

Abstract
Functionally annotating genetic variations is an essential yet challenging topic in human genetics research. As large consortia including ENCODE and Roadmap Epigenomics Project continue to generate high-throughput transcriptomic and epigenomic data, many computational frameworks have been developed to integrate these experimental data to predict functionality of genetic variations in both protein-coding and noncoding regions. Here, we compare a number of recently developed annotation frameworks for noncoding regions through enrichment analysis on genome-wide association studies (GWASs). We also compare several different strategies to quantify enrichment using GWAS summary statistics. Our analyses highlight the importance of jointly modeling context-specific annotations with genome-wide data in providing statistically powerful and biologically interpretable enrichment for complex disease associations. Our findings provide insights into when and how computational genome annotations may benefit future complex disease studies on the genome-wide scale.
Keywords: noncoding genome annotation, genome-wide association study, enrichment analysis
Introduction
Facilitated by reliable, high-throughput microarray technology, genome-wide association studies (GWASs) have become a popular and relatively economical method to study complex disease genetics. Over the past decade, GWASs have greatly accelerated the processes of identifying trait-associated single-nucleotide polymorphisms (SNPs) [1], mapping causal gene candidates [2], and uncovering biological pathways involved in disease etiology [3–5]. To date, tens of thousands of SNP–trait associations have been identified and replicated for numerous complex diseases and traits. Despite the early and continuing success of GWAS, many issues remain unresolved. First, evidence has emerged that the genetic basis of complex traits consists of a large number of regulatory elements [5], while only a small proportion of heritability can be attributed to the statistically significant genetic loci identified by GWASs [3]. Second, the complex structure of linkage disequilibrium (LD) in the genome hinders us from distinguishing biologically functional risk variants from neutral SNPs in LD. In addition, the majority of identified susceptibility loci are located in noncoding regions in the human genome [6]. Without accurate noncoding genome annotation, it remains challenging to interpret GWAS associations.
In contrast to protein-coding genes, which are generally better annotated with information regarding transcriptional activity and protein structure, annotating the noncoding genome is largely limited by our lack of comprehensive understanding of the genome’s regulatory machinery and a lack of gold-standard training and validation data. However, overwhelming evidence has suggested a critical role of noncoding DNA elements in complex disease etiology [7]. Consequently, there is pressing need for accurate annotation of noncoding genome functionality. As publicly accessible, high-throughput transcriptomic and epigenomic annotation data become available [8, 9], numerous statistical and computational methods have been developed to characterize noncoding genome functionality through integration of annotation data. These frameworks delineate the functional potential of DNA variants with different levels of specificity. Most existing methods produce a single score to quantify the general functionality for each DNA variation [10–13]. As data on various cell types are made available, metrics that summarize variants’ functional potential on the single tissue and cell-type level have emerged [14, 15]. In addition to different levels of specificity, these frameworks also vary in their input data, modeling techniques and the interpretation of the output metrics. Commonly used annotations include molecular data that pinpoint various transcriptional regulations and evolutionary conservation metrics measured by aligning different species’ DNA sequences [16]. Most frameworks exploit annotations from multiple categories yet with different focuses. The models also vary in their supervised or unsupervised nature. Supervised-learning models’ performance and generalizability depend heavily on the quality of the labeled training data [10, 11]. Unsupervised-learning models, on the other hand, are more difficult to train [12–15]. This is because they generally require a wider range of informative features for the learning to perform the classification tasks. Finally, the interpretation of output metrics varies across different methods and so far there is no gold standard in assessing the quality and utility of these annotation tools. Although some of these computational genome annotations can potentially be evaluated and compared through functional genomics experiments, such work is in many cases low-throughput, undesirably costly and labor-intensive.
Integration of GWAS results with functional genome annotations has achieved some success and provided biological insights into the genetic basis of disease etiology. Many statistical and computational methods that explicitly model functional annotations have been proposed and implemented to address challenging problems in human genetics research, including functional variant fine-mapping [17–21], estimating annotation-stratified trait heritability [22, 23], pleiotropy analysis [24] and genetic risk prediction [25–27]. However, there is no consensus among researchers regarding which annotation to use under different contexts. In this article, we compare different annotation frameworks through enrichment analysis using summary statistics from 15 GWASs. We evaluate the ability of these annotations in providing consistent and significant heritability enrichment across complex diseases and traits. In addition, we examine whether the heritability enrichments are biologically interpretable to biologists and disease specialists by investigating the consistency between the patterns of heritability enrichment with what was reported in the existing literature. Our analyses provide insights into the specific context where the annotations may be applied to benefit GWAS-based analyses, and provide guidance for designing integrative models in future studies.
Methods
GWAS data sets
We downloaded summary statistics for 15 GWAS traits, including Crohn’s disease (CD), multiple sclerosis (MS), rheumatoid arthritis (RA), bipolar disorder (BPD), schizophrenia (SCZ), Alzheimer’s disease (ALZ), body mass index (BMI), height (HT), waist–hip ratio adjusted for BMI (WHR), high- and low-density lipoprotein cholesterol (HDL and LDL), systolic blood pressure (SBP), coronary artery disease (CAD), cognitive performance (CP) and education attainment (EDU). All summary statistics are publicly accessible (URLs provided in Table 1). These data sets represent a spectrum of immune, psychiatric, neurological, anthropometric, cardiovascular and metabolic traits. In addition, it has been shown that GWAS sample size is critical in enrichment analysis using LD score regression [15, 23]. Thus, we selected studies with relatively large sample sizes, so that we could have meaningful comparisons of different annotation tools and analysis strategies.
Table 1.
List of 15 complex diseases and traits
Functional annotations
We considered seven recently developed computational annotation methods with precalculated scores readily available (Table 2). Leveraging various types of annotation data, methods such as CADD, GWAVA, GenoCanyon and EIGEN can predict the general functionality of genetic variations in the human genome [10–13]. CADD and GWAVA incorporated a supervised-learning approach. CADD was implemented as a support vector machine classifier that integrates conservation metrics, regulatory information and transcript information as predictive features. GWAVA is based on a modified random forests algorithm. It provides three different versions of prediction scores, corresponding to three different control data sets in the training process: a random selection of SNPs from across the genome (i.e. GWAVA-unmatched), a collection of SNPs matched for distance to the nearest transcription start site (TSS) with the disease-implicated variants (i.e. GWAVA-TSS) and a collection of all 1000 Genomes variants in the 1 kb surrounding each of the disease-implicated variants (i.e. GWAVA-region). We compared all three sets of scores in our analysis. The performance of supervised-learning models is subject to the quality and the abundance of the labeled training data. In contrast, GenoCanyon and EIGEN, two recent methods based on unsupervised-learning methods, are less affected by the limitations of training data. Both tools can produce integrative metrics measuring the functional potential of genetic variations from a collection of conservation scores and epigenomic annotations. We compared the distribution of different annotation scores. Spearman’s correlation was used to quantify similarities between annotations. We also studied the overlap between dichotomized annotations under different quantile cutoffs.
Table 2.
List of computational genome annotations
| Annotation | Version | Year | Reference | URL |
|---|---|---|---|---|
| CADD | 1.3 | 2014 | Kircher et al. [10] | http://cadd.gs.washington.edu |
| GWAVA | 1.0 | 2014 | Ritchie et al. [11] | https://www.sanger.ac.uk/sanger/StatGen_Gwava |
| GenoCanyon | 1.0.3 | 2015 | Lu et al. [12] | http://genocanyon.med.yale.edu |
| EIGEN | 1.0 | 2015 | Ionita-Laza et al. [13] | http://www.columbia.edu/∼ii2135/eigen.html |
| fitCons | 1.01 | 2015 | Gulko et al. [42] | http://compgen.cshl.edu/fitCons/ |
| GenoSkyline | 1.0.1 | 2016 | Lu et al. [14] | http://genocanyon.med.yale.edu/GenoSkyline |
| GenoSkyline-Plus | 1.0.0 | 2017 | Lu et al. [15] | http://genocanyon.med.yale.edu/GenoSkyline |
In addition to these annotation frameworks that predict the general functional potential of genetic variants, context-specific functional annotations have been developed to exploit the rich epigenomic and transcriptomic data from diverse tissue and cell types. GenoSkyline is able to predict functional regions for seven broadly defined tissues, namely, brain, gastrointestinal tract, lung, heart, blood, muscle and epithelium [14]. With further improved specificity, GenoSkyline-Plus predicts context-specific functionality to 127 tissue and cell types assayed in the Roadmap Epigenomics Project [15]. Another tool, fitCons, defines the functional potential for point mutations from an evolutionary perspective. It measures the potential of genetic variations in influencing the fitness by contrasting the patterns of polymorphism and quantifying the divergence of genomic sites with nearby neutral sites [42]. fitCons provides precalculated scores for three ENCODE cell lines, i.e. human umbilical vein epithelial cells (HUVECs), H1 human embryonic stem cells (H1 hESCs) and lymphoblastoid cells (GM12878). An integrated score that combines information from all three cell types is also provided. Context-specific annotations may not only increase the statistical power to prioritize risk variants from GWAS associations but also shed light on the biological interpretation of GWAS findings. To comparatively evaluate the performance of the integrative functional annotations, we also calculated enrichment solely based on H3K4me3, a well-studied histone mark highly enriched near active promoters and TSSs [43].
Precomputed scores for all these annotations are publicly available (Table 2). We extracted annotation scores for all SNPs in the 15 GWASs using hg19 coordinates. Scores saved in BED format are matched to variant coordinates using tabix [44], and scores saved in bigWig format are extracted using bigWig utilities [45].
Enrichment calculation
To make enrichment results comparable across different annotations, we dichotomized each annotation metric using the upper 30% quantile as the cutoff. We also performed a sensitivity analysis and tested the robustness of enrichment results to different cutoffs (Supplementary Figure S1). Quantiles were calculated based on SNPs with minor allele count >5 in samples with European ancestry in the 1000 Genomes Project as described in [23]. Genetic variations with annotation scores exceeding the cutoff were labeled as ‘functional’ in general or specific to a cell type, depending on the annotation method, whereas the rest were labeled as ‘non-functional’. We calculated enrichment using heritability estimated from GWAS summary statistics using annotation-stratified LD score regression [23] (implemented software available at https://github.com/bulik/ldsc/). Finucane et al. provided a set of 52 baseline annotations including several histone marks, chromatin states, promoter and enhancer regions and conservation metrics (Supplementary Table S1). We followed the recommended protocol of LDSC and always included these baseline annotations in the model to achieve better model fitting. Then, we added one integrative annotation at a time and studied the heritability enrichment in each annotation category. Enrichment was defined as the ratio of the proportion of heritability explained by the annotated SNPs and the proportion of SNPs covered by that annotation:
GenoSkyline-Plus predicts the functionality of genetic variations in 127 tissue and cell types. Genetic variants predicted to be functional in one cell type may also be functional in other related cell types. Given consideration to the overlap among these annotation tracks, we compared two analysis strategies: (1) incorporate 127 cell-type-specific annotations one track at a time along with the 52 baseline annotations in the model; and (2) incorporate all 127 context-specific annotation tracks and baseline annotations in the model. In the full joint model with the complete set of annotations, we studied the tissue-specific enrichment controlling for baseline annotations as well as functional regions in all other tissues.
Results
General functional annotations
Spearman’s correlations between general functional annotations are shown in Figure 1. All annotations were positively correlated. GWAVA-unmatched and GWAVA-TSS (), CADD and EIGEN () and GWAVA-unmatched and GenoCanyon () had stronger correlations than other pairs, whereas GenoCanyon and GWAVA-region had the weakest correlation ().
Figure 1.
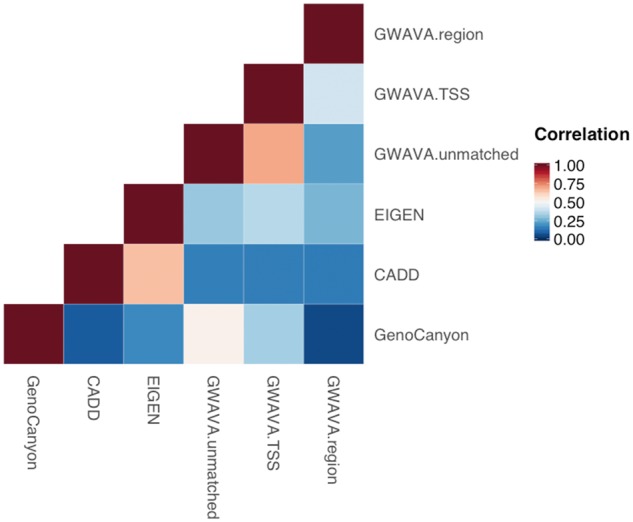
Spearman’s correlations between functional annotations.
Using each annotation’s upper 30% quantile as a cutoff, regions annotated as functional by a pair of dichotomized annotations covered 10–20% of the genome (Supplementary Table S2). The odds ratios (ORs) of proportions of concordant classification and inconsistent classification between each pair of dichotomized annotations ranged from 1.34 to 11.59. Consistent with rank correlations, more concordant classifications were seen between dichotomized GWAVA-unmatched and GWAVA-TSS (OR = 11.59), CADD and EIGEN (OR = 8.35) and GWAVA-unmatched and GenoCanyon (OR = 5.01). The least concordant dichotomized annotations were GenoCanyon and GWAVA-region (OR = 1.34).
In addition to the descriptive exploration of similarities between annotations, we evaluated the disease heritability enrichments for different annotations using GWAS summary statistics. We first tested the robustness of enrichment results to different choices of dichotomization cutoffs. Supplementary Figure S1 suggests that for different annotations, the enrichment significance patterns change by cutoffs. GenoCanyon showed more significant enrichments for lower quantile cutoffs across traits. CADD and EIGEN, in contrast, showed stronger evidence of enrichments for higher quantile cutoffs. GWAVA annotations showed no systematic pattern for different dichotomization cutoffs. For example, within anthropometric traits, GWAVA-unmatched had more significant enrichments for higher quantile cutoffs in BMI, contrary pattern in HT, and showed the most significant enrichment when using the upper 30% quantile as the cutoff in WHR. Our findings suggest that the optimal cutoff may differ by the genetic basis of traits and diseases. One may use different quantile cutoffs for single-trait studies; however, one consistent dichotomization cutoff is needed for comparison across various annotations. Notice that (1) despite that the strength of the enrichment evidence varied by dichotomization cutoffs, the significantly enriched annotations were relatively robust to different cutoff choices, and (2) among all cutoffs, the upper 30% quantile cutoff preserved most of the significant enrichments; we thus selected the upper 30% quantile as the dichotomization cutoff to make further detailed comparisons.
Different annotations showed distinct patterns of enrichment for disease heritability (Figure 2). GenoCanyon showed significant heritability enrichments in all traits and diseases. GWAVA annotations were frequently significantly enriched across traits. In particular, GWAVA-unmatched was significantly enriched for ALZ and HDL. Heritability enrichments in other traits were comparable among three GWAVA tracks. EIGEN and CADD, on the other hand, showed significant enrichments in considerably fewer traits. EIGEN was consistently significantly enriched in psychiatric disorders (BPD and SCZ), anthropometric traits (BMI, HT and WHR), cardiovascular traits (SBP and CAD) and social traits (CP and EDU), but was lack of performance in immune diseases and metabolic traits. CADD was typically significantly enriched for traits that were highly polygenic (e.g. HT and EDU) but showed no significant result in any of the immune diseases, neuropsychiatric disorders and metabolic traits.
Figure 2.

Heritability enrichments for general annotations (GenoCanyon, EIGEN, CADD and GWAVA). Asterisks indicate significant enrichment after Benjamini–Hochberg correction with false discovery rate controlled at 0.01.
Context-specific functional annotations
GenoSkyline was powerful to identify tissues that were relevant to a complex trait or disease. In addition, the top enriched GenoSkyline annotation tracks often showed more significant enrichments than general functional annotations (Figure 3). For example, GenoSkyline-blood was strongly associated with immune diseases (CD , MS and RA ) as well as neurodegenerative disease (ALZ ) in trait heritability. GenoSkyline-brain is the most significantly enriched annotation for heritability of psychiatric disorders (BPD and SCZ ), BMI () and social traits (CP and EDU ). Another approach, fitCons, provided insight into the association between traits and three specific cell types. Overall, all fitCons tracks were significantly enriched for SCZ, anthropometric traits (BMI, HT and WHR) and CAD. In particular, fitCons-GM12878 was highly enriched for immune diseases, neuropsychiatric disorders and cognitive social traits; fitCons-H1hESC was also significantly associated with neuropsychiatric disorders and cognitive social traits but showed no significant enrichment in immune diseases, and fitCons-HUVEC was consistently enriched for immune diseases and metabolic/cardiovascular traits. The fitCons-multicell integrated information from all three cell types. However, fitCons-multicell did not show apparent superior performance than its context-specific annotations.
Figure 3.

Heritability enrichments for context-specific annotations (GenoSkyline and fitCons). Asterisks indicate significant enrichment after Benjamini–Hochberg correction with false discovery rate controlled at 0.01.
Functional annotation for 127 tissue and cell types
For functional annotations with even higher specificity, given that annotation tracks may overlap, we compared the functionally relevant cell types identified from the marginal model and the joint model (see ‘Methods’ section). We then contrasted the enrichment patterns of GenoSkyline-Plus annotations and a single histone mark H3K4me3.
Using annotation-stratified LD score regression, we first estimated the marginal association between a trait and each annotation track adjusting for 52 baseline annotations. The marginal model effectively identified a collection of relevant tissues and cell types for each trait (Figure 4; Supplementary Figure S2). Furthermore, jointly modeling all 127 cell type-specific and 52 baseline annotations allows us to control for the potential confounding effect among annotations and thus was superior in pinpointing specific cell types that were significantly enriched for the trait. The heritability enrichments partitioned by tissue and cell types are generally consistent with what has been reported in the literature [15, 17, 23]. Take the enrichment pattern of GenoSkyline-Plus annotations in CD as an example. In the marginal model, almost all blood cells (T cells, B cells and hematopoietic stem cells) were significantly enriched for CD, suggesting that blood is a critically related tissue for CD, a fact that has been extensively reported [28], [46]. However, these results could not highlight any specific T cells or B cells for CD, thus providing limited biological insights. In the joint model, four cell tracks, namely, primary T helper 17 cells PMA-I stimulated, primary monocytes from peripheral blood, primary natural killer cells from peripheral blood and monocytes-CD14+ RO01746 primary cells stood out from other immune-related cell types and were highly enriched for CD. Conditional on functionality in all annotation tracks, the joint model based on GWAS summary statistics provided a neater enrichment pattern and hinted at specific immune cell types that can be potentially validated in follow-up experiments. In addition to investigating cell types highly enriched for each single trait, we also looked into cell types that are significantly enriched across traits. For example, brain anterior caudate was highlighted for SCZ, BPD, CP and EDU, implying that psychiatric disorders and cognitive traits may share a genetic component in the trait development and acquisition.
Figure 4.

Comparison of enrichment patterns of GenoSkyline-Plus annotations based on different analytical strategies. We present only blood-, brain- and heart-related annotations for clarity. The enrichment figure of complete annotations and all 15 traits can be found in Supplementary Figure S2. The left panel is based on a marginal model, and the right panel is based on a full joint model with complete set of annotations. Grouping of annotation tracks was based on the order of tissue and cell types in the Roadmap Epigenomics Project. Asterisks indicate significant enrichment after Benjamini–Hochberg correction with false discovery rate controlled at 0.01.
In comparison with integrative cell-type-specific annotations generated by GenoSkyline-Plus, we also applied a joint model to the raw ChIP-seq peaks for histone mark H3K4me1. Single histone mark annotations were able to relate immune diseases to blood cells and SCZ to brain cells (Figure 5). However, we observed substantially fewer significantly enriched tracks based on only one histone mark (Supplementary Figure S2 right panel; Supplementary Figure S3). For example, no significant enrichment in heart was identified for SBP in single histone mark tracks, whereas four GenoSkyline-Plus tracks relating to heart, namely, fetal heart, right atrium, left ventricle and aorta, were significantly enriched for SBP. These results highlight the benefits of integrating various types of epigenomic and transcriptomic information to improving the statistical power in enrichment analysis.
Figure 5.

Heritability enrichment in H3K4me1 ChIP-seq peaks based on a joint model using complete set of annotations. We present only blood-, brain- and heart-related annotations for clarity. The enrichment figure of complete annotations and all 15 traits can be found in Supplementary Figure S3. Asterisks indicate significant enrichment after Benjamini–Hochberg correction with the false discovery rate controlled at 0.01.
Discussion
Emerging evidence suggests a crucial role of regulatory elements in the genetic basis and etiology of complex traits and diseases [4, 5]. Across all 15 studies considered, over 90% of significant variants are located in the regulatory regions (Supplementary Table S1). To better understand GWAS associations located in the regulatory regions, there is a great need to develop and exploit integrative noncoding functional annotations. Accurate functional annotation of DNA variations in the noncoding genome can provide insights into the genetic architecture of complex human traits. Through exploratory analysis of both continuous and dichotomized annotations, we demonstrated that GWAVA-unmatched and GWAVA-TSS, CADD and EIGEN, and GWAVA-unmatched and GenoCanyon were more similar than other pairs of annotations. In addition, we evaluated different annotations through comprehensive enrichment analysis on 15 complex traits. For supervised-learning approaches, GWAVA presented more significant enrichments than CADD in trait heritability, particularly in psychiatric disorders and metabolic traits. Comparing the three sets of GWAVA scores trained using different benign variants, enrichment patterns were comparable among three tracks, while GWAVA-unmatched was superior in providing unique significant heritability enrichments in ALZ and HDL. These results reveal that the choice of training data set may have considerable impacts on the performance of supervised-learning methods. For unsupervised approaches, GenoCanyon showed more significant enrichments than EIGEN across different data sets, which suggested its better performance in GWAS-related applications. As an alternative approach to evaluate functional annotations through estimating disease heritability enrichment, we may also calculate the enrichment solely based on GWAS top hits. The idea was to evaluate how well functional annotations capture and overlap with GWAS top hits. For illustration, we investigated how annotation tools with different levels of specificity annotated the BIN1 region, a major risk locus for ALZ. Supplementary Figure S4 demonstrates that context-specific annotations better align with top significant SNP–trait associations and thus are better than general annotations in prioritizing GWAS signals. However, the LD structure at identified risk loci may impede us from providing valid estimation and biological meaningful interpretations for the enrichments. The significant evidence might be spurious if the functional annotations happen to overlap with top hits in strong LD [47]. This situation is almost inevitable and there is no effective and straightforward way of accounting LD in this context. In comparison, estimating heritability enrichment using complete GWAS summary statistics through LD score regression exploits more information from the GWAS data and incorporates the LD structure into the model, and thus is superior in providing more accurate and interpretable enrichment estimations.
As high-throughput annotation data continue to expand, tissue-specific annotations have been developed to take full advantage of the abundant information. Tissue-specific annotations surpassed general functional annotations in two ways. First, they established the connection between tissues and complex traits, and therefore added to our knowledge about the biological mechanism for the traits. Second, enrichment in biologically relevant tissues was more significant than the enrichment in general, nonspecific annotations, showing improved statistical power. GenoSkyline annotations identified known functionally relevant tissue types for complex traits. Brain was the most enriched tissue for SCZ, BPD, BMI, CP and EDU, while blood was significantly enriched for immune diseases such as CD, MS and RA. Despite context-specific, fitCons scores were only available for three cell lines. Whether it can also provide similar insights about complex traits remains to be investigated using data from primary human tissues. With even higher specificity, GenoSkyline-Plus provides functional predictions for 127 tissue and cell types. Regarding different analytical strategies, jointly modeling all context-specific and baseline annotation tracks was preferred in the sense that it reduced confounding effects because of the substantial overlap among annotations and thus efficiently pinpointed tissue and cell types most relevant to the traits. For example, almost all blood cells were significantly enriched for immune diseases (CD, MS and RA) in the marginal analysis, making the results difficult to interpret. The joint analysis, in contrast, highlighted three cell types, namely, primary T helper cells PMA-I stimulated, primary monocytes from peripheral blood and primary natural killer cells from peripheral blood for immune diseases, providing biological insights and guidance to the follow-up validations. Comparing integrative annotations with raw ChIP-seq peaks, GenoSkyline-Plus identified considerably more significantly enriched tracks than that based on H3K4me3 only. No significant enrichment in heart was identified for SBP in single histone mark tracks, whereas four heart-related tracks in GenoSkyline-Plus annotations, namely, fetal heart, right atrium, left ventricle and aorta, were significantly enriched for SBP. In summary, the computational algorithms, modeling techniques and the composition of various input data jointly contribute to the comprehensive performance of functional annotations in heritability enrichment analysis. Our results suggest that jointly modeling various types of annotation data, including histone marks, DNase-I hypersensitive sites and transcription activity, increases power in enrichment analysis while maintaining the biological interpretability.
It is worth noting that unremitting efforts have been made to predict genome functionality through integrating epigenomic and transcriptomic data, and innovative methods continue to emerge. Chen et al. [48] recently developed DIVAN, a novel approach that predicts genetic variations’ disease-specific functionality. Lee et al. [49] developed deltaSVM that estimates the functional effect of DNA variations in a given regulatory element active in cell-type-dependent activities. An almost concurrent model DeepSEA, proposed by Zhou et al. [50] leverages information on sequence context and predicts chromatin effects of genomic variations. The most recent method LINSIGHT, developed by Huang et al. [51], adopted an evolutionary model and predicts deleterious noncoding variations from functional genomic data and conservation scores. These methods focus on different aspects of noncoding genome functionality and have different levels of cell type specificity. Along with the annotations we discussed in our analysis, many of them may be integrated into GWAS downstream analysis and benefit complex disease research in the future.
We evaluated state-of-the-art noncoding genome annotations through heritability enrichment analysis using GWAS summary statistics for 15 complex diseases and traits. We exploited several different strategies to quantify heritability enrichments and compared annotations with difference level of specificity. However, our study has several limitations. First, analyzing GWAS summary statistics as opposed to using individual-level data may lead to information loss. However, in our analysis, we applied LD score regression to estimate heritability enrichment. This method has been shown to provide comparable heritability estimated with individual-level data when GWAS sample size is large [23]. Therefore, when summary statistics are available in large sample populations, the benefits may outweigh limitations arising from the inaccessibility of individual-level data [52]. Second, our analysis focused on common SNPs in GWAS. It is possible that some functional annotations and analysis strategies are more suitable for the analysis of rare variants or structural variations. Those questions will need to be further investigated using sequencing data in the future. Third, we used enrichment of trait heritability to assess the effectiveness of functional annotations. This strategy cannot be used to study disease-specific functional annotations, e.g. DIVAN [48], as GWAS information may have been embedded in the model training. Nevertheless, our comparative analysis covers a large collection of recently proposed noncoding functional annotations and provides insights into the selection of annotations and analysis strategies in future studies. Finally, we note that no statistical method could be guaranteed to work well on all GWASs, and neither does our analysis. This could be because of many reasons, including sample size, genetic architecture and data quality issues. We note, however, as GWAS summary statistics become increasingly accessible, it is now possible to apply a method to a large number of independent GWAS data sets to demonstrate the consistent performance. In the past few years, this has become a popular approach to justify the effectiveness of a method [17, 53–55]. In this article, we analyzed a variety of complex traits from 15 GWASs, so that consistently superior performance of certain annotations provided convincing evidence to our conclusion.
Taken together, overwhelming evidence has shown that context-specific information obtained from high-throughput genomic experiments and comparative sequence analysis is beneficial for genetic studies. It not only improves the prediction accuracy of context-specific functional regions in the genome but also provides biological insights into the etiology of complex diseases and traits. With the context-specific annotations, different tissues and cell types are highlighted for different traits, which will add to our knowledge about these traits. With more comprehensive epigenomic and transcriptomic data being generated and made publicly available, the breadth and resolution of the annotation will be improved, and the integrative analyses of these genomic data will lead to more accurate and informative prediction of the context-specific functionality of genetic variations, reliable and applicable to more complex diseases and traits.
Supplementary Material
Acknowledgements
The authors thank all consortia and investigators for providing publicly accessible GWAS summary statistics. The authors also thank Yiming Hu for discussions and helpful comments regarding this work.
Funding
The National Institutes of Health (grant number R01 GM59507); the VA Cooperative Studies Program of the Department of Veterans Affairs, Office of Research and Development; and the Yale World Scholars Program sponsored by the China Scholarship Council.
Biographies
Boyang Li is a doctoral student in the Department of Biostatistics, Yale School of Public Health.
Qiongshi Lu is an Assistant Professor in the Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison.
Hongyu Zhao is Ira V. Hiscock Professor of Biostatistics, Professor of Genetics, and Professor of Statistics and Data Science at Yale University.
Key Points
Among general functional annotations, GWAVA-unmatched and GWAVA-TSS, CADD and EIGEN, and GWAVA-unmatched and GenoCanyon are more similar than other pairs of annotations.
The enrichment patterns of annotations differ by different analytical strategies and across traits.
Among general annotations, GWAVA shows considerably more significant enrichments than CADD in supervised approaches, and GenoCanyon presents more significant enrichments than EIGEN in unsupervised approaches. Comparing GWAVA tracks, GWAVA-unmatched is preferred in providing unique significant enrichments in ALZ and HDL.
Jointly modeling all context-specific and baseline annotation tracks is preferable to reduce confounding effects because of substantial overlap among annotations and highlight tissue and cell types most relevant to the traits.
Compared with general functional and single histone mark annotations, context-specific annotations can identify considerably more significantly enriched and biologically interpretable tissue and cell types to complex traits.
References
- 1. Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 2014;42:D1001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016;48:481–7. [DOI] [PubMed] [Google Scholar]
- 3. Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet 2012;90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boyle EA, Li YI, Pritchard JK.. An expanded view of complex traits: from polygenic to omnigenic. Cell 2017;169:1177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009;106:9362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ritchie GR, Dunham I, Zeggini E, et al. Functional annotation of noncoding sequence variants. Nat Methods 2014;11:294–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu Q, Hu Y, Sun J, et al. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci Rep 2015;5:10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ionita-Laza I, McCallum K, Xu B, et al. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet 2016;48:214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu Q, Powles RL, Wang Q, et al. Integrative tissue-specific functional annotations in the human genome provide novel insights on many complex traits and improve signal prioritization in genome-wide association studies. PLoS Genet 2016;12:e1005947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu Q, Powles RL, Abdallah S, et al. Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. PLoS Genet 2017;13:e1006933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kellis M, Wold B, Snyder MP, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci USA 2014;111:6131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet 2014;94:559–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kichaev G, Yang WY, Lindstrom S, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet 2014;10:e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kichaev G, Pasaniuc B.. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am J Hum Genet 2015;97:260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu Q, Yao X, Hu Y, et al. GenoWAP: GWAS signal prioritization through integrated analysis of genomic functional annotation. Bioinformatics 2016;32:542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y, Kellis M.. Joint Bayesian inference of risk variants and tissue-specific epigenomic enrichments across multiple complex human diseases. Nucleic Acids Res 2016;44:e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gusev A, Lee SH, Trynka G, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 2014;95:535–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Finucane HK, Bulik-Sullivan B, Gusev A, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 2015;47:1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chung D, Yang C, Li C, et al. GPA: a statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLoS Genet 2014;10:e1004787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Speed D, Balding DJ.. MultiBLUP: improved SNP-based prediction for complex traits. Genome Res 2014;24:1550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu Y, Lu Q, Powles R, et al. Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput Biol 2017;13:e1005589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang H, Wheeler W, Hyland PL, et al. A powerful procedure for pathway-based meta-analysis using summary statistics identifies 43 pathways associated with type II diabetes in European populations. PLoS Genet 2016;12:e1006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010;42:1118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium, Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011;43:977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014;46:1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shungin D, Winkler TW, Croteau-Chonka DC, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015;518:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ehret GB, Ferreira T, Chasman DI, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet 2016;48:1171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rietveld CA, Esko T, Davies G, et al. Common genetic variants associated with cognitive performance identified using the proxy-phenotype method. Proc Natl Acad Sci USA 2014;111:13790–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okbay A, Beauchamp JP, Fontana MA, et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 2016;533:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gulko B, Hubisz MJ, Gronau I, et al. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genet 2015;47:276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guenther MG, Levine SS, Boyer LA, et al. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007;130:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 2011;27:718–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kent WJ, Zweig AS, Barber G, et al. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 2010;26:2204–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khor B, Gardet A, Xavier RJ.. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011;474:307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang K, Dickson SP, Stolle CA, et al. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet 2010;86:730–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen L, Jin P, Qin ZS.. DIVAN: accurate identification of non-coding disease-specific risk variants using multi-omics profiles. Genome Biol 2016;17:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee D, Gorkin DU, Baker M, et al. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet 2015;47:955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou J, Troyanskaya OG.. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015;12:931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang YF, Gulko B, Siepel A.. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat Genet 2017;49:618–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pasaniuc B, Price AL.. Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet 2017;18:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pickrell JK, Berisa T, Liu JZ, et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 2016;48:709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi H, Kichaev G, Pasaniuc B.. Contrasting the genetic architecture of 30 complex traits from summary association data. Am J Hum Genet 2016;99:139–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mancuso N, Shi H, Goddard P, et al. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am J Hum Genet 2017;100:473–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.