

Abstract
Extracellular vesicles (EVs) are promising natural nanocarriers for delivery of various types of therapeutics. Earlier engineered EV-based formulations for neurodegenerative diseases and cancer are reported. Herein, the use of macro-phage-derived EVs for brain delivery of a soluble lysosomal enzyme tripeptidyl peptidase-1, TPP1, to treat a lysosomal storage disorder, Neuronal Ceroid Lipo-fuscinoses 2 (CLN2) or Batten disease, is investigated. TPP1 is loaded into EVs using two methods: i) transfection of parental EV-producing macrophages with TPPI-encoding plasmid DNA (pDNA) or ii) incorporation therapeutic protein TPP1 into naive empty EVs. For the former approach, EVs released by pretransfected macrophages contain the active enzyme and TPPI-encoding pDNA. To achieve high loading efficiency by the latter approach, sonication or permeabilization of EV membranes with saponin is utilized. Both methods provide proficient incorporation of functional TPP1 into EVs (EV-TPP1). EVs significantly increase stability of TPPI against protease degradation and provide efficient TPP1 delivery to target cells in in vitro model of CLN2. The majority ofEV-TPP1 (≈70%) is delivered to target organelles, lysosomes. Finally, a robust brain accumulation of EV carriers and increased lifespan is recorded in late-infantile neuronal ceroid lipofuscinosis (LINCL) mouse model following intraperitoneal administration of EV-TPP1.
Keywords: Batten disease, brain, drug delivery systems, EVs, lysosomal storage diseases
1. Introduction
The neuronal ceroid lipofuscinoses (NCLs) or Batten disease is a group of severe neurodegenerative diseases that primarily affect children and are characterized by the intracellular accumulation of storage material in neural tissues and progressive neurodegeneration. Morphologically, they are identified by loss of neurons, predominantly in the cerebellar cortices, and near-ubiquitous accumulation of NCL-specific lipopigments. Core symptoms of these conditions typically include epilepsy, cognitive decline and visual failure. CLN2 disease is one of a group of lysosomal storage disorders (LSDs) that results from mutations in the TPP1 gene that cause an insufficiency or complete lack of the soluble lysosomal enzyme tripeptidyl peptidase-1 (TPP1). Without functional TPP1, neurons develop inclusions of abnormal storage material; the retina and central nervous system (CNS) undergo progressive degeneration[1] resulting in loss of neurological functions and vision.[2] Thus, the successful delivery of functional TPP1 to CNS is of great importance.
One of the major reasons for the failure of current therapeutic agents in treating neurodegenerative disorders is the existence of multiple biological barriers that prohibit effective drug delivery to target cells. In particular, the blood brain barrier (BBB) remains a seemingly insurmountable obstacle to the routine use of systemically administered macromolecules—including TPP1. In this regard, recently emerged field of nanotechnology, and specifically development of different nanoformulations that may improve drug transport across the BBB attracted significant efforts of the research community.[3,4] Regrettably, these nanoparticles are rapidly cleared from the bloodstream by mononuclear phagocyte system (MPS).[5] Thus, there is an unmet clinical need to develop new drug delivery systems for treatment of different neurodegenerative disorders, and in particular, lysosomal storage diseases.[6,7]
To circumvent this problem, we propose using EVs as bio-compatible nanocarriers for delivery therapeutic material. EVs are naturally occurring biological vesicles in the size range of 60–500 nm that are known to specialize in cell–cell communication and therefore can provide unprecedented opportunities for delivering drugs to target cells. Furthermore, we suggest to utilize EVs derived from inflammatory-response cells, monocytes and macrophages, as drug delivery vehicles. It was reported that besides intrinsic neuronal defects, most LSDs patients show signs of neurodegeneration and brain inflammation.[8] The development of lysosomal inclusions results in microglial and astrocyte activation that is a hallmark of many LSDs. The inflammatory process affects the CNS, which often precedes and predicts regions where eventual neuron loss will occur. This provides the opportunity for site-specific delivery of therapeutic enzymes using inflammatory response cell-based platform, specifically, macrophage-derived EVs.
There is a growing interest in the use of EVs as nanocarriers; EVs were suggested as drug delivery vehicles for different small molecular weight therapeutics, such as an anti-inflammatory agent, cur cumin, [9,10] or anticancer agents, doxorubicin[11,12] and paclitaxel.[13–15] Moreover, EVs were harnessed for systemic delivery of exogenous nucleic acids across the biological barriers,[16–25] as well as adeno-associated viral vectors[26] and superparamagnetic iron oxide nanoparticles (SPIONS).[27] The incorporation of therapeutic agents into EVs increased the circulation time, preserved drug therapeutic activity, and improved their transport to the disease site. Furthermore, several reports indicate that EVs may improve pharmacokinetics and preserve activity of incorporated therapeutics. Furthermore, EVs have low immunogenicity due to the expression of CD47 receptor[12,28,29] that interacts with signal regulatory protein α (SIRPα) to produce a “don’t eat me” signal in phagocytes.[30,31] Finally, EVs can exert unique biological activity reflective of their origin. Thus, macrophage-derived EVs may preferentially interact with inflamed tissues and accomplish targeted delivery of therapeutics to the disease site. These exceptional features make EVs an attractive option for use as a drug delivery vehicle for Batten disease treatment, and should work in concert to dramatically improve the therapeutic efficacy of current treatment strategies utilizing TPP1.
We demonstrated earlier that macrophages pretransfected with catalase-, or glial cell line-derived neurotrophic factor (GDNF)-encoding plasmid DNA (pDNA) ex vivo, release EVs with the encoded therapeutic protein, and improve transport of the drug to target cells of neurovascular unit.[32–34] This resulted in significant therapeutic effects in mouse models of Parkinson’s disease (PD) and lipopolysaccharide (LPS)-induced encephalitis. We also reported that naïve macrophage-derived EVs can be loaded with therapeutic proteins, catalase or brain-derived neurotrophic factor (BDNF), ex vivo, and deliver their therapeutic payload to the brain resulting in the increased neuronal survival in different models of neurodegenerative disorders.[35,36]
Herein, we developed a novel biomimetic EV-based delivery system capable of TPP1 transfer to the CNS. TPP1 was incorporated into macrophage-derived EVs by transfection of parental cells with TPP1-encoding pDNA (EV-TPP1-t), or loading of therapeutic protein, TPP1, into naïve (empty) EVs (EV-TPP1-l). The last approach utilized the permeabilization of EVs membranes with saponin, or sonication in the presence of TPP1. The obtained EV-TPP1 formulations were evaluated for morphology, drug loading efficiency, TPP1 enzymatic activity, and stability. As expected, EVs provided a potent protection of TPP1 against protease degradation, and efficient transport of functional enzyme to target cells in in vitro model of CLN2. Furthermore, prolonged brain accumulation of EVs was demonstrated in mouse model of Batten disease, LINCL mice, in which CLN2 is disrupted by gene targeting.[37] Importantly, treatment of LINCL mice with EV-based formulations of TPP1 significantly increased their lifespan. We suggest that EVs secreted by immunocytes offer distinct advantages that uniquely position them as natural biocompartible and highly effective drug nanocarriers for systemic delivery of the lysosomal enzyme, TPP1, to the brain to treat NCLs.
2. Results and Discussion
2.1. EVs Secreted by Pretransfected Macrophages Contain Functional TPP1 and TPP1-Encoding DNA
In one approach, TPP1-transfected EVs (EV-TPP1-t) were produced by transfection of IC21 macrophages with TPP1-encoding pDNA, followed by isolation of EVs from the conditioned media. The optimal transfection conditions that provide for high levels and duration of therapeutic protein expression in macrophages[32] were used. First, we assessed the TPP1 levels in cell lysates and EVs at different time points using ELISA (Figure 1A). Similar to our previous reports regarding macrophages transfected with catalase- and GDNF-encoding pDNA,[32,34] the optimal time for highest TPP1 expression levels in cells and EVs was between second and fifth day after transfection (Figure 1A). Next, the TPP1 enzymatic activity was measured with a TPP1 substrate, AF-AMC (ala-ala-pheny-lalanine-7-amido-4-methylcoumarin) (Figure 1B). In this assay the maximal TPP1 activity in cells was observed at day 4 and 5. Based on the activity measurements and the particle number quantification by Nanoparticle Tracking Analysis (NTA) at day 5, 1011 EVs contained ≈10 μg of active TPP1, while the enzyme activity in nontransfected EVs was about one order of magnitude less. Interestingly, according to NTA, the average size of EVs isolated from TPP1-transfected macrophages was slightly greater (133.8 ± 4.1 nm) than that of EVs released from nontransfected parental cells (106.3 ± 9.3 nm) at the same time point (day 5). The TPP1 in EVs displayed increased stability against protease degradation compared to the free enzyme (Figure 1C). Thus, the TPP1 enzymatic activity was preserved for at least 25 h upon the treatment of EV-TPP1-t with the mixture pronases from Streptomyces Griseus, compared to TPP1 alone that was significantly inactivated by this time point (Figure 1C). EV-TPP1-t retained the round morphology as demonstrated by AFM (Figure 1D).
Figure 1.

Characterization of EVs isolated from TPP1-transfected macrophages (EV-TPP1-t). TPP1-transfected macrophages and their EVs display elevated levels of A) TPP1 protein and B) enzymatic activity between second and seventh day post-transfection. IC21 macrophages were transfected with TPP1-encoding pDNA (2 μg mL−1 pDNA with GenePorter 3K for 4 h), and (A) TPP1 protein expression and (B) TPP1 enzymatic activity were assessed in the parental cells (white bars) and EVs (black bars) using (A) ELISA and (B) a TPP1 substrate, AF-AMC (400 μM), respectively. (A) TPP1 protein or (B) activity levels in nontransfected cells (dashed line) or EVs secreted by them (solid line) were also recorded. For EVs’ activity, the levels are normalized to the number of cells used to isolate these EVs. C) Increased stability of TPP1 in EVs (solid line) compared to EV-free TPP1 (dashed line) upon incubation with pronase protease from Streptomyces Greseus (4 × 10−5 M). D) Round morphology of EVs released by empty-transfected macrophages and TPP1-transfected macrophages. E) Quantitative PCR analysis indicated a significant amount of TPP1-encoding pDNA incorporated in EVs from pretransfected macrophages. (A–C) Statistical significance *p < 0.05, or **p < 0.005 compared to TPP1 levels in nontransfected cells or (A,B) EVs, or (E) EV-free TPP1.
Previous reports suggested that EVs released by catalase-and GDNF-transfected macrophages contained pDNA and mRNA encoding the respective proteins.[32,34] Therefore, we examined by quantitative real-time PCR (qPCR), whether the TP P1-transfected macrophages can also release EVs with nucleic acids encoding TPP1 (Figure 1E). As expected, TPP1 pDNA was detected in the EVs isolated from TPP1-transfected macrophages, but not in the control EVs released by empty-transfected cells (cont. EVs). According to the obtained calibration curve (Figure S1, Supporting Information), one million EVs carried ≈1955 copies or 1 × 10–5 ng of TPPl-encoding pDNA. Noteworthy, no TPP1-encoding mRNA was detected in EVs released by either TPP1- or empty-transfected macrophages by RT-PCR, although significant levels were found in the control HeLa cell as a positive control (Figure S2, Supporting Information).
2.2. Loading Naïve EVs with TPP1 Ex Vitro
In another approach, TPP1 protein was loaded into naïve macrophage-derived EVs. Two different loading protocols were used to cause disruption/healing of EVs membranes during incubation with TPP1: a) sonication in water bath, or b) permeabilization with saponin. The TPP1 loaded EVs (EV-TPP1-l) were purified from nonincorporated enzyme by gel-filtration chromatography (Figure S3, Supporting Information). Based on NTA, EV-TPP1-l were slightly larger than the naïve EVs (Figure 2A), but retained the round morphology as demonstrated by AFM (Figure 2B). Catalytically active TPP1 was efficiently incorporated into EVs in both procedures, albeit sonication produced slightly better results than the saponin permeabilization, as measured by the enzyme activity assay (Figure 2A). Overall, 1011 EVs loaded by sonication contained ≈70 μg TPP1, while EVs loaded by saponin permeabilization contained ≈50 μg TPP1 by enzymatic activity. This is respectively, ≈7- and ~5-times more than the TPP1 content determined in EV-TPP1-t isolated from transfected cells. Noteworthy, TPP1-loaded formulations showed presence of proteins that are specific for EVs: CD63, TSG101, and HSP90, according to western blot analysis (Figure S4, Supporting Information).
Figure 2.
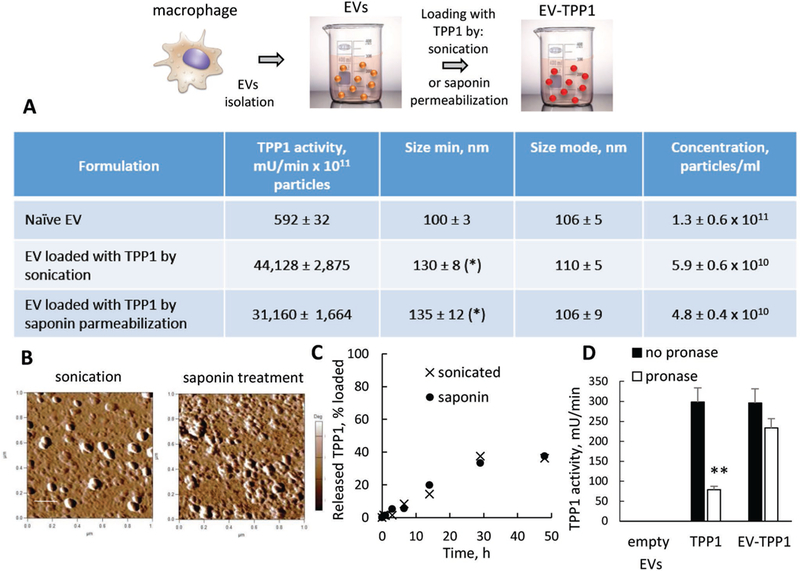
Characterization of EV-TPP1-l obtained by TPP1 loading into naïve EVs by either sonication or saponin permeabilization: A) TPP1 enzymatic activity and NTA parameters, B) morphology by AFM, C) TPP1 release, and D) TPP1 stability in the presence of pronase protease from Streptomyces Creseus (4 × 10−5 M) for sonicated EV-TPP1 versus free TPP1. (A–D) IC21 macrophage-derived EVs (1011 particles mL−1) were loaded with TPP1 (20 μg/100 μL) by sonication in water bath, or saponin permeabilization (0.4 mg mL−1). (B) The bar: 200 nm. (D) TPP1 activity was measured using AF-AMC (400 × 10−6 M) as a substrate. Statistical comparisons of (A) loaded EV-TPP1 versus naive EVs or (D) treated versus untreated with pronase TPP1 formulations: *p < 0.05, **p < 0.005 (n = 4).
The enzyme was slowly released from EV-TPP1-l for over 24 h upon dialysis using membranes with a cutoff 2000 kDa (Figure 2C). Furthermore, the incorporation of the enzyme into EVs led to TPP1 stabilization against proteases digestion (Figure 2D), as well as considerable increase in the enzyme stability upon storage. Specifically, EV-TPP1-l retained at least ≈40% activity in water solution at 4 °C for over a month, while the free TPP1 was completely inactivated during this time (Figure S5, Supporting Information).
2.3. EVs Facilitate Active TPP1 Transport into CLN2 Cells
To exert therapeutic activity, TPP1 needs to be delivered to the lysosomes of the cells. This can be facilitated in the format of EVs that display at their surface tetraspanins and integrins enhancing their cellular attachment and accumulation.[38–40] In these studies, we used CLN2 cells that are deficient in TPP1 enzyme, as an in vitro model representative of enzyme deficiency in Batten disease. These cells were exposed to either EV-TPP1-t or EV-TPP1-l and the levels of TPP1 were determined in the lysates as presented in Figure 3. The TPP1 levels in CLN2 cells treated with EV-TPP1-t at various time points were significantly higher than those for untreated cells, although not significantly different from the cells treated by “empty” EVs from macrophages transfected with GDNF-encoding pDNA (Figure 3A). We posit that the empty EVs from IC21 macrophages have some endogenous TPP1 that was thereby delivered to CLN2 cells exposed to these EVs. This is consistent with spectrophotometry measurements indicating very low, but detectable TPP1 enzymatic activity in naïve EVs released by IC21 macrophages (Figure 2A). Interestingly, the TPP1 levels in the EV-treated CLN2 cells decreased over time (Figure 3A) suggesting that the enzyme was degraded over several days. Therefore, we used a shorter exposure time point for the EV-TPP1-l (loaded using sonication or saponin permeabilization methods) that contained greater amount of TPP1 than EV-TPP1-t. In this case, CLN2 cells treated with EV-TPP1-l displayed much higher levels of TPP1 protein and enzyme activity than either untreated cells, or cells treated with empty EVs containing some endogenous TPP1 (Figure 3B,C). Overall, EVs loaded with TPP1 protein delivered more enzyme to CLN2 cells than the EVs isolated from TPP1-transfected macrophages. Consistent with the loading results, the EV-TPP1-l loaded by sonication delivered 1.5 times more active TPP1 than the EV-TPP1-l loaded by saponin permeabilization.
Figure 3.
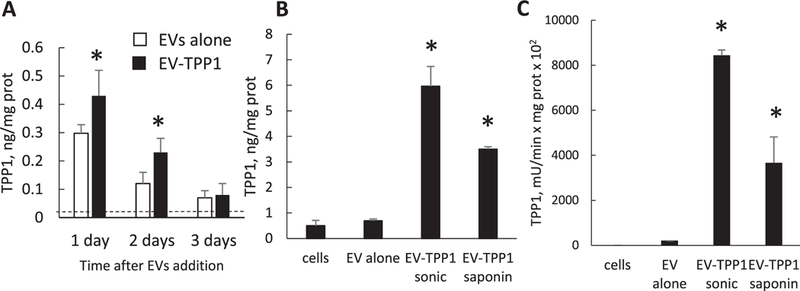
Delivery ofTPPl to CLN2 cells with EVs. CLN2 cells were treated with A) either EV-TPP1-t (black bars) or EVs from sham macrophages (white bars) for various time intervals, or B,C) EV-TPP1-l produced using sonication or saponin permeabilization for 6 h. Following treatments, CLN2 cells were lysed and the A,B) TPP1 protein levels were determined by ELISA or C) TPP1 activity was measured using AF-AMC (400 × 10−6 M) as a substrate. Dashed line—TPP1 protein levels in untreated CLN2 cells (A). Treatments with all EV-TPP1 formulations resulted in significant increases in the enzyme levels in CLN2 cells. Statistical comparisons of (A) EV-TPP1-t treated versus untreated cells or (B,C) cells treated with EV-TPP1-l versus empty EVs: *p < 0.05, (n = 4). (A) Difference between EV-TPP1-t and empty EV treated cells is not significant.
To address the intracellular localization of the EVs after delivery to neurons we performed, confocal studies in PC12 neuronal cells (Figure 4). In the described above experiments that were focused on TPP1 levels in target cells, we utilized deficient in the neuronal ceroid-lipofuscinoses CLN2 cells that originated from human skin CLN2 fibroblasts. Low basic levels of TPP1 in these cells were crucial for assessing EV-mediated TPP1 delivery. However, to study intracellular distribution of EVs nanocarriers, and targeting lysosomal compartments, we consider that neuronal P12 cells would more accurately represent target neurons than fibroblast cells. Therefore, we used neuronal PC12 cells for intracellular localization studies. These studies revealed that the EVs following internalization into cells are predominantly localized in the lysosomal compartments. Specifically, after 1 and 4 h incubation of fluorescently labeled EVs with the PC12 cells, 74.2 ± 15.2% and 68.4 ± 16.7% EVs, respectively, were colocalized with the lysosomes (Figure 4A,B). This suggests that EVs are suitable vehicles for TPP1 delivery to lysosomes in neurons that are the presumable target for the therapeutic delivery of the TPP1. Noteworthy, similar targeting of lysosomal compartments was also found in CLN2 cells (Figure S6, Supporting Information).
Figure 4.
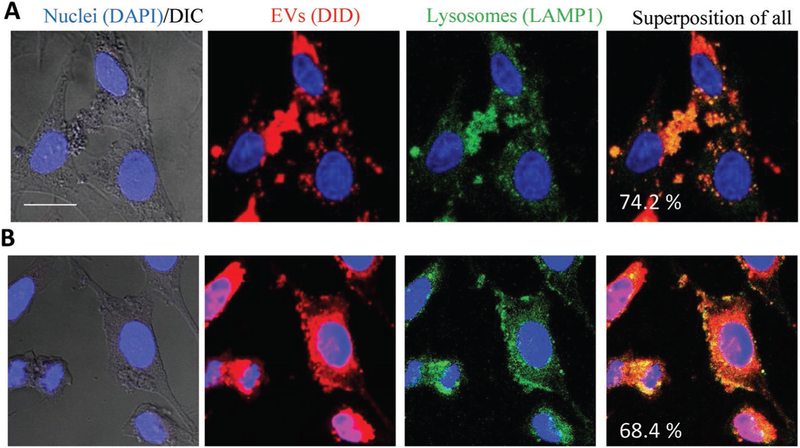
EVs target lysosomal compartments in PC12 neuronal cells. The cells were incubated with DID-labeled EVs (1010 particles/ml) for A) 1 h or B) 4 h, and then stained with FITC-LAMP1 antibodies for lysosomes and DAPI for nuclei. Colocalization of EVs (red) and lysosomes (green) is manifested in yellow. The bar: 20 μm.
2.4. Brain Accumulation of EVs in In Vivo Model of Batten Disease
To examine the ability of EVs to reach the brain tissues and deliver their payload, infrared spectroscopy (IVIS) studies were conducted in two months old LINCL mice. Specifically, macrophage-derived EVs were labeled with near-infrared lipophilic fluorescent dye DiR, and administered i.p. to LINCL mice (Figure 5). Fluorescent and light images of dorsal planes of the injected animals taken at various times showed significant accumulation of EVs in the brain (Figure 5A). Quantitative analysis of the in vivo imaging indicated that maximal brain accumulation of EVs was at 72 h. EVs were slowly cleared from the brain for over 3 weeks (Figure 5B) that was confirmed by the postmortem imaging of main organs (Figure 5C). The same pattern was showed for TPP1-loaded EVs (Figure S7, Supporting Information). Noteworthy, at the endpoint, the animals were sacrificed and perfused before the organs imaging to remove any blood contents from them. As expected for i.p. administration, the highest EVs accumulation was detected in the liver, spleen, and lungs of CLN2 mice (Figure 5D). Fluorescent and light images of ventral planes of the injected animals taken at various times are presented on Figure S8 (Supporting Information).
Figure 5.
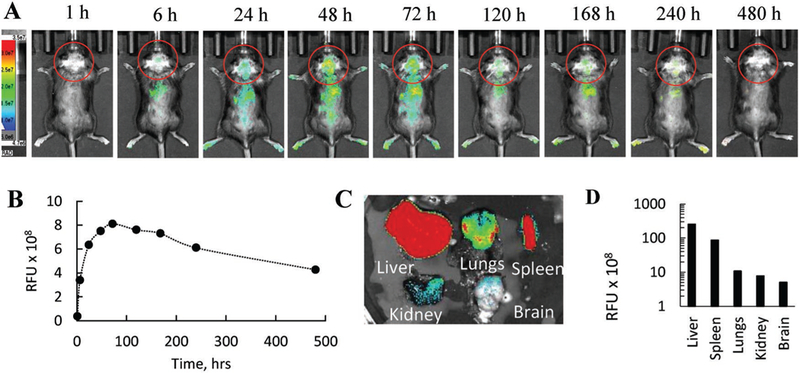
Biodistribution of EVs in LINCL mice by IVIS. LINCL mice (1 month old) were injected with DiR-labeled EVs through i.p. route. Animals were imaged over 20 d by IVIS. A) Representative images from N = 4 mice per group (dorsal planes) taken at various time points demonstrate prolonged brain accumulation of EVs in LINCL mice. B) Quantitative analysis revealed maximal brain accumulation at day 3 with slow decrease of the EVs fluorescence levels over 3 weeks. C,D) Postmortem imaging of organs indicate significant EV accumulation in the organs decreasing in order: liver > spleen > lungs > kidney > brain.
Using live imaging does not allow distinguishing between EVs that are present in the blood stream or in the brain parenchyma. To eliminate this factor, LINCL mice were injected with fluorescently labeled EVs, then sacrificed and perfused according to standard protocol to eliminate EVs in the blood stream. The accumulation of EVs in the brain was evaluated by confocal microscopy. Furthermore, the age-related changes in brain accumulation of EVs carriers were examined. 1 week old and 2 months old animals were injected with DiD-labeled EVs (red) through intraperitoneal (i.p.) route (Figure S9,Supporting Information). The early treatment of 1 week old animals (Figure S9A, Supporting Information) resulted in a greater amount of EVs in the brain, compared to the two months old mice (Figure S9B, Supporting Information). No fluorescence was found in control mice injected with saline (Figure S9C, Supporting Information).
Next, we compared the brain accumulation of DiD-labeled EVs and the same amount of DiD-labeled liposomes in 1 week old LINCL mice (Figure 6). Mice were sacrificed at 4 and 24 h following i.p. injections and perfused to wash out possible contamination with nanocarriers in the blood. Confocal images revealed a significant amount of EVs throughout the brain tissues (Figure 6A), and low, if any, liposomes at these time points (Figure 6B). Finally, we investigated brain accumulation of fluorescently labeled Alexa 555-TPP1, which was loaded into EVs by sonication (Figure S10, Supporting Information). EV-Alexa 555-TPP1 was injected into LINCL mice through i.p. route, mice were sacrificed at 24 h following injections, perfused, and brain slides were examined by confocal microscopy. LINCL mice injected with TPP1 alone were used as controls. In agreement with the EVs distribution pattern (Figure 6), significant fluorescence of Alexa 555-TPP1-EVs was detected in the brain of 1 week old and two months old animals (Figure S10A,B, respectively, Supporting Information). In contrast, low if any TPP1 fluorescence was detected, when the enzyme was administered alone (Figure S10C, Supporting Information).
Figure 6.
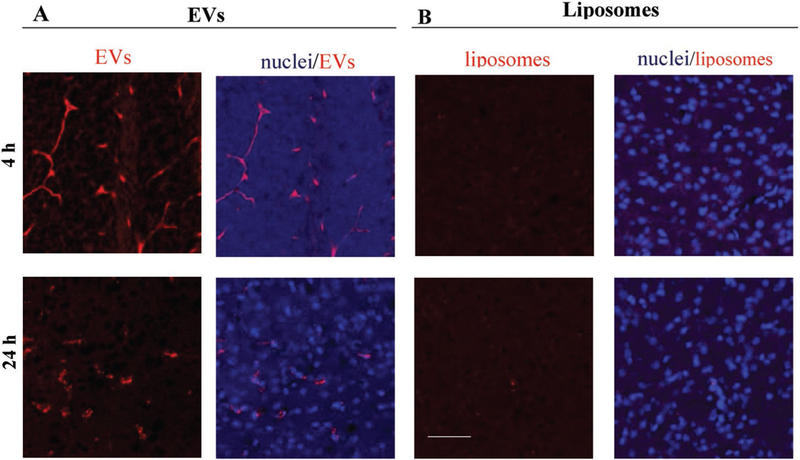
Brain distribution of DiD-labeled A) EVs and B) liposomes in knock-out LINCL mice. Confocal images showed a strong fluorescence in different brain areas in case of DiD-EV, and low, if any, fluorescence in case of DiD-liposomes. 1 week old knock-out mice were injected i.p. with DiD-labeled (A) EVs or (B) liposomes (1010 particles/100 μL/mouse). Animals were sacrificed 4 h or 24 h after injections and perfused. Brain slides were processed and examined by confocal microscopy. Nuclei were stained by DAPI (blue). The bar: 50 μm. EVs were isolated from macrophages concomitant media, and labeled with fluorescent dye, DiD (red). Liposomes were prepared and labeled with DiD (red).
2.5. Therapeutic Efficacy of EV-TPP1-l in In Vivo Model of Batten Disease
Based on the results of biodistribution studies, 1 week old LINCL mice were injected through intraperitoneal route with EV-TPP1-l formulation obtained by sonication in water bath (4.3 × 1012 particles mL−1, 150 μ.L per mouse, 15 mg kg−1) once per week, three weeks. LINCL mice and wild type (wt) mice treated with saline were used as positive and negative controls, respectively. LINCL mice injected with empty EVs were used in another control group. The survival was recorded for over three months (Figure 7). Administration of EV-TPP1-l formulation resulted in a significantly greater lifespan compared to control LINCL mice treated with saline. Treatment with naïve EVs produced subtle, but not significant increase in life span of LINCL mice. This confirms the therapeutic efficacy of systemically administered EV-based TPP1 formulation.
Figure 7.
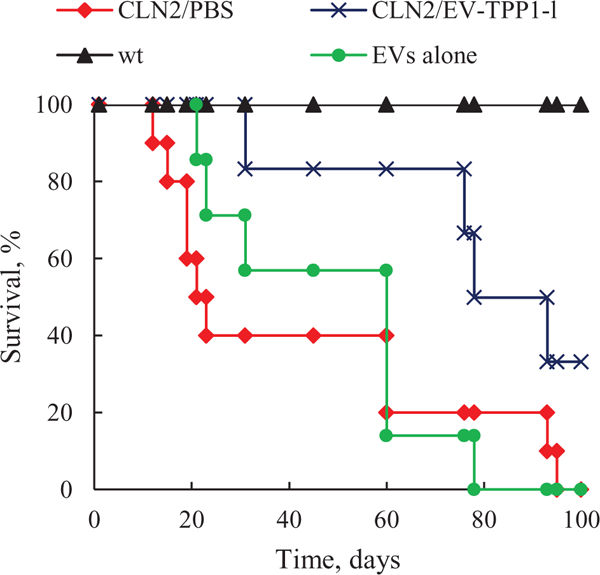
EV-TPP1-l treatment increased lifespan in LINCL mice. 1 week old LINCL mice were injected i.p. with EV-TPP1-l three times every week (crosses) or EVs alone (circles). Control LINCL mice (diamonds) and wild type animals (triangles) were injected with saline. A survival of treated animals was recoded over three months. A significantly greater lifespan (p < 0.05) was demonstrated in EV-TPP1-l treatment group compared to CLN2 mice treated with saline (N = 6).
Finally, systemic administration of EV-TPP1-l decreased neuroinflammation in LINCL mice compared to LINCL mice treated with saline (Figure 8). The obtained data indicate that LINCL mice treated with EV-TPP1-l has significantly lower astrocytosis (Figure 8B,D) compared to LINCL mice treated with saline (Figure 8A,D) that were near to the levels of healthy littermates (Figure 8C,D).
Figure 8.

EV-TPP1-l treatment decreases astrocytosis in LINCL mice. 1 week old LINCL mice were injected i.p. with A) saline, or B) EV-TPP1-l once per week, three weeks. C) Control wild type animals were injected with saline. Animals were sacrificed at day 100, and brain slides were stained with antibody to GFAP, a marker for activated astrocytes. The obtained confocal images (A–C) and quantification of astrocyte staining (D) indicate significant decreases in astrocytosis in the brain of LINCL mice upon EV-TPP1-l treatment (B) compared to LINCL mice treated with saline (A). *p < 0.05. The bar: 100 μm.
3. Conclusions
Batten disease is a lysosomal storage disease that typically begins in childhood. There are more than forty different lysosomal storage disorders (LSDs) resulting from deficiencies in lysosomal enzymes that lead to accumulation of various lysosomal substrates and subsequent neuronal death. In many cases the CNS is dramatically affected because of high vulnerability of neurons.[8] As such, targeted and efficient delivery of active enzymes across the BBB is of great importance for treatment of these conditions. Indeed, intracranial infusions of a therapeutically active lysosomal enzyme that is deficient in each LSD may provide protection of neurons. Unfortunately, this invasive procedure carries a high risk of adverse effects. Furthermore, a limited diffusion of the injected therapeutic enzyme through the brain tissues diminishes drug efficacy. In contrast, a systemic administration enables direct access to the BBB and uniform brain distribution through the brain capillaries. Regrettably, most potent therapeutic proteins failed to cross the BBB following systemic administration. In this respect, development of unconventional biocompatible and clinically applicable drug delivery systems may help to solve this challenging task. Thus, systemic coadministration of recombinant TPP1 along with a trans-acting peptide mediator (K16ApoE) was shown to significantly reduce brain lysosomal storage, increase lifespan and improve neurological function in LINCL mice.[41]
We report here a new EV-based technology for lysosomal enzyme, TPP1. Two different techniques for manufacturing EV-TPP1 were developed: transfection of parent cells, macrophages, with TPP1-encoding pDNA (EV-TPP1-t), and loading naïve EVs with TPP1 enzyme ex vitro using sonication or saponin permeabilization (EV-TPP1-l). These methods were utilized earlier in our lab for loading different therapeutic proteins and low molecular chemotherapeutics.[13,14,32,34–36,42–44] The size of EV-TPP1-t and EV-TPP1-l was slightly increased by 30–35% compared to empty nanocarriers. Both methods secured manufacture of EV-TPP1 with active enzyme. For the first method, 1011 EVs carriers contained approximately 10 μg TPP1. Noteworthy, EV-TPP1-t also contained TPP1-encoding pDNA, suggesting that along with brain delivery of the enzyme, treatment with this formulation may result in transfection of brain tissues and de novo synthesis of TPP1. Markedly, no TPP1-encoding mRNA was detected in EVs released by pretransfected macrophages, although we reported earlier the presence of GDNF- and catalase-encoding mRNA in EVs released by pretransfected with these therapeutic proteins plasmids macrophages. We speculate that it might be related to the specifics of these different proteins.
Regarding the second approach, both sonication and saponin permeabilization provided substantial TPP1 loading into EVs, although sonication was slightly more efficient. We hypothesized that the extensive reformation and reshaping of EVs membranes upon sonication enabled TPP1 diffusion across relatively tight and highly structured lipid bilayers. In case of latter approach, saponin may selectively remove membrane-bound cholesterol of EVs, creating holes/pores in the EVs lipid bilayers and thus, promoting TPP1 loading. Interestingly, saponin permeabilization resulted in 1.5× times lesser loading efficiency compared to sonication method. Significantly, the incorporation of TPP1 in EVs ensued the efficient preservation of TPP1 enzymatic activity against proteases degradation, and prolonged sustained release over 24 h for both EV-TPP1-t and EV-TPP1-l formulations. Finally, these methods for drug incorporation into EVs may be specific not only for TPP1 enzyme, but can be applied to other therapeutic and imaging agents.
Regarding the delivery of TPP1, this study demonstrated the ability of EVs to accumulate within target cells and specifically in lysosomal compartments. Thus, we demonstrated that both EV-TPP1-t and EV-TPP1-l delivered considerable amount of enzymatically active TPP1 in the in vitro model of Batten disease, CLN2 cells. As expected, EV-TPP1-l formulations, and especially, EVs loaded by sonication, provided the most efficient enzyme transport into target cells. Furthermore, confocal images revealed that ≈70% of fluorescently labeled EVs were accumulated in lysosomes in PC12 neural cells as well as CLN2 cells in vitro. Indeed, EVs are known to enter cells by endocytosis[45] and then accumulate in lysosomes, the target organelles for lysosomal storage disorders. This suggests that EVs carriers should be able to accomplish passive targeted delivery of TPP1 to lysosomes that is crucial for the treatment of LSDs. Further investigations will reveal mechanism and intracellular trafficking of various EVs formulations in target cells.
The main question remains, whether EVs carriers could deliver TPP1 to CNS. To address this issue, we investigated brain accumulation of fluorescently labeled EVs, as well as fluorescently labeled TPP1 in mouse model of Batten disease, LINCL mice. This is the model of early infant and childhood disease, therefore, we investigated two groups of LINCL mice, at early stages (1 week old), and late stages (2 months old) of the development. IVIS studies followed by confocal investigations demonstrated that EVs, as well as TPP1-loaded EVs efficiently accumulated in the mouse brain upon i.p. administration, and remained in the brain tissues for more than 3 weeks. Note-worthy, confocal images indicated that brain bioavailability for EVs and TPP1 is greater in younger 1 week old animals, suggesting that the treatment of LINCL mice should be initiated at earlier stages of development. Consequently, i.p. injections of EV-TPP1-l of 1 week old LINCL mice significantly increased their lifespan compared to control LINCL mice treated with saline.
It is known that many LSDs are accompanied with neuro-inflammation, which leads to further neuronal damage and death.[18,46–49] We demonstrated earlier that macrophages,[32–34,44] as well as macrophage-derived EVs target brain inflammation in animal models of PD and LPS-induced encephalitis.[35,36] The enhanced transport across the BBB was mediated by interactions between LFA-1 protein expressed on the membranes of macrophages and macrophage-derived EVs, and ICAM-1 receptor[36] that is known to be overexpressed in the inflamed endothelium. Therefore, neuroinflammation that is known to be present in lysosomal storage diseases[8] could increase brain influx rate and brain accumulation of macrophage-derived EVs in case of Batten disease. To this point, we demonstrated significant reduction of astrocytosis in the brain of LINCL mice treated with EV-TPP1-l, compared to control LINCL mice treated with saline. Overall, EVs nanocarriers efficiently accumulate in lysosomes, which are the target organelles for delivery of the depleted lysosomal enzymes. This suggests that macrophage-derived EVs may be a promising drug delivery platform for the enzyme replacement therapy to treat different LSDs.
4. Experimental Section
Reagents:
Recombinant Human TPP1 protein (lot #BIQE03) was a generous gift from BioMarin Pharmaceutical Inc. (Novato, CA, USA). GenePORTER 3000 transfection agent was purchased from AMS Biotechnology (Milton, Abingdon, UK). Lipophilic fluorescent dyes, 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindodicarbocyanine Perchlorate (DID) and 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Invitrogen (Carlsbad, CA, USA) and Sigma-Aldrich (St. Louis, MO, USA), respectively. FITC-conjugated mouse antibodies to LAMP1 were purchased from BD Biosciences (San Diego, CA, USA). Cell culture medium and fetal bovine serum (FBS) were purchased from Gibco Life Technologies, (Grand Island, NY, USA). Human TPP1/CLN2 ELISA Kit was obtained from LifeSpan BioSciences, Inc. (Seattle, WA, USA). TPP1-encoding pDNA (pSJG-JaTl-hTPP1opt-myc-spa) was a generous gift from Dr. Steven J. Gray (Department of Ophthalmology Gene Therapy Center, UNC). TPP1 substrate, AF-AMC and Triton X-100 were obtained from Sigma-Aldrich. Neuronal growth factor (NGF) was obtained from Gemini Bio-Products (West Sacramento, CA, USA).
Cells:
IC21 cell line derived by transformation of normal C57BL/6 mouse peritoneal macrophages with SV40, and neuronal PC12 rat adrenal pheochromocytoma cell line were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, South Logan, UT, USA) supplemented with 10% FBS, and 1% (v/v) of both penicillin and streptomycin. Regarding PC12 neuronal cells, they were differentiated with neuronal growth factor (NGF, 100 ng mL−1) for 4 d before the experiments. Human skin CLN2 fibroblasts deficient in the neuronal ceroid-lipofuscinoses (in vitro model of Batten disease), IC21 cells, and PC12 cells were grown in an incubator with optimal culture conditions of 37 °C and 5% CO2, and the medium was routinely replaced every 2–3 d.
Isolation of EVs:
Conditioned media from IC21 macrophages grown on 75T flasks (20 × 106 cells per flask) was collected, and EVs were isolated using gradient centrifugation.[38] In brief, the culture supernatants were cleared of cell debris and large vesicles by sequential centrifugation at 300 g for 10 min, 1000 g for 20 min, and 10 000 g for 30 min, followed by filtration using 0.2 μm syringe filters. Then, the cleared sample was spun at 100 000 g for 4 h to pellet the EVs, and supernatant was collected. The collected EVs (1011–1012 EV per flask) were washed twice with phosphate-buffered saline (PBS). Specifically, the obtained pellet was resuspended in high volume PBS (about 10 mL), and then centrifuge it again at 120 000 × g for 70 min. Additional step with resuspension in high volume of PBS resulted in dissolution of all proteins and small nonvesicular contaminants that were precipitated initially in low volume, and allowed us to obtain pure EVs fraction with narrow size distribution. To avoid contamination by the FBS-derived EV, FBS was spun at 100 000 g for 2 h to remove EVs before the experiment. The recovery of was estimated by measuring the protein concentration using the Bradford assay and by Nanoparticle Tracking Analysis (NTA). NTA analysis presented in Figure S11 (Supporting Information) showed a narrow size distribution with average mean 122.0 ± 2.4 nm, and Mode 105.6 ± 4.6 nm. The obtained EVs fraction was re-suspended in PBS (500 μL, 1 mg mL−1 total protein), and characterized for size and concentration.
Transfection of Macrophages with TPP1-Encoding pDNA: Manufacture of Therapeutic Gene-Encoding pDNA:
The TPP1-encoding pDNA (pSJG-JaTl-hTPP1opt-myc-spa) was expanded in DH5α E. coli and then isolated using Qiagen endotoxin-free plasmid Giga-prep kits (Qiagen, Valencia, CA, USA) according to the supplier’s protocol. The quantification of the manufactured pDNA was accessed by Nanodrop 2000/2000c spectrophotometer (Thermo Scientific) and verified by gel electrophoresis (Figure S12, Supporting Information). Totally, 4.5 mg TPP1-encoding pDNA was manufactured.
Transfection of Parental Cells.
IC21 macrophages were incubated with a mixture of 2 μg mL−1 TPP1-encoding pDNA and GenePORTER 3000 in serum free media for four h. Following incubation, the cells were washed with PBS and cultured for additional 1–10 d in the complete media containing 10% FBS. The transfected cells were collected, and lysed using four freeze-thaw cycles. The levels of TPP1 in the cell lysates, EV, and in conditioned media were assessed at different time points by TPP1/CLN2 ELISA according to manufacturer’s protocol.
Loading Naive EVs with TPP1:
Two methods for TPP1 incorporation into macrophage-derived EVs were evaluated: the sonication of EVs in water bath, or permeabilization of EVs membranes with saponin in the presence of TPP1. For drug loading by sonication, 500 μL EVs suspension (1011 particles mL−1) were supplemented with 5 μL TPP1 (20 μg/100 μL) and sonicated in water bath at room temperature (RT) for 30 min. In case of a saponin treatment, 500 μL EVs suspension (1011 particles mL−1) were supplemented with 5 μL TPP1 (20 μg/100μL) and 10 μL saponin solution to final concentration 0.4 mg mL−1, and incubate at RT for 30 min. TPP1-loaded EVs were purified by size-exclusion chromatography on Sepharose CL 4B (Sigma) (Figure S13, Supporting Information). EVs recovery was 90–95%.
Characterization of EVs:
The particle concentration and size were determined by NTA using NanoSight 500 Version 2.2 (Wiltshire, UK). For the size measurements EVs were dispersed at concentration ≈3 × 1010 particles mL−1 in phosphate-buffered saline (PBS). The atomic force microscopy (AFM) was used to assess the morphology of EVs. Briefly, a drop of the sample was placed on a glass slide, dried under an argon flow, and imaged as described earlier.[43] The levels of proteins constitutively expressed in EVs (CD63, TSG101, and HSP90) were identified in EV-TPP1 formulations by western blot analysis using Wes (ProteinSimple, San Jose, California, USA). Protein concentrations were determined using BCA kit (Pierce Biotechnology, Rockford, IL, USA). The protein bands were detected with CD63 primary monoclonal antibodies (Novus Biologicals, Centennial, CO, USA; 1:1000 dilution, #NPB2-67425), or TSG101 monoclonal antibodies (Novus Biologicals, #NPB2-67884), or HSP90 monoclonal antibodies (Novus Biologicals, #NPB2-67395), and secondary HRP-conjugated rabbit antigoat IgG-HRP (Santa Cruse, CA, USA; 1:5000 dilution.
To measure the TPP1 enzymatic activity, 20 μL of EVs or free TPP1 were first added to 60 μL of activation buffer (50 × 10−3 M acetate, 100 × 10−3 M NaCL, 0.1% Triton X100, 0.4 mg mL−1 saponin, pH 3.5) and incubated for 1 h at 37 °C. Then, 10 μL of these solutions were mixed with 40 μL of the assay buffer (50 × 10–3 M acetate, 100 × 10−3 M NaCL, 0.1% Triton X100, pH 5.0) and 50 [L of AF-AMC in the assay buffer (substrate final concentration 400 × 10−6 M, EVs final concentration 4.5 × 1011 particles mL−1, or free TPP1 final concentration 3 × 10−9 M). The reaction rate was determined by recoding AMC fluorescence (λex = 380 nm, λem = 460 nm) using Spectramax for 10 min and employing the AMC calibration curve. The same assay was used to determine the enzyme stability against proteases degradation. For these studies, 4 μL of proteases from Streptomyces Greseus (4 × 10−5 M, Sigma) was added to 400 μL EVs (0.2 × 1010 particles) containing TPP1 or free TPP1 (3 × 10−9 M) and incubated for 3 h at 37 °C. Data were expressed as the ratio of the residual versus initial TPP1 activities. The initial TPP1 activity was ≈850 pmoles min−1 μg−1.
qPCR Analysis:
RNA content in EVs from TPP1-transfected macrophages was determined by reverse transcriptase polymerase-chain reaction (RT-PCR). Briefly, the EVs were lysed, treated with DNase, reverse transcribed into copy DNA (cDNA) using SuperScript III CellsDirect cDNA Synthesis System (Invitrogen, cat # 18080200) according to manufacturer’s protocol, and then analyzed by real-time PCR (qPCR). For DNA determination, the EVs were lysed using the same protocol as for RNA and then analyzed by qPCR. In both cases qPCR was performed on cDNA or lysed EVs, 1 μL each of 20 × 10−6 m forward and reverse primers, and 2X PowerUp SYBR Green Master Mix (Applied Biosystems) in a total volume of 25 μL. qPCR assay was performed using primers specific to GAPDH or the TPP1 gene (the forward sequence CTGTCCTCATCACCGCATTT, the reverse sequence CGGTTGGAAACGACCCAATA) on a StepOnePlus Real-Time PCR System (Applied Biosystems). GAPDH was used as a control gene and RNA isolated from HeLa cells (included in CellsDirect cDNA synthesis kit above) was used as a cellular RNA control. For absolute quantification of pDNA, a standard curve was generated using gel-extracted pDNA quantified with a Nanodrop (Thermo).
Intracellular Localization of EVs in PC12 Cells by Confocal Microscopy:
To study intracellular localization, IC21 macrophages (20 × 106 cells per flask) were cultured for 3 d in DMEM supplemented with 10% FBS, then conditioned media was collected, and EVs were isolated by gradient centrifugation as described above. The isolated EVs (1010 particles mL−1 total protein) were labeled with a fluorescent dye DID (2 μmol)[33] and added to the differentiated neuronal PC12 cells for 1 and 4 h at 37° C. Following incubation, the cells were washed with PBS and incubated with FITC-LAMP1 antibodies. Then, the staining solution was removed, and cells were washed 2x PBS, fixed, and stained for nuclei with DAPI prior to the imaging.[42] The same setup was used to study intracellular distribution of EVs in CLN2 cells in vitro. Labeled cells were examined by a confocal fluorescence microscopic system ACAS-570 (Meridian Instruments, Okimos, MI) with argon ion laser and corresponding filter set. Digital images were obtained using the CCD camera (Photometrics) and Adobe Photoshop software. Quantification of immunostaining was performed with ImageJ software, utilizing JACoP plugins to calculate Pearson’s colocalization coefficients.[50] A comparison was performed on 30–40 sets of images acquired with the same optical settings.
LINCL Mice:
LICL mice with mutations of the CLN2 gene encoding a soluble lysosomal enzyme TPP1 were used as in vivo model of Batten disease.[37] The TETRA-ARMS design (Figure S12A, Supporting Information) was used for CLN2 genotyping. Specifically, mutant: 266 bp; wild-type (WT): 493 bp; and locus: 704 bp bands were visualized to identify mutant KO and wild-type mice with inner primers that bind to either the wild-type or mutant (KO) sequence (Figure S12B, Supporting Information). Animals were treated in accordance to the Principles of Animal Care outlined by National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Bioimaging and IVIS:
To reduce fluorescence quenching by fur and autofluorescence from solid diet, CLN2 mice were shaved and kept on liquid diet for 48 h prior to the imaging studies. Macrophage-derived naive EVs and EV-TPP1-l were labeled with DiR (Invitrogen) that is a lipophilic, near-infrared fluorescent cyanine dye (emission peak of 790) according to manufacturer’s instructions. Then, EVs or EV-TPP1-l were administered i.p. CLN2 (one mo. old) mice (2 × 1011 per mouse in 200 μL saline). For background fluorescence level evaluation, all animals were imaged before the injections in the IVIS 200 Series imaging system (Caliper, Xenogen Co., Life Sciences). Injected animals were imaged at various time points (1–20 d) post-treatment. At the endpoint of the experiment (22 d), mice were sacrificed, perfused according to the protocol; organs were isolated and imaged by IVIS. Quantitative analysis of the levels of fluorescence in the brain was performed using spectral instruments imaging ADL Aura software (Biocompare, Tuscon, AZ).
Brain Accumulation Studies:
Average life span for LINCL mice is about 13 weeks. Therefore, two groups of LINCL mice were utilized: 1 week old and 2 months old. The mice were i.p. injected with DiD-labeled EVs (obtained as described above) or DID-liposomes (1 × 1010 particles/100 μL per mouse). LINCL mice injected with saline were used as controls. Liposomes were prepared as described in ref. .[35] According to NTA, the average size of liposomes was 98 ± 4 nm. In another experiment, TPP1 was labeled with Alexa 555 according to manufacturer’s protocol, and loaded into nonlabeled EVs using sonication method. LINCL mice injected with TPP1 alone were used as controls. 4 and 24 h later, mice were sacrificed, perfused according to standard protocol, the brain slides were washed 3x PBS/Tween 5 min/wash ddH2O, and covered using Vectashield Hardset mounting media with DAPI. The images of brain tissues were examined by a confocal fluorescence microscopic system ACAS-570 and corresponding filter set.
Therapeutic Efficacy of EV-TPP1 in LINCL Mice:
1 week old mice were injected with EV-TPP1-l loaded by sonication in water bath (4.3 × 1012 particles/150 μL saline per mouse, 15 mg kg−1 TPP1) once a week over 3 weeks. LINCL mice as well as wild type littermates were injected with saline as controls. A lifespan in all groups was recorded over three months (N = 6).
Immunohistochemical Analyses:
At the end-point (100 d) all treated animals were sacrificed, perfused; brains were removed, washed, postfixed, and immunohistochemical analysis was performed in 30 μm thick consecutive coronal brain sections.[51] Levels of astrocyte activation were determined by fluorescent analysis of GFAP expression. For this purpose, the tissue slices were permeabilized with Triton X100, and incubated with primary antibody anti-GFAP (Abcam ab7260, 1:500 dilution), and secondary antibody goat antirabbit IgG H+L (Alexa Flour 488, Invitrogen A-11008) for 1 h at RT in the dark. Quantification of the fluorescence levels of astrocytes was performed as the function of the positive area by ImageJ software (free access provided by the National Institute of Health).
Statistical Analysis:
For the all experiments, data were presented as the mean ± SEM. Tests for significant differences between the groups in in vitro experiments investigating transfection of macrophages were performed using a one-way ANOVA with multiple comparisons (Fisher’s pairwise comparisons) using GraphPad Prism 5.0 (GraphPad software, San Diego, CA, USA). A standard T-test was performed when only two groups (for example, for the evaluation of the expression levels of TPP1 by ELISA) were compared. A minimum p value of 0.05 was chosen as the significance level for all tests.
For analysis of statistical significance of therapeutic effects assessed by immunohistochemistry for astrocytisis, the endpoint values were compared by group (LINCL mice injected with EV-TPP1, or saline, as well as wild type mice injected with saline). The endpoint for this assessment was at 90 d. If the differences between the groups were significant at the 0.05 level then pairwise tests were conducted using a Bonferroni correction for multiple comparisons. This was then analyzed in the same way as the endpoint analysis. The sample size for all in vitro experiments, as well as in vivo therapeutic efficacy experiments, was N = 6.
Supplementary Material
Acknowledgements
M.J.H. and N.L.K. contributed equally to this work. This study was supported by generous gift from Mr. and Mrs. Lehrman, and Eshelman Institute for Innovation EII UNC-29202 grant. The authors are grateful to Drs. Steven J. Gray and Erik Lykken (Department of Ophthalmology Gene Therapy Center, UNC) for providing a breeding pair of CLNL mice, and TPPl-encoding pDNA, as well as various invaluable comments and suggestions. The authors are also thankful to BioMarin Pharmaceutical Inc. for their generous gift of recombinant human TPP1 protein. Additionally, the study was supported by NIH Predoctoral Training Program T32CA196589 (EBH), and Russian Foundation for Basic Research grants 17-54-33027 (NLK, AVK) and 18-29-09154 (NLK).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Matthew J. Haney, Center for Nanotechnology in Drug Delivery and Carolina, Institute for Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Natalia L. Klyachko, Center for Nanotechnology in Drug Delivery and Carolina, Institute for Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Deparment of Chemical Enzymology, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow 119991, Russia.
Emily B. Harrison, Center for Nanotechnology in Drug Delivery and Carolina, Institute for Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Yuling Zhao, Center for Nanotechnology in Drug Delivery and Carolina, Institute for Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Alexander V. Kabanov, Center for Nanotechnology in Drug Delivery and Carolina, Institute for Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA; Deparment of Chemical Enzymology, Faculty of Chemistry, M.V. Lomonosov Moscow State University, Moscow 119991, Russia.
Elena V. Batrakova, Center for Nanotechnology in Drug Delivery and Carolina, Institute for Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill Chapel Hill, NC 27599, USA.
References
- [1].Kohan R, Cismondi IA, Oller-Ramirez AM, Guelbert N, Anzolini TV, Alonso G, Mole SE, de Kremer DR, de Halac NI, Curr. Pharm. Biotechnol. 2011, 12, 867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Whiting RE, Jensen CA, Pearce JW, Gillespie LE, Bristow DE, Katz ML, Exp. Eye Res. 2016, 146, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Silva GA, Ann. N. Y Acad. Sci. 2010, 1199, 221. [DOI] [PubMed] [Google Scholar]
- [4].Tsou YH, Zhang XQ, Zhu H, Syed S, Xu X, Small 2017, 13, 1701921. [DOI] [PubMed] [Google Scholar]
- [5].Peng Q, Zhang S, Yang Q, Zhang T, Wei XQ, Jiang L, Zhang CL, Chen QM, Zhang ZR, Lin YF, Biomaterials 2013, 34, 8521. [DOI] [PubMed] [Google Scholar]
- [6].Desnick RJ, Schuchman EH, Nat. Rev. Genet. 2002, 3, 954. [DOI] [PubMed] [Google Scholar]
- [7].Urayama A, Grubb JH, Sly WS, Banks WA, Proc. Natl. Acad. Sci. USA 2004, 101, 12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bosch ME, Kielian T, Front. Neurosci. 2015, 9, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, Ju S, Mu J, Zhang L, Steinman L, Miller D, Zhang HG, Mol. Ther. 2011, 19, 1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, Barnes S, Grizzle W, Miller D, Zhang HG, Mol. Ther. 2010, 18, 1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jang SC, Kim OY, Yoon CM, Choi DS, Roh TY, Park J, Nilsson J, Lotvall J, Kim YK, Gho YS, ACS Nano 2013, 7, 7698. [DOI] [PubMed] [Google Scholar]
- [12].Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, Wei J, Nie G, Biomaterials 2014, 35, 2383. [DOI] [PubMed] [Google Scholar]
- [13].Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, Inskoe E, Piroyan A, Sokolsky M, Okolie O, Hingtgen SD, Kabanov AV, Batrakova EV, Nanomedicine 2016, 12, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim MS, Haney MJ, Zhao Y, Yuan D, Deygen I, Klyachko NL, Kabanov AV, Batrakova EV, Nanomedicine 2018, 14, 195. [DOI] [PubMed] [Google Scholar]
- [15].Yang T, Martin P, Fogarty B, Brown A, Schurman K, Phipps R, Yin VP, Lockman P, Bai S, Pharm. Res. 2015, 32, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pan Q, Ramakrishnaiah V, Henry S, Fouraschen S, de Ruiter PE, Kwekkeboom J, Tilanus HW, Janssen HL, van der Laan LJ, Gut 2012, 61, 1330. [DOI] [PubMed] [Google Scholar]
- [17].Y Lee S Andaloussi El, Wood MJ, Hum. Mol. Genet. 2012, 21, R125. [DOI] [PubMed] [Google Scholar]
- [18].El Andaloussi S, Lakhal S, Mager I, Wood MJ, Adv. Drug Delivery Rev 2013, 65, 391. [DOI] [PubMed] [Google Scholar]
- [19].Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T, Erkan EP, Fan JB, Breakefield XO, Saydam O, Mol. Ther.–Nucleic Acids 2012, 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil YV, Filatov MV, Cell Commun. Signaling 2013, 11, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wahlgren J, De LKT, Brisslert M, Vaziri Sani F, Telemo E, Sunnerhagen P, Valadi H, Nucleic Acids Res. 2012, 40, e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Munoz JL, Bliss SA, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P, Mol. Ther.–Nucleic Acids 2013, 2, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T, Gotoh N, Kuroda M, Mol. Ther. 2013, 21, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xin H, Li Y Buller B, Katakowski M, Zhang Y, Wang X, Shang X, Zhang ZG, Chopp M, Stem Cells 2012, 30, 1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Katakowski M, Buller B, Zheng X, Lu Y, Rogers T, Osobamiro O, Shu W, Jiang F, Chopp M, Cancer Lett. 2013, 335, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maguire CA, Balaj L, Sivaraman S, Crommentuijn MH, Ericsson M, Mincheva-Nilsson L, Baranov V, Gianni D, Tannous BA, Sena-Estev M, Breakefield XO, Skog J, Mol. Ther. 2012, 20, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hood JL, Scott MJ, Wickline SA, Anal. Biochem. 2014, 448, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lai RC, Yeo RW, Tan KH, Lim SK, Biotechnol. Adv. 2013, 31, 543. [DOI] [PubMed] [Google Scholar]
- [29].Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M, Biochim. Biophys. Acta 2014, 7846, 75. [DOI] [PubMed] [Google Scholar]
- [30].Long KB, Beatty GL, Oncolmmunology 2013, 2, e26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Koh E, Lee EJ, Nam GH, Hong Y, Cho E, Yang Y, Kim IS, Biomaterials 2017, 121, 121. [DOI] [PubMed] [Google Scholar]
- [32].Haney MJ, Zhao Y, Harrison EB, Mahajan V, Ahmed S, He Z, Suresh P, Hingtgen SD, Klyachko NL, Mosley RL, Gendelman HE, Kabanov AV, Batrakova EV, PLoS One 2013, 8, e61852. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [33].Haney MJ, Zhao Y, Li S, Higginbotham SM, Booth SL, Han HY, Vetro JA, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV, Nanomedicine 2011, 6, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao Y, Haney MJ, Gupta R, Bohnsack JP, He Z, Kabanov AV, Batrakova EV, PLoS One 2014, 9, e106867. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [35].Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, Patel T, Piroyan A, Sokolsky M, Kabanov AV, Batrakova EV, J. Controlled Release 2015, 207, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yuan D, Zhao Y, Banks WA, Bullock KM, Haney M, Batrakova E, Kabanov AV, Biomaterials 2017, 142, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, Passini MA, Davidson BL, Stewart GR, Lobel P, J. Neurosci. 2004, 24, 9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thery C, Amigorena S, Raposo G, Clayton A, Curr. Protoc. Cell Biol. 2006, 30, 3.22.1. [DOI] [PubMed] [Google Scholar]
- [39].Thery C, Ostrowski M, Segura E, Nat. Rev. Immunol. 2009, 9, 581. [DOI] [PubMed] [Google Scholar]
- [40].Rana S, Yue S, Stadel D, Zoller M, Int. J. Biochem. Cell Biol. 2012, 44, 1574. [DOI] [PubMed] [Google Scholar]
- [41].Meng Y, Sohar I, Sleat DE, Richardson JR, Reuhl KR, Jenkins RB, Sarkar G, Lobel P, Mol. Ther. 2014, 22, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Haney MJ, Suresh P, Zhao Y, Kanmogne GD, Kadiu I, Sokolsky-Papkov M, Klyachko NL, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV, Nanomedicine 2012, 7, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao Y, Haney MJ, Klyachko NL, Li S, Booth SL, Higginbotham SM, Jones J, Zimmerman MC, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV, Nanomedicine 2011, 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhao Y, Haney MJ, Mahajan V, Reiner BC, Dunaevsky A, Mosley RL, Kabanov AV, Gendelman HE, Batrakova EV, J. Nanomed. Nanotechnol. 2011, S4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McKelvey KJ, Powell KL, Ashton AW, Morris JM, McCracken SA, J. Circ. Biomarkers 2015, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Macauley SL, Roberts MS, Wong AM, McSloy F, Reddy AS, Cooper JD, Sands MS, Ann. Neurol. 2012, 71, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Platt FM, Boland B, van der Spoel AC, J. Cell Biol. 2012, 199, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Baudry M, Yao Y, Simmons D, Liu J, Bi X, Exp. Neurol. 2003, 184, 887. [DOI] [PubMed] [Google Scholar]
- [49].Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES, Nature 2009, 458, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bolte S, Cordelieres FP, J. Microsc. 2006, 224, 213. [DOI] [PubMed] [Google Scholar]
- [51].Brynskikh AM, Zhao Y, Mosley RL, Li S, Boska MD, Klyachko NL, Kabanov AV, Gendelman HE, Batrakova EV, Nanomedicine 2010, 5, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.