edits in nontarget cells.
In this Account, we will address the current status of in vivo CRISPR/Cas delivery with both synthetic and viral vectors and will focus on the differences in delivery methods in terms of on-target genome editing efficiency and off-target effects. In addition, we will discuss ways how immunogenicity via bacterial Cas9 in humans can be diminished.1
CRISPR/Cas Mechanism of Action and the Minimal Components for Genome Editing
Guide RNA (gRNA) and CRISPR-associated (Cas) proteins are key components of a bacterial defense system based around clustered regularly interspaced palindromic repeats (CRISPR). Together, they enable prokaryotes to develop adaptive immune responses against invading mobile genetic elements, such as bacteriophages. This CRISPR/Cas system has been engineered into a two-part system to enable therapeutic genome editing in eukaryotic cells: a single guide RNA (sgRNA) and a Cas endonuclease together form the active ribonucleoprotein (RNP) complex. The most commonly used Cas endonuclease is Cas9, although other variants have been discovered for gene editing purposes since then, such as Cpf1.2 The sgRNA sequence consists of two domains: the spacer sequence, which consists of 20 nucleotides targeting the RNP complex to the DNA, and a backbone sequence anchoring it to the protein.3
Therapeutic gene editing is achieved through induction of a double-strand break (DSB) at the DNA locus, directed by the sgRNA. This process requires a specific nucleotide sequence, the protospacer-adjacent motif (PAM), to be present on the target strand in order for the Cas protein to be activated. The active complex cleaves the two DNA strands upstream of the PAM. Different Cas proteins require different PAM sequences, for example 5′-NGG for Cas9 derived from Streptococcus pyogenes (spCas9) or 5′-TTTN for Cpf1. Different Cas proteins also have different cleavage patterns. SpCas9, for example, induces a blunt DSB 3 nucleotides upstream of the PAM. A DSB can be induced near any PAM site specific to the chosen Cas protein by changing the 20nt guide RNA sequence. This makes CRISPR/Cas a more appealing method for gene editing than the previously used Zinc-finger nucleases and TAL-effector nucleases, which rely on the engineering of Fok1 endonuclease to induce double-strand breaks.1,4 Cas9 can also be engineered to induce a single-strand nick (Cas9 nickase, nCas9) or to simply bind the DNA without endonuclease activity (inactive Cas9, dCas9). The latter can be fused to other active regulatory components, such as base-editors.5,6
There are several formats in which the sgRNA and Cas protein can be delivered into the cell to achieve therapeutic gene editing. These have been summarized in Figure 1A. The endonuclease is problematic to deliver, due to the high molecular weight of the protein (158.9 kDa for spCas9) and the gene length (around 4 kb). The gene can be delivered either as an expression plasmid or by viral vectors which need to be imported into the nucleus for transcription. Additionally, it can be delivered as mRNA which is directly translated in the cytosol. sgRNA can be delivered as synthetic oligonucleotides, or expressed through plasmids or viral vectors. The combination of Cas protein and gRNA can be delivered as a single plasmid, viral vector(s), or as preformed RNP complexes which only need to localize to the nucleus. An HDR template for specific repair can finally be delivered as single strand DNA (suited for small mutational corrections) or as large DNA plasmids (suited knock-in of large sequences or whole genes). HDR template sequences contain the corrected gene and two flanking homology arms (HA) to improve affinity around the site of the DSB (1,4,8). After the induction of a DSB, the broken DNA ends are recognized by proteins belonging to the DNA repair machinery, leading to activation of DNA repair. This is achieved through one of several different repair pathways, which are more extensively reviewed elsewhere.7 The most relevant pathways are nonhomologous end joining (NHEJ), homology directed repair (HDR), and microhomology mediated repair (MMR). NHEJ is imperfect and often leads to small insertions or deletions (indels) in the genome. This can be exploited for gene knockout by introduction of premature STOP-codons or shifts of the genetic reading frame. Gene correction and knock-in can be achieved through HDR, by addition of a template DNA strand, thereby leading to repair complementary to the provided template.7 These are shown in Figure 1B.
Figure 1.

Schematic summary of CRISPR/Cas endonuclease concepts. (A) Different formats in which Cas protein, gRNA, and HDR templates can be used to achieve gene editing. (B) The active RNP complex acts by cleaving 2 DNA strands at the sgRNA target site in the prescence of a PAM sequence (red). Three repair mechanisms can occur: (1) NHEJ, which can induce gene knockout by random indel formation; (2, 3) HDR using a ssDNA or dsDNA template, respectively.7
Direct Delivery of CRISPR/Cas
While CRISPR/Cas mediated therapeutic gene knockout and correction have many potential applications, the practical execution is not straightforward. Multiple components need to be delivered into the nuclei of target cells for the desired therapeutic effect. Delivery of genetic material or proteins can be done by directly disrupting the barriers between a drug and its target, while barely interacting with the therapeutic cargo. These methods are used extensively in vitro to study the effects of CRISPR/Cas systems on the genome because they are economical and often easy to implement on cell lines. While most direct methods of delivery are difficult to utilize in vivo, they can be used to introduce CRISPR/Cas components ex vivo to cells harvested from patients, before reintroducing them into the patient. Notable examples are hematopoietic cells for treatment of sickle-cell anemia, chimeric antigen receptor (CAR) T cells, and germline cells. The main delivery barriers in these cases are the target cell membrane, potentially endosomal release, and nuclear localization of the active complex.8,9
Traditional methods of direct transfection have first been investigated. The main advantage of these techniques is that the uptake mechanism is independent of the cell. Microinjection of single fast-dividing cells has been used to generate a great variety of knockout and transgenic animals by directly injecting zygotes with CRISPR components into the nucleus. While this technique is very effective, it has the distinct disadvantage of cells requiring individual manipulation.10 Electroporation, by which pores are formed in cell membranes upon application of a high voltage, can be used to directly transfect cells ex vivo as well as some in vivo tissues. This has, for example, been used to transfect human B-cells with CRISPR/Cas RNP to induce production of therapeutic proteins, after differentiation into plasma cells.11 Electroporation can be very toxic, however, due to this technique harming the cell membrane. In some cases this leads to permanent permeabilization of the membrane.12
Two novel techniques to deliver CRISPR/Cas RNPs into cells are through induction of transmembrane internalization assisted by membrane filtration (TRIAMF) and induced transduction by osmocytosis and propane betaine (iTOP). In TRIAMF, cells are extruded through a membrane, which has smaller pores than the cell diameter, thereby inducing transient pore formation in the cell membrane. This method was used to deliver RNPs in hematopoietic stem/progenitor cells (HSPCs), which generally exhibit low endocytic uptake and require more direct methods of transfection. They achieved a similar efficiency compared to electroporation techniques, while observing less cytotoxicity.13 In iTOP, hypertonic sodium chloride is added to the outside milieu of the cells along with propane-betaine NDSB-201. These components cause the formation of endosomes through macropinocytosis, which allow uptake of proteins and subsequent release by disrupting the endosomal membrane.14
While these direct delivery methods are promising to alter specific cells ex vivo, they are limited in their application as not all tissues are suitable for ex vivo manipulation. Other delivery methods are therefore needed to deliver CRISPR/Cas directly in vivo. This can be done either intravenously or through local administration, for example, intramuscularly for Duchenne’s muscular dystrophy. The latter has the distinct advantage of achieving a high dose in the target tissue and thus a high likelihood of gene editing.15 Intravenous administration has the relative advantage of reaching a wider target, such as whole organs or systemic targets like vascular endothelium. The optimal route of administration needs to be determined for each tissue individually.
Viral Delivery Methods
The ultimate goal in CRISPR therapy is to genetically correct cells directly in the human body and thereby curing a debilitating genetic disease. This requires sophisticated carrier systems that ideally target cells with high specificity, combined with minimal cytotoxicity, and rapid clearing of the CRISPR system after successful gene modification. However, none of the currently available delivery methods fulfill all of the above criteria. Viral vectors have been used as a first resort to solve the delivery problem of CRISPR/Cas gene editing system. The most widely studied vectors include lentiviral, adeno-associated viral, and adenoviral vectors. A comparison of their main properties is given in Table 1.
Table 1. Comparison of the Main Properties, Advantages and Disadvantages of Commonly Used Viral Vectorsa.
| vector type | packaging capacity | diameter | genome type | advantages | disadvantages | current examples |
|---|---|---|---|---|---|---|
| AAV | <4.4 kB | 20–22 nm | ssDNA | large variety of target tissues, low immunogenicity on first injection | low packaging capacity | (16) |
| AV | >8 kB | 80–100 nm | dsDNA | large packaging capacity, transient Cas expression | pre-existing antibodies, high immunogenicity | (17) |
| LV | <8.5 kB | 80–120 nm | ssRNA | large packaging capacity | potential insertional mutagenesis | (18−21) |
References of current examples are given for future reading.
Adeno-associated viruses (AAVs) combine low immunogenicity upon first injection with serotype-related target cell specificity and relatively long expression of the gene without the necessity for genome integration. However, the packaging capacity is limited and, as a consequence, the genetic material encoding the most frequently used spCas9 (4.2 kB) leaves limited space for necessary regulatory elements, such as promoter and polyadenylation signal sequences. This can be solved by splitting spCas9 into two fragments that can recombine inside the cell so that the truncated genes will fit the AAV vector, but this comes at the cost of efficiency in terms of delivery as well as target DNA cutting.16
Adenoviral vectors (AVs) can easily contain all elements for genome editing due to their high packaging capacity, expressing both the Cas protein as well as one or multiple sgRNAs from a single vector. In addition, large donor DNA sequences to mediate homology-directed repair can be codelivered as well. The advantage of this is that sgRNA and Cas protein are consistently expressed in the same cell at a fixed ratio and since AVs are nonintegrating, Cas expression is transient in dividing cells. AVs have been successfully used for in vivo genome editing in mice, although immune-related toxicities were observed.17
Lentiviral vectors (LVs) are at present the most widely used viral vectors for clinical gene therapy applications in which long-lasting expression of a gene is required. The advantage of LVs is the relatively safe genomic integration of the gene construct and the capacity to transduce both dividing and nondividing cells with high efficiency. However, the feature that makes this vector suitable for gene delivery (stable and long-lasting expression) is counterproductive for gene editing purposes. Long-lasting expression of the Cas protein is considered to be unfavorable for the on-target/off-target ratio of indel formation.18−20 Indeed, a direct comparison of frequencies of indel formation at three potential genomic off-target sites by spCas9 delivered as mRNA, pDNA, RNP, or lentivirus showed highest off-target frequencies with the lentiviral delivery method.21 To counteract this, self-inactivating constructs have been designed in which the lentiviral vector encodes for Cas9 protein and two sgRNAs: one against the target sequence of choice and one against the Cas9 gene.22 In this way, transient expression of Cas9 from an integrating lentiviral vector can be obtained.
Immunogenicity associated with the use of viral vectors for gene editing is often downplayed by assuming single injections will be enough to obtain gene correction and thereby cure of a disease. As long as pre-existing antibodies are absent, this single-shot approach could indeed be effective in isolated cases. However, for many monogenic diseases a certain threshold of gene-correction is required to revert the disease phenotype. For example, to cure hemophilia B, it is estimated that the levels of FIX activity should be increased from <2% of normal activity to at least 25–100% (0.25–1.00 IU/ml). Current gene therapy applications can reach levels of 0.12 IU/ml, which is enough to revert severe hemophilia into a mild form, but not enough to completely stop prophylactic FIX treatment.23 Given the low gene correction efficiencies currently obtained through HDR in vivo such a threshold can only be obtained in case multiple injections of the viral vector are feasible to accumulate enough gene corrections to revert the disease. At present, this is not possible as high dose systemic delivery of viral vectors will prime the immune system to generate large quantities of neutralizing antibodies upon concomitant exposure, even under an immunosuppressive regimen.24
Nonviral Delivery Methods
The disadvantages of viral systems, such as a limited packaging capacity and immune activation, have led to the development of synthetic delivery vectors. Synthetic materials are often well characterized and controlled, do not rely on a viral genome and are tunable through chemical modification. Notable properties have been summarized in Figure 2. Disadvantages include possible problematical biocompatibility and toxicity, immunogenic potential, and problems with therapeutic cargo release. A variety of materials can be used to create these particles and address these problems, some efforts of which will be discussed here.
Figure 2.
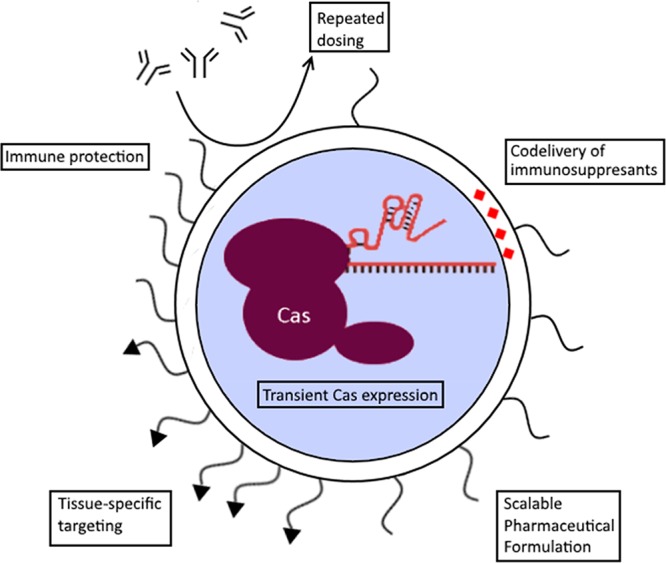
Advantages of synthetic vectors for CRISPR/Cas delivery using a lipid nanoparticle as example. The active RNP complex can be encapsulated by synthetic vectors, leading to a transient expression of the Cas protein. Addtionally, there is less risk of immune activation compared to viral vectors which allows for repeated dosing regimens, to potentially achieve cumulative gene editing.28 Most particles incorporate an inert component which shields the particle from immune detection, such as polyethylene glycol (PEG). These chains can be functionalized to target specific tissues or cells of interest using targeting ligands. Other cargoes can be codelivered as well, such as immune suppresant drugs. Finally, the chemical nature of the particle formation and modification allows for upscaling of the pharmaceutical production compared to biological production methods for viral particles.
The simplest synthetic delivery method is by direct conjugation of an excipient molecule to an active substance. This can, for example, be done by conjugation of cell-penetrating peptides (CPPs) to gRNA and Cas protein. By doing so, Ramakrishna et al. have shown effective gene editing in HEK293T cells. The conjugation lead to 6,2% editing efficacy for RNP and 7,2% for plasmids, measured by knockout of a reporter gene. However, it is unlikely that these CPP conjugates will circumvent all delivery barriers outlined in the introduction.25 Sophisticated delivery platforms such as nanoparticles can be engineered to do just that.
Lipid materials are well characterized to create nanocarrier systems. Recent development of liposomal systems has given rise to lipid nanoparticles (LNPs) based on ionizable cationic lipids, which exhibit a cationic charge in the lowered pH of late endosomes to induce endosomal escape, because of the tertiary amines in their structure.26 While these LNPs were initially developed for use with RNA interference (RNAi) components such as Onpattro, they can also be used for CRISPR/Cas delivery.27
One such application was examined by Wang et al. Briefly they show that using biodegradable cationic lipid nanoparticles, one can deliver CRISPR/Cas RNP into cells and induce effective gene knockout.29 The use of a disulfide chain in the lipid would then act as a release mechanism by leading to degradation of the particle in the endosome, which may also contribute to endosomal release.30 An example of in vivo delivery of CRISPR/Cas is the LNP platform developed by Finn et al. They used an ionizable lipid along with cholesterol, DSPC, and a PEGylated lipid to create nanoparticles for delivery of Cas9 mRNA and sgRNA to rat livers. They targeted the gene for transthyretin, after which they showed a decrease of >97% of serum transthyretin levels.28 Interestingly, they demonstrated that multiple injections with these LNPs with weekly or monthly intervals led to cumulative gene editing. This will be relevant for correcting genetic defects that require high levels of gene correction in order to revert the disease phenotype. A comparison of the mentioned cationic lipids has been given in Figure 3.
Figure 3.

Key lipid structures of the formulations in the main text. D-Lin-MC3-DMA and LP01 are ionizable lipids used in Onpattro and the formulation of Finn et al., respectively.28 8-O14B is the biodegradable cationic lipid outlined by Wang et al.29
Polymer based particles can be used for CRISPR/Cas delivery in a similar manner as lipids. Materials which have been used for delivery of other nucleic acids have also been investigated for CRISPR/Cas delivery. Cationic polymers such as polyethylenimine (PEI) can be complexed to nucleic acids and can induce endosomal uptake and release, similarly to cationic lipids. Zhang et al. have for example formulated particles consisting of PEI-β-cyclodextrin to deliver plasmids coding for sgRNA and Cas9 in HeLa cells, achieving gene knockout.31 Sun et al. have also used PEI in their formulation, in which they utilized DNA as a nanomaterial for encapsulation of CRISPR/Cas vectors. These particles were coated by PEI to improve endosomal release. They injected these particles directly into tumors expressing EGFP in mice and found phenotypes exhibiting efficient EGFP knockout.32 Dendrimeric structures of poly(amido-amine) (PAMAM) can also be used for transfection. These particles consist of a core, from which the polymer branches and they exhibit cationic primary amines on their surface, which can complex to nucleic acids. Kretzmann et al., for example, used dendrimers to deliver CRISPR/dCas9 plasmids to MCF-7, a human breast adenocarcinoma cell line. They showed effective transfection while maintaining low cytotoxicity.33
Inorganic materials are currently being studied to encapsulate CRISPR/Cas components as well. Alsaiari et al. have, for example, formulated a network based on zinc to aid cross-linking of imidazole. The low pH of late endosomes would then, after uptake, result in cationic charges due to dissolution of the zeolitic imidazole frameworks (ZIF), after which the CRISPR-Cas components are released into the cytosol. These ZIFs have been used to successfully deliver Cas9-based RNPs into CHO cells. They showed endosomal release of the RNP’s and cell viability for at least 12 h after transfection.34 Lee et al. showed successful delivery of RNP and HDR template using colloidal gold nanoparticles in a mouse model for the treatment of Duchenne muscular dystrophy. They induced HDR to repair a single nucleotide mutation which caused knockout of the active dystrophin. They showed that 5.4% of expression was restored compared to the expression in wild-type mice, which was sufficient to restore the musculature to a healthy phenotype.35
The main properties and stage of development of the described formulations have been summarized in Table 2. In addition, it is poorly understood how an HDR template can be delivered into nuclei using synthetic vectors, especially for slow or nondividing cells where the nuclear envelope is rarely or not disrupted for mitosis. Viral vectors are innately able to do so and often exploit active transport pathways through the nuclear pore complex. A mixture of particles containing different cargo may be used to overcome these issues. One example is the combination of LNPs for delivery of Cas9 mRNA along with an AAV containing both sgRNA and HDR template sequences. The rationale is that the sgRNA and HDR template are needed in the nucleus while the mRNA is needed in the cytosol. Yin et al. showed successful delivery and phenotypic repair in a knockout mouse model of hereditary tyrosinemia type I.36 This example shows promise for the utilization of multiple particles in vivo for liver targeting. A disadvantage of such an approach is the requirement of uptake of both particles into the same tissue at roughly the same time to ensure intracellular RNP formation and HDR-mediated repair.
Table 2. Summary of the Specific Synthetic Delivery Systems Outlined in the Main Texta.
| particle material | investigated cargo format | reported advantages | reported stage of development | route of administration | ref |
|---|---|---|---|---|---|
| cationic lipids | RNP | high endosomal escape, biodegradable | in vivo reporter model in mouse brain | intravenous | (29) |
| ionizable lipids (LNPs) | mRNA | cumulative gene editing upon repeated dosing in vivo | in vivo disease model for tyrosinemia | intravenous | (26, 28) |
| PEI polyplexes | Plasmid DNA | easily characterizable | in vitro | not yet applicable | (31) |
| PEI-coated DNA nanoclews | Plasmid DNA | high efficacy upon local administration in a reporter system | in vivo reporter model | intratumoral injection | (32) |
| PAMAM dendrimers | Plasmid DNA | high loading efficiency | in vitro | not yet applicable | (33) |
| ZIF-8 | RNP | high loading capacity, biodegradable | in vitro | not yet applicable | (34) |
| CRISPR Gold | RNP | low immunogenicity locally, in vivo proof of concept in relevant disease model (Duchenne’s muscular dystrophy) | in vivo disease model for Duchenne’s muscular dystrophy | intramuscular | (35) |
The cargo formats and some advantages and disadvantages are given.
For direct in vivo application, current trends seem to favor use of synthetic particles to deliver the CRISPR/Cas components either as mRNA or as RNP complexes. Lipid, polymeric, and inorganic particles have all been tested in vivo and seem able to deliver CRISPR/Cas components. Of these, LNP based formulations seem the most promising for in vivo gene delivery as their low toxicity was already examined for siRNA formulations earlier. Currently, the most advanced CRISPR/Cas study has been performed by Finn et al. using LNPs in mice, which targets the liver. This tissue exhibits fenestrated capillary endothelia, through which the LNPs can pass. In addition to this passive targeting, these LNPs are opsonized by apolipoprotein E in the bloodstream which then acts as a targeting ligand due to overexpression of the low density lipoprotein receptor on hepatocytes.37 More research needs to be done examining other target tissues to confirm if LNP based delivery is more generally applicable and can achieve the desired effects in a clinical setting.
Immune Responses and Immunogenicity
Since the CRISPR system is of bacterial origin, an immune reaction against one of its components is likely to occur when it is administered for direct in vivo genome editing.38 Moreover, the type of delivery vector used may fortify this immune response and should therefore be carefully chosen. The mode of delivery (e.g., as gene construct, mRNA, or RNP) will also influence the overall immunogenicity of the gene editing system as longevity of Cas protein expression generally favors antigen presentation and thus potential activation of adaptive immune responses.39,40
A distinction should be made between innate and adaptive immune responses. Innate immune responses can be triggered by the nucleic acid cargo, especially when formulated in as nanoparticles.41 It has been reported that exogenous mRNA as well as siRNA delivered by lipid nanoparticles activate innate immune responses through activation of various pattern recognition receptors, specifically toll-like receptors. Pseudouridine modification of the in vitro transcribed mRNA or 2′OMe or 2′MOE modifications of the siRNA can ameliorate such responses. Furthermore, CRISPR guide RNAs consist of hairpins that are known to be good activators of such receptors, like TLR3, PKR, and RIG-I. This should be considered when CRISPR/Cas components are delivered as mRNA or ribonucleoproteins. Pharmacological inhibition of these innate immune responses would be an option to prevent undesired immunological effects against CRISPR/Cas.42−44 For example, toll-like receptor antagonists or drugs inhibiting the downstream signaling pathways (e.g., NfkB or MyD88) could help in dampening innate immune responses against CRISPR/Cas components, although full inhibition of immune responses is most likely difficult to achieve.
Adaptive responses can be directed against the Cas protein or against components of the delivery system. Viral vectors (in particular adenoviral vectors) are immunogenic, especially at the high doses that are often needed for effective transduction in humans.42−44 Synthetic vectors can also mount adaptive immune responses. For lipid-based systems with grafted PEG polymers to enhance circulation times, anti-PEG antibodies have been described although clinical effects of such antibodies are under dispute.45,46 Antivector antibodies may prevent repeated dosing to boost the overall level of gene editing that may be needed for a therapeutic effect.
Adaptive immune responses against the Cas proteins are common. In fact, several studies have demonstrated that both anti-Cas antibodies and Cas-specific cellular responses pre-exist in the human population due to exposure via the microbiome.47−49 This pre-existing immunity has important implications for clinical applications of CRISPR/Cas as it may influence the effectiveness of the gene editing therapy but may also cause serious safety problems. Antibody-responses can be partly mitigated by mRNA delivery of Cas instead of RNPs or by encapsulation of the Cas RNP into nanocarriers to shield the immunogenic protein from neutralizing antibodies. Conversely, Cas proteins could be immuno-engineered to remove B and T cell epitopes without losing activity or one could revert to Cas variants from microorganisms that are not common to humans, such as the recently discovered CasX.50 Such strategies would at most lead to reduction rather than elimination of immunogenicity. More troublesome are the cellular responses that could potentially lead to cell killing after gene correction, thereby nullifying the therapeutic effect. Like gene therapy with viral vectors, CRISPR/Cas will most likely require coadministration of immunosuppressants, a proven method to prevent immune responses against often very immunogenic proteins. The downside is that most immunosuppressant regimens are systemic, resulting in an increased vulnerability of the patient against infectious diseases during treatment. Recent developments in antigen-specific tolerization might be further explored to avoid the need of systemic immunosuppression.51
Off-Target Events and the Influence of Cargo format
While the on-target efficiency of therapeutic gene editing is important to optimize, we also need to recognize the risk of gene editing outside the target locus. This can potentially lead to gene knockout of other genes. Several bioinformatic tools predict off-target sites based on homology to the target sequence, which can be used to choose sgRNA with minimal off-target effects, for example the Cas-OFFinder tool.52 Occurred off-target events can be confirmed experimentally in a biased (based on predicted off-target sites) or unbiased (whole genome) manner.53,54 The variety of techniques can make direct comparisons between experiments difficult, as there are conflicting variables, such as sensitivity and different on-target efficiencies, between experiments. In addition, the choice of Cas protein is significant to reduce off-target events. For example, Shen et al. have shown reduced generation of off-target events using Cas9 nickases in mice, possibly due to the requirement of two cleavage events instead of one.6 In addition, Anderson et al. have shown, for example, that using higher fidelity Cas proteins significantly reduce the generation of off-target editing events.55 Guide RNAs can be engineered as well, to improve targeting specificity by chemical or structural modifications and DNA replacements. Modifications such as phosphorothiolates to the ribose-phosphate backbone of gRNA have been shown to improve editing efficiency on-target.56,57 Internal 2′-O-methyl-3′-phosphonacetate modifications lead to fewer off-target events.57 Additionally, Yin et al. demonstrated that partial replacement of RNA nucleotides with DNA nucleotides can lead to higher on-target efficiency and reduce off-target cleavage.58
To theoretically reduce the risk of off-target events, one can minimize the exposure time to the active RNP complex. This can, for example, be achieved by fusing Cas9 to a FKBP12-like domain, which marks Cas9 for intracellular degradation unless a specific ligand is bound to that domain. This ligand can then be codelivered, which achieves a period of Cas9 activity while also lowering the half-life.18,19 Alternatively, the CRISPR/Cas complex can be directly inhibited by the peptide AcrllA4, which is able to bind active RNP complexes and directly compete on the PAM recognition site. Using this inhibitory peptide, Shin et al. have shown that there is an ideal time window for Cas9 with mostly on target cutting in the first 6 h followed by off-target events later on.59 The exposure time can also be lowered by choosing more transiently active cargo formats. Kim et al. showed that treatment with RNPs reduced the generation of off-target mutations up to 10-fold compared to delivered plasmids coding for Cas9 and sgRNA. They also showed that Cas9 exhibits a maximum activity after 1 day of exposure when delivered as RNP compared to 3 days when delivered as plasmid, proposing that these kinetic differences contribute to the perceived off-target frequencies.60 Kouranova et al. compared Cas9 delivered as protein, DNA vector, or mRNA along with sgRNA in two cell lines. They found the highest on-target efficiency and lowest off-target events in normal cells treated with RNPs or cells stably expressing Cas9 treated with sgRNA.61 Finally, Lattanzi et al. showed by using a deep-sequencing assay on known off-target sites that a lentiviral vector produced more off-target editing compared to mRNA, plasmid, or RNP delivery, while not reaching the same on-target effects as RNP or mRNA delivery.21
Based on the current body of data, delivery of RNPs using bioinformatics inspired sgRNA design and an optimized Cas protein seems to be the most rational method to minimize the risk of off-target effects. However, the influence of exposure time and dose-dependency on off-target editing needs further elucidation, preferably using unbiased whole-genome screening. In addition, the main focus in the literature is on the off-target editing events in targeted cells. The unwanted targeting of other cells can also be considered as off-target events, even if the genomic target is correct. This can be caused by usage of viral vectors with an undesired tropism, or by the poor ability of synthetic vectors to target certain cell types. For example, the majority of synthetic vectors are accumulated in the liver and spleen after intravenous injection and this may not be desired if a genetic disease is manifested outside these organs.
Concluding Remarks
CRISPR/Cas genome editing is less than a decade old but has already reached the stage of clinical development. CTX001 from CRISPR Therapeutics and Vertex Pharma is the first ex vivo CRISPR therapy for beta thalassemia in clinical development and more are ongoing in China. These initial applications of CRISPR/Cas in the clinic are treating diseases in which the affected cells are readily accessible and can be edited ex vivo. This avoids the ongoing challenge of tissue and cell type specific delivery in vivo and mitigates two main hurdles that CRISPR/Cas systems are currently facing: immunogenicity and off-target editing effects. These pioneering clinical trials are being watched with much anticipation but may also reveal some unanticipated side effects. While every effort is being taken to ensure effectiveness and safety, such potential side effects can only be disclosed by performing human trials.
The ultimate goal would be to cure debilitating (mono)genetic diseases with a single injection of CRISPR/Cas. We are still far from this goal and to achieve this several shortcomings of the CRISPR/Cas system need to be addressed.
First, we should have better insights into the frequency and clinical impact of off-target events. Although the algorithms to predict off-target sites are getting better over time, as well as the design of the gRNAs, unbiased whole genome approaches have revealed several sites that have remained under the radar of such algorithms. Additionally, the clinical consequences of such off-target mutagenesis are unclear. Engineering Cas proteins to make them more potent to specific sites or to induce point mutations without the need of introducing double strand breaks are being explored and may in fact be the way forward for safe gene editing. Another approach to increase the on-target/off-target ratio is to reduce exposure time of the genomic DNA to Cas proteins. Prolonged expression seems to favor increased off-target frequency and strategies to limit or control exposure times are being explored. Moreover, targeted delivery is also crucial to limit unnecessary exposure of nontarget tissue to the Cas nucleases. Although we are still far from such a magic bullet, several delivery systems have been developed that show good targeting to hepatocytes in the liver. As such it is expected that the first applications of direct in vivo genome editing will focus on liver diseases in which gene knockout is enough to revert the disease phenotype. With all of these potential reductions of off-target events in mind, it will still be nearly impossible to fully eliminate the probability of off-target events, let alone prove that no off-target events have occurred.
By far the biggest hurdle for widespread in vivo application of CRISPR/Cas is the immunogenicity of the CRISPR/Cas components. Although encapsulation of the components in nanocarrier systems might temporarily cause protection against antibody binding and neutralization, eventually the components need to be released to exert their gene editing action. Cellular responses against cells expressing Cas9 have been described, which pose a serious threat to the success and safety of in vivo gene editing. Strategies to mitigate such immune responses, including coadministration of immunosuppressive drugs, should therefore be explored.
Despite the challenging tasks ahead, the first steps toward direct in vivo application of CRISPR/Cas gene editing have been made and the preclinical results look promising. Intellia Therapeutics has developed a lipid nanoparticle (LNP) platform for the delivery of CRISPR/Cas to the liver, in particular to hepatocytes. With their delivery platform they have reached >97% knock down of serum transthyretin (TTR) levels in healthy mice with a single injection. Moreover, knock down was effective for at least one year.28
These encouraging results will spur other in vivo applications with CRISPR/Cas. One that might be very interesting is the targeted integration of gene expression constructs for long-term in situ expression of biopharmaceuticals. Increasing numbers of patients require lifelong treatment with biopharmaceuticals that often need frequent injections either i.v. or s.c. Examples are anti-TNF alpha antibody therapies and enzyme replacement therapies. These treatments are expensive and inconvenient for the patient. Targeted insertion of gene constructs in long-lived liver hepatocytes could in principle provide prolonged (up to years) expression without the need of frequent injections. However, this will only become a reality in case we can fully guarantee the safety of in vivo genome editing. Whatever the application, it is important to balance the medical benefit with the risks that come from the treatment. With this in mind, it is likely that CRISPR will eventually realize its potential to cure a wide range of diseases.
Acknowledgments
The authors thank J. A. W. Jong for his significant contributions toward preparing Figure 3.
Biographies
Danny Wilbie completed his Pharmacy B.Sc. and M.Sc. degrees at Utrecht University in 2016 and 2018, respectively. Currently, he is working as a Ph.D. candidate in the group of Enrico Mastrobattista, where he is currently studying the delivery barriers for therapeutic CRISPR/Cas gene editing.
Johanna Walther received her bachelor’s degree in biochemistry at Martin-Luther-University, Halle (Saale), Germany, in 2016. Since 2017 she is doing her master’s at Utrecht University in Drug Innovation, within which she worked in the group of Enrico Mastrobattista on delivery of CRISPR/Cas as gene therapy.
Enrico Mastrobattista obtained his Ph.D. in Advanced Drug Delivery from Utrecht University in 2001. He currently leads a research group that develops biomimetic drug delivery systems for the targeted delivery of therapeutic proteins, peptides, and nucleic acids. His main areas of expertise are drug delivery, pharmaceutical biotechnology, and nanobiotechnology with a focus on the intracellular delivery of nucleic acids and genetic vaccines.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Nanomedicine and Beyond”.
References
- Doudna J.; Charpentier E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Zetsche B.; Gootenberg J. S.; Abudayyeh O. O.; Regev A.; Koonin E. V.; Zhang F. Cpf1 Is a Single RNA-Guided Endonuclease of a Article Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A.; Cong L.; Yan W. X.; Scott D. A.; Gootenberg J. S.; Kriz A. J.; Zetsche B.; Shalem O.; Wu X.; Makarova K. S.; Koonin E. V.; Sharp P. A.; Zhang F. In Vivo Genome Editing Using Staphylococcus Aureus Cas9. Nature 2015, 520, 186–191. 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Blenke E.; Evers M. J. W.; Mastrobattista E.; van der Oost J. CRISPR-Cas9 Gene Editing: Delivery Aspects and Therapeutic Potential. J. Controlled Release 2016, 244, 139–148. 10.1016/j.jconrel.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Wu W. Y.; Lebbink J. H. G.; Kanaar R.; Geijsen N.; Van Der Oost J. Genome Editing by Natural and Engineered CRISPR-Associated Nucleases. Nat. Chem. Biol. 2018, 14, 642–651. 10.1038/s41589-018-0080-x. [DOI] [PubMed] [Google Scholar]
- Shen B.; Zhang W.; Zhang J.; Zhou J.; Wang J.; Chen L.; Wang L.; Hodgkins A.; Iyer V.; Huang X.; Skarnes W. C. Efficient Genome Modification by CRISPR-Cas9 Nickase with Minimal off-Target Effects. Nat. Methods 2014, 11, 399–402. 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- Salsman J.; Masson J.-Y.; Orthwein A.; Dellaire G. CRISPR/Cas9 Gene Editing: From Basic Mechanisms to Improved Strategies for Enhanced Genome Engineering In Vivo. Curr. Gene Ther. 2018, 17, 263–274. 10.2174/1566523217666171122094629. [DOI] [PubMed] [Google Scholar]
- Foss D. V.; Hochstrasser M. L.; Wilson R. C. Clinical Applications of CRISPR-Based Genome Editing and Diagnostics. Transfusion 2019, 59, 1389. 10.1111/trf.15126. [DOI] [PubMed] [Google Scholar]
- Sürün D.; von Melchner H.; Schnütgen F. CRISPR/Cas9 Genome Engineering in Hematopoietic Cells. Drug Discovery Today: Technol. 2018, 28, 33–39. 10.1016/j.ddtec.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Xu W. Microinjection and Micromanipulation: A Historical Perspective. Methods Mol. Biol. 2019, 1874, 1–16. 10.1007/978-1-4939-8831-0_1. [DOI] [PubMed] [Google Scholar]
- Hung K. L.; Meitlis I.; Hale M.; Chen C. Y.; Singh S.; Jackson S. W.; Miao C. H.; Khan I. F.; Rawlings D. J.; James R. G. Engineering Protein-Secreting Plasma Cells by Homology-Directed Repair in Primary Human B Cells. Mol. Ther. 2018, 26, 456–467. 10.1016/j.ymthe.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S.-W.Overview of Drug Delivery and Alternative Methods to Electroporation BT - Electroporation Protocols: Preclinical and Clinical Gene Medicine. In Electroporation Protocols. Methods in Molecular Biology; Li S., Ed.; Humana Press: Totowa, NJ, 2008; pp 91–107. [DOI] [PubMed] [Google Scholar]
- Yen J.; Fiorino M.; Liu Y.; Paula S.; Clarkson S.; Quinn L.; Tschantz W. R.; Klock H.; Guo N.; Russ C.; Yu V. W. C.; Mickanin C.; Stevenson S. C.; Lee C.; Yang Y. TRIAMF: A New Method for Delivery of Cas9 Ribonucleoprotein Complex to Human Hematopoietic Stem Cells. Sci. Rep. 2018, 8, 16304. 10.1038/s41598-018-34601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Astolfo D. S.; Pagliero R. J.; Pras A.; Karthaus W. R.; Clevers H.; Prasad V.; Lebbink R. J.; Rehmann H.; Geijsen N. Efficient Intracellular Delivery of Native Proteins. Cell 2015, 161, 674–690. 10.1016/j.cell.2015.03.028. [DOI] [PubMed] [Google Scholar]
- Tabebordbar M.; Zhu K.; Cheng J. K. W.; Chew W. L.; Widrick J. J.; Yan W. X.; Maesner C.; Wu E. Y.; Xiao R.; Ran F. A.; Cong L.; Zhang F.; Vandenberghe L. H.; Church G. M.; Wagers A. J. In Vivo Gene Editing in Dystrophic Mouse Muscle and Muscle Stem Cells. Science 2016, 351 (6271), 407–411. 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B.; Volz S. E.; Zhang F. A Split-Cas9 Architecture for Inducible Genome Editing and Transcription Modulation. Nat. Biotechnol. 2015, 33, 139–142. 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Mou H.; Li S.; Li Y.; Hough S.; Tran K.; Li J.; Yin H.; Anderson D. G.; Sontheimer E. J.; Xue W.; et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum. Gene Ther. 2015, 26, 432–442. 10.1089/hum.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski L. A.; Chen L.-c.; Maynard-Smith L. A.; Ooi A. G. L.; Wandless T. J. A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell 2006, 126, 995–1004. 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senturk S.; Shirole N. H.; Nowak D. G.; Corbo V.; Pal D.; Vaughan A.; Tuveson D. A.; Trotman L. C.; Kinney J. B.; Sordella R. Rapid and Tunable Method to Temporally Control Gene Editing Based on Conditional Cas9 Stabilization. Nat. Commun. 2017, 8, 14370. 10.1038/ncomms14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.; Lee N.; Cho S.; Cho B. K. Targeted Genome Editing Using DNA-Free RNA-Guided Cas9 Ribonucleoprotein for CHO Cell Engineering. Methods Mol. Biol. 2018, 1772, 151–169. 10.1007/978-1-4939-7795-6_8. [DOI] [PubMed] [Google Scholar]
- Lattanzi A.; Meneghini V.; Pavani G.; Amor F.; Ramadier S.; Felix T.; Antoniani C.; Masson C.; Alibeu O.; Lee C.; Porteus M. H.; Bao G.; Amendola M.; Mavilio F.; Miccio A. Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Mol. Ther. 2019, 27, 137–150. 10.1016/j.ymthe.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merienne N.; Vachey G.; Longprez L.; De; Perrier A. L.; du Pasquier R.; Deglon N.; et al. The Self-Inactivating KamiCas9 System for the Editing of CNS Disease Genes Resource. Cell Rep. 2017, 20, 2980–2991. 10.1016/j.celrep.2017.08.075. [DOI] [PubMed] [Google Scholar]
- Miesbach W.; Meijer K.; Coppens M.; Kampmann P.; Klamroth R.; Schutgens R.; Tangelder M. Gene Therapy with Adeno-Associated Virus Vector 5 – Human Factor IX in Adults with Hemophilia B. Blood 2018, 131, 1022–1031. 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haasteren J.; Hyde S. C.; Gill D. R. Lessons Learned from Lung and Liver In-Vivo Gene Therapy: Implications for the Future. Expert Opin. Biol. Ther. 2018, 18, 959–972. 10.1080/14712598.2018.1506761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna S.; Kwaku Dad A. B.; Beloor J.; Gopalappa R.; Lee S. K.; Kim H. Gene Disruption by Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA. Genome Res. 2014, 24, 1020–1027. 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Ashwanikumar N.; Robinson E.; Duross A.; Sun C.; Murphy-Benenato K. E.; Mihai C.; Almarsson Ö.; Sahay G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated MRNA. Nano Lett. 2017, 17, 5711–5718. 10.1021/acs.nanolett.7b02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi E. P.; Mire C. E.; Lee A. C. H.; Geisbert J. B.; Ursic-Bedoya R.; Agans K. N.; Robbins M.; Deer D. J.; Cross R. W.; Kondratowicz A. S.; Fenton K. A.; MacLachlan I.; Geisbert T. W. SiRNA Rescues Nonhuman Primates from Advanced Marburg and Ravn Virus Disease. J. Clin. Invest. 2017, 127 (12), 4437–4448. 10.1172/JCI96185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn J. D.; Smith A. R.; Patel M. C.; Shaw L.; Youniss M. R.; van Heteren J.; Dirstine T.; Ciullo C.; Lescarbeau R.; Seitzer J.; Shah R. R.; Shah A.; Ling D.; Growe J.; Pink M.; Rohde E.; Wood K. M.; Salomon W. E.; Harrington W. F.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- Wang M.; Zuris J. A.; Meng F.; Rees H.; Sun S.; Deng P.; Han Y.; Gao X.; Pouli D.; Wu Q.; Georgakoudi I.; Liu D. R.; Xu Q. Efficient Delivery of Genome-Editing Proteins Using Bioreducible Lipid Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 2868–2873. 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Chen X.; Glass Z.; Gao F.; Mao L.; Wang M.; Xu Q. Integrating Combinatorial Lipid Nanoparticle and Chemically Modi Fi Ed Protein for Intracellular Delivery and Genome Editing. Acc. Chem. Res. 2019, 52, 665–675. 10.1021/acs.accounts.8b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Wan T.; Chen Y.; Chen Y.; Sun H.; Cao T.; Songyang Z.; Tang G.; Wu C.; Ping Y.; Xu F. J.; Huang J. Cationic Polymer-Mediated CRISPR/Cas9 Plasmid Delivery for Genome Editing. Macromol. Rapid Commun. 2019, 40, 1800068. 10.1002/marc.201800068. [DOI] [PubMed] [Google Scholar]
- Sun W.; Ji W.; Hall J. M.; Hu Q.; Wang C.; Beisel C. L.; Gu Z. Self-Assembled DNA Nanoclews for the Efficient Delivery of CRISPR-Cas9 for Genome Editing. Angew. Chem., Int. Ed. 2015, 54, 12029–12033. 10.1002/anie.201506030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzmann J. A.; Ho D.; Evans C. W.; Plani-Lam J. H. C.; Garcia-Bloj B.; Mohamed A. E.; O’Mara M. L.; Ford E.; Tan D. E. K.; Lister R.; Blancafort P.; Norret M.; Iyer K. S. Synthetically Controlling Dendrimer Flexibility Improves Delivery of Large Plasmid DNA. Chem. Sci. 2017, 8, 2923–2930. 10.1039/C7SC00097A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaiari S. K.; Patil S.; Alyami M.; Alamoudi K. O.; Aleisa F. A.; Merzaban J. S.; Li M.; Khashab N. M. Endosomal Escape and Delivery of CRISPR/Cas9 Genome Editing Machinery Enabled by Nanoscale Zeolitic Imidazolate Framework. J. Am. Chem. Soc. 2018, 140, 143–146. 10.1021/jacs.7b11754. [DOI] [PubMed] [Google Scholar]
- Lee K.; Conboy M.; Park H. M.; Jiang F.; Kim H. J.; Dewitt M. A.; Mackley V. A.; Chang K.; Rao A.; Skinner C.; Shobha T.; Mehdipour M.; Liu H.; Huang W.-C.; Lan F.; Bray N. L.; Li S.; Corn J. E.; Kataoka K.; et al. Nanoparticle Delivery of Cas9 Ribonucleoprotein and Donor DNA in Vivo Induces Homology-Directed DNA Repair. Nat. Biomed. Eng. 2017, 1, 889–901. 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Song C. Q.; Dorkin J. R.; Zhu L. J.; Li Y.; Wu Q.; Park A.; Yang J.; Suresh S.; Bizhanova A.; Gupta A.; Bolukbasi M. F.; Walsh S.; Bogorad R. L.; Gao G.; Weng Z.; Dong Y.; Koteliansky V.; Wolfe S. A.; et al. Therapeutic Genome Editing by Combined Viral and Non-Viral Delivery of CRISPR System Components in Vivo. Nat. Biotechnol. 2016, 34, 328–333. 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; Querbes W.; De S.; Qin J.; Frank-kamenetsky M.; Jayaprakash K. N.; Jayaraman M.; Rajeev K. G.; Cantley W. L.; Dorkin J. R.; Butler J. S.; Qin L.; Racie T.; Sprague A.; Fava E.; Zeigerer A.; Hope M. J.; Zerial M.; Sah D. W. Y.; et al. Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew W. L. Immunity to CRISPR Cas9 and Cas12a Therapeutics. WIREs Syst. Biol. Med. 2018, 10, e1408. 10.1002/wsbm.1408. [DOI] [PubMed] [Google Scholar]
- Karikó K.; Muramatsu H.; Welsh F. A.; Ludwig J.; Kato H.; Akira S.; Weissman D. Incorporation of Pseudouridine Into MRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering R.; Real C. I.; John M. J.; Jahn-Hofmann K.; Ickenstein L. M.; Kleinehr K.; Paul A.; Gibbert K.; Dittmer U.; Gerken G.; Schlaak J. F. Chemical Modifications on SiRNAs Avoid Toll-Likereceptor-Mediated Activation of the Hepatic Immune System in Vivo and in Vitro. Int. Immunol. 2014, 26, 35–46. 10.1093/intimm/dxt040. [DOI] [PubMed] [Google Scholar]
- Kedmi R.; Ben-arie N.; Peer D. The Systemic Toxicity of Positively Charged Lipid Nanoparticles and the Role of Toll-like Receptor 4 in Immune Activation. Biomaterials 2010, 31, 6867–6875. 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Colamonici O. R.; Domanski P.; Sweitzer S. M.; Larner A.; Buller R. M. L. Vaccinia Virus B18R Gene Encodes a Type I Interferon-Binding Protein That Blocks Interferon Alpha Transmembrane Signaling. J. Biol. Chem. 1995, 270, 15974–15978. 10.1074/jbc.270.27.15974. [DOI] [PubMed] [Google Scholar]
- Kanzler H.; Barrat F. J.; Hessel E. M.; Coffman R. L. Therapeutic Targeting of Innate Immunity with Toll-like Receptor 4 (TLR4) Antagonists. Nat. Med. 2007, 13, 552–559. 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S.; Wang W.; Tamaki Z.; Shi B.; Yeldandi A.; et al. Pharmacological Inhibition of Toll-Like Receptor-4 Signaling by TAK242 Prevents and Induces Regression of Experimental Organ Fibrosis. Front. Immunol. 2018, 9, 2434. 10.3389/fimmu.2018.02434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh Y.; Wang H.; Lin W.; Roffler S. R.; Cheng T.; Su Y. Pre-Existing Anti-Polyethylene Glycol Antibody Reduces the Therapeutic Efficacy and Pharmacokinetics of PEGylated Liposomes. Theranostics 2018, 8, 3164–3175. 10.7150/thno.22164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier P.; Viana I. M. d. O.; Lima E. M.; Bertrand N. Anti-Polyethylene Glycol Antibodies Alter the Protein Corona Deposited on Nanoparticles and the Physiological Pathways Regulating Their Fate in Vivo. J. Controlled Release 2018, 287, 121–131. 10.1016/j.jconrel.2018.08.022. [DOI] [PubMed] [Google Scholar]
- Simhadri V. L.; McGill J.; McMahon S.; Wang J.; Jiang H.; Sauna Z. E. Prevalence of Pre-Existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther.--Methods Clin. Dev. 2018, 10, 105–112. 10.1016/j.omtm.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth C. T.; Deshpande P. S.; Dever D. P.; Camarena J.; Lemgart V. T.; Cromer M. K.; Vakulskas C. A.; Collingwood M. A.; Zhang L.; Bode N. M.; Behlke M. A.; Dejene B.; Cieniewicz B.; Romano R.; Lesch B. J.; Gomez-ospina N.; Mantri S.; Pavel-dinu M.; Weinberg K. I.; et al. Identification of Preexisting Adaptive Immunity to Cas9 Proteins in Humans. Nat. Med. 2019, 25, 249–255. 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner D. L.; Amini L.; Wendering D. J.; Burkhardt L.; Akyüz L.; Reinke P.; Volk H.; Schmueck-henneresse M. High Prevalence of Streptococcus Pyogenes Cas9- Reactive T Cells within the Adult Human Population. Nat. Med. 2019, 25, 242–248. 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- Liu J.-J.; Orlova N.; Oakes B. L.; Ma E.; Spinner H. B.; Baney K. L. M.; Chuck J.; Tan D.; Knott G. J.; Harrington L. B.; Al-Shayeb B.; Wagner A.; Brötzmann J.; Staahl B. T.; Taylor K. L.; Desmarais J.; Nogales E.; Doudna J. A. CasX Enzymes Comprise a Distinct Family of RNA-Guided Genome Editors. Nature 2019, 566, 218–223. 10.1038/s41586-019-0908-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübbers J.; Rodríguez E.; van Kooyk Y. Modulation of Immune Tolerance via Siglec-Sialic Acid Interactions. Front. Immunol. 2018, 9, 1–13. 10.3389/fimmu.2018.02807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S.; Park J.; Kim J. S. Cas-OFFinder: A Fast and Versatile Algorithm That Searches for Potential off-Target Sites of Cas9 RNA-Guided Endonucleases. Bioinformatics 2014, 30, 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel A.; Fine E. J.; Bao G.; Porteus M. H. Quantifying On- and off-Target Genome Editing. Trends Biotechnol. 2015, 33, 132–140. 10.1016/j.tibtech.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F.; Sánchez-Hernández S.; Gutiérrez-Guerrero A.; Pinedo-Gomez J.; Benabdellah K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int. J. Mol. Sci. 2016, 17, 1507. 10.3390/ijms17091507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. R.; Haeussler M.; Watanabe C.; Janakiraman V.; Lund J.; Modrusan Z.; Stinson J.; Bei Q.; Buechler A.; Yu C.; Thamminana S. R.; Tam L.; Sowick M. A.; Alcantar T.; O’Neil N.; Li J.; Ta L.; Lima L.; Roose-Girma M.; et al. CRISPR Off-Target Analysis in Genetically Engineered Rats and Mice. Nat. Methods 2018, 15, 512–514. 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Zeng C.; Dong Y. Design and Assessment of Engineered CRISPR-Cpf1 and Its Use for Genome Editing. Nat. Protoc. 2018, 13, 899–914. 10.1038/nprot.2018.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D. E.; Taussig D.; Steinfeld I.; Phadnis S. M.; Lunstad B. D.; Singh M.; Vuong X.; Okochi K. D.; McCaffrey R.; Olesiak M.; Roy S.; Yung C. W.; Curry B.; Sampson J. R.; Bruhn L.; Dellinger D. J. Improving CRISPR-Cas Specificity with Chemical Modifications in Single-Guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. 10.1093/nar/gkx1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Song C.-Q.; Suresh S.; Kwan S.-Y.; Wu Q.; Walsh S.; Ding J.; Bogorad R. L.; Zhu L. J.; Wolfe S. A.; Koteliansky V.; Xue W.; Langer R.; Anderson D. G. Partial DNA-Guided Cas9 Enables Genome Editing with Reduced off-Target Activity. Nat. Chem. Biol. 2018, 14, 311–316. 10.1038/nchembio.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.; Jiang F.; Liu J.-J.; Bray N. L.; Rauch B. J.; Baik S. H.; Nogales E.; Bondy-Denomy J.; Corn J. E.; Doudna J. A. Disabling Cas9 by an Anti-CRISPR DNA Mimic. Sci. Adv. 2017, 3, e1701620. 10.1126/sciadv.1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Kim D.; Cho S. W.; Kim J.; Kim J. S. Highly Efficient RNA-Guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouranova E.; Forbes K.; Zhao G.; Warren J.; Bartels A.; Wu Y.; Cui X. CRISPRs for Optimal Targeting: Delivery of CRISPR Components as DNA, RNA, and Protein into Cultured Cells and Single-Cell Embryos. Hum. Gene Ther. 2016, 27, 464–475. 10.1089/hum.2016.009. [DOI] [PMC free article] [PubMed] [Google Scholar]