

Abstract
Chemerin receptor (CMKLR1) is a G protein-coupled receptor (GPCR) implicated in macrophage-mediated inflammation and in several forms of human arthritis. Analogous to other GPCR, CMKLR1 is likely regulated by G protein-coupled receptor kinase (GRK) phosphorylation of intracellular domains in an activation-dependent manner, which leads to recruitment and termination of intracellular signaling via desensitization and internalization of the receptor. The ubiquitously expressed GRK family members include GRK2, GRK3, GRK5, and GRK6, but it is unknown which GRK regulates CMKLR1 cellular and signaling functions. Our data show that activation of CMKLR1 by chemerin in primary macrophages leads to signaling and functional outcomes that are regulated by GRK6 and β-arrestin 2. We show that arrestin recruitment to CMKLR1 following chemerin stimulation is enhanced with co-expression of GRK6. Further, internalization of endogenous CMKLR1, following the addition of chemerin, is decreased in inflammatory macrophages from GRK6- and β-arrestin 2-deficient mice. These GRK6- and β-arrestin 2-deficient macrophages display increased migration toward chemerin and altered AKT and Extracellular-signal Related Kinase (ERK) signaling. Our findings show that chemerin-activated CMKLR1 regulation in inflammatory macrophages is largely GRK6 and β-arrestin mediated, which may impact innate immunity and have therapeutic implications in rheumatic disease.
Keywords: chemerin, G protein-coupled receptor kinase (GRK), arrestin, arthritis, macrophage, G protein-coupled receptor (GPCR)
1. Introduction
The chemokine-like receptor 1 (CMKLR1), also known as ChemR23 or the chemerin receptor, was first cloned in 1996 and classified as a G protein-coupled receptor (GPCR) (1,2). It is a 42 kDa, seven-transmembrane domain protein structurally similar to complement component 3a receptor (C3aR), C5a receptor (C5aR), and other chemoattractant receptors (1,2). CMKLR1 is expressed on various leukocyte subsets, including monocytes, macrophages, natural killer cells, immature plasmacytoid dendritic cells (pDCs), as well as adipocytes (2–5). In 2003, a CMKLR1 ligand, chemerin, was identified and was classified as both an adipokine and a chemoattractant peptide (5,6). Chemerin is secreted into the plasma as pro-chemerin and requires proteolytic processing for activation (6). Previous reports have demonstrated CMKLR1-mediated migration ex vivo of both human and mouse leukocytes, such as pro-inflammatory macrophages and pDCs (6–9).
Chemerin and CMKLR1 are implicated in various inflammatory diseases and metabolic syndromes, and in particular, rheumatic disease. Elevated levels of bioactive chemerin are found in rheumatoid arthritis (RA), osteoarthritis (OA), psoriatic arthritis (PA), and lupus nephritis (5,6,10–18). In contrast, Resolvin E1, another natural ligand of CMKLR1, has anti-inflammatory properties (19,20).
RA is a chronic, inflammatory autoimmune disease that affects more than 1 million people in the United States (21). It is characterized by inflammation coupled with leukocyte migration to the joint in a chemokine-dependent manner followed by articular destruction. Within the RA synovium, CMKLR1 is expressed by articular chondrocytes (12), macrophages, immature dendritic cells (DCs), and fibroblast-like synoviocytes (FLS) (13). In addition, chemerin was found in vitro to enhance secretion of pro-inflammatory proteins, including TNF-α, IL1-β, IL-6, IL-8, MMP-1 and MMP-8 by articular chondrocytes, IL-6 and MMP-3 by FLS, and to increase phosphorylation of AKT and ERK1/2 (p44/42 MAPK) in both chondrocytes and FLS (12,13). Circulating plasmacytoid dendritic cells have been shown to express CMKLR1 and migrate toward chemerin in transendothelial migration assays (7), which further implicates chemerin and CMKLR1 in RA disease pathogenesis.
Analogous to the established activation paradigm of other GPCRs, it is likely that CMKLR1 undergoes a conformational change after binding chemerin. Activation of GPCR typically leads to dissociation of intracellular heterotrimeric G proteins (Gα and Gβγ subunits) and subsequent downstream signaling via second messengers (22). G protein-coupled receptor kinases (GRKs) regulate GPCRs via phosphorylation of their cytoplasmic domains in an activation-dependent manner, which leads to termination of receptor signaling via desensitization and subsequent internalization of the receptor (23). Previous reports have identified 12 serine/threonine residues within the intracellular domains of CMKLR1 that are putative sites for GRK phosphorylation (2). Site-directed mutagenesis of CMKLR1 Ser343, one of the predicted GRK phosphorylation sites, results in loss of receptor internalization after stimulation by chemerin (24). Although not all GPCRs employ this mechanism after agonist activation, desensitization by GRKs is often followed by β-arrestin recruitment to the receptor, which targets the receptor for internalization (25). There are two ubiquitously expressed, non-visual arrestin isoforms in mammals, β-arrestin 1 (also called arrestin-2) and β-arrestin 2 (arrestin-3), as well as two “visual arrestins” (arrestin-1 and arrestin-4), which are restricted to the eye (26). Previous work by De Henau, et al. has shown that chemerin binding to CMKLR1 can induce the recruitment of both β-arrestin-1 and β-arrestin-2, but that downstream signaling to ERK1/2 requires β-arrestin-2, but not β-arrestin-1 (27). To our knowledge, specific GRK-mediated regulation of chemerin/CMKLR1 functions has not been previously defined.
There are seven known GRK isoforms (GRK1 – 7) further sub-divided into three families, all of which have a high degree of sequence homology, particularly within sub-families (28,29). GRK2, GRK3, GRK5, and GRK6 are expressed ubiquitously (28,30,31), whereas the expression patterns of GRK1, GRK4 and GRK7 are more tissue restricted (32–35). GRK2 and GRK3 are members of the same subfamily (share 85% sequence homology) (28) and GRK5 and GRK6 comprise another subfamily (share 80% sequence homology) (29). The GRK2/GRK3 subfamily are cytosolic proteins that require a C-terminal pleckstrin homology (PH) domain for receptor ligand-induced translocation to the membrane (36). In contrast, the GRK5/GRK6 subfamily are predominately localized to the membrane via C-terminal post-translational modifications and N-terminal Phosphatidylinositol 4,5-bisphosphate (PIP2) binding regions and lack the PH domain (36).
While similar expression patterns and shared sequence homology suggest overlapping GRK function, there is growing evidence that specific GRKs can 1) selectively regulate different GPCRs through differential phosphorylation patterns and/or recruitment, and 2) can be selectively activated by specific ligands (37–41). In addition, recent studies have suggested that GPCRs can initiate G protein independent signaling (e.g., GRK/β-arrestin-dependent signaling), leading to biased physiologic outcomes (25,42–45), suggesting that not only do GRKs regulate GPCRs directly, but also influence and control downstream signals.
Given the relevance of chemerin/CMKLR1 in inflammatory macrophages and in the pathogenesis of rheumatic diseases, such as RA (13,15), we sought to better understand the regulatory role of GRK isoforms 2, 3, 5 and 6 after receptor activation by pro-inflammatory chemerin. These investigations have identified specific regulation by GRK6 and β-arrestin 2 that is important to chemerin/CMKLR1-mediated cellular functions, which could, in turn, better inform the development and utilization of future therapeutics for rheumatic diseases.
2. Materials and Methods
2.1. Mice.
All mouse primary cells used in ex vivo experiments were harvested from C57BL/6J mice (WT) or mice deficient in Grk6 (GRK6−/−) or β-arrestin 2 (Barr2−/−) backcrossed (>12 generations) onto the C57BL/6J background. All mice were cared for under the Institutional Animal Care and Use Committee (IACUC) approved protocols at the University of North Carolina at Chapel Hill.
2.2. Cells.
Human fibroblast-like synoviocytes (FLS) were generated from de-identified, IRB-exempt samples isolated from surgical explanted tissue of patients with RA, OA, or normal synovium. Murine white blood cells were harvested 5–7 days post-treatment with injection of 3% Brewer thioglycollate (Sigma-Aldrich, St. Louis, MO) into the peritoneal cavity. Contaminating red blood cells were lysed. This procedure resulted in a heterogeneous white blood cell population where the only population of CMKLR1 expressing cells were F4/80+ pro-inflammatory murine peritoneal elicited monocyte/macrophages (referred to as “pro-inflammatory macrophages” in text).
2.3. Quantitative PCR (qRT-PCR).
RNA was isolated from human FLS using the Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany) and cDNA synthesized using BioRad iScript™ cDNA synthesis kit (BioRad, Hercules, California, USA) and the Eppendorf Mastercycler pro S (Eppendorf, Hamburg, Germany). qRT-PCR was performed using BioRad SsoAdvanced™ Universal SYBR Green Supermix on the BioRad CFX96™ Real-Time System. Total volume for each reaction was 20 µL (10.0 µL Supermix 2x, 1.0 µL primers at 20 µM, 8.0 µL RNase-free H2O, and 1 µL template cDNA at 25 ng/ µL). The qRT-PCR was carried out using the following thermocycling conditions: 95°C for 30 sec, 95°C for 5 sec, 55°C for 30 sec, plate-read and repeat for 39 cycles, followed by melt-curve analysis. The following published human primers were used: IDUA forward (5’-CTC GGG CCA CTT CAC TGA C-3’), IDUA reverse (5’-CAG TCC GTA CCT ACC GAT GTA T-3’), GRK2 forward (5’-ACT TCA GCG TGC ATC GCA T-3’), GRK2 reverse (5’-GCT TTT TGT CCA GGC ACT TCA T-3’), GRK3 forward (5’-AGC TGT AGA ACA CGT ACA AAG TC-3’), GRK3 reverse (5’-ATG TCA CCT CGA AGG CTT TCA-3’), GRK6 forward (5’-TAG CGA ACA CGG TGC TAC TC-3’), GRK6 reverse (5’-GCT GAT GTG AGG GAA CTG GA-3’) (46), and CMKLR1 forward (5’-ACC TGC ATG GGA AAA TAT CCT-3’), CMKLR1 reverse (5’-GAG GTT GAG TGT GTG GTA GGG-3’) (15). IDUA served as our reference gene for delta Ct (ΔCt) analysis, as per our previously published protocol (47).
2.4. Modified Tango assay of β-arrestin mobilization.
A modified Tango assay was used to measure β-arrestin 2 recruitment to chemerin-stimulated receptors, based on the previously reported technique (48). HTLA cells were transfected with a CMKLR1-TCS-tTA receptor construct (48). This construct has the C-terminal tail of the V2 vasopressin receptor removed to ensure GRK effects were CMKLR1-specific (Figure 2A). A previous report indicated that removal of the V2 tail from the CMKLR1 construct increased the ligand-induced responses of the receptor in the Tango assay (49). The following plasmids were used for GRK over-expression as obtained from Origene (Rockville, MD, USA): pCMV6-XL5 GRK2, pCMV6-XL5 GRK3, pCMV6-XL5 GRK5, pCMV6-XL5 GRK6 transcript 1, and pCMV6-XL5 vector as a negative control. Equivalent overexpression of the GRK proteins was confirmed by Western blot (Supplemental Figure 2). A separate expression vector encoding yellow-fluorescent protein (YFP) was simultaneously transfected for use as a transfection control and epifluorescence detection was consistently >70%. HTLA cells were transiently transfected with 6.5 μg of total plasmid DNA per 10-cm plate (3 μg of CMKLR1-Tango, 0.5 μg of YFP, and either 3 μg of empty-vector control, GRK2, GRK3, GRK5 or GRK6) via calcium-phosphate precipitation and arrestin-recruitment and data normalization was performed as previously described (50). Normalized data was combined and graphed using a log (agonist) vs response (three-parameter) dose-response curve. An extra sum-of-squares F test was performed comparing the Emax (Top) and the LogEC50 for each GRK over-expression curve compared to control curve. Our p-value was set at 0.001.
FIGURE 2.
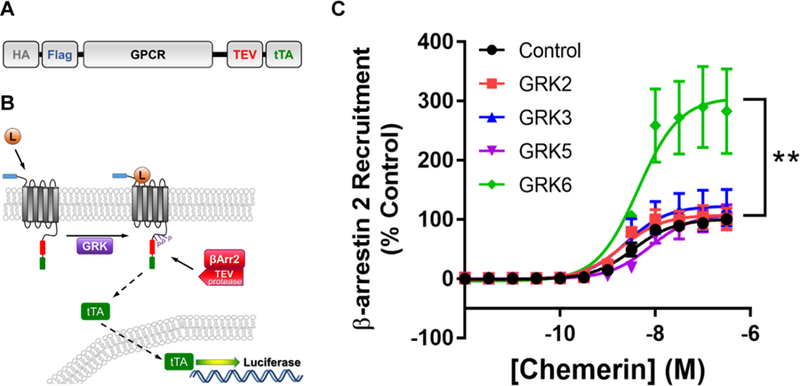
GRK6 overexpressing HTLA cells show enhanced recruitment of β-arrestin 2 after stimulation with chemerin. The CMKLR1 Tango construct (A) consists of hemagglutinin (HA), flag epitope tag, CMKLR1 (or other GPCR), TEV protease, and tetracycline transactivator (tTA). To ensure GRK effects of β-arrestin recruitment were CMKLR1-specific, this construct removed the C-terminal tail of the V2 vasopressin receptor (that promotes β-arrestin recruitment) that was present in the original construct design (49). (B). The CMKLR1 Tango assay is initiated by the chemerin ligand (L) binding to the CMKLR1 GPCR (construct shown in A), followed by G protein-coupled receptor kinase (GRK) phosphorylation of the native GPCR cytoplasmic domains and recruitment of a modified β-arrestin (with attached TEV protease). TEV protease cleavage releases the tTA transcription factor which translocates into the nucleus to activate transcription of the luciferase reporter gene, providing a luminescence assay readout. (C). HTLA cells were transfected with 3 µg of CMKLR1-Tango plasmid, 0.5 µg YFP, and 3 µg of either pCMV6-XL5 empty vector (control), or GRK2, GRK3, GRK5, and GRK6 DNA for a Tango arrestin recruitment assay and stimulated with up to 300 nM of chemerin. Shown are dose-response curves of chemerin stimulation. Error bars represent SEM. An extra sum-of-squares F test was done on the log(agonist) vs response (three-parameter) dose-response curves with significance set at p = 0.001. HTLA cells overexpressing GRK6 showed a significantly higher Emax (Top) compared to control cells (p < 0.0001). There was no statistical difference in Emax between GRK2, 3, or 5 overexpressing cells and Control cells. In addition, there was no statistical difference in EC50 in any condition tested compared to Control cells. n = 3–6
2.5. Receptor internalization assay.
Internalization of CMKLR1 upon activation by chemerin was measured by flow cytometry using pro-inflammatory macrophages from WT (control), Grk6−/−, and Barr2−/− mice. Cells were aliquoted (2.5 × 105 cells/ tube), starved for 1 hour in serum-free RPMI 1640, and then stimulated with either 6.25 nM or 200 nM chemerin (R&D systems) for 30 sec, 1 min, 5 min, and 10 min at 37 °C + 5 % CO2. Cells were not stimulated at the 0 minute time point in order to determine maximum receptor expression before addition of ligand. At each time point, receptor internalization was arrested with 4°C buffer (HBSS + 5% FBS + 2 mM EDTA) and by placing cells immediately on ice. Cells were then stained for CMKLR1 (anti-mouse CMKLR1 PE, eBioscience, 12-7582, clone BZ194, discontinued, or anti-mouse CMKLR1, Miltenyi, 130-106-902, clone REA461) and F4/80 (rat anti-mouse F4/80:APC, AbC serotec, MCA497APCT, clone CI:A3-1) as previously described (51). CMKLR1 surface expression was measured via flow cytometry by gating on F4/80 positive cells to confirm the myeloid lineage and measuring mean fluorescence intensity (MFI) of CMKLR1. Data was normalized as a percent relative to the MFI at 0 min for each cell type.
2.6. Real-time fluorophore-based detection of cell migration.
Cell migration to chemerin was examined using the Falcon™ HTS FluoroBlok 96-Multiwell Insert System (3 µm pore-size) (BD Biosciences, Bedford, MA) and pro-inflammatory macrophages from WT (control), Grk6−/−, and Barr2−/− mice obtained as previously described. Cells were starved in RPMI 1640 + 1% BSA at 37°C for one hour and labeled with calcein (485 nm/ 527 nm). Cells were washed and suspended in RPMI 1640 + 1% BSA + 10 mM HEPES, and 1 × 105 cells were loaded into FalconTM HTS FluoroBlok 96-Multiwell Insert System chemotaxis chambers (Corning, NY). Cells were allowed to migrate to RPMI/BSA/HEPES medium alone or to medium with addition of 6.25 nM chemerin (R&D Systems, Minneapolis, MN). Fluorescence was measured at 2 minute intervals over the course of 100 minutes at 37°C using a Fluoroskan Ascent Microplate Fluorometer (Thermo Scientific, Waltham, MA). Data was normalized by subtracting time 0 from each subsequent time point for each condition and graphed as fluorescence vs time for the first 100 minute interval. Analysis was done using linear mixed models and tested for a significant group effect using likelihood ratio test.
2.7. Western Immunoblotting.
Serum starved pro-inflammatory macrophages from WT, Grk6−/−, and Barr2−/− mice either were not stimulated (0 min) or were stimulated with 6.25 nM chemerin (R&D Systems) for 1, 5, 10, 15, and 20 minutes, and then lysed in ice cold RIPA buffer (plus protease and phosphatase inhibitors) to arrest signaling. Protein lysates were normalized using a BCA assay (Thermo Scientific, Rockford, IL, USA) and run on a Mini-PROTEAN TGXTM gel (Any kD™, 15-well comb, 15 µl well volume) (Bio-Rad, Hercules, CA, USA). Between 2–4 µg total cellular protein was run per independent experiment. Lysates from Barr2−/− and GRK6−/− samples were always run on the same gel as the WT control lysates for direct comparison by densitometry of signaling strength and duration. Antibodies used were p44/42 MAPK (ERK1/2) (137F5) Rabbit mAb (1:2,000), phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E) XP™ Rabbit mAb (1:2,000), phospho-AKT (Ser473) (D9E) XP™ Rabbit mAb (1:2,000), pan-AKT (C67E7) Rabbit mAb (1:2000), and anti-rabbit IgG, HRP-linked Antibody (1:5,000) (Cell Signaling Technology, Danvers, MA, USA). Images were scanned at 600 DPI resolution and imported into ImageJ where images were converted into an 8-bit black and white image. Quantification of band density was performed and graphs represent the ratio of phospho-protein to total-protein (relative density).
2.8. Statistics.
Linear mixed models were used to determine differences between control and Barr 2−/−, or GRK6−/− migration curves of primary inflammatory macrophage/monocytes to chemokine ligand stimulation over time according to our previously published methodology (40,52). Briefly, quadratic terms were included to account for the curve of the data and likelihood ratio tests (LRT) with three degrees of freedom (df) were used to test for significant group differences over time. Statistical significance was established at 0.05. All analyses were carried out using SAS, version 9.3 (Cary, NC).
3. Results
CMKLR1, GRK2, GRK5, and GRK6 are expressed in human fibroblast-like synoviocytes.
Differential expression of GRK isoforms have been reported in various diseases and disease models, including RA (50,53–55). In addition, previous reports have detected variable CMKLR1 within RA and OA synovial tissue (13). Therefore, we chose to look at mRNA expression levels of CMKLR1 and GRKs that could potentially regulate chemerin-specific GPCR responses in human fibroblast-like synoviocytes (FLS). Our results confirm that FLS from normal, OA, and RA synovial tissue express CMKLR1. The data further indicate slight increases in CMKLR1 transcript expression observed in FLS cells from diseased tissue compared to normal FLS (Figure 1A). In addition, we show that GRK2, −5, and −6 isoform transcripts are expressed in all FLS, independent of arthritis, but that GRK3 is minimally expressed compared to the other isoforms (Figure 1B–E).
FIGURE 1.
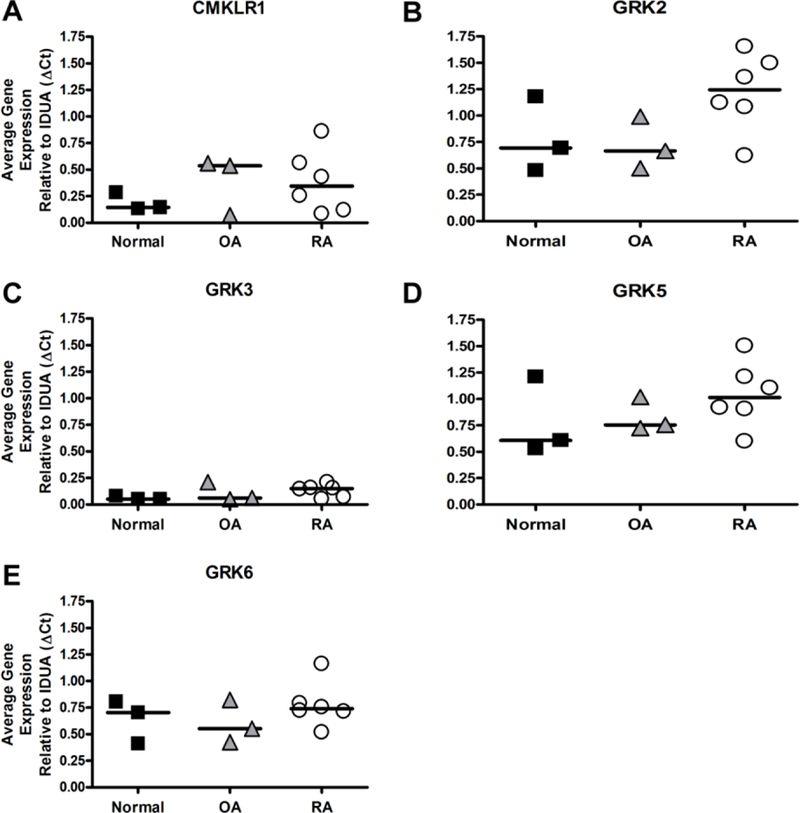
Normal, Osteoarthritis (OA), and Rheumatoid arthritis (RA)-derived fibroblast-like synoviocytes (FLS) express CMKLR1, GRK2, GRK5, and GRK6. Human FLS from normal, OA, and RA samples were cultured, and gene expression of (A) CMKLR1, (B) GRK2, (C) GRK3, (D) GRK5, and (E) GRK6 was determined via qRT-PCR. Data was analyzed relative to the housekeeping gene IDUA (ΔCt). Horizontal bar represents the median expression for each group. All sample types tested expressed CMKLR1, GRK2, GRK5, and GRK6. Minimal expression of GRK3 was observed in this cell type.
GRK6 recruits β-arrestin 2 to CMKLR1 after chemerin stimulation.
RA FLS express CMKLR1, GRK2, GRK5, and GRK6, suggesting that these kinases may play a role in CMKLR1 regulation. We used a modified Tango assay (described in Figure 2A–B and Experimental Procedures) to compare the effects of GRKs on β-arrestin 2 recruitment to chemerin-stimulated CMKLR1 (48). Our CMKLR1 modified Tango assay system behaves similar to other previous reports (48) with an EC50 to chemerin of 3.5 nM in control cells. HTLA cells transiently co-transfected with the cDNA of CMKLR1 and GRK2, GRK3, GRK5 or GRK6 did not exhibit significantly altered EC50 values for chemerin response as compared to control cells (Figure 2C). However, HTLA cells transfected with GRK6 resulted in up to 3-fold increase in reporter gene activity compared to control (100%) in response to chemerin (Figure 2C). This increase in GRK6-mediated β-arrestin 2 recruitment was significant (p <0.001), whereas cells transfected with GRK2, GRK3, or GRK5 did not show a significant increase in reporter gene activity compared to control at any concentration tested (Figure 2C). These data show that GRK6 specifically increases β-arrestin 2 recruitment to CMKLR1 following stimulation with chemerin.
CMKLR1 internalization in pro-inflammatory myeloid cells following chemerin stimulation is mediated by β-arrestin 2.
Phosphorylation of the GPCR C-terminus and intracellular loops by GRKs after ligand stimulation desensitizes the receptor and leads to β-arrestin recruitment and subsequent internalization of the GPCR (24,56). Pro-inflammatory macrophages are invasive hematopoietic cells that invade synovial tissue in RA (57,58) and expresses high levels of CMKLR1 (6,9) and GRK6 (55). To examine the role of β-arrestin and GRK6 in the regulation of CMKLR1 surface expression, we examined endogenous CMKLR1 internalization using pro-inflammatory macrophages. CMKLR1 receptor internalization tested after stimulation by 6.25 nM chemerin (near EC50) resulted in >50 % internalization after 1 minute and >70 % after 10 minutes in wild type control cells. However, β-arrestin 2 deficient (Barr2−/−) pro-inflammatory macrophages had decreased internalization compared to controls at all time points (Figure 3A). The difference between CMKLR1 surface expression on Barr2−/− deficient cells compared to WT cells was also statistically significant when cells were treated with 200 nM (supersaturating range). Stimulation with this high concentration of chemerin resulted in >70% CMKLR1 internalization by 1 minute and >80% in wild type control cells. Again, Barr2−/− cells had significantly less internalization for each time point (Figure 3B).
FIGURE 3.

β-arrestin-2 deficient (Barr2−/−) and GRK6 deficient (GRK6−/−) pro-inflammatory macrophages show delayed CMKLR1 internalization. (A) Wild type control, GRK6−/−, and Barr2−/− pro-inflammatory macrophages were stimulated with 6.25 nM chemerin ex vivo. Cell surface expression of CMKLR1 protein on F4/80 positive myeloid cells was determined using flow cytometry. (B) Control, GRK6−/−, and Barr2−/− pro-inflammatory macrophages were stimulated with 200 nM chemerin ex vivo. Data are graphed by using the ratio mean fluorescence intensity (MFI) of surface CMKLR1 compared to unstimulated cells (0 min) as a percent of maximum. Error bars represent SEM. A 2-way ANOVA was performed for statistical analysis. n = 4–6. N.S. = not significant; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001
GRK6 is known to have strong regulatory roles in arthritis inflammation (41,55,59). Because of this and our Tango data suggesting a prominent role for GRK6 in β-arrestin 2 recruitment, we focused on the mechanistic effects of GRK6 on CMKLR1 internalization. GRK6-deficient (GRK6−/−) pro-inflammatory macrophages exhibited decreased internalization of CMKLR1 over time compared to control cells after 6.25 nM chemerin stimulation (Figure 3A), though this difference was not significant. The impact of GRK6 deficiency was lost when stimulating macrophages with high dose chemerin (200 nM) (Figure 3B). Taken together, these data show that internalization of CMKLR1 by pro-inflammatory macrophages is strongly dependent upon β-arrestin 2, and while GRK6 appears to affect internalization at physiologically functional chemerin concentrations, it may be unnecessary or overwhelmed at high concentrations of chemerin.
β-arrestin 2 and GRK6 negatively regulate migration of pro-inflammatory macrophages toward chemerin.
Evidence suggests that chemerin is involved in recruiting pro-inflammatory macrophage/monocytes to synovial tissue in arthritis (6,7,13); thus, we examined how deletion of GRK6 and β-arrestin 2 affected the migration of CMKLR1-expressing pro-inflammatory macrophages to chemerin. Our hypothesis was that increased migration would be observed in cells deficient in either GRK6 or β-arrestin 2, which we have established as components of the CMKLR1 desensitization/internalization machinery. Barr2−/−, GRK6−/−, and wild type controls all showed significant increases in macrophage migration toward chemerin over media alone (Figure 4). In addition, Barr2−/− macrophage chemotaxis to chemerin was significantly enhanced above that seen by control macrophages to chemerin. GRK6−/− macrophage migration toward chemerin was significantly increased over both control and Barr2−/− macrophages (Figure 4 and Supplemental Figure 1). These results support GRK6 and β-arrestin 2 involvement in chemerin/CMKLR1 regulation of cell migration.
FIGURE 4.
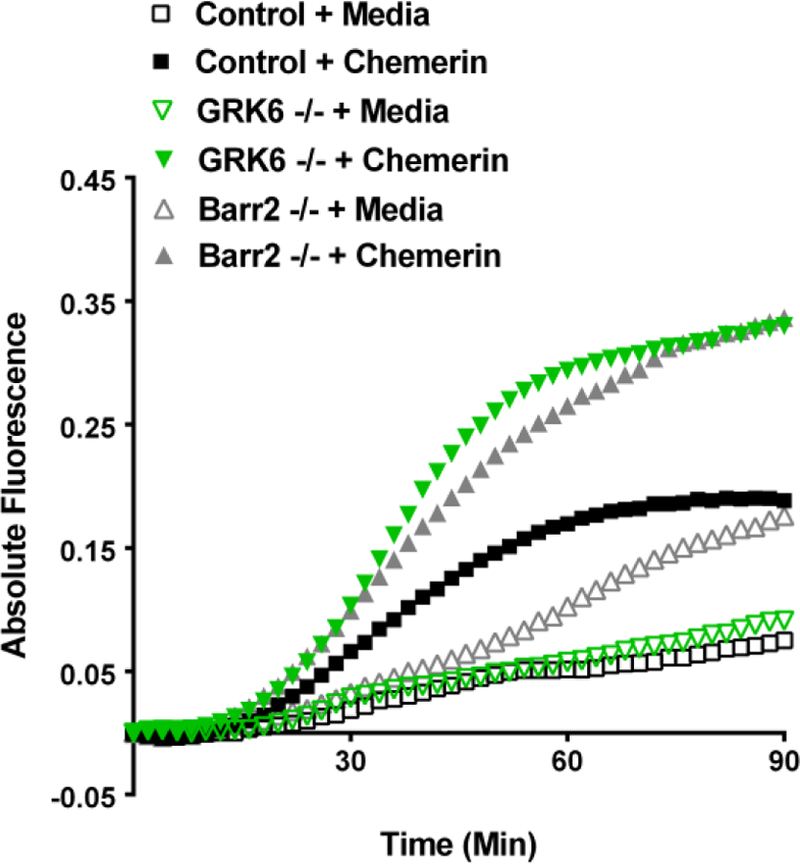
β-arrestin-2 deficient (Barr2−/−) and GRK6 deficient (GRK6−/−) pro-inflammatory macrophages show enhanced migration to chemerin. Control, Barr2−/−, and GRK6−/− pro-inflammatory monocytes/macrophages were fluorescently labeled with calcein and loaded into the upper chamber wells of a Falcon™ HTS FluoroBlok 96-Multiwell Insert System with either 6.25 nM chemerin in the lower chamber to stimulate migration or medium alone as a control. Shown is mean fluorescence intensity (MFI) (measured at 2 minute intervals) of 3–5 independent experiments. Error bars are omitted for clarity. Data was normalized by setting 0 absolute fluorescence as the start point for each curve. The effect of chemerin was examined using an ANCOVA for statistical analysis (Supplemental Figure 1). GRK6−/−macrophage migration was significantly enhanced compared to Barr2−/− and Control (p<0.0001). Barr2−/−macrophage migration was also significant compared to Control (p<0.01).
β-arrestin 2 and GRK6 function as negative regulators of CMKLR1 signaling.
Barr2−/− and GRK6−/− pro-inflammatory macrophages show decreased CMKLR1 internalization and enhanced migration to chemerin; therefore, to determine whether CMKLR1 signaling is also altered, we first examined the Ras-Raf-MEK-ERK signaling cascade, which is known to be a downstream effector of G protein and β-arrestin 2 activation. We hypothesized that loss of either Barr2 or GRK6 would result in prolonged and/or enhanced ERK1/2 signaling (60,61). Upon stimulation with 6.25 nM chemerin, Barr2−/− pro-inflammatory macrophages showed significantly enhanced signaling over time compared to control (Figure 5A and 5B). GRK6−/−macrophages appear to have a similar trend of enhanced signaling to ERK1/2 that persists throughout the time course shown (Figure 5C). However, densitometric analysis of the Western blot data show that this difference is not significant (Figure 5D).
FIGURE 5.

Barr2−/− pro-inflammatory macrophages show increased ERK1/2 phosphorylation after activation by chemerin. Control versus Barr2−/− (A, B) or GRK6−/− (C, D), C57BL/6J peritoneal macrophages were either left unstimulated (0 minutes) or stimulated with 6.25 nM chemerin ex vivo for 1, 5, 10, 15, or 20 minutes. Total protein was normalized using a BCA assay. Western blot analysis was performed on cell lysates probed using phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (Phospho-ERK1/2) and p44/42 MAPK (ERK1/2) (Total-ERK1/2). Representative blots (A, C) of 3 (Barr2−/−) or 4 (GRK6−/−) independent experiments are shown. Quantification shown in (B, D) was done by densitometry using ImageJ software and is shown as a mean ratio of relative density ± SEM (Barr2−/− n=3; GRK6−/− n=4).
Because GPCR signaling through the PI3K/AKT pathway can potentially influence cellular functions that contribute to inflammation (e.g. migration, survival), the activation of this pathway was also examined in Barr2−/− and GRK6−/− macrophages stimulated with 6.25 nM chemerin by Western blot for phospho-AKT (Figure 6). We again hypothesized that loss of either Barr2 or GRK6 would result in prolonged and/or enhanced AKT signaling. Chemerin stimulation (6.25 nM) of Barr2−/− pro-inflammatory macrophages did not appear to initially enhance phosphorylation of AKT and the observed prolonged activation of this pathway over time was not statistically significant (Figures 6A and 6B). In contrast, GRK6−/− macrophages stimulated with 6.25 nM chemerin showed a significant increase in AKT phosphorylation that appears to degrade at a similar rate to that seen in wild type controls (Figures 6C and 6D). Taken together, these data suggest non-redundant roles for Barr2 and GRK6 in regulating chemerin/CMKLR1 signals to the ERK1/2 and AKT pathways.
FIGURE 6.

GRK6−/− pro-inflammatory macrophages show enhanced AKT phosphorylation after activation by chemerin. Control versus Barr2−/− (A, B) or GRK6−/− (C, D), C57BL/6J peritoneal macrophages were either left unstimulated (0 minutes) or stimulated with 6.25 nM chemerin ex vivo for 1, 5, 10, 15, or 20 minutes. Total protein was normalized using a BCA assay. Western blot analysis was performed on cell lysates probed using Phospho-AKT (Ser473) and Total-AKT. Representative blots (A, C) of 4 independent experiments are shown. Quantification shown in (B, D) was done by densitometry using ImageJ software and is shown as a mean ratio of relative density ± SEM (n=4).
4. Discussion
Chemerin is a known chemotactic agent for CMKLR1-expressing antigen-presenting cells (APCs), such as immature DCs, plasmacytoid DCs, and macrophages (6,7), which are important to the pathogenesis of RA (57,62,63). In addition, high levels of chemerin are found in arthritic synovial fluid (6) and chemerin protein has been detected in RA and OA synovial tissue (13). Taken together, these findings suggest that chemerin/CMKLR1 signaling promotes inflammation in RA by recruiting inflammatory cells, such as macrophages, to the joint. Therefore, elucidation of the specific cellular machinery underpinning the regulation of chemerin/CMKLR1 signaling should provide a better understanding of RA pathogenesis, as well as highlight potential therapeutic targets. A previous study has shown that CMKLR1 undergoes internalization after chemerin stimulation via a non-clathrin-mediated pathway, and that this pathway is likely via caveolae (24), which is generally considered to be GRK/arrestin independent (64,65). However, a previous study by Rourke, et al. states that β-arrestin could be involved in CMKLR1 internalization, which our results confirm (66). Our data show that using the modified Tango assay to monitor arrestin mobilization, expression of GRK6 enhances β-arrestin 2 recruitment to chemerin-activated CMKLR1 (Figure 2). In addition, endogenously-expressed CMKLR1 internalization in inflammatory macrophages was β-arrestin 2-dependent. CMKLR1 internalization was not completely abolished, suggesting a possible role for β-arrestin 1 or contributions of a non-clathrin mediated mechanism of internalization (e.g. caveolae) (24,67). Further, internalization, cellular migration, and downstream signaling to ERK1/2 and AKT was altered in macrophages deficient in β-arrestin 2 and GRK6, suggesting that these are likely the predominant receptor-proximal regulators of CMKLR1 in response to chemerin stimulation (Figures 3–6), though to differing degrees.
To better define the functional effects of GRKs on chemerin stimulated CMKLR1, we interrogated GRK-mediated, β-arrestin 2 recruitment using a modified Tango assay to measure GRK specificity. As noted in the Experimental Procedures, this assay requires an 18–24-hour incubation after stimulation for reporter expression and irreversible cleavage of the transcription factor from the receptor. These caveats of the TANGO assay (and our modified version) do not strictly imitate the rapid time scale or catalytic activity of arrestins (68). However, the modified TANGO assay used does recapitulate concentration-dependent responses and broad-level regulatory functions by GRKs. Our results show that GRK6 was the only GRK isoform, of those ubiquitously expressed, that increased β-arrestin 2 recruitment to chemerin-stimulated CMKLR1 receptor (Figure 2). As expected from these data, GRK6-deficient macrophages also exhibited enhanced migration (Figure 4), and increased phosphorylation of Akt (Figures 6) with chemerin stimulation. Additionally, the more pronounced defects in internalization observed by β-arrestin 2 deficient cells, as compared to GRK6, suggest that there may be GRK redundancy or GRK-independent mechanisms of CMKLR1 regulation and provide evidence of underlying signaling bias that is known to exist with many GPCR (22,65,69).
Our results show that FLS cells express CMKLR1, GRK2, GRK5, and GRK6, but very low levels of GRK3 by comparison (Figure 1). Although some trends are observed in the differential expression of CMKLR1, GRK2 and GRK5 in FLS from arthritic patients, the small sample size limited statistical comparisons. Previous studies have observed decreased expression of GRK2 and GRK6 in hematopoietic cells of patients with active RA, suggesting these two kinases could be relevant in GPCR function specifically in inflammatory arthritis (55,59), but further investigation is needed to determine the roles of GRK regulation of CMKLR1 in these resident fibroblast-like synoviocytes. While data in arthritis models suggest a pro-inflammatory role for chemerin in joint synovium (6,11–13), a lung disease model has shown an anti-inflammatory role for chemerin using CMKLR1 knockout mice (8,70). Therefore, understanding the regulatory mechanisms of CMKLR1 signaling could help characterize the role of the chemerin/CMKLR1 axis in inflammatory disease states.
GRK6−/− mice in a K/BxN model of acute RA showed an increase in disease severity compared to wild type mice, further implicating the role of GRK6 in migration and inflammation (41). Our observations using real-time fluorophore-labeled cell migration paralleled these findings in that we found absence of β-arrestin 2 or GRK6 caused enhanced migration of pro-inflammatory macrophages to chemerin (Figure 4), thereby supporting a regulatory role of these two proteins in normal CMKLR1 desensitization and chemotaxis. In addition to enhanced chemotaxis of Barr2−/− cells to chemerin, we observed significant migration to medium alone in these cells (p < 0.0001), which is possibly the result of chemokinesis that has been described previously in migration assays utilizing β-arrestin 2 deficient cells (71,72).
To explore if the functional phenotypes observed were the result of changes in downstream signaling, we examined the MAPK and AKT signaling cascades. Previous results by the Lefkowitz group have shown differential effects of GRK knockdown on receptor internalization and that ERK1/2 phosphorylation are GRK- and receptor-dependent (69,73,74). Based on our own previously published results (40), we hypothesized that cells lacking either β-arrestin 2 or GRK6 would exhibit prolonged signaling due to sustained CMKLR1 surface expression and signaling after stimulation when compared to wild type control cells. Indeed, Barr2−/− pro-inflammatory macrophages exhibited enhanced signaling of ERK1/2 compared to control cells with maximum ERK1/2 phosphorylation at 5 minutes (Figure 5), similar to other published reports (24). β-arrestin 2 is known to directly transduce signals through the ERK1/2 pathway following receptor ligation as part of a bifurcated GPCR signaling pathway (75); therefore, it is possible that the increased ERK1/2 signaling in Barr2−/− cells is due to an indirect mechanism predicated on delayed CMKLR1 internalization in these deficient cells. Chemerin stimulation increased AKT signals in GRK6−/− which could also be explained by the delayed internalization of CMKLR1 seen in these cells (Figure 3A) or by another, unknown mechanism that impacts alternate signaling cascades. Taken together, these data suggest non-redundant or partially-overlapping roles for Barr2 and GRK6 in regulating chemerin/CMKLR1 CMKLR1 signals to the ERK1/2 and AKT pathways. Our findings are consistent with results previously published by others (61) and recent work by Grundmann, et al. that elegantly dissect the dependence of arrestin-mediated ERK signaling on G protein function (76). Their study also highlights the importance of considering whether or not absolute absence of expression (“zero functional”) is achieved when interpreting results. While the macrophages used in our experiments are Grk6−/− and Barr2−/− genetic knockouts, we were unable to directly test the potential confounding effects of other Grk or arrestin isoforms that might be present in these cells.
In conclusion, this present study provides evidence that GRK6 and β-arrestin 2 are involved in desensitization, internalization, and migration of primary inflammatory macrophages, which are relevant to RA pathogenesis. These functional outcomes are similar in Barr2−/− and GRK6−/− macrophages, but appear to be based on unique signaling characteristics. This work provides a foundation to better define the role of CMKLR1 in inflammatory arthritis, as well as highlights potential therapeutic targets for treatment within the chemerin/CMKLR1 signaling axis.
Supplementary Material
Highlights.
A model for CMKLR1 receptor regulation on inflammatory macrophages is proposed
GRK6 helps recruit β-arrestin 2 to CMKLR1 after chemerin stimulation
CMKLR1 internalization in pro-inflammatory myeloid cells following chemerin stimulation is mediated by β-arrestin 2
β-arrestin 2 and GRK6 negatively regulate migration of pro-inflammatory macrophages toward chemerin
β-arrestin 2 and GRK6 have non-redundant function as negative regulators of CMKLR1 signaling
Acknowledgements.
Dr. Bryan Roth for expertise and reagents in the Tango assay. We would also like to acknowledge funding sources R03AR059286 (TKT), K01AI091863 (MJB), and a Rheumatoid Arthritis Pilot Award (to DPS) under U54GM104942.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest.
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1.Gantz I, Konda Y, Yang YK, Miller DE, Dierick HA, and Yamada T (1996) Molecular cloning of a novel receptor (CMKLR1) with homology to the chemotactic factor receptors. Cytogenetics and cell genetics 74, 286–290 [DOI] [PubMed] [Google Scholar]
- 2.Samson M, Edinger AL, Stordeur P, Rucker J, Verhasselt V, Sharron M, Govaerts C, Mollereau C, Vassart G, Doms RW, and Parmentier M (1998) ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. European journal of immunology 28, 1689–1700 [DOI] [PubMed] [Google Scholar]
- 3.Vermi W, Riboldi E, Wittamer V, Gentili F, Luini W, Marrelli S, Vecchi A, Franssen JD, Communi D, Massardi L, Sironi M, Mantovani A, Parmentier M, Facchetti F, and Sozzani S (2005) Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. The Journal of experimental medicine 201, 509–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, Communi D, Parmentier M, Majorana A, Sironi M, Tabellini G, Moretta A, and Sozzani S (2007) The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 109, 3625–3632 [DOI] [PubMed] [Google Scholar]
- 5.Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, Walder K, and Segal D (2007) Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 148, 4687–4694 [DOI] [PubMed] [Google Scholar]
- 6.Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, Brezillon S, Tyldesley R, Blanpain C, Detheux M, Mantovani A, Sozzani S, Vassart G, Parmentier M, and Communi D (2003) Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. The Journal of experimental medicine 198, 977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zabel BA, Silverio AM, and Butcher EC (2005) Chemokine-like receptor 1 expression and chemerin-directed chemotaxis distinguish plasmacytoid from myeloid dendritic cells in human blood. Journal of immunology 174, 244–251 [DOI] [PubMed] [Google Scholar]
- 8.Luangsay S, Wittamer V, Bondue B, De Henau O, Rouger L, Brait M, Franssen JD, de Nadai P, Huaux F, and Parmentier M (2009) Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. Journal of immunology 183, 6489–6499 [DOI] [PubMed] [Google Scholar]
- 9.Zabel BA, Ohyama T, Zuniga L, Kim JY, Johnston B, Allen SJ, Guido DG, Handel TM, and Butcher EC (2006) Chemokine-like receptor 1 expression by macrophages in vivo: regulation by TGF-beta and TLR ligands. Experimental hematology 34, 1106–1114 [DOI] [PubMed] [Google Scholar]
- 10.Huss RS, Huddleston JI, Goodman SB, Butcher EC, and Zabel BA (2010) Synovial tissue-infiltrating natural killer cells in osteoarthritis and periprosthetic inflammation. Arthritis and rheumatism 62, 3799–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang K, Du G, Li L, Liang H, and Zhang B (2012) Association of chemerin levels in synovial fluid with the severity of knee osteoarthritis. Biomarkers : biochemical indicators of exposure, response, and susceptibility to chemicals 17, 16–20 [DOI] [PubMed] [Google Scholar]
- 12.Berg V, Sveinbjornsson B, Bendiksen S, Brox J, Meknas K, and Figenschau Y (2010) Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin(21–157). Arthritis research & therapy 12, R228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaneko K, Miyabe Y, Takayasu A, Fukuda S, Miyabe C, Ebisawa M, Yokoyama W, Watanabe K, Imai T, Muramoto K, Terashima Y, Sugihara T, Matsushima K, Miyasaka N, and Nanki T (2011) Chemerin activates fibroblast-like synoviocytes in patients with rheumatoid arthritis. Arthritis research & therapy 13, R158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao L, Yamaguchi Y, Sharif S, Du XY, Song JJ, Lee DM, Recht LD, Robinson WH, Morser J, and Leung LL (2011) Chemerin158K protein is the dominant chemerin isoform in synovial and cerebrospinal fluids but not in plasma. The Journal of biological chemistry 286, 39520–39527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisinger K, Bauer S, Schaffler A, Walter R, Neumann E, Buechler C, Muller-Ladner U, and Frommer KW (2012) Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Experimental and molecular pathology 92, 90–96 [DOI] [PubMed] [Google Scholar]
- 16.Albanesi C, Scarponi C, Bosisio D, Sozzani S, and Girolomoni G (2010) Immune functions and recruitment of plasmacytoid dendritic cells in psoriasis. Autoimmunity 43, 215–219 [DOI] [PubMed] [Google Scholar]
- 17.Albanesi C, Scarponi C, Pallotta S, Daniele R, Bosisio D, Madonna S, Fortugno P, Gonzalvo-Feo S, Franssen JD, Parmentier M, De Pita O, Girolomoni G, and Sozzani S (2009) Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. The Journal of experimental medicine 206, 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Palma G, Castellano G, Del Prete A, Sozzani S, Fiore N, Loverre A, Parmentier M, Gesualdo L, Grandaliano G, and Schena FP (2011) The possible role of ChemR23/Chemerin axis in the recruitment of dendritic cells in lupus nephritis. Kidney international 79, 1228–1235 [DOI] [PubMed] [Google Scholar]
- 19.Aoki H, Hisada T, Ishizuka T, Utsugi M, Kawata T, Shimizu Y, Okajima F, Dobashi K, and Mori M (2008) Resolvin E1 dampens airway inflammation and hyperresponsiveness in a murine model of asthma. Biochem Biophys Res Commun 367, 509–515 [DOI] [PubMed] [Google Scholar]
- 20.Seki H, Fukunaga K, Arita M, Arai H, Nakanishi H, Taguchi R, Miyasho T, Takamiya R, Asano K, Ishizaka A, Takeda J, and Levy BD (2010) The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. Journal of immunology 184, 836–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH, and National Arthritis Data, W. (2008) Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis and rheumatism 58, 15–25 [DOI] [PubMed] [Google Scholar]
- 22.Rajagopal S, Rajagopal K, and Lefkowitz RJ (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature reviews. Drug discovery 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lefkowitz RJ (1998) G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. The Journal of biological chemistry 273, 18677–18680 [DOI] [PubMed] [Google Scholar]
- 24.Zhou JX, Liao D, Zhang S, Cheng N, He HQ, and Ye RD (2014) Chemerin C9 peptide induces receptor internalization through a clathrin-independent pathway. Acta pharmacologica Sinica 35, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefkowitz RJ, and Shenoy SK (2005) Transduction of receptor signals by beta-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 26.Smith JS, and Rajagopal S (2016) The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of biological chemistry 291, 8969–8977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Henau O, Degroot GN, Imbault V, Robert V, De Poorter C, McHeik S, Gales C, Parmentier M, and Springael JY (2016) Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. PloS one 11, e0164179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benovic JL, Onorato JJ, Arriza JL, Stone WC, Lohse M, Jenkins NA, Gilbert DJ, Copeland NG, Caron MG, and Lefkowitz RJ (1991) Cloning, expression, and chromosomal localization of beta-adrenergic receptor kinase 2. A new member of the receptor kinase family. The Journal of biological chemistry 266, 14939–14946 [PubMed] [Google Scholar]
- 29.Haribabu B, and Snyderman R (1993) Identification of additional members of human G-protein-coupled receptor kinase multigene family. Proceedings of the National Academy of Sciences of the United States of America 90, 9398–9402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunapuli P, and Benovic JL (1993) Cloning and expression of GRK5: a member of the G protein-coupled receptor kinase family. Proceedings of the National Academy of Sciences of the United States of America 90, 5588–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benovic JL, DeBlasi A, Stone WC, Caron MG, and Lefkowitz RJ (1989) Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science 246, 235–240 [DOI] [PubMed] [Google Scholar]
- 32.Hisatomi O, Matsuda S, Satoh T, Kotaka S, Imanishi Y, and Tokunaga F (1998) A novel subtype of G-protein-coupled receptor kinase, GRK7, in teleost cone photoreceptors. FEBS letters 424, 159–164 [DOI] [PubMed] [Google Scholar]
- 33.Weller M, Virmaux N, and Mandel P (1975) Light-stimulated phosphorylation of rhodopsin in the retina: the presence of a protein kinase that is specific for photobleached rhodopsin. Proceedings of the National Academy of Sciences of the United States of America 72, 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andresen BT (2010) Characterization of G protein-coupled receptor kinase 4 and measuring its constitutive activity in vivo. Methods in enzymology 484, 631–651 [DOI] [PubMed] [Google Scholar]
- 35.Premont RT, Macrae AD, Stoffel RH, Chung N, Pitcher JA, Ambrose C, Inglese J, MacDonald ME, and Lefkowitz RJ (1996) Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. The Journal of biological chemistry 271, 6403–6410 [DOI] [PubMed] [Google Scholar]
- 36.Pitcher JA, Freedman NJ, and Lefkowitz RJ (1998) G protein-coupled receptor kinases. Annual review of biochemistry 67, 653–692 [DOI] [PubMed] [Google Scholar]
- 37.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, and Benovic JL (2010) Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. The Journal of biological chemistry 285, 7805–7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z, Gaudreau R, Le Gouill C, Rola-Pleszczynski M, and Stankova J (2004) Agonist-induced internalization of leukotriene B(4) receptor 1 requires G-protein-coupled receptor kinase 2 but not arrestins. Molecular pharmacology 66, 377–386 [DOI] [PubMed] [Google Scholar]
- 39.Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, and Richardson RM (2012) The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. Journal of immunology 189, 2824–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarrant TK, Billard MJ, Timoshchenko RG, McGinnis MW, Serafin DS, Foreman O, Esserman DA, Chao NJ, Lento WE, Lee DM, Patel D, and Siderovski DP (2013) G protein-coupled receptor kinase-3-deficient mice exhibit WHIM syndrome features and attenuated inflammatory responses. Journal of leukocyte biology 94, 1243–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarrant TK, Rampersad RR, Esserman D, Rothlein LR, Liu P, Premont RT, Lefkowitz RJ, Lee DM, and Patel DD (2008) Granulocyte chemotaxis and disease expression are differentially regulated by GRK subtype in an acute inflammatory arthritis model (K/BxN). Clin Immunol 129, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibrahim IA, and Kurose H (2012) beta-arrestin-mediated signaling improves the efficacy of therapeutics. Journal of pharmacological sciences 118, 408–412 [DOI] [PubMed] [Google Scholar]
- 43.DeWire SM, Ahn S, Lefkowitz RJ, and Shenoy SK (2007) Beta-arrestins and cell signaling. Annual review of physiology 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 44.Luttrell LM, and Gesty-Palmer D (2010) Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacological reviews 62, 305–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watari K, Nakaya M, and Kurose H (2014) Multiple functions of G protein-coupled receptor kinases. Journal of molecular signaling 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balabanian K, Levoye A, Klemm L, Lagane B, Hermine O, Harriague J, Baleux F, Arenzana-Seisdedos F, and Bachelerie F (2008) Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. The Journal of clinical investigation 118, 1074–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarrant TK, Liu P, Rampersad RR, Esserman D, Rothlein LR, Timoshchenko RG, McGinnis MW, Fitzhugh DJ, Patel DD, and Fong AM (2012) Decreased Th17 and antigen-specific humoral responses in CX(3) CR1-deficient mice in the collagen-induced arthritis model. Arthritis and rheumatism 64, 1379–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, and Lee KJ (2008) The genetic design of signaling cascades to record receptor activation. Proceedings of the National Academy of Sciences of the United States of America 105, 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, Sciaky N, and Roth BL (2015) PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nature structural & molecular biology 22, 362–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Billard MJ, Fitzhugh DJ, Parker JS, Brozowski JM, McGinnis MW, Timoshchenko RG, Serafin DS, Lininger R, Klauber-Demore N, Sahagian G, Truong YK, Sassano MF, Serody JS, and Tarrant TK (2016) G Protein Coupled Receptor Kinase 3 Regulates Breast Cancer Migration, Invasion, and Metastasis. PloS one 11, e0152856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rama D, Esendagli G, and Guc D (2011) Expression of chemokine-like receptor 1 (CMKLR1) on J744A.1 macrophages co-cultured with fibroblast and/or tumor cells: modeling the influence of microenvironment. Cellular immunology 271, 134–140 [DOI] [PubMed] [Google Scholar]
- 52.Giguere PM, Billard MJ, Laroche G, Buckley BK, Timoshchenko RG, McGinnis MW, Esserman D, Foreman O, Liu P, Siderovski DP, and Tarrant TK (2013) G-protein signaling modulator-3, a gene linked to autoimmune diseases, regulates monocyte function and its deficiency protects from inflammatory arthritis. Molecular immunology 54, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakaya M, Tajima M, Kosako H, Nakaya T, Hashimoto A, Watari K, Nishihara H, Ohba M, Komiya S, Tani N, Nishida M, Taniguchi H, Sato Y, Matsumoto M, Tsuda M, Kuroda M, Inoue K, and Kurose H (2013) GRK6 deficiency in mice causes autoimmune disease due to impaired apoptotic cell clearance. Nature communications 4, 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bychkov ER, Ahmed MR, Gurevich VV, Benovic JL, and Gurevich EV (2011) Reduced expression of G protein-coupled receptor kinases in schizophrenia but not in schizoaffective disorder. Neurobiology of disease 44, 248–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, and Heijnen CJ (1999) Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 13, 715–725 [DOI] [PubMed] [Google Scholar]
- 56.Kohout TA, and Lefkowitz RJ (2003) Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Molecular pharmacology 63, 9–18 [DOI] [PubMed] [Google Scholar]
- 57.Kinne RW, Stuhlmuller B, and Burmester GR (2007) Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis research & therapy 9, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JS, An H, Rieter WJ, Esserman D, Taylor-Pashow KM, Sartor RB, Lin W, Lin W, and Tarrant TK (2009) Multimodal optical and Gd-based nanoparticles for imaging in inflammatory arthritis. Clinical and experimental rheumatology 27, 580–586 [PubMed] [Google Scholar]
- 59.Lombardi MS, Kavelaars A, Cobelens PM, Schmidt RE, Schedlowski M, and Heijnen CJ (2001) Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. Journal of immunology 166, 1635–1640 [DOI] [PubMed] [Google Scholar]
- 60.Roskoski R Jr. (2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacological research : the official journal of the Italian Pharmacological Society 66, 105–143 [DOI] [PubMed] [Google Scholar]
- 61.Luo J, Busillo JM, and Benovic JL (2008) M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Molecular pharmacology 74, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lande R, Giacomini E, Serafini B, Rosicarelli B, Sebastiani GD, Minisola G, Tarantino U, Riccieri V, Valesini G, and Coccia EM (2004) Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. Journal of immunology 173, 2815–2824 [DOI] [PubMed] [Google Scholar]
- 63.Santiago-Schwarz F, Anand P, Liu S, and Carsons SE (2001) Dendritic cells (DCs) in rheumatoid arthritis (RA): progenitor cells and soluble factors contained in RA synovial fluid yield a subset of myeloid DCs that preferentially activate Th1 inflammatory-type responses. Journal of immunology 167, 1758–1768 [DOI] [PubMed] [Google Scholar]
- 64.Barki-Harrington L, and Rockman HA (2008) Beta-arrestins: multifunctional cellular mediators. Physiology (Bethesda) 23, 17–22 [DOI] [PubMed] [Google Scholar]
- 65.Pierce KL, and Lefkowitz RJ (2001) Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nature reviews. Neuroscience 2, 727–733 [DOI] [PubMed] [Google Scholar]
- 66.Rourke JL, Dranse HJ, and Sinal CJ (2015) CMKLR1 and GPR1 mediate chemerin signaling through the RhoA/ROCK pathway. Molecular and cellular endocrinology 417, 36–51 [DOI] [PubMed] [Google Scholar]
- 67.Gong Q, Huntsman C, and Ma D (2008) Clathrin-independent internalization and recycling. J Cell Mol Med 12, 126–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Violin JD, Dewire SM, Barnes WG, and Lefkowitz RJ (2006) G protein-coupled receptor kinase and beta-arrestin-mediated desensitization of the angiotensin II type 1A receptor elucidated by diacylglycerol dynamics. The Journal of biological chemistry 281, 36411–36419 [DOI] [PubMed] [Google Scholar]
- 69.Zidar DA, Violin JD, Whalen EJ, and Lefkowitz RJ (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proceedings of the National Academy of Sciences of the United States of America 106, 9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bondue B, Vosters O, de Nadai P, Glineur S, De Henau O, Luangsay S, Van Gool F, Communi D, De Vuyst P, Desmecht D, and Parmentier M (2011) ChemR23 dampens lung inflammation and enhances anti-viral immunity in a mouse model of acute viral pneumonia. PLoS pathogens 7, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, and Patel DD (2002) Defective lymphocyte chemotaxis in beta-arrestin2- and GRK6-deficient mice. Proceedings of the National Academy of Sciences of the United States of America 99, 7478–7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma D, Malik A, Lee E, Britton RA, and Parameswaran N (2013) Gene dosage-dependent negative regulatory role of beta-arrestin-2 in polymicrobial infection-induced inflammation. Infect Immun 81, 3035–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, and Lefkowitz RJ (2005) Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proceedings of the National Academy of Sciences of the United States of America 102, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ren XR, Reiter E, Ahn S, Kim J, Chen W, and Lefkowitz RJ (2005) Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proceedings of the National Academy of Sciences of the United States of America 102, 1448–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, and Lefkowitz RJ (2006) beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. The Journal of biological chemistry 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 76.Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, Ruttiger N, Ziegler N, Benkel T, Schmitt NK, Ishida S, Muller I, Reher R, Kawakami K, Inoue A, Rick U, Kuhl T, Imhof D, Aoki J, Konig GM, Hoffmann C, Gomeza J, Wess J, and Kostenis E (2018) Lack of beta-arrestin signaling in the absence of active G proteins. Nature communications 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.