

Abstract
Proteins on the cell surface are frequently glycosylated, and they are essential for cells. Surface glycoproteins regulate nearly every extracellular event, but compared with global analysis of proteins, comprehensive and site-specific analysis of surface glycoproteins is much more challenging and dramatically understudied. Here, combining metabolic labeling, click-chemistry and enzymatic reactions, and mass spectrometry-based proteomics, we globally characterized surface glycoproteins from eight popular types of human cells. This integrative and effective method allowed for the identification of 2172 N-glycosylation sites and 1047 surface glycoproteins. The distribution and occurrence of N-glycosylation sites were systematically investigated, and protein secondary structures were found to have a dramatic influence on glycosylation sites. As expected, most sites are located on disordered regions. For the sites with the motif N-!P-C, about one-third of them are located on helix structures, while those with the motif N-!P-S/T prefer strand structures. There is almost no correlation between the number of glycosylation sites and protein length, but the number of sites corresponds well with the frequencies of the motif. Quantification results reveal that besides cell-specific glycoproteins, the uniqueness of each cell type further arises from differential expression of surface glycoproteins. The current research indicates that multiple surface glycoproteins including their abundances need to be considered for cell classification rather than a single cluster of differentiation (CD) protein normally used in conventional methods. These results provide valuable information to the glycoscience and biomedical communities and aid in the discovery of surface glycoproteins as disease biomarkers and drug targets.
Graphical abstract
Proteins located on the cell surface are normally modified with carbohydrates.1 These surface glycoproteins play vital roles in nearly every extracellular event, including cell−cell communication, cell−matrix interactions, and cellular response to environmental cues.2,3 Many surface glycoproteins function as ion channels and transporters for molecules across the plasma membrane, while others are receptors, such as G-protein-coupled receptors, that sense and mediate cellular responses to extracellular stimuli.4,5 Enzymes and binding proteins located on the cell surface are also commonly glycosylated.3
Surface glycoproteins frequently reflect the developmental and diseased statuses of cells. A number of surface glycoproteins serve as disease biomarkers and for cell-type classification.6−8 Moreover, these surface glycoproteins are often the targets of macromolecular drugs such as antibodies or enzymes in the emerging immunotherapy field.9 Comprehensive and site-specific analysis of cell-surface glycoproteins will aid in a better understanding of glycoprotein functions and cellular activities. Immunophenotyping of surface glycoproteins has been performed using flow cytometry.10 This technique, however, requires prior knowledge of the proteins of interest or the cell type. The availability and the specificity of antibodies and the low throughput could also be an issue. Modern mass spectrometry (MS)-based proteomics provides a unique opportunity for global and site-specific analysis of proteins and their modifications.11−20 However, it is extraordinarily challenging to comprehensively and site-specifically analyze glycoproteins located only on the cell surface because of the following reasons. First, many glycoproteins occur in very low abundance, and their analysis is hampered by highly abundant proteins.21,22 Second, the heterogeneity of the glycans further complicates the analysis.23,24 In addition, surface glycoproteins have to be effectively separated before MS analysis.
Previously, Zhang et al. developed an innovative MS-based method to identify surface N-glycoproteins by first oxidizing the glycans with NaIO4, and the resulting aldehyde groups on the glycoproteins were used for the enrichment with hydrazide beads.22 A few years later, Wollscheid et al. designed a beautiful cell surface-capturing (CSC) method to identify and quantify cell surface N-glycoproteins. This was based on glycan oxidation with NaIO4 and biocytin hydrazide tagging prior to the enrichment with streptavidin beads.25 Despite the importance of glycoproteins on the cell surface, their comprehensive analysis is much understudied compared with global analysis of proteins.
In this work, we systematically studied glycoproteins on the surface of eight types of commonly used human cells. Surface glycoproteins were metabolically labeled with a functionalized sugar and then tagged with biotin through the strain-promoted copper-free click chemistry reaction. Surface glycopeptides with biotin were selectively enriched and subsequently deglycosylated with PNGase F in heavy-oxygen water for site-specific analysis using MS. This approach allowed for global and site-specific identification of >2000 N-glycosylation sites from >1000 surface glycoproteins, with an average of 683 glycosylation sites and 354 surface glycoproteins per cell type. We also quantified glycoproteins using label-free quantification and discovered that only a small portion of the proteins are cell-specific, while many were differentially expressed across the cell types. Furthermore, different groups of proteins were more highly expressed in one cell line than in the others and served particular functions depending on the cell type. Benefiting from site-specific analysis, we explored the behaviors and occurrence of the glycosylation sites, including the solvent accessibility of the sites and the effect of protein structures on the sites. The current results lead to a better understanding of cell-surface glycoproteins and provide vital information in discovering new biomarkers and drug targets.
EXPERIMENTAL SECTION
Cell Culture, Metabolic Labeling, and Click-Chemistry Reaction.
Adherent cell lines, including HEK293T, HeLa, HepG2, MCF7, and PANC1 cells, were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM, Sigma-Aldrich) containing 10% fetal bovine serum (FBS, Corning). Suspension cell lines (i.e., Jurkat, K562, and U266 cells) were maintained in RPMI-1640 medium (Sigma-Aldrich) containing 10% FBS. All cells were grown in a humidified incubator at 37 °C with 5% carbon dioxide (CO2). While different media were used for these cells, they are considered as standard media for culturing each cell line. Therefore, the cells should be in their normal states for general biological experiments.
When adherent cells reached ~50% confluency, they were labeled with 100 μM N-azidoacetylgalactosamine-tetraacylated (Ac4GalNAz). Suspension cells were cultured until the cell density was ~7 × 105 cells/mL and then labeled similarly to the adherent cells. After 24 h of metabolic labeling, cells were tagged with 100 μM dibenzocyclooctyne (DBCO)-biotin for 1h. The reaction was quenched with 10 mM dithiothreitol (DTT). A more detailed description is in Supporting Information.
Protein Extraction and Peptide Purification.
Cells were lysed, and proteins were extracted. Proteins were reduced with 5 mM DTT at 56 °C for 25 min and subsequently alkylated with 14 mM iodoacetamide (Sigma-Aldrich) for 30 min in the dark. The alkylation reaction was quenched by incubating with DTT to the final concentration of 5 mM in the dark for another 15 min.26 Proteins were purified and digested with trypsin for 16 h. The peptides were desalted using a Sep-Pak Vac tC18 cartridge. The detailed information is included in Supporting Information.
Enrichment of Cell-Surface Glycopeptides.
Glycopep-tides tagged with biotin were enriched with high-capacity NeutrAvidin agarose resin (Thermo Scientific) according to the manufacturer’s protocol. The enriched peptides were eluted three times with 200 μL 8 M guanidine hydrochloride (pH = 1.5, Promega) at 56 °C for 2 min each. The eluates were pooled, desalted, and dried in a vacuum concentrator overnight. Glycopeptide deglycosylation was performed with 3 units of PNGase F (Sigma-Aldrich) in 40 μL of 40 mM ammonium bicarbonate (pH = 9, Sigma-Aldrich) in heavy-oxygen water (H 182O, Isoflex) at 37 °C for 3 h with shaking. The reaction was quenched with formic acid (FA, Fisher Scientific) to the final concentration of 1%. The peptides were desalted with StageTips and eluted into three fractions with 20%, 50%, and 80% ACN containing 1% acetic acid (Sigma-Aldrich).27 The eluates were dried again in a vacuum concentrator.
LC-MS/MS Analysis and Database Searching.
The peptides were analyzed by an online LC-MS system. They were separated by reversed-phase liquid chromatography (LC), and the LC is coupled to an LTQ Orbitrap Elite Hybrid Mass Spectrometer (Thermo Scientific). MS/MS analysis was performed with a data-dependent Top20 method. The detailed information is in Supporting Information.
Raw MS files were analyzed by MaxQuant (version1.6.2.3).28 MS spectra were searched against the human proteome database downloaded from UniProt using the integrated Andromeda search engine.29 All default parameters were left unchanged, except adding variable modification for glycosylation site identification (+2.9883 Da) and 3 maximum missed cleavages. Label-free quantification was also enabled with the LFQ min ratio count of 1, the match-between-runs option was enabled, and the iBAQ option was enabled. The false discovery rates (FDRs) were kept at 0.01 at the peptide spectrum match, protein, and site decoy fraction levels.
Bioinformatic Analysis.
Data analyses were performed with Perseus30 and Excel. Glycopeptides were filtered to only contain the sequences N-X-S/T/C (X is any amino acid except proline) for N-linked glycosylation. Human membrane protein information was extracted from UniProt database. For those whose membrane information is not available, further sequence analyses were performed using Phobius (phobius.sbc.su.se).31 SecretomeP (cbs.dtu.dk/services/SecretomeP) was used to further predict protein secretion through nonclassical secretory pathways.32 Gene ontology (GO)-based enrichment analysis was performed on Gene Ontology Consortium Web site (http://www.geneontology.org). Residue solvent accessibility and structure were predicted using NetsurfP (version 1.1).33 For the quantification, the glycopep-tide LFQ intensity was extracted from the peptides.txt table and limited to only glycopeptides. iBAQ was used to estimate the absolute protein abundance ranking.34 Shannon’s entropy was calculated the same way as the previous report.35 More information is in Supporting Information. Cell-surface glycoprotein interactions and pathway analysis were performed, and the results are included in Supporting Information
RESULTS AND DISCUSSION
Global Analysis of Cell-Surface Glycoproteins in Human Cells.
Sugar analogues containing a biologically inert but chemically functional group have been proven to be powerful labeling reagents for glycoproteomic studies.36,37 We previously demonstrated that labeling with Ac4GalNAz resulted in the highest coverage of cell-surface N-glycoproteins compared with N-azidoacetylglucosamine-tetraacetylated (Ac4GlcNAz) and N-azidoacetylmannosamine-tetraacetylated (Ac4ManNAz),38 and thus, Ac4GalNAz is used in this study. Cells incorporated GalNAz into the glycans on glycoproteins, including those located on the cell surface. These surface glycoproteins were selectively tagged through the strain-promoted, copper-free click chemistry reaction between the azido group and dibenzocyclooctyne (DBCO)-biotin in flask under very mild conditions.39 They are then enriched with NeutrAvidin beads at the peptide level, deglycosylated with PNGase F in H 182O to generate a common tag, and analyzed with LC-MS/MS (Figure 1A).
Figure 1.

Overview of global and site-specific analysis of cell-surface glycoproteins from eight popular types of human cells. (A) A diagram showing the experimental procedure. (B) Numbers of cell-surface glycoproteins and glycosylation sites identified from each cell type. The error bars represent one standard deviation from two biological duplicate experiments. (C) Number of cell-surface glycoproteins identified from multiple cell types. (D) Types of the identified surface glycoproteins. Types I−IV for single-pass membrane protein types I−IV, TM for transmembrane domain, SP for signal peptide, and NC for proteins entering the non-classical secretory pathway. (E) Single-pass types I and II suface glycoproteins from K562 cells are aligned against TM. Yellow dots represent the identified glycosylation sites. (F) Protein clustering of all identified surface glycoproteins based on biological process and molecular function. The number in the parentheses show the −logP values.
Using this approach, a total of 1047 glycoproteins and 2172 N-glycosylation sites were identified with an average of 354 glycoproteins and 683 sites from each cell type (Figure 1B, Table S1). The average posterior error probability of the peptide identification is 0.005. Compared with the previously reported results, including total glycoproteomic analysis,40−42 we identified 349 new glycosylation sites. Protein occurrence analysis showed that the number of glycoproteins identified in only one cell line is the highest, and as the number of cell types increases, the occurrence decreases (Figure 1C). Biological duplicate experiments revealed that, on average, over 70% of glycoproteins and glycosylation sites were identified in both experiments, showing high reproducibility of the approach (Figure S1). The conditions for tagging cell surface glycoproteins are mild, which do not stimulate cellular response, or harm the cells because copper or oxidizing reagents are not employed. This allows site-specific quantification and dynamic studies of cell-surface glycoproteins.38,43,44 Even though we globally analyzed surface glycoproteins in cultured cells, Spiciarich et al. recently employed metabolic labeling with ManNAz for the identification of sialoglycoproteins from the proteomes of human prostate cancer and normal tissues. The authors identified 972 proteins from both samples with about 50% of the proteins localized on the plasma membrane.45 This is very promising and shows the efficiency of metabolic labeling for tissue samples. Sugar analogues can also be fed to animals, such as zebrafish46 and mouse.47 Therefore, this method is applicable to study surface glycoproteins in tissue samples and model animals.
Classification of Identified Surface Glycoproteins.
Among the identified surface glycoproteins (1047), over 800 belong to membrane proteins (P = 4.51 × 10−128) according to gene ontology analysis based on cellular component. Compared with UniProt subcellular location information, on average, 75% of the glycoproteins identified from all cell types are classified into single-pass types I−IV, multipass, and peripheral membrane proteins (Figure 1D). For those without membrane protein classification information available, Phobius was employed to predict if they have either a transmembrane domain (TM) and/or a signal peptide (SP).31 SecretomeP 2.0 was also used to predict whether they may be secreted through the nonclassical secretory (NC) pathways and then located on the cell surface (Table S2).32 Eventually, only 5.9% of the identified glycoproteins were left without information supporting their localization on the cell surface. These proteins may not be annotated or discovered at the cell surface yet. Another possibility is that their identifications could be due to nonspecific binding of some peptides during the enrichment. In spite of that, the approach specifically targets proteins on the extracellular side because the cells were tagged with DBCO directly in the flask without affecting the cell integrity. These sites on types I and II single-pass membrane proteins from K562 cells are displayed as yellow dots in Figure 1E, with the X-axis representing the transmembrane region and the Y-axis showing how far away the glycosylation sites are from the transmembrane region. No glycosylation sites inside the cells were identified.
The biological functions of the identified proteins from GO enrichment analysis correspond very well with the known functions of cell-surface glycoproteins (Figure 1F), including biological adhesion (P = 2.36 × 10−76), cell surface receptor signaling pathway (P = 2.30 × 10−50), locomotion (P = 2.24 × 10−43), and cell communication (P = 2.19 × 10−30). Proteins with binding activities, such as growth factor binding (P = 3.64 × 10−19), collagen binding (for cell-matrix adhesion, P = 5.23 × 10−10), and calcium, copper, and chloride ion bindings were also enriched. Many membrane enzymes with signaling receptor activity (P = 7.06 × 10−49) were identified, including those involved in the regulation of protein kinase B signaling, phosphatidylinositol 3-kinase signaling, and MAP kinase activity. Other less famous functions of cell surface glycoproteins, such as the regulation of cell size (P = 7.09 × 10−15), the regulation of body fluid level (P = 3.24 × 10−7), ossification (P = 5.86 × 10−7), and learning or memory (P =5.31 × 10−5) were also found. Interestingly, we identified proteins involved in DNA-binding transcription factor activity, such as vasculin and alpha-enolase, which were reported to localize in both the plasma membrane and the nucleus.48,49 Therefore, some proteins without the membrane information could still be localized at the surface.
Distribution and Occurrence of N-Linked Glycosylation Sites on Surface Glycoproteins.
Benefiting from the deglycosylation reaction with PNGase F in H 182O to generate a mass tag on glycosylation sites, we confidently localized the sites with an average probability of 0.97. N-glycosylation has a well-known N-!P-S/T canonical sequences, and here, the N-!PC motif was also included because of the previous reports.50,51 The majority of the identified proteins contains 1 or 2 sites (Figure 2A), and some have many more such as 25 sites identified on LRP1 (prolow-density lipoprotein receptor-related protein 1). The protein is made of over 4000 amino acid residues and has 75 N-!P-S/T/C motifs. The total number of glycosylation sites does not depend on the number of glycosylation motifs (Figure S2A). In our data set, 39% of all glycosylation motifs were glycosylated. For example, both neural cell adhesion molecule 1 (L1CAM) and lymphocyte antigen 75 (LY75) have 22 glycosylation motifs but 19 and 2 glycosylation sites were identified, respectively. More than half of the identified glycosylation sites have the N-!P-T sequence despite the similar occurrence of the N-!P-S and N-!P-T motifs (Figure 2B), agreeing with the previous results.50 Only 2% of the sites have the N-!P-C sequence when this motif occurs at 10% of the total motifs. As the protein length increases, the number of motifs also increases with an acceptable R2 value of0.71. However, this does not translate into a higher number of glycosylation sites (R2 = 0.02) (Figure S2B). Because the surface glycoproteome is relatively small, the number of glycosylation sites from a large-scale study by Xiao et al.,40 which identified one of the largest experimental data sets for N-glycosylation, was also evaluated, and the result is the same (Figure S2C).
Figure 2.

Site-specific analysis shows the distribution and occurrence of N-glycosylation sites and the motifs of cell-surface glycoproteins. (A) Number of protein glycosylation sites from each cell type. (B) Relative abundance of the glycosylation motifs and the identified glycosylation sites.(C) Solvent accessibility prediction of all asparagine residues. (D) Distributions of the predicted structure at each N-glycosylation motif and glycosylation site. (E) The occurrence of the identified glycosylated sites, the glycosylation motifs, and total sites from GO surface proteins extracted from UniProt when each protein length is divided into 100 bins. (F) The correlation between the number of identified glycosylation sites and the number of the motifs in each bin.
The relatively large and hydrophilic N-glycans play critically important roles in the regulation of protein folding and structures. Accordingly, the location of the asparagine residues in each protein was predicted using NetsurfP algorithm (Table S3).33 It was found that the asparagine residues are, as expected, exposed to the solvent with only a small fraction buried inside proteins (0.79 and 0.21, respectively) (Figure 2C). A closer look into these residues shows that those containing a glycosylation motif are even more exposed to the solvent than those without one (0.87 and 0.76, respectively). This observation also applies to the glycosylated residues. We also determined the solvent accessibility for each of the N-!PS/T/C sequons. The fractions of N-!P-S and N-!P-T exposed to the solvent are very similar (0.87 and 0.89), compared to0.80 for N-!P-C. Overall, the fractions of the solvent accessibility for the glycosylated sites are similar to those for the motifs (Figure S2D).
We then investigated the predicted structure of these sites in proteins using the results from NetsurfP (Table S3). Because of its structural role, N-glycosylation sites are usually located on loops and turns.52 The majority of the asparagine residues are on coils (loops and turns), and fewer on helices and strands, respectively (Figure S2E). The fractions of the identified sites and the motifs are very similar for each structure. For the helix structure, the fractions of identified sites and residues with the glycosylation motifs are half of those without the motif and the total, but for the strand structure they are twice, even though both are ordered structures. Surprisingly, a higher fraction of the N-!P-C motifs is located on coils than the N-!P-S and N-!P-T motifs, but a lower fraction is actually glycosylated (Figure 2D). Glycosylation with the N-!P-C motif is, however, more preferred at the helices even though the fraction of helical N-!P-C is smaller than that of coiled N-!P-C. The prediction was compared with the solved structures of some proteins, and the results agree very well.
To further investigate the occurrence of glycosylation sites along the protein length, each protein length was divided into 100 bins, and the number of glycosylation sites in each bin was counted (Figure 2E). Generally, fewer identified glycosylation sites on the N- and C-termini were observed. For the N-termini, it may be due to the inability of the accepting site near the signal peptide sequence to reach the active site of oligosaccharyltransferase (OST) before the signal peptide is cleaved.53 For the C-termini, it may be because of the steric hindrance from the secondary structures formed in the near-complete nascent polypeptide or the ineffciency of N-linked glycosylation when the polypeptide translation is terminated before the glycosylation accepting residue reaches the active site of OST.54,55 Not only is the frequency of glycosylation sites lower at both termini, but also the number of the glycosylation motifs themselves are different in each bin and lower at both termini. A similar pattern of the glycosylation motifs and the glycosylation sites was observed (Figure 2E). Even though we showed earlier in Figure S2B that the number of glycosylation sites does not depend on the protein length, the correlation between the number of glycosylation sites and the number of the motifs is reasonably high with R2 of 0.73 when their position along the protein length is considered (Figure 2F). We compared this with proteins annotated with GO surface proteins (Figure 2E) and those from Xiao et al.,40 and we also found a similar correlation (Figure S2F,G).
Some glycopeptides might be present but were not identified by the mass spectrometer. For example, tryptic glycopeptides that are too short or too long may not be identified. Other post-translational modifications on the same glycopeptide may also hinder its identification. Therefore, some peptides with a particular glycosylation site could be missing from the analysis. Nevertheless, our approach is very sensitive as 87 proteins containing only one glycosylation motif (maximally one glycosylation site) were identified. It should also be noted that these sites are pooled from all cell types, so some sites may not be found in a specific cell type.
The coverage of surface glycoproteins may be further improved. For example, previously, different sugar analogues were used for labeling different subsets of surface glycoproteins such as ManNAz for sialoglycoproteins,36,38,46 and thus a combination of sugar analogues may further increase the coverage. Many surface glycoproteins are also present in low abundance. Therefore, more cells used in the experiments will allow us to more effectively identify low-abundance glyco-proteins on the surface.
Label-Free Quantification of Cell-Surface Glycoproteins.
The identification of cell-surface glycoproteins from eight types of cells does not provide quantitative information. Therefore, we quantified these cell-surface glycoproteins between cell types with label-free quantification (LFQ) using MaxQuant.28 Based on the LFQ results, 784 surface glycoproteins were quantified (Table S4). The reproducibility of LFQ quantification was first evaluated between two biological duplicate experiments because the abundance of cell-surface glycoproteins is intrinsically low compared to intracellular proteins, which may affect the quantification precision between runs. The log2 intensities from both runs are in an acceptable agreement with an average R2 of 0.81 (Figure 3A). Proteins with zero intensities were excluded because the log values cannot be determined. Conversely, a comparison between different cell types showed a weaker correlation with an average R2 of 0.45 (Figures 3B and S3A). These results differ from the previous proteome experiments where the correlations from biological replicate experiments are very similar (0.83) while the correlation between the results from different cell lines were much higher (0.74).56 These results clearly demonstrate that the method is reasonably reproducible, and compared with intracellular proteins, surface glycoproteins are much more unique to the cell type.
Figure 3.

Label-free quantification of surface glycoproteins. (A) Correlation of LFQ intensity from biological duplicate experiments of each cell type. (B) Correlation and hierarchical clustering of LFQ intensity between cell lines. R2 values are displayed in the figure. The log2-transformed average LFQ intensity of the two duplicate experiments were used when calculating the correlation. (C) The numbers of proteins that are cell-specific, are in 2−7 cell lines, and are globally expressed from each cell line from the quantification with LFQ. (D) Ranking of absolute protein abundance by iBAQ from K562 cells. Data points from global and cell-specific proteins were shifted by +5 and −5, respectively, to clearly show their positions. (E) A similar plot as Figure 3E. However, the iBAQ intensity of proteins in the yellow square of K562 cells of Figure 3F are plotted as orange circles against total proteins from K562. Data points were shifted by +5 to more clearly show their positions. (F) A Z-score transformed heat map of log2 LFQ protein intensity showing relative protein expression of surface glycoproteins. Missing values were imputed with a normal distribution (width = 0.3, shift = 1.8).
The identified proteins are categorized into globally expressed and cell-specific surface glycoproteins. Here, we define globally expressed surface glycoproteins as those quantified across all cell lines without any missing values while the cell-specific ones are quantified in a single cell type. Shannon’s entropy was also applied to the quantified proteins to show the expression of each protein across the cell types.35 Generally, a higher entropy value means a more uniform expression across cell types or that the protein is quantified with valid abundance values in more cell types, while a lower entropy indicates differential expression of the protein or it is expressed in particular cell types (Figure S3B,C and Table S4). We quantified 104 globally expressed surface glycoproteins (Figure 3C), with Shannon’s entropy in the range of 0.71−2.04 (Table S4). These proteins are involved in biological processes such as adhesion, cell-surface receptor signaling pathway, and cell migration. Four-hundred and eighty-three proteins were found in 2−7 cell lines. Surprisingly, a total of 197 proteins were cell-specific, corresponding to an average of 25 proteins or 7% of all quantified proteins per cell type (examples are included in Table S6). The proteins have the Shannon’s entropy values in the range of 1.76 × 10−4 − 8.26 × 10−8. The number is different from that in Figure 1C because the match-between-runs option was enabled during the quantification, and some proteins were assigned to more than one cell line. Protein clustering analysis did not yield much valuable information about these proteins for a specific cell type due to the low number of proteins. Nevertheless, the functions enriched from HEK293T-specific proteins are consistent with relevant biological processes to kidney cells including kidney development and nervous system development.
The absolute abundance of these surface glycoproteins was also estimated using intensity-based absolute quantification (iBAQ). The log2-transformed intensity plots show a normal S-shaped distribution and varies within 5 orders of magnitude (Figure S3D and Table S5). Surface glycoproteins from each cell type are categorized as high or low abundance if their intensities fall within the fourth or the first quartiles, respectively. The 104 glycoproteins that are globally expressed have higher absolute abundance while the cell-specific ones are present in relatively low abundance. The estimated absolute abundance of 77% globally expressed proteins falls into the third and fourth quartiles while 83% of those K562-specific proteins are in the first and second quartiles (Figure 3D). There are also 12 high-abundance glycoproteins that were quantified in all cell types. These high-abundance proteins are mostly transporters or adhesion molecules, including integrin beta-1 (ITGB1), basigin (BSG), and neutral amino acid transporter (SLC1A5). No common low-abundance proteins were found in all cell types.
We then performed a hierarchical clustering analysis with the LFQ intensities to compare the surface glycoprotein expression between cell types. The heat map shows differential expression of proteins across the cell types (Figure 3F). ANOVA test was also performed and the expression of over half of the proteins were statistically different in at least one cell type (65% without missing value imputation and 71% with the imputation). Because of the intrinsic low abundance of cell-surface glycoproteins, it is expected that many values would not pass the cutoff for statistical significance, and thus, the percentages may be underestimated. The first, closest expression pattern from hierarchical clustering analysis arose from MCF7 and HEK293T while the second was from Jurkat and K562 cells. There are also groups of glycoproteins that are more abundant in a specific cell type than in the others. These glycoproteins are responsible for particular processes regarding the cell type. For example, those from K562 cells (in the yellow frame of Figure 3F) are involved in the processes such as neutrophil degranulation, while those from Jurkat include positive regulation of T cell activation. Interestingly, the estimated absolute abundances of these proteins span a wide range in that cell line, as shown in Figure 3E for K562 cells, but their relative abundances are higher than the corresponding ones in the other cell lines if expressed.
Overall, the current results demonstrate that the majority of commonly identified surface glycoproteins across the cell types usually have higher absolute abundance. Most of these proteins are necessary for normal cellular functions and cell survival. In biomedical research, cell lines are often chosen on the basis of specific protein expression on the surface that are appropriate for the experiment. We observed, however, that only a small portion of quantified surface glycoproteins from each cell type are cell-specific, and their absolute abundances are quite low. While we cannot disregard that these cell-specific proteins define the cell type, the difference among the cell types further arises from their differential expression. The cell-specific proteins cannot be excluded in other cell types. If the detection limit is lower, some of these proteins may be detected in another different cell type, but the relative abundance is still different. In this case, quantification information will be more meaningful. In addition, proteins responsible for specific functions of the cell type are expressed at any absolute abundance, but the relative abundances of these proteins are normally higher than in other cell types. It also illustrates that not only the expression but also the abundance of the protein of interest should be taken into consideration when choosing a cell type for a specific experiment.
Classification of Cell Types by Cluster of Differentiation.
As of December 2018, 451 cluster of differentiation (CD) molecules were listed on UniProt (https://www.uniprot.org/docs/cdlist), among which 396 are proteins. Here, we identified a total of 155 CD proteins with an average of 76 proteins per cell type. With LFQ intensity and the MBR option enabled, we quantified 148 CD proteins. Twenty-nine CDs were globally expressed in all cell lines, and most of these proteins function in the response to stimulus process. On average, 26 of the CD proteins were cell-specific, such as CD7 protein that was identified and quantified only in Jurkat cells.
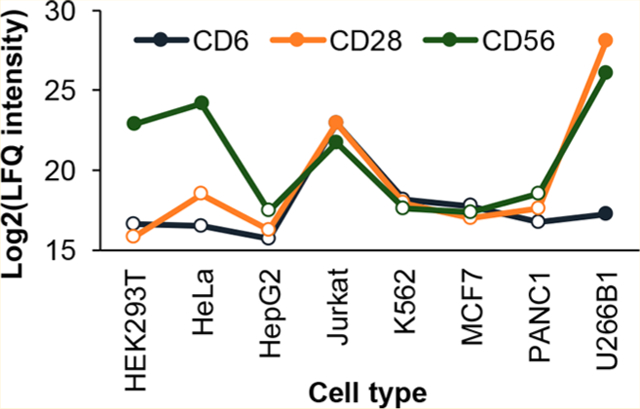
Similar to the surface glycoproteome results, we performed hierarchical clustering analysis and observed differential expression with specific groups of proteins being more abundant in specific cell types (Figure 4A). For example, proteins highlighted in the yellow box in the figure are highly expressed in Jurkat cells. These are proteins involved in specific T-cell processes, such as CD3D, CD5, and CD6. Despite the use of CDs as markers for a specific cell type, we noticed that some CD proteins can be expressed in other types of cells. For example, CD28 was detected in both Jurkat and U266B1 (Figure 4B), and the relative abundance of CD28 in U266B1 is even greater than that in Jurkat cells. Similarly, CD6 was also detected in both cell lines. The estimated absolute abundance of CD28 in both types of cells is high, and that of CD6 is high in Jurkat, but is low in U266B1 (Figure S4C). Another example is CD56, a phenotype marker for natural killer cells, which was also found in HEK293T, HeLa, Jurkat, and U266B1 at different expression levels (Figure 4B). Previous studies found that CD56 could be expressed in different cell types, including T cells, dendritic cells, and monocytes.57 There are also some published cases where a specific CD molecule was discovered in other different cell lines.58,59
Figure 4.

Cluster of differentiation (CD) proteins are differentially expressed in different cell types. (A) A heat map with hierarchical clustering showing relative expression of CD proteins. Missing values were imputed similar to that in Figure 3F. (B) Relative expression of CD6, CD28, and CD56. Missing values were imputed and are indicated by blank data points.
With the differential expression of CD proteins, the abundances of these CD molecules might need to be taken into consideration when using them to classify cell types. There is an effort to determine the expression of CD molecules through the CDmaps project.8 A combination of surface CD molecule identification with their abundances may help increase the accuracy of the classification considering that many of CD proteins are expressed with different abundances in different cell types.
CONCLUSIONS
Cells are normally covered with glycans, and almost all proteins on the surface are glycosylated. Surface glycoproteins are essential for cells and regulate nearly every extracellular event, and aberrant protein glycosylation on the cell surface is often related to human diseases. Compared with global analysis of proteins, comprehensive analysis of surface glycoproteins is much more understudied despite their importance. It is extraordinarily challenging to globally characterize surface glycoproteins because of the low abundance of many surface glycoproteins, the heterogeneity of glycans, and the requirement of selective separation of glycoproteins only located on the cell surface. In this work, we comprehensively analyzed cell-surface glycoproteins from eight types of commonly used human cells. The distribution and occurrence of N-glycosylation sites were systematically investigated, and it was found that protein secondary structures have a dramatic influence on N-glycosylation sites. Quantification results reveal that besides cell-specific surface proteins, the relative expression of surface glycoproteins also contributes to the uniqueness of each type. Our results suggested that it is better to consider multiple surface glycoproteins including their abundances for cell classification, rather than a single CD protein normally used in conventional methods. Global analysis of cell-surface glycoproteins facilitates a better understanding of protein glycosylation and cellular properties, and their quantitative analysis may lead to the identification of important surface glycoproteins as effective disease biomarkers and drug targets.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM118803.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.9b01447.
Methods for cell culture; metabolic labeling and click-chemistry reaction; protein extraction and purification; LC-MS/MS analysis and database searching; bioinformatic analysis; cell-surface glycoprotein interaction and pathway analyses; examples of cell-specific surface glycoproteins (Table S6); reproducibility of the identification (Figure S1); site-specific analysis (Figure S2); label-free quantification (Figure S3); protein interaction and pathway analyses (Figure S4); protein interaction and KEGG analyses (Figure S5 and S6) (PDF)
Identified glycosylation sites (Table S1) (XLSX)
Prediction of membrane proteins (Table S2) (XLSX)
Prediction of structure and solvent accessibility (Table S3) (XLSX)
Calculated entropy values and LFQ intensities of proteins (Table S4) (XLSX)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Stanley P; Schachter H; Taniguchi N N-Glycans. In Essentials of Glycobiology, 2nd ed.; Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2009. [Google Scholar]
- (2).Ekblom P; Vestweber D; Kemler R Annu. Rev. Cell Biol 1986, 2, 27–47. [DOI] [PubMed] [Google Scholar]
- (3).Gahmberg CG; Tolvanen M Trends Biochem. Sci 1996, 21(8), 308–311. [PubMed] [Google Scholar]
- (4).Rosenbaum DM; Rasmussen SG; Kobilka BK Nature 2009, 459 (7245), 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).le Coutre J; Kaback HK Biopolymers 2000, 55 (4), 297–307. [DOI] [PubMed] [Google Scholar]
- (6).Drake PM; Cho W; Li B; Prakobphol A; Johansen E; Anderson NL; Regnier FE; Gibson BW; Fisher SJ Clin. Chem 2010, 56 (2), 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kailemia MJ; Park D; Lebrilla CB Anal. Bioanal. Chem 2017, 409 (2), 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Engel P; Boumsell L; Balderas R; Bensussan A; Gattei V; Horejsi V; Jin BQ; Malavasi F; Mortari F; Schwartz-Albiez R; Stockinger H; van Zelm MC; Zola H; Clark GJ Immunol. 2015, 195 (10), 4555–4563. [DOI] [PubMed] [Google Scholar]
- (9).Kimiz-Gebologlu I; Gulce-Iz S; Biray-Avci C Mol. Biol. Rep 2018, 45 (6), 2935–2940. [DOI] [PubMed] [Google Scholar]
- (10).Craig FE; Foon KA Blood 2008, 111 (8), 3941–3967. [DOI] [PubMed] [Google Scholar]
- (11).Witze ES; Old WM; Resing KA; Ahn NG Nat. Methods 2007, 4 (10), 798–806. [DOI] [PubMed] [Google Scholar]
- (12).Zhu ZK; Desaire H Carbohydrates on proteins: site-specific glycosylation analysis by mass spectrometry In Annual Review of Analytical Chemistry; Cooks RG, Pemberton JE, Eds.; Annual Reviews: Palo Alto, 2015; Vol. 8, pp 463–483. [DOI] [PubMed] [Google Scholar]
- (13).Wu RH; Haas W; Dephoure N; Huttlin EL; Zhai B; Sowa ME; Gygi SP Nat. Methods 2011, 8 (8), 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yang Y; Liu F; Franc V; Halim LA; Schellekens H; Heck AJ R. Nat. Commun 2016, 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shi TJ; Fillmore TL; Sun XF; Zhao R; Schepmoes AA; Hossain M; Xie F; Wu S; Kim JS; Jones N; Moore RJ; Pasa-Tolic L; Kagan J; Rodland KD; Liu T; Tang KQ; Camp DG; Smith RD; Qian WJ Proc. Natl. Acad. Sci. U. S. A 2012, 109 (38), 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wu J; Xie XL; Liu YS; He JT; Benitez R; Buckanovich RJ; Lubman DM J. Proteome Res 2012, 11 (9), 4541–4552. [DOI] [PubMed] [Google Scholar]
- (17).Yu C; Mao HB; Novitsky EJ; Tang XB; Rychnovsky SD; Zheng N; Huang L Nat. Commun 2015, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zheng JN; Xiao HP; Wu RH Angew. Chem., Int. Ed 2017, 56 (25), 7107–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang XS; Yuan ZF; Fan J; Karch KR; Ball LE; Denu JM; Garcia BA Mol. Cell. Proteomics 2016, 15 (7), 2462–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Banazadeh A; Veillon L; Wooding KM; Zabetmoghaddam M; Mechref Y Electrophoresis 2017, 38 (1), 162–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bausch-Fluck D; Hofmann A; Bock T; Frei AP; Cerciello F; Jacobs A; Moest H; Omasits U; Gundry RL; Yoon C; Schiess R; Schmidt A; Mirkowska P; Hartlova A; Van Eyk JE; Bourquin JP; Aebersold R; Boheler KR; Zandstra P; Wollscheid B PLoS One 2015, 10 (3), No. e0121314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang H; Li XJ; Martin DB; Aebersold R Nat. Biotechnol 2003, 21 (6), 660–666. [DOI] [PubMed] [Google Scholar]
- (23).Rillahan CD; Paulson JC Glycan microarrays for decoding the glycome In Annual Reviews of Biochemistry; Kornberg, Raetz RD, Rothman CRH, Thorner JE, W. J, Eds.; Annual Reviews: Palo Alto, 2011; Vol. 80, pp 797–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zauner G; Deelder AM; Wuhrer M Electrophoresis 2011, 32 (24), 3456–3466. [DOI] [PubMed] [Google Scholar]
- (25).Wollscheid B; Bausch-Fluck D; Henderson C; O’Brien R; Bibel M; Schiess R; Aebersold R; Watts JD Nat. Biotechnol 2009, 27 (4), 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Suttapitugsakul S; Xiao H; Smeekens J; Wu R Mol. BioSyst 2017, 13 (12), 2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rappsilber J; Mann M; Ishihama Y Nat. Protoc 2007, 2 (8), 1896–1906. [DOI] [PubMed] [Google Scholar]
- (28).Cox J; Mann M Nat. Biotechnol 2008, 26 (12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- (29).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann MJ Proteome Res. 2011, 10 (4), 1794–1805. [DOI] [PubMed] [Google Scholar]
- (30).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J Nat. Methods 2016, 13 (9), 731–740. [DOI] [PubMed] [Google Scholar]
- (31).Kall L; Krogh A; Sonnhammer EL J. Mol. Biol 2004, 338(5), 1027–1036. [DOI] [PubMed] [Google Scholar]
- (32).Bendtsen JD; Jensen LJ; Blom N; Von Heijne G; Brunak S Protein Eng., Des. Sel 2004, 17 (4), 349–356. [DOI] [PubMed] [Google Scholar]
- (33).Petersen B; Petersen TN; Andersen P; Nielsen M; Lundegaard C BMC Struct. Biol 2009, 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Schwanhausser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Nature 2011, 473 (7347), 337–342. [DOI] [PubMed] [Google Scholar]
- (35).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villen J; Haas W; Sowa ME; Gygi SP Cell 2010, 143 (7), 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mahal LK; Yarema KJ; Bertozzi CR Science 1997, 276 (5315), 1125–1128. [DOI] [PubMed] [Google Scholar]
- (37).Chen WX; Smeekens JM; Wu RH Chem. Sci 2015, 6(8), 4681–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Xiao H; Tang GX; Wu R Anal. Chem 2016, 88 (6), 3324–3332. [DOI] [PubMed] [Google Scholar]
- (39).Baskin JM; Prescher JA; Laughlin ST; Agard NJ; Chang PV; Miller IA; Lo A; Codelli JA; Bertozzi CR Proc. Natl. Acad. Sci. U. S. A 2007, 104 (43), 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Xiao H; Chen W; Smeekens JM; Wu R Nat. Commun 2018, 9, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Deeb SJ; Cox J; Schmidt-Supprian M; Mann M Mol. Cell. Proteomics 2014, 13 (1), 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sun S; Shah P; Eshghi ST; Yang W; Trikannad N; Yang S; Chen L; Aiyetan P; Hoti N; Zhang Z; Chan DW; Zhang H Nat. Biotechnol 2016, 34 (1), 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Xiao H; Wu R Chem. Sci 2017, 8 (1), 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xiao HP; Suttapitugsakul S; Sun FX; Wu RH Acc. Chem. Res 2018, 51 (8), 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Spiciarich DR; Nolley R; Maund SL; Purcell SC; Herschel J; Iavarone AT; Peehl DM; Bertozzi CR Angew. Chem., Int. Ed 2017, 56 (31), 8992–8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Laughlin ST; Baskin JM; Amacher SL; Bertozzi CR Science 2008, 320 (5876), 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Xiong DC; Zhu JJ; Han MJ; Luo HX; Wang C; Yu Y; Ye YQ; Tai GH; Ye XS Org. Biomol. Chem 2015, 13 (13), 3911–3917. [DOI] [PubMed] [Google Scholar]
- (48).Feo S; Arcuri D; Piddini E; Passantino R; Giallongo A FEBS Lett. 2000, 473 (1), 47–52. [DOI] [PubMed] [Google Scholar]
- (49).Hsu LC; Liu S; Abedinpour F; Beech RD; Lahti JM; Kidd VJ; Greenspan JA; Yeung CY Mol. Cell. Biol 2003, 23(23), 8773–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zielinska DF; Gnad F; Wisniewski JR; Mann M Cell 2010, 141 (5), 897–907. [DOI] [PubMed] [Google Scholar]
- (51).Zajonc DM; Striegl H; Dascher CC; Wilson IA Proc. Natl. Acad. Sci. U. S. A 2008, 105 (46), 17925–17930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lam PV; Goldman R; Karagiannis K; Narsule T; Simonyan V; Soika V; Mazumder R Genomics, Proteomics Bioinf. 2013, 11 (2), 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chen X; VanValkenburgh C; Liang H; Fang H; Green NJ Biol. Chem 2001, 276 (4), 2411–2416. [DOI] [PubMed] [Google Scholar]
- (54).Walmsley AR; Hooper NM Biochem. J 2003, 370 (1), 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Nilsson I; von Heijne GJ Biol. Chem 2000, 275 (23), 17338–17343. [DOI] [PubMed] [Google Scholar]
- (56).Geiger T; Wehner A; Schaab C; Cox J; Mann M Mol. Cell. Proteomics 2012, 11 (3), M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Van Acker HH; Capsomidis A; Smits EL; Van Tendeloo VF Front. Immunol 2017, 8, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bernstein HB; Plasterer MC; Schiff SE; Kitchen CM; Kitchen S; Zack JA J. Immunol 2006, 177 (6), 3669–3676. [DOI] [PubMed] [Google Scholar]
- (59).Soma L; Wu D; Chen X; Edlefsen K; Fromm JR; Wood B Cytometry, Part B 2014, 88 (2), 145–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.