

Heart failure (HF) is a disease of epidemic portions in the United States affecting over 6 million people. In slightly over one‐half of affected individuals, function of the heart is reduced, as demonstrated by a decrease in ejection fraction (heart failure with reduced ejection fraction; HFrEF), and the left ventricle is dilated. New drugs that target pathways critical to progression of HF, along with implantable cardiac defibrillators and resynchronization devices, have been introduced over the past 3 decades. However, both the morbidity and mortality associated with HFrEF remains at unacceptable levels, with as many as 50% of affected individuals dying within 5 years of diagnosis. This has led investigators to evaluate the role of gene therapy in mitigating or curing HFrEF by increasing the amount of a specific protein in the heart.
The concept that a noninfectious viral vector could carry a gene of interest into a cell in the cardiovascular system was first demonstrated almost 2 decades ago by 2 laboratories in the United States. Betsy and Gary Nabel at the University of Michigan showed that retroviral vectors could transfer DNA into the arterial wall,1 whereas Jeffrey Isner at St. Elizabeth's Medical Center in Boston used a plasmid containing the human vascular endothelial growth factor gene applied to the hydrogel polymer coating an angioplasty balloon to achieve the same result.2, 3 More recently, investigators have tested the ability of gene therapy to change the cardiac phenotype of both animal models and patients with left ventricular (LV) dysfunction. In this review, we will briefly discuss contemporary methods for gene therapy and then focus on the specific cardiac proteins that are currently being evaluated as therapeutic targets, including: adenylyl cyclase (AC) 6 (AC6), S100A1, β‐adrenergic receptor kinase‐ct (βARKct), sarco/endoplasmic reticulum (SR) Ca2+‐ATPase (SERCA2a), urocortins, and B‐cell lymphoma 2 (Bcl2)‐associated anthanogene‐3 (BAG3; Figure).
Figure 1.
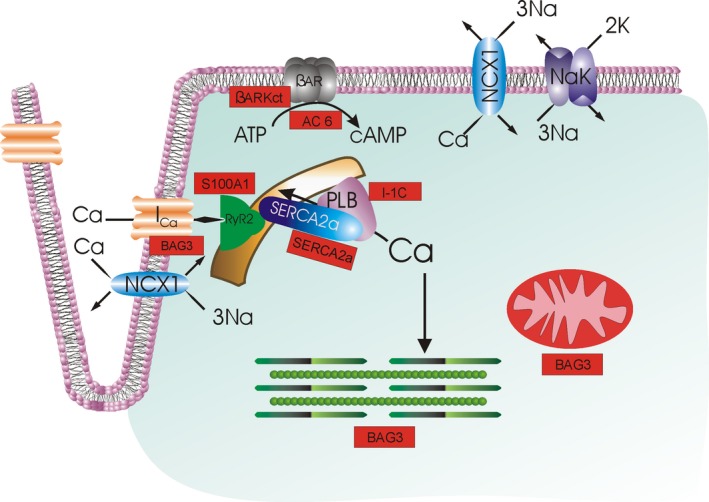
Current heart failure gene therapy approaches targeted to cardiac excitation‐contraction coupling. With depolarization, extracellular Ca2+ enters by L‐type Ca2+ channels (IC a), triggering Ca2+ release from the ryanodine receptor (RyR2) in the sarcoplasmic reticulum (SR). Ca2+ in the sarcoplasm binds to troponin to initiate contraction. During diastole, Ca2+ is resequestered in the SR by SR Ca2+‐ATPase (SERCA2a), whose activity is regulated by phospholamban (PLB). The amount of Ca2+ that has entered during systole is largely extruded by Na+/Ca2+ exchanger (NCX1; utilizing the electrochemical gradient established by Na+‐K+‐ATPase NaK) and, to a much smaller extent, the sarcolemmal Ca2+‐ATPase (not shown). When β‐adrenergic receptor (βAR) is stimulated, cAMP is generated, which activates protein kinase A (PKA), which, in turn, increases IC a and RyR2 activities and Ca2+ sensitivity of myofilaments, thereby enhancing contractility. PKA also phosphorylates PLB, thereby relieving its inhibition on SERCA2a, resulting in enhanced SR Ca2+ uptake, which improves both contraction (larger SR Ca2+ content leading to larger intracellular Ca2+ transients) and relaxation (faster SR Ca2+ sequestration during diastole). Current gene therapy products (shown in rectangular red boxes) target βAR (AC6, βARKct), SR Ca2+ uptake (SERCA2a), PLB (I‐1c), and Ca2+ cycling by RyR2 and SERCA2a (S100A1). BAG3 has multiple downstream effectors, including IC a, myofilaments, and mitochondria; not shown are autophagy, nuclear envelope integrity, and cell‐to‐cell communication (connexin43), also positively regulated by BAG3. Urocortins effect mainly vasodilation and are not shown here. SERCA2a indicates sarcoplasmic/endoplasmic reticulum calcium ATPase 2a.
Gene Transduction of the Heart
Both viral‐ and no‐viral‐based vectors have been used to successfully deliver genetic material to the heart. Viral vectors are generally more efficient at nucleic acid delivery, can carry relatively robust‐sized genes, and have the ability to provide long‐term gene expression. In fact, to date, only viral vectors have been utilized in clinical trials. Therefore, in this review, we will focus exclusively on the viral vectors that either have already been used or are presently under active investigation either in animal models or in humans for altering the expression of cardiac proteins with the aim of improving the function of the failing heart.
The Viral Vector
The viral vector is responsible for carrying exogenous genetic material from the site where it is introduced into the vasculature or directly into cells to the nucleus of a target cell. The viral genome is packaged in a protein coat called a capsid. Some capsids are surrounded by a lipid bilayer or envelope that contains proteins that facilitate coupling to targeted cells. After coupling with a specific cell‐surface receptor, the virus is carried across the cell membrane and the genetic material is then trafficked to the nucleus.4 Because viral vectors are more efficient than nonviral vectors in crossing the cell membrane, they have become the method of choice for introducing genetic material into the heart. Pathogenic genes of the parent virus have been deleted so that the viral vectors do not cause disease. Use of a viral vector, however, is not without significant challenges. Manufacturing of viral vectors is complex and requires careful oversight, and immune responses to the viral capsid can limit use in individual patients. Three types of viral vectors are presently under evaluation in the United States for targeting the cardiovascular system: lentiviral vectors, adenoviral vectors (AVs), and adeno‐associated viral vectors (AAVs).
Lentiviral Vectors
Lentiviruses are enveloped, single‐stranded RNA vectors that are able to transduce both dividing and nondividing cells. They display relatively robust transduction efficiency. In fact, a lentiviral vector driving transduction of SERCA2a delivered by direct injection into the hearts of mice with diminished LV function secondary to a single injection of the anticancer drug, doxorubicin, or infusion into hearts of rats with LV dysfunctions 2 weeks after a myocardial infarction (MI) by hypothermic intracoronary delivery, increased SERCA2a expression and favorably affected function and remodeling.5, 6 In addition, LV‐mediated gene therapy resulted in sustained expression of β‐glucuronidase for up to 12 months in a mouse model of mucopolysaccharidosis type VII.7 Unfortunately, lentivirus vectors have a characteristic feature that limits their usefulness for the cardiovascular system. Specifically, lentivirus vectors stably integrate into the genome their cargoes randomly integrate into the genome of the target cell with a preference for targeting to the coding regions of genes. Thus, they carry an intrinsic risk of mediating insertional oncogenesis.8 Theoretically, risk of insertional oncogenesis might be lower in a postmitotic and: terminally differentiated cell, such as a cardiac myocyte; however, as we will see later in this discussion, no gene delivery system or promoter is 100% cardiac specific, and, for that reason, lentiviral vectors are not being used in any HF clinical trials nor, to the best of our knowledge, are they presently being investigated.
Adenoviral Vectors
AVs are nonenveloped viruses that consist of an icosahedral capsid and a linear, nonintegrating, double‐stranded DNA. The vector enters the cell by clathrin‐mediated endocytosis after binding with coxsackie‐adenovirus receptors on the cell surface. There are 7 species of AVs and more than 50 different serotypes.9 Production of first‐generation AVs was complicated by the presence of replication‐competent adenovirus contaminants resulting in viral‐like symptoms.10 Third‐generation AVs or “gutless” vectors are devoid of all viral coding regions; however, helper particles are required during vector production and must be removed before use in humans.11 AVs have advantages: (1) The double‐stranded DNA is transported to the nucleus, providing efficient transduction12, 13, 14; (2) third‐generation AVs can hold large transgenes (up to 35 kb)15; (3) transgene expression occurs within hours to days after transduction16; (4) AVs are nonintegrating and therefore do not pass to daughter cells during cell division; and (5) they achieve high levels of cardiac transduction.17 However, AVs have significant limitations: (1) Transgene expression is transient, lasting between 2 and 4 weeks16; (2) AVs may require direct intramyocardial injection17; and (3) AVs can activate the innate immune system with significant dose‐related toxicity, and preexisting antibodies of the adaptive immune system can limit the effectiveness of AVs.18, 19 In fact, enthusiasm was limited for AVs after the death of a young man with ornithine transcarbamylase deficiency who developed a systemic inflammatory response to AV.20
Adeno‐Associated Viral Vectors
AAVs are single‐stranded DNA vectors that are small and nonenveloped with icosahedral capsids. The 4.7‐kb genome is flanked by 2 viral inverted terminal repeats that consist of 2 genes: rep and cap. Because the rep protein facilitates the integration of the AAV into the S1 site of the host chromosome, it is often eliminated from therapeutic viral vectors.21 In the absence of rep, the recombinant AAV vectors do not integrate, yet are able to impart long‐term episomal persistence in postmitotic tissues. Most commonly, they exist as large head‐to‐tail circularized multimeric concatemer structures. Generation of concatemers may involve recombination and a rolling circle‐type DNA replication mechanism. Most important, AAVs can provide stable transgene expression.22
Tissue specific expression can be achieved by use of a tissue‐specific promoter; however, when a nontarget organ is exposed to between 1011 and 1013 viral particles, there is the theoretical risk that the exogenous DNA could be randomly colocalized with a strong and promiscuous enhancer that could initiate transcription of the gene in an unwanted location. Because this sporadic transduction occurs within a single cell, it is impossible to determine that the event has occurred or prognosticate that it might occur. Therefore, all gene‐therapy clinical protocols must include long‐term follow‐up and ongoing surveillance for possible malignancy.
Twelve strains of AAVs have been identified.23, 24, 25 The different serotypes share similar structures, size, and organization; however, they have different tropism based on their capsid protein structure.26 AAV1, AAV6, AAV8, and AAV9 are the most cardiotropic serotypes27; however, there is a consensus that AAV9 is the most effective serotype for cardiac gene delivery.28, 29, 30, 31 AAV9 has a >200‐fold increase in transduction efficiency in the mouse as compared with AAV1 whereas AAV8 transduced myocardium at ≈20‐fold higher levels than AAV1.25 AAV9 is best delivered by coronary infusion32 but can also be delivered by direct myocardial injection.33 To the best of our knowledge, there are no head‐to‐head comparisons of viral vectors in pigs or nonhuman primates, owing, in large part, to the cost of such experiments. Nonetheless, AAV9 has been shown to facilitate effective transduction into the myocardium of a variety of cardiac genes.34, 35, 36 Much like a stealth bomber, AAV9 is less immunogenic because of smoothing out the edges of the AAV9 capsid.37 Efficiency of AAV9 is also enhanced by including terminal repeats that are taken from AAV2. The designation of this variant is AAV2/9.
The primary limiting factor to the use of gene therapy is that despite optimization of design, AAV vectors still demonstrate some level of immunogenicity that can lead to decreased transcription, limited biologic effects, or even development of myocarditis.38, 39, 40, 41 The problem is not inconsequential given that preexisting neutralizing anti‐AAV antibodies exist in 30% to 50% of the population38, 42 and cross‐reactivity can occur between the different serotypes.43 The adaptive immune system can also alter the efficacy of a gene therapy approach because the antibodies to the AAV can eliminate the vector itself, thereby blunting its ability to transduce the cell. Some investigators have proposed a counterintuitive method for limiting the effectiveness of the adaptive immune system: using empty vectors to flood the system. This approach, however, requires further careful investigation. Animal models may not predict treatment outcomes in humans, despite the fact that nonhuman primates can be infected by AAVs that are closely related to those in humans.44, 45 This is explained by the finding that preexisting T‐cell responses in nonhuman primates are distinct from those in humans.45 Preclinical animal models have been developed in which to study immune responses to AAV vectors; however, all of the models have limitations and inconsistencies. For example, aggressive immunosuppressive therapy in a pig model was not effective in reducing seroconversion of preexisting neutralizing antibodies, but immune suppression has been effective in humans who have developed inflammation. Therefore, immune responsiveness to a specific vector‐gene combination is best determined in early‐phase clinical trials.
The complexity of the AAVs is illustrated by 2 recent publications that identified toxicity with intrathecal administration of AAV9 carrying the survival motor neuron 1 gene.46, 47 The authors of the 2 reports urged great caution in using AAV9 in clinical studies of children with skeletal muscle atrophy (floppy baby syndrome). Subsequent work, however, showed that the toxicity was species‐strain and dose dependent.46, 47 Proof of safety and efficacy, however, came from the clinical outcomes. Replacement of the survival motor neuron gene in 15 patients with disease using either low dose (6.7×1013 viral particles [vp] per kg of body weight; n=3) or high dose (2.0×1014 vp/kg; n=12) resulted in all 15 patients surviving at 20 months versus a historical survival rate of 8%. The patients also showed significant and marked improvements in motor function. Serum aminotransferase levels were elevated in 4 patients, but were attenuated with prednisolone.48 Nonetheless, the most prudent approach might be to exclude individuals with high AAV antibody titers to the specific AAV serotype, careful follow‐up of patients during both the early and late phases of therapy, including the use of echocardiography, electrocardiography, and biomarkers for inflammation, and development of an algorithm prestudy for treatment of inflammation should it occur.
Alternative Methods for Gene Transduction
To obviate the major disadvantage of viral‐mediated gene transduction, specifically the immune response to the viral capsids, some laboratories have pursued nonviral gene transfer approaches. These have included circular plasmid DNA vectors and synthetic modified mRNA.49 These approaches also allow for larger genes to be carried into cells and can be administered repeatedly. However, their low efficiency of gene delivery has obviated there use in either investigational studies or in therapeutic programs at present.
Methods for Gene Delivery
The methodology for delivering viral vectors to the heart has transitioned over the past decade, albeit not in a linear fashion. Initial efforts to transduce the heart utilized direct intramyocardial injection in both small50 and large animals.51 Whereas direct myocardial injection was able to alter levels of the target protein and, in some cases, alter cardiac function, there was general agreement that the only region of the heart in which levels of the target gene were modified was along the track of the needles. This led investigators to inject adenovirus and adeno‐associated viruses into the aortic root proximal to the placement of a transaortic clamp: a technique that worked in mice and rats, but was certainly problematic for large animals or humans.52, 53 However, early work by Pacak et al demonstrated, in multiple animal models, that recombinant adeno‐associated virus, in particular serotype 2/9, could effectively transduce the heart in mice when delivered intravenously,29 and a similar approach could be used with an adenovirus.54 Nevertheless, there was a persistent belief that AAV vectors, in particular serotype 9, were only effective at modifying cardiac gene expression when administered by retrograde infusion into the anterior interventricular vein.55, 56 Alternatively, effective gene delivery was also thought to be optimized by using molecular cardiac surgery with recirculating delivery.57 Most recently, preclinical studies, both in mice and large animals, have demonstrated the efficacy of antegrade intracoronary injection of viral vectors in combination with intracoronary nitroprusside and other modifying agents that improve gene transfer (eg, substance P). These studies are detailed below. However, in the absence of an approved gene therapy program for heart failure, a definitive “best practice” for gene delivery has yet to be defined.
Preclinical and Clinical Evaluation of Gene Therapy for the Failing Heart
In parallel with studies aimed at creating the optimal viral vector, cardiovascular investigators have pursued studies in animal models of HF to clarify delivery systems, optimize promoter selection, and rationalize vector selection. A group of targets have been studied using algorithms that are generally similar: target identification, gene delivery in murine models of HF, proof of concept in a large animal model (most commonly a pig HF model), toxicology, and, finally, first‐in‐man studies. Only 2 therapeutic options have traversed this pathway from start to finish, and no gene therapy options are presently approved for human use. Nonetheless, a review of both successful and unsuccessful targets (Figure) is highly informative for designing and interpreting future studies.
Adenylyl Cyclase VI
Background
No cardiac signaling pathway has been more thoroughly studied then AC‐cAMP signaling. The role of cAMP in modulating cardiac contractility in response to epinephrine was first reported by Earl Sutherland and his colleagues in the mid‐1960s.58 That AC could serve as a target for therapy of patients with HFrEF was demonstrated in a series of studies over 2 decades by H. Kirk Hammond and colleagues at the Veteran's Administration Hospital in San Diego. In 1998, they reported that the amount of AC6 limited the ability of adrenergic agonists to stimulate the production of cAMP.59 Subsequent studies showed that there were 2 highly homologous isoforms of AC (AC5 and AC6).60, 61 The presence of multiple variants of both AC5 and AC6 made identification of the unique properties of the 2 isoforms initially challenging.62 However, a series of studies by the Hammond laboratory showed that levels of AC6 decreased during HF, making it a potential target for gene therapy.61
Proof of Concept
The first proof of the concept that AC6 could serve as a target for the therapy of HFrEF came in 1999 when Gao et al showed that transgenic overexpression of AC increased adrenergic responsiveness without altering β‐AR receptor density, G‐protein content, or basal levels of cAMP.63 When AC6‐overexpressing mice were crossed with mice with HFrEF secondary to overexpression of Gq, cardiac function improved.64 By contrast, overexpression of AC5 increased AC activity, but did not alter adrenergic responsiveness, and led to a decrease in cardiac function.65
Intuitively, overexpression of AC6 might have an adverse effect on the heart during ischemia because increased cAMP generation might increase the oxygen demand/oxygen supply ratio. Surprisingly, mice with cardiac‐restricted overexpression of AC6 showed a 45% reduction in mortality 7 days after an MI when compared with WT mice.66 One explanation for this surprising outcome was that overexpression of AC6 reduced phospholamban expression secondary to upregulation of activating transcription factor 3, a transcriptional suppressor that binds to and inhibits the phospholamban promoter.67, 68, 69 In aggregate, these studies led investigators to suggest that AC‐cAMP might serve as a target for HFrEF therapy.70
In subsequent large animal studies, Lai et al demonstrated that a single intracoronary infusion of Ad‐CMV‐AC6 (1.4×1012 vp) in a normal pig increased peak LV dP/dt and cardiac output: changes that were evident 6 days after gene transfer and persisted for at least 57 days.54 When investigators administered Ad‐CMV‐AC6 to pigs with reduced LV function secondary to rapid ventricular pacing, AC6 improved LV hemodynamics.71 cAMP levels were increased 1.7‐fold and brain natriuretic peptide (BNP) levels were significantly reduced at 14 days.
Clinical Proof of Concept
From July 2010 to October 2014, 56 adult patients with HFrEF were randomized to an ascending dose study of the intracoronary administration of adenovirus serotype 5‐cytomegalovirus‐adenylyl cyclase 6 (Ad5‐CMV‐hAC6) or placebo.72 Cardiovascular evaluations were performed at baseline and again at weeks 4 and 12. In subjects in the 2 high‐dose groups (3.2×1011 or 1×1012 vp) Ad5‐CMV‐hAC6 effected a 36% increase in LV ejection fraction (LVEF) at week 4 but not at week 12. By contrast, no change in ejection fraction (EF) was observed in patients who had received placebo. Serum troponin levels were elevated in 3 patients, 1 of whom received placebo. There was no association between prerandomization anti‐Ad5 titer and any efficacy end point.
A phase 3 clinical trial testing the effects of intracoronary administration of Ad5‐CMV‐hAC6 (RT‐100; 1×1012 vp) or placebo in 536 HFrEF patients is expected to start in 2018.73 The primary end point of the study will be the reduction in the event rate of all (first and repeat) HF hospitalizations between baseline and 12 months. The study has received a fast‐track designation from the US Food and Drug Administration.
Limitations and Future Studies
The major limitation of adenovirus‐driven gene therapy is that the virus is not entrapped in the nucleus and is therefore cleared within ≈4 weeks. Despite efforts to delete the offending epitopes, adenoviruses remain substantially more immunogenic then other vectors and their use in patients with preexisting antibodies can lessen the effect of the viral vectors through neutralization. An AV backbone was used with AC6 because it is too large to fit into an AAV vector. However, recent studies have shown that portions of the catalytic domain of AC6 can reverse cardiac dysfunction caused by sustained β‐adrenergic receptor (β‐AR) stimulation.74 If this or other AC6 domains show efficacy, they hold promise for successful packaging into an AAV delivery system.
G‐Protein–Coupled Receptor Kinase 2 Inhibitor—βARK‐ct
Background
The classic signaling program that initiates an increase in cardiac contractility is composed of the β‐AR, the stimulatory G protein (Gs), AC, and protein kinase A (PKA).75 This complex signaling pathway is regulated by a number of different proteins, including a family of G‐protein–coupled receptor kinases or GRKs (also referred to as β‐AR receptor kinases or β‐ARK), and β‐arrestins, small proteins with great plasticity that desensitize G‐protein–coupled receptors. Activation of the pathway occurs when an agonist (eg, norepinephrine) couples with the β‐AR, which elicits a conformational change in the receptor allowing it to interact with the membrane‐bound Gs protein.76 Occupancy of the receptor by an agonist results in disassociation of the heterotrimeric G protein into Gα and Gβγ subunits. In the case of norepinephrine, Gsα stimulates AC to produce cAMP.
The heart is protected from continuous activation of the adrenergic receptors by internalization and downregulation of the β‐ARs.77, 78 Desensitization of adrenergic signaling also occurs through the actions of a family of serine/threonine kinases known as β‐ARKs or GRKs.79 GRKS selectively phosphorylate G‐protein–coupled receptors, which leads to desensitization of the receptor.80 In the heart, 3 GRKs regulate β‐adrenergic signaling: GRK2 (β‐ARK1), GRK3 (β‐ARK2), and GRK5. The cytosolic GRK must bind to Gβγ in order to translocate to the membrane, where it phosphorylates the activated receptor.81 In the presence of continuous activation of the adrenergic receptors, desensitization requires not only a GRK, but also β‐arrestins that prevent Gs from coupling to the agonist bound β‐AR.82 HF in animal models as well as in the failing human heart is characterized by multiple abnormalities in the β‐AR/G‐protein/AC signaling cascade, including an increase in GRK2 (β‐ARK1).77, 83, 84
Proof of Concept
To counteract the effects of increased expression of GRK2 in the heart, Koch et al generated a peptide consisting of the last 195 amino acids of GRK2 that contained the Gβγ binding site and was cardiac targeted (β‐ARKct).84 To evaluate the effect of GRK2 inhibition on cardiac function, Rockman et al crossed mice with HF secondary to homozygous deletion of the muscle LIM protein (MLP−/−) with mice overexpressing β‐ARKct or the β2‐AR. Overexpression of the β2‐AR exacerbated HF whereas the GRK2 inhibitor (β‐ARKct) restored adrenergic responsiveness in a variety of murine models of HF and prevented progression of disease.85, 86 When transgenic mice with varying levels of β‐ARKct underwent transverse aortic constriction for 12 weeks, mice with high levels of β‐ARKct demonstrated significantly less cardiac dysfunction then WT mice.87
Large Animal Studies
In the first study to evaluate the efficacy of β‐ARKct gene therapy in a large animal model, investigators tested the ability of adenoviral‐mediated delivery of β‐ARKct into ventricle of rabbits with diminished LV function secondary to coronary occlusion.88 Contractility, relaxation and end‐diastolic pressure were less impaired in rabbits that had been pretreated with β‐ARKct when compared with controls. In a second study, AAV6/β‐ARKct was administered by direct injection into ventricular myocardium of rats with HF secondary to cryoablation of the left anterior descending coronary artery.50 Controls included HF rats that received AAV6‐GFP (green fluorescent protein), AAV6/β‐ARKct+metoprolol, AAV6‐GFP+metoprolol or saline. Both short‐ and long‐term follow‐up demonstrated that β‐ARKct effected an increase in contractility, a decrease in adrenergic signaling, and a reversal of the maladaptive cardiac remodeling that characterizes HFrEF. Finally, pigs with HF secondary to 2 hours of occlusion of the circumflex coronary artery were treated by retrograde infusion with either AAV6/β‐ARKct or AAV6‐luciferase as a control.55 Pigs underwent echocardiography and invasive hemodynamics 2 and 6 weeks after gene delivery. AAV6/β‐ARKct infusion led to significant improvements in LV function as compared with pigs that received AAV6‐luciferase control. In addition, AAV6/β‐ARKct increased adrenergic responsiveness and decreased adrenergic activation as evidenced by decreased levels of norepinephrine.
Clinical Development
Several issues have delayed or limited the development of β‐ARKct. The first is that the long‐term effects of enhanced adrenergic activation have not yet been evaluated in normal large animals. This is important in view of the fact that long‐term activation of the β‐AR by simply including norepinephrine in drinking water can result in cardiac dysfunction. More important, the patent for β‐ARKct has a very short runway, and thus the path to clinical development of AAV/β‐ARKct remains unclear. Coupling β‐ARKct with a unique promoter or creating an inducible promoter might result in renewed patent life, making it a more‐attractive target for commercial development.
S100A1 Protein
Background
S100A1 is a dimeric Ca2+ binding protein that is expressed predominantly in the heart and, to a lesser extent, in skeletal muscle.89 Located in the sarcoplasmic reticulum, myofilaments, and mitochondria,90, 91, 92 S100A1 is thought to play the role of a Ca2+ sensing protein that undergoes a conformational change, enabling it to recognize downstream target proteins.93 Initial studies led to the observation that S100A1 regulates cardiac and skeletal muscle contractility.94, 95 This effect on contraction is mediated by enhancing intracellular Ca2+ transients through increased SR Ca2+ cycling because there is no change in activity of either the L‐type Ca2+ channel or Na+/Ca2+ exchanger (NCX1).93, 94, 95 To enhance cardiac excitation‐contraction coupling, S100A1 interacts with both the ryanodine receptor (ryanodine receptor type 2 [RyR2]—increased Ca2+ induced SR Ca2+ release) and SERCA2a (increased SR Ca2+ reuptake). In addition, S100A1 can decrease diastolic SR Ca2+ leak through attenuated Ca2+ spark frequency, suggesting a biphasic Ca2+‐dependent modulation of the RyR2 through interaction with S100A1.96
Levels of S100A1 are substantially decreased in the failing human heart at both the transcriptional and translational levels.97 Similarly, S100A1 protein and mRNA levels were significantly decreased in rats with decreased LV function after ligation of the left anterior descending coronary artery. Intracoronary injection of ADV‐S100A1 restored normal levels of S100A1 and improved SR Ca2+ cycling. Taken together, these adaptive changes in Ca2+‐related LV performance reversed the maladaptive remodeling that characterizes failing myocardium.98 These results suggested that replenishment of S100A1 could enhance SR Ca2+‐ATPase activity, thereby improving function and activating adaptive remodeling.
Proof of Concept
To confirm effectiveness of S100A1in a large animal model, Pleger et al assessed the functional consequences and safety of rAAV9‐S100A1 administered retrograde by the great cardiac vein.56 Two weeks after inducing a proximal circumflex coronary artery MI, German farm pigs were randomized to receive saline, a rAAV9‐luc control virus, or rAAV9‐CMV‐S100A1 by retrograde infusion of the great cardiac vein. Twelve weeks later, the EF of rAAV9‐S100A1 pigs was significantly greater than that of pigs that received rAAV9‐luc or those that received saline, but was not different from sham pigs. Similarly, heart rate, ratio of heart weight to body weight, LV end‐diastolic volume, and level of BNP mRNA were elevated in saline and rAAV9‐luc groups, but not in rAAV9‐S100A1 groups. At the cellular level, rAAV9‐S100A1 therapy rescued the decrease in Ca2+ transient amplitude and SR Ca2+ uptake. Importantly, the investigators did not include a SHAM‐rAAV9‐S100A1 group to control for any deleterious effects of enhanced SR Ca2+ cycling in the normal heart.
In a second set of experiments, the same investigators evaluated safety and efficacy of “high” myocardial expression levels of S100A1 in a pig model of HF.34 High‐level S100A1 myocardial overexpression after AAV6‐S100A1 gene delivery was compared with low‐level S100A1 control samples obtained from the previous study using AAV9‐S100A1–treated pigs. By contrast with the earlier study, a CMV‐MLC promoter was used to drive expression and the AAV was serotype 6. The vector was again delivered by retrograde infusion of the anterior cardiac vein, which resulted in a 95‐fold increase in S100A1 protein over control. AAV6‐S100A1 did not demonstrate a robust decrease in BNP, had a smaller effect on EF, and had no effect on heart rate or dP/dt. In myocytes isolated from postinfarct pigs, AAV6‐S100A1 had no effect on levels of RyR2, SERCA2a, or NCX1. Furthermore, studies in isolated myocytes showed that increasing doses of AAV6‐S100A1 actually decreased Ca2+ transient amplitude.
Preclinical Development
A plausible explanation for the failure to see benefits in the high‐dose study in pigs could be explained by the exuberant overexpression of S100A1 (95‐fold increase) that may give rise to the phenomenon of “molecular torture” in which high levels of protein expression cause a nonspecific and toxic phenotype.99 More work will be required to clarify the dose, select a single vector and promoter, and understand the ambiguity in the negative dose relationship with Ca2+ transient amplitude before undertaking investigational new drug–enabling studies.
SR Ca2+‐ATPase
Background
SERCA2a regulates both contraction (SR Ca2+ content is a major determinant of [Ca2+]i transient amplitude) and relaxation (SR Ca2+ reuptake during diastole). A universal observation in the failing heart in both animal models and in humans is a reduction in expression of SERCA2a at both the mRNA and protein level.100, 101, 102, 103, 104, 105, 106 This led to efforts in both rats and pigs to assess the effectiveness of increased levels of SERCA2a in ameliorating the HF phenotype.
Proof of Concept
Ad‐CMV‐SERCA2a was introduced into the coronary arteries of rats with LV dysfunction secondary to transverse aortic constriction by injecting into the aortic root after cross‐clamping the aorta. Rats that had undergone sham surgery and received Ad/β‐galactosidase (β‐gal) served as controls. Two to 3 days postinjection, Ad/β‐gal mice demonstrated a significant decrease in SERCA2a activity when compared with nonfailing sham controls. In addition, the +dP/dt decreased from 8.43 in sham rats to 4.83 mm Hg/s in the Ad/β‐gal mice.104
Preclinical Development
The first investigational new drug–enabling study was performed in Yorkshire–Landrace pigs with LV dysfunction and LV dilatation secondary to percutaneous dissection of the chordae tendinea of the mitral apparatus.107 Two months after the surgical intervention, pigs received an intracoronary injection of either AAV1‐CMV‐SERCA2a (1012 vp) or saline. No significant differences were found between the rAAV1‐SERCA2a group and the saline group in heart rate, peak LV pressure, and end‐diastolic pressure. By contrast, SERCA2a effected a modest improvement in LVEF (61.5±7.4% versus 70.6±3.5%; P<0.05) and dP/dtmax (1396±383 mm versus 1822±317 mm Hg/s; P<0.05). Infection with rAAV1‐SERCA2a also increased SERCA2a protein and mRNA levels and decreased BNP levels, but had no effect on phospholamban levels.
Clinical Development
In the pig studies, the inverted terminal repeat sequence of AAV2 replaced the corresponding sequence in AAV1.108 The CMV enhancer/promoter drove expression of the transgene. The buffer without vector served as the placebo, and the construct was delivered using standard antegrade epicardial coronary artery Infusion. The results of the initial 9 patients to receive open‐labeled AAV1‐SERCA2a who were part of the first‐in‐human phase ½ trial were reported in 2009.40 Patients received a single intracoronary infusion of AAV1‐SERCA2a. Several subjects showed improvements from baseline to 6 months whereas 2 patients who failed to improve had preexisting anti‐AAV1 neutralizing antibodies. These results supported transition to a phase 2 double‐blind, placebo‐controlled study.
The results of the phase 2 trial with AAV‐SERCA were reported in 2011.41 The primary end point was prospectively defined and was based on patients meeting group‐level, individual‐level, and outcome end points.40 A detailed discussion of the intricate and complex analytic methodology used in this study is outside of the scope of this article, but the interested reader is referred to the 2 CuPID publications.40, 41 In brief, only the high‐dose group met the prespecified criteria for success at the group‐level, individual‐level, and outcome analyses (cardiovascular hospitalizations) at 6 and 12 months. However, there were obvious differences between baseline characteristics for the placebo and AAV1‐SERCA2a groups and variations in the timing of the entry of a group of placebo patients that might have biased the result, even though the investigators assiduously, but retrospectively, stratified all patients based on prestudy demographics and the 2 entry groups. These concerns are moot because the phase IIb study (CuPID 2) was discontinued by the sponsor because of futility.109 The most important information to come out of CuPID and CuPID 2 was that there were no adverse safety signals across all studies and all doses of AAV1‐SERCA2a. Furthermore, the CUPID studies demonstrate the importance of comparable baseline demographics in the control and treatment groups regardless of study size in order to avoid statistical bias.
Cardiac I‐1c
Background
As noted above, a key constituent in the homeostatic regulation of Ca2+ is SERCA2a, which is negatively regulated by phospholamban.110 Phosphorylation of phospholamban, through either activation of PKA or calcium‐calmodulin kinase II (CaMKII), relieves the inhibitory effects of phospholamban on SERCA2a, thereby facilitating the uptake of Ca2+ from the SR. Phospholamban levels are decreased in animal models of HF111 and in humans, and levels return to normal with restitution of normal cardiac function.100 The decrease in phosphlamban in failing hearts appear to be related to the degree of ventricular dysfunction given that modest changes in LV function do not appear to alter levels of phospholamban.112 Phospholamban expression is regulated by a diverse group of signaling events.113, 114 Recent attention has focused on 1 of its most important regulators—protein phosphatase‐1 (PP1). PP1 dephosphorylates phospholamban, resulting in inhibition of SERCA2a. PP1 activity is elevated in HF; however, it can be inhibited by the naturally occurring regulatory protein—the inhibitor of protein phosphatase‐1 or (I‐1). The increase in PP1 activity is attributed, in part, to a reduction in levels and activity of I‐1.115, 116, 117
Proof of Concept
Transgenic mice with either chronic or inducible expression of constitutively phosphorylated and truncated inhibitor‐1 (I‐1c) increased phosphorylation of phospholamban, enhanced contractility, and was cardioprotective in pressure overload, chronic adrenergic activation, and ischemia‐reperfusion injury.118, 119, 120 Furthermore, overexpression of I‐1c showed benefits in both rat and pig HFrEF models by reducing remodeling and/or scar size.118, 119 However, it should be noted that in mice subjected to transverse aortic constriction, administration of an AAV9‐vector–expressing I‐1c under control of a human troponin T‐promoter (AAV9‐I‐1c; 2.8×1012 vp), as compared with a control vector, effected LV hypertrophy as well as reduced contractility. Pressure‐volume loops revealed significant impairment of contractility after AAV9‐I‐1c. At the molecular level, transverse aortic constriction mice showed hyperphosphorylation of phospholamban and augmented cardiac contractility, but I‐1c failed to protect against remodeling in response to stress.121 These results were consistent with an earlier study showing that I‐1c improved LV function in young mice was associated with the development of a dilated cardiomyopathy in old mice and led to lethal catecholamine‐associated ventricular tachycardia.122 In that study, however, I‐1c was overexpressed in the absence of endogenous I‐1.
Preclinical Development
In an initial study, Yorkshire pigs with post‐MI LV dysfunction received an intracoronary injection of either AAV9‐CMV‐I‐1c (2.5×1012 vp) or saline.123 One month after MI, pigs had a significant reduction in EF (46.4%) and impaired dP/dtmax and dP/dtmin. Control animals demonstrated a small reduction in EF whereas AAV9‐CMV‐I‐1c–treated animals demonstrated a modest increase in EF. However, the difference between the decrease in controls and increase in the active treatment group resulted in an overall modest increase in EF. Long‐term electrocardiographic monitoring showed 2 episodes of ventricular tachycardia in each group. Expression in the border zone and scar tissue of the heart were not substantially greater than that observed in liver or lung, and only a small amount of expression was observed in the right ventricle. The vector was made by the investigators using a well‐characterized human constitutively active I‐1c that had been truncated to remove the S67 and T35D domains.
In a second study, Yorkshire pigs with HFrEF secondary to a large anterior MI were injected with I‐1c transgene driven by a novel cardiotropic viral vector consisting of the BNP116 vector packaging the CMV enhance/promoter and I‐1c at either high or low dose.124 The EF increased from 38.8% to 40.4% in the high‐dose group and fell from 43.3% to 40.0% in the low‐dose group. The EF fell from 37.2% to 34.5% in the control. The investigators converted the change in EF in the high‐dose group to an increase of 5.7% and the decrease in control to a decrease of 7%—which together equaled an “increase” of nearly 15%. The high dose of the vector showed relative expression that was comparable with that observed with AAV9 in an earlier study.125 The investigators also showed very high expression of AAV9 in the liver—something that is not routinely observed with the AAV2/9 vector.
Future Clinical Development
Because of the modest size of the increase in EF clinical evaluation may require a substantial number of patients to see a statistical outcome. Therefore, unless subsequent studies demonstrate a more robust effect of I‐1c gene therapy it is unlikely that sponsors would support further clinical evaluation. None the less, a modest increase in EF may be safer and more effective long term than a robust effect on EF given that the inotropic effect will theoretically continue throughout the life of each cell.
The Urocortins
Background
The urocortins are an endogenous peptide hormone group comprising urocortin1, urocortin2, and urocortin3 that are regulated by corticotropin‐releasing factor. Corticotropin‐releasing factor and urocortin1 can induce cellular injury through release of the proinflammatory cytokine, tumor necrosis factor‐alpha (TNF‐α), and mast cell proteases.126, 127 By contrast, urocortin2 and urocortin3 have inotropic, lusitropic, and chronotropic effects attributed predominantly to their robust vasodilatory effects.128 Preclinical studies in which the agents were infused in large animals as well as evaluation in patients with acute decompensated HF have demonstrated beneficial effects of urocortin2 and urocortin3 on LV function.129, 130 The major impediment to the translation of laboratory evaluation and short‐term administration to patients with acute decompensation has been that both urocortin2 and urocortin3 have very short half‐lives in the peripheral circulation (minutes). Furthermore, urocortin2, but not urocortin3, significantly decreased fasting glucose levels, suggesting that urocrotin2 might be most effective in patients with diabetes mellitus whereas urocortin3 could be more useful in patients with normal glucose metabolism. Because of its short half‐life, investigators hypothesized that levels of urocortin2 and/or urocortin3 could be modified using gene therapy.
Proof of Concept
In an initial group of experiments, mice were treated with either AAV serotype‐8‐chicken‐β‐actin‐urocortin3 (AAV8‐CBA‐mUCn3), AAV8‐CBA‐mUCn2, or a saline control.131 Both urocortin2 and urocortin3 significantly decreased systolic blood pressure, diastolic blood pressure, mean arterial pressure, and LVEF as well as decreasing the end‐diastolic dimension, and the end‐systolic dimension without a change in heart rate. However, urocortin2, but not urocortin3, gene transfer reduced fasting glucose and increased glucose disposal. Urocortin gene therapy was also associated with significant changes in calcium homeostasis without a change in markers of HF.
In a second group of experiments, mice with LV dysfunction secondary to cryoinjury of the left ventricle received either intravenous administration of AAV8‐CBA‐mUCn3 (1.9×1013 genome copies/kg—5×1011 genome copies/mouse) or a saline control.132 Mice randomized to urocortin3 gene therapy demonstrated increased plasma urocortin3 (from 0.1±0.01 versus 5.6±1.1 ng/mL) and a modest increase in the LVEF (28±1 versus 37±1). However, there was no significant difference in end‐diastolic dimension, end‐systolic dimension, or heart rate. The increase in contractility was not associated with significant changes in HF markers, including phospholamban, ryanodine receptor, or phosphorylated troponin I or protein kinase A. Urocortin3 gene therapy had no effect on amount of fibrosis or on common markers of hypertrophy. Levels of atrial natriuretic peptide and BNP mRNA, markers of adaptive cardiac remodeling, decreased significantly with AAV8‐mUCn3.
Clinical Development
Although the effects of urocortin2 and 3 on cardiac function is intriguing, the hypoglycemic effects of urocortin2 and the fact that gene therapy cannot be titrated make its use in nondiabetics or diabetics potentially problematic. In the case of urocortin3, preliminary studies in mice suggest that the primary mechanism responsible for the salutary effects of urocortin is improved LV Ca2+ handling and potentially increased expression of LV SERCA2a. Therefore, additional safety and efficacy studies in a large animal model would facilitate moving the therapy from the laboratory to the clinic.
Bcl2‐Associated Anthanogene 3
Background
B‐cell lymphoma 2 (Bcl2)‐associated athanogene 3 (BAG3) is a 575‐amino‐acid protein that is expressed predominantly in heart, skeletal muscle, brain, and in many cancers (see previous reviews133, 134). It is highly conserved in nature, with homologs being found in fruit flies, invertebrates, silkworms, and even plants. The protein carries out multiple functions in the cell. Specifically, BAG3 mediates chaperone‐assisted autophagy by serving as a cochaperone with Hsc70/Hsp70, blocks apoptosis by coupling with Bcl‐2, preserves integrity of the sarcomere by coupling the actin filaments with the Z disc, and enhances β‐agonist–stimulated excitation‐contraction by linking the β‐adrenergic receptor and the L‐type Ca2+ channel.135, 136, 137 Recent studies suggest that BAG3 also binds with lamin B to support the integrity of the nuclear envelope and protects the heart from ischemia‐reperfusion injury. This panoply of diverse effects is facilitated by presence of multiple protein‐protein binding domains within the BAG3 protein.
BAG3 levels are significantly reduced by ≈50% in hearts with diminished LV function, including: mice with LV dysfunction secondary to a MI or transaortic constriction; mice with LV dysfunction attributed to homozygous deletion of the MLP gene; pigs with cardiac dilation after an MI; and humans with both ischemic and nonischemic dilated cardiomyopathy.136, 138, 139, 140, 141 That decreased levels of BAG3 can be causative of LV dysfunction has been shown by the fact that truncation or deletion mutations in BAG3 that excise the BAG region of the protein segregate with affected family members and are associated with haploinsufficiency of BAG3.141, 142 More recently, an epidemiological study from the European Genetic Cardiomyopathies Initiative found that dilated cardiomyopathy attributed to BAG3 variants was characterized by high penetrance in carriers aged >40 years and a progression in the disease that was more aggressive than that observed in patients without BAG3 mutations.143 Recently, phenotypic refinement of HF in a national biobank found BAG3 as a genetic signal that independently of clinical HF risk factors is associated with subclinical LV dysfunction, thereby corroborating earlier data implicating BAG3 as a bona‐fide disease susceptibility gene for dilated cardiomyopathy. Although not “causing” a dilated cardiomyopathy, loss‐of‐function missense mutations can alter the HF phenotype.144
Proof of Concept
Mice with LV dysfunction secondary to a previous left coronary artery ligation were injected retro‐orbitally with either AAV9‐CMV‐BAG3 or AAV9‐CMV‐GFP.145 Controls consisted of sham‐operated animals that then received either AAV9‐CMV‐BAG3 or AAV9‐CMV‐GFP. MI‐GFP mice demonstrated a significant decrease in LVEF at 1 week post‐MI with a progressive diminution in contractility over time. By contrast, mice that suffered MI and were injected with AAV9‐BAG3 had a significantly higher LVEF at 9 days postinjection with continued improvement at 3 weeks postinjection. Importantly, injection of AAV9‐BAG3 had no effect on LVEF in sham mice when compared with sham mice that received rAAV9‐GFP. In a second set of experiments, mice with LV dysfunction secondary to deletion of 1 allele of BAG3 and haploinsufficiency were injected with AAV9‐CMV‐BAG3 or AAV9‐CMV‐GFP.144 The EF decreased gradually over time in the haploinsufficient BAG3+/− mice when compared with BAG3+/+ controls. Injection of AAV9‐BAG3 into the retro‐orbital plexus of BAG3+/− mice markedly improved LV function consistent with that seen in BAG3+/+ mice.
It is interesting to note that unlike previous gene therapy approaches which directly (S100A1, SERCA2a, and I‐1c) or indirectly (βARKct, AC6) target Ca2+ cycling proteins, BAG3 has only a modest effect Ca2+ homeostasis (L‐type Ca2+ channel) that is only apparent in isolated myocytes in the presence of isoproterenol. The dominant processes that influence cardiac function and survival after BAG3 expression are critical cellular processes, such as apoptosis, autophagy, sarcomere integrity, nuclear envelope integrity, and cell‐to‐cell communication, all of which are altered in HF. Viewed in this context, the multiple effects of BAG3 may confer a theoretical advantage over other gene therapy approaches which target single pathways. In addition, by targeting individuals with haploinsufficiency of BAG3 secondary to a deletion or truncation, BAG3 stands as the only gene therapy for pure dilated cardiomyopathy presently undergoing development that focuses on replacing the output of a missing allele or gene “replacement” therapy. For example, gene therapy for the cardiovascular complications of Danon disease focus on treatment of hypertrophic cardiomyopathy. And, finally, AAV9‐BAG3 appears to be unique, in that therapy of WT mice does not result in increased contractility suggesting that long‐term therapy in patients with haploinsufficiency would be safe.
Future Clinical Development
AAV9‐BAG3 requires large animal efficacy and safety studies (including toxicology) to provide the requisite data for an investigational new drug.
Summary
Gene therapy for patients with HF is now on the cusp of being successfully translated from the laboratory to the clinical arena. It is no longer a theoretical vision, but a realistic opportunity for the future treatment of patients with HFrEF. This transition is attributed to an increased understanding of the optimization of the various AAV serotypes, a better understanding of the efficacy of the various cardiac‐restricted promoters, and clarification of the most effective delivery systems. Although recent studies in humans did not prove efficacious, they were of great importance because they demonstrated the safety of gene therapy and defined the methods for gene delivery. Nonetheless, challenges remain. First, the development of AAVs that are nonimmunogenic will allow treatment of patients with preexisting AAV antibodies and will obviate, or at least lessen, the risk of the development of adaptive immunity. Second, the construction of AAVs that can package large genes will facilitate the generation of new therapies for individuals with mutations that are not currently approachable (eg, titin and lamin) or that require indirect approaches. Third, the identification of new HFrEF‐causing mutations in large‐scale genomic studies, such as the recently initiated All of Us study, will expand the number of patients who can benefit from gene therapy. And, finally, development of highly cardiac trophic vectors may allow therapy to be delivered intravenously rather than intracoronary with comparable or greater efficiency of transduction.
Sources of Funding
This work was supported, in part, by NIH grants HL91799 (Feldman) and HL123093 (Cheung and Feldman).
Disclosures
Dr Feldman is the co‐founder and the chair of the board pf directors of Renovacor, Inc, a biotechnology start‐up focused on developing gene therapy for both inherited and noninherited forms of dilated cardiomyopathy associated with BAG3 haploinsufficiency. Drs Feldman, Cheung, and Myers have significant equity in Renovacor. Dr Feldman reports having a patent pending for BAG3 therapy for patients with heart failure, a patent pending for a BAG3 composition and methods, a patent pending for the role of BAG3 in ischemia/reperfusion injury, and a patent pending for BAG3 mechanisms of action. The patents are owned by Temple University: Renovacor, Inc has an exclusive option to license the BAG3 patents. The remaining authors have no disclosures to report.
J Am Heart Assoc. 2019;8:e012239. DOI: 10.1161/JAHA.119.012239.
References
- 1. Nabel EG, Plautz G, Nabel GJ. Site‐specific gene expression in vivo by direct gene transfer into the arterial wall. Science. 1990;249:1285–1288. [DOI] [PubMed] [Google Scholar]
- 2. Isner JM, Pieczek A, Schainfeld R, Blair R, Haley L, Asahara T, Rosenfield K, Razvi S, Walsh K, Symes JF. Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet. 1996;348:370–374. [DOI] [PubMed] [Google Scholar]
- 3. Yla‐Herttuala S, Baker AH. Cardiovascular gene therapy: past, present, and future. Mol Ther. 2017;25:1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petrus I, Chuah M, VandenDriessche T. Gene therapy strategies for hemophilia: benefits versus risks. J Gene Med. 2010;12:797–809. [DOI] [PubMed] [Google Scholar]
- 5. Mattila M, Koskenvuo J, Soderstrom M, Eerola K, Savontaus M. Intramyocardial injection of SERCA2a‐expressing lentivirus improves myocardial function in doxorubicin‐induced heart failure. J Gene Med. 2016;18:124–133. [DOI] [PubMed] [Google Scholar]
- 6. Niwano K, Arai M, Koitabashi N, Watanabe A, Ikeda Y, Miyoshi H, Kurabayashi M. Lentiviral vector‐mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther. 2008;16:1026–1032. [DOI] [PubMed] [Google Scholar]
- 7. Derrick‐Roberts AL, Pyragius CE, Kaidonis XM, Jackson MR, Anson DS, Byers S. Lentiviral‐mediated gene therapy results in sustained expression of beta‐glucuronidase for up to 12 months in the gus(mps/mps) and up to 18 months in the gus(tm(L175F)Sly) mouse models of mucopolysaccharidosis type VII. Hum Gene Ther. 2014;25:798–810. [DOI] [PubMed] [Google Scholar]
- 8. Papayannakos C, Daniel R. Understanding lentiviral vector chromatin targeting: working to reduce insertional mutagenic potential for gene therapy. Gene Ther. 2013;20:581–588. [DOI] [PubMed] [Google Scholar]
- 9. Goncalves MA, de Vries AA. Adenovirus: from foe to friend. Rev Med Virol. 2006;16:167–186. [DOI] [PubMed] [Google Scholar]
- 10. Haddada H, Cordier L, Perricaudet M. Gene therapy using adenovirus vectors. Curr Top Microbiol Immunol. 1995;199(Pt 3):297–306. [DOI] [PubMed] [Google Scholar]
- 11. Alba R, Bosch A, Chillon M. Gutless adenovirus: last‐generation adenovirus for gene therapy. Gene Ther. 2005;12(suppl 1):S18–S27. [DOI] [PubMed] [Google Scholar]
- 12. Wasala NB, Shin JH, Duan D. The evolution of heart gene delivery vectors. J Gene Med. 2011;13:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parker AL, Nicklin SA, Baker AH. Interactions of adenovirus vectors with blood: implications for intravascular gene therapy applications. Curr Opin Mol Ther. 2008;10:439–448. [PubMed] [Google Scholar]
- 14. Du L, Dronadula N, Tanaka S, Dichek DA. Helper‐dependent adenoviral vector achieves prolonged, stable expression of interleukin‐10 in rabbit carotid arteries but does not limit early atherogenesis. Hum Gene Ther. 2011;22:959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Otto‐Wilhelm M, Gaillet B. Viral vectors for gene therapy and gene modification approaches. Biochem Eng J. 2016;108:98–115. [Google Scholar]
- 16. Wright MJ, Wightman LM, Lilley C, de Alwis M, Hart SL, Miller A, Coffin RS, Thrasher A, Latchman DS, Marber MS. In vivo myocardial gene transfer: optimization, evaluation and direct comparison of gene transfer vectors. Basic Res Cardiol. 2001;96:227–236. [DOI] [PubMed] [Google Scholar]
- 17. Vassalli G, Bueler H, Dudler J, von Segesser LK, Kappenberger L. Adeno‐associated virus (AAV) vectors achieve prolonged transgene expression in mouse myocardium and arteries in vivo: a comparative study with adenovirus vectors. Int J Cardiol. 2003;90:229–238. [DOI] [PubMed] [Google Scholar]
- 18. Rincon MY, VandenDriessche T, Chuah MK. Gene therapy for cardiovascular disease: advances in vector development, targeting, and delivery for clinical translation. Cardiovasc Res. 2015;108:4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schiedner G, Morral N, Parks RJ, Wu Y, Koopmans SC, Langston C, Graham FL, Beaudet AL, Kochanek S. Genomic DNA transfer with a high‐capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat Genet. 1998;18:180–183. [DOI] [PubMed] [Google Scholar]
- 20. Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–158. [DOI] [PubMed] [Google Scholar]
- 21. Musayev FN, Zarate‐Perez F, Bishop C, Burgner JW II, Escalante CR. Structural insights into the assembly of the adeno‐associated virus type 2 Rep68 protein on the integration site AAVS1. J Biol Chem. 2015;290:27487–27499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Athanasopoulos T, Fabb S, Dickson G. Gene therapy vectors based on adeno‐associated virus: characteristics and applications to acquired and inherited diseases (review). Int J Mol Med. 2000;6:363–375. [DOI] [PubMed] [Google Scholar]
- 23. Rutledge EA, Halbert CL, Russell DW. Infectious clones and vectors derived from adeno‐associated virus (AAV) serotypes other than AAV type 2. J Virol. 1998;72:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno‐associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mori S, Wang L, Takeuchi T, Kanda T. Two novel adeno‐associated viruses from cynomolgus monkey: pseudotyping characterization of capsid protein. Virology. 2004;330:375–383. [DOI] [PubMed] [Google Scholar]
- 26. Tilemann L, Ishikawa K, Weber T, Hajjar RJ. Gene therapy for heart failure. Circ Res. 2012;110:777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE. Comparative cardiac gene delivery of adeno‐associated virus serotypes 1‐9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin Transl Sci. 2010;3:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, Nakai H. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, Zolotukhin I, Tarantal AF, Byrne BJ. Recombinant adeno‐associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. [DOI] [PubMed] [Google Scholar]
- 30. Sarkar R, Mucci M, Addya S, Tetreault R, Bellinger DA, Nichols TC, Kazazian HH Jr. Long‐term efficacy of adeno‐associated virus serotypes 8 and 9 in hemophilia a dogs and mice. Hum Gene Ther. 2006;17:427–439. [DOI] [PubMed] [Google Scholar]
- 31. Vandendriessche T, Thorrez L, Acosta‐Sanchez A, Petrus I, Wang L, Ma L, L DEW, Iwasaki Y, Gillijns V, Wilson JM, Collen D, Chuah MK. Efficacy and safety of adeno‐associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost. 2007;5:16–24. [DOI] [PubMed] [Google Scholar]
- 32. Fang H, Lai NC, Gao MH, Miyanohara A, Roth DM, Tang T, Hammond HK. Comparison of adeno‐associated virus serotypes and delivery methods for cardiac gene transfer. Hum Gene Ther Methods. 2012;23:234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prasad KM, Smith RS, Xu Y, French BA. A single direct injection into the left ventricular wall of an adeno‐associated virus 9 (AAV9) vector expressing extracellular superoxide dismutase from the cardiac troponin‐T promoter protects mice against myocardial infarction. J Gene Med. 2011;13:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weber C, Neacsu I, Krautz B, Schlegel P, Sauer S, Raake P, Ritterhoff J, Jungmann A, Remppis AB, Stangassinger M, Koch WJ, Katus HA, Muller OJ, Most P, Pleger ST. Therapeutic safety of high myocardial expression levels of the molecular inotrope S100A1 in a preclinical heart failure model. Gene Ther. 2014;21:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lipskaia L, Chemaly ER, Hadri L, Lompre AM, Hajjar RJ. Sarcoplasmic reticulum Ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woitek F, Zentilin L, Hoffman NE, Powers JC, Ottiger I, Parikh S, Kulczycki AM, Hurst M, Ring N, Wang T, Shaikh F, Gross P, Singh H, Kolpakov MA, Linke A, Houser SR, Rizzo V, Sabri A, Madesh M, Giacca M, Recchia FA. Intracoronary cytoprotective gene therapy: a study of VEGF‐B167 in a pre‐clinical animal model of dilated cardiomyopathy. J Am Coll Cardiol. 2015;66:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DiMattia MA, Nam HJ, Van Vliet K, Mitchell M, Bennett A, Gurda BL, McKenna R, Olson NH, Sinkovits RS, Potter M, Byrne BJ, Aslanidi G, Zolotukhin S, Muzyczka N, Baker TS, Agbandje‐McKenna M. Structural insight into the unique properties of adeno‐associated virus serotype 9. J Virol. 2012;86:6947–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Calcedo R, Wilson JM. Humoral immune response to AAV. Front Immunol. 2013;4:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bass‐Stringer S, Bernardo BC, May CN, Thomas CJ, Weeks KL, McMullen JR. Adeno‐associated virus gene therapy: translational progress and future prospects in the treatment of heart failure. Heart Lung Circ. 2018;27:1285–1300. [DOI] [PubMed] [Google Scholar]
- 40. Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ; Calcium Up‐Regulation by Percutaneous Administration of Gene Therapy In Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first‐in‐human phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ; Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+‐ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arruda VR, Xiao W. It's all about the clothing: capsid domination in the adeno‐associated viral vector world. J Thromb Haemost. 2007;5:12–15. [DOI] [PubMed] [Google Scholar]
- 44. Wahl SE, McLane LE, Bercury KK, Macklin WB, Wood TL. Mammalian target of rapamycin promotes oligodendrocyte differentiation, initiation and extent of CNS myelination. J Neurosci. 2014;34:4453–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rozas G, Guerra MJ, Labandeira‐Garcia JL. An automated rotarod method for quantitative drug‐free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc. 1997;2:75–84. [DOI] [PubMed] [Google Scholar]
- 46. Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, Wilson JM. Severe toxicity in nonhuman primates and piglets following high‐dose intravenous administration of an adeno‐associated virus vector expressing human SMN. Hum Gene Ther. 2018;29:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. The neurotropic properties of AAV‐PHP.B are limited to C57BL/6J mice. Mol Ther. 2018;26:664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mendell JR, Al‐Zaidy S, Shell R, Arnold WD, Rodino‐Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, Kissel JT, Nagendran S, L'Italien J, Sproule DM, Wells C, Cardenas JA, Heitzer MD, Kaspar A, Corcoran S, Braun L, Likhite S, Miranda C, Meyer K, Foust KD, Burghes AHM, Kaspar BK. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 49. Liu LC, Su CH, Wang HC, Tsai CW, Chang WS, Ho CY, Wu CI, Li FJ, Lin CH, Lane HY, Bau DT. Significant association of caveolin‐1 (CAV1) genotypes with breast cancer in Taiwan. Anticancer Res. 2011;31:3511–3515. [PubMed] [Google Scholar]
- 50. Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, Koch WJ. Myocardial adeno‐associated virus serotype 6‐betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McTiernan CF, Mathier MA, Zhu X, Xiao X, Klein E, Swan CH, Mehdi H, Gibson G, Trichel AM, Glorioso JC, Feldman AM, McCurry KR, London B. Myocarditis following adeno‐associated viral gene expression of human soluble TNF receptor (TNFRII‐Fc) in baboon hearts. Gene Ther. 2007;14:1613–1622. [DOI] [PubMed] [Google Scholar]
- 52. Roth DM, Lai NC, Gao MH, Drumm JD, Jimenez J, Feramisco JR, Hammond HK. Indirect intracoronary delivery of adenovirus encoding adenylyl cyclase increases left ventricular contractile function in mice. Am J Physiol Heart Circ Physiol. 2004;287:H172–H177. [DOI] [PubMed] [Google Scholar]
- 53. Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE, Koch WJ. Stable myocardial‐specific AAV6‐S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–2515. [DOI] [PubMed] [Google Scholar]
- 54. Lai NC, Roth DM, Gao MH, Fine S, Head BP, Zhu J, McKirnan MD, Kwong C, Dalton N, Urasawa K, Roth DA, Hammond HK. Intracoronary delivery of adenovirus encoding adenylyl cyclase VI increases left ventricular function and cAMP‐generating capacity. Circulation. 2000;102:2396–2401. [DOI] [PubMed] [Google Scholar]
- 55. Raake PW, Schlegel P, Ksienzyk J, Reinkober J, Barthelmes J, Schinkel S, Pleger S, Mier W, Haberkorn U, Koch WJ, Katus HA, Most P, Muller OJ. AAV6.betaARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur Heart J. 2013;34:1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, Schinkel S, Leuchs B, Ludwig J, Qiu G, Weber C, Raake P, Koch WJ, Katus HA, Muller OJ, Most P. Cardiac AAV9‐S100A1 gene therapy rescues post‐ischemic heart failure in a preclinical large animal model. Sci Transl Med. 2011;3:92ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Katz MG, Fargnoli AS, Swain JD, Tomasulo CE, Ciccarelli M, Huang ZM, Rabinowitz JE, Bridges CR. AAV6‐betaARKct gene delivery mediated by molecular cardiac surgery with recirculating delivery (MCARD) in sheep results in robust gene expression and increased adrenergic reserve. J Thorac Cardiovasc Surg. 2012;143:720–726.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sutherland EW. Studies on the mechanism of hormone action. Science. 1972;177:401–408. [DOI] [PubMed] [Google Scholar]
- 59. Gao M, Ping P, Post S, Insel PA, Tang R, Hammond HK. Increased expression of adenylyl cyclase type VI proportionately increases beta‐adrenergic receptor‐stimulated production of cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci USA. 1998;95:1038–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ishikawa Y, Sorota S, Kiuchi K, Shannon RP, Komamura K, Katsushika S, Vatner DE, Vatner SF, Homcy CJ. Downregulation of adenylylcyclase types V and VI mRNA levels in pacing‐induced heart failure in dogs. J Clin Invest. 1994;93:2224–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ping P, Anzai T, Gao M, Hammond HK. Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am J Physiol. 1997;273:H707–H717. [DOI] [PubMed] [Google Scholar]
- 62. Lai HL, Lin TH, Kao YY, Lin WJ, Hwang MJ, Chern Y. The N terminus domain of type VI adenylyl cyclase mediates its inhibition by protein kinase C. Mol Pharmacol. 1999;56:644–650. [DOI] [PubMed] [Google Scholar]
- 63. Gao MH, Lai NC, Roth DM, Zhou J, Zhu J, Anzai T, Dalton N, Hammond HK. Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation. 1999;99:1618–1622. [DOI] [PubMed] [Google Scholar]
- 64. Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. Cardiac‐directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099–3102. [DOI] [PubMed] [Google Scholar]
- 65. Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. [DOI] [PubMed] [Google Scholar]
- 66. Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WY, Clopton P, Hammond HK. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–396. [DOI] [PubMed] [Google Scholar]
- 67. Gao MH, Tang T, Guo T, Miyanohara A, Yajima T, Pestonjamasp K, Feramisco JR, Hammond HK. Adenylyl cyclase type VI increases Akt activity and phospholamban phosphorylation in cardiac myocytes. J Biol Chem. 2008;283:33527–33535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gao MH, Tang T, Guo T, Sun SQ, Feramisco JR, Hammond HK. Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J Biol Chem. 2004;279:38797–38802. [DOI] [PubMed] [Google Scholar]
- 69. Phan HM, Gao MH, Lai NC, Tang T, Hammond HK. New signaling pathways associated with increased cardiac adenylyl cyclase 6 expression: implications for possible congestive heart failure therapy. Trends Cardiovasc Med. 2007;17:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feldman AM. Adenylyl cyclase: a new target for heart failure therapeutics. Circulation. 2002;105:1876–1878. [DOI] [PubMed] [Google Scholar]
- 71. Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, Spellman M, Clopton P, Hammond HK. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation. 2004;110:330–336. [DOI] [PubMed] [Google Scholar]
- 72. Hammond HK, Penny WF, Traverse JH, Henry TD, Watkins MW, Yancy CW, Sweis RN, Adler ED, Patel AN, Murray DR, Ross RS, Bhargava V, Maisel A, Barnard DD, Lai NC, Dalton ND, Lee ML, Narayan SM, Blanchard DG, Gao MH. Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: a randomized clinical trial. JAMA Cardiol. 2016;1:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Penny WF, Henry TD, Watkins MW, Patel AN, Hammond HK. Design of a phase 3 trial of intracoronary administration of human adenovirus 5 encoding human adenylyl cyclase type 6 (RT‐100) gene transfer in patients with heart failure with reduced left ventricular ejection fraction: the FLOURISH Clinical Trial. Am Heart J. 2018;201:111–116. [DOI] [PubMed] [Google Scholar]
- 74. Gao MH, Lai NC, Giamouridis D, Kim YC, Tan Z, Guo T, Dillmann WH, Suarez J, Hammond HK. Cardiac‐directed expression of adenylyl cyclase catalytic domain reverses cardiac dysfunction caused by sustained beta‐adrenergic receptor stimulation. JACC Basic Transl Sci. 2016;1:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bristow MR, Port JD, Hershberger RE, Gilbert EM, Feldman AM. The beta‐adrenergic receptor‐adenylate cyclase complex as a target for therapeutic intervention in heart failure. Eur Heart J. 1989;10(suppl B):45–54. [DOI] [PubMed] [Google Scholar]
- 76. Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Model systems for the study of seven‐transmembrane‐segment receptors. Annu Rev Biochem. 1991;60:653–688. [DOI] [PubMed] [Google Scholar]
- 77. Bristow MR, Minobe WA, Raynolds MV, Port JD, Rasmussen R, Ray PE, Feldman AM. Reduced beta 1 receptor messenger RNA abundance in the failing human heart. J Clin Invest. 1993;92:2737–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Feldman AM, Cates AE, Veazey WB, Hershberger RE, Bristow MR, Baughman KL, Baumgartner WA, Van Dop C. Increase of the 40,000‐mol wt pertussis toxin substrate (G protein) in the failing human heart. J Clin Invest. 1988;82:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hausdorff WP, Caron MG, Lefkowitz RJ. Turning off the signal: desensitization of beta‐adrenergic receptor function. FASEB J. 1990;4:2881–2889. [PubMed] [Google Scholar]
- 80. Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Beta‐adrenergic receptor kinase: primary structure delineates a multigene family. Science. 1989;246:235–240. [DOI] [PubMed] [Google Scholar]
- 81. Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Role of beta gamma subunits of G proteins in targeting the beta‐adrenergic receptor kinase to membrane‐bound receptors. Science. 1992;257:1264–1267. [DOI] [PubMed] [Google Scholar]
- 82. Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta‐Arrestin: a protein that regulates beta‐adrenergic receptor function. Science. 1990;248:1547–1550. [DOI] [PubMed] [Google Scholar]
- 83. Bristow MR, Feldman AM. Changes in the receptor‐G protein‐adenylyl cyclase system in heart failure from various types of heart muscle disease. Basic Res Cardiol. 1992;87(suppl 1):15–35. [DOI] [PubMed] [Google Scholar]
- 84. Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the beta‐adrenergic receptor kinase or a beta ARK inhibitor. Science. 1995;268:1350–1353. [DOI] [PubMed] [Google Scholar]
- 85. Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J Jr, Lefkowitz RJ, Koch WJ. Expression of a beta‐adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene‐targeted mice. Proc Natl Acad Sci USA. 1998;95:7000–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Harding VB, Jones LR, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac beta ARK1 inhibition prolongs survival and augments beta blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci USA. 2001;98:5809–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tachibana H, Naga Prasad SV, Lefkowitz RJ, Koch WJ, Rockman HA. Level of beta‐adrenergic receptor kinase 1 inhibition determines degree of cardiac dysfunction after chronic pressure overload‐induced heart failure. Circulation. 2005;111:591–597. [DOI] [PubMed] [Google Scholar]
- 88. Tevaearai HT, Walton GB, Keys JR, Koch WJ, Eckhart AD. Acute ischemic cardiac dysfunction is attenuated via gene transfer of a peptide inhibitor of the beta‐adrenergic receptor kinase (betaARK1). J Gene Med. 2005;7:1172–1177. [DOI] [PubMed] [Google Scholar]
- 89. Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech. 2003;60:540–551. [DOI] [PubMed] [Google Scholar]
- 90. Haimoto H, Kato K. S100a0 (alpha alpha) protein in cardiac muscle. Isolation from human cardiac muscle and ultrastructural localization. Eur J Biochem. 1988;171:409–415. [DOI] [PubMed] [Google Scholar]
- 91. Kato K, Kimura S. S100ao (alpha alpha) protein is mainly located in the heart and striated muscles. Biochem Biophys Acta. 1985;842:146–150. [DOI] [PubMed] [Google Scholar]
- 92. Most P, Boerries M, Eicher C, Schweda C, Volkers M, Wedel T, Sollner S, Katus HA, Remppis A, Aebi U, Koch WJ, Schoenenberger CA. Distinct subcellular location of the Ca2+‐binding protein S100A1 differentially modulates Ca2+‐cycling in ventricular rat cardiomyocytes. J Cell Sci. 2005;118:421–431. [DOI] [PubMed] [Google Scholar]
- 93. Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Borries M, Niroomand F, Pieske B, Janssen PM, Eschenhagen T, Karczewski P, Smith GL, Koch WJ, Katus HA, Remppis A. S100A1: a regulator of myocardial contractility. Proc Natl Acad Sci USA. 2001;98:13889–13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Most P, Remppis A, Pleger ST, Loffler E, Ehlermann P, Bernotat J, Kleuss C, Heierhorst J, Ruiz P, Witt H, Karczewski P, Mao L, Rockman HA, Duncan SJ, Katus HA, Koch WJ. Transgenic overexpression of the Ca2+‐binding protein S100A1 in the heart leads to increased in vivo myocardial contractile performance. J Biol Chem. 2003;278:33809–33817. [DOI] [PubMed] [Google Scholar]
- 95. Kettlewell S, Most P, Currie S, Koch WJ, Smith GL. S100A1 increases the gain of excitation‐contraction coupling in isolated rabbit ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:900–910. [DOI] [PubMed] [Google Scholar]
- 96. Volkers M, Loughrey CM, Macquaide N, Remppis A, DeGeorge BR Jr, Wegner FV, Friedrich O, Fink RH, Koch WJ, Smith GL, Most P. S100A1 decreases calcium spark frequency and alters their spatial characteristics in permeabilized adult ventricular cardiomyocytes. Cell Calcium. 2007;41:135–143. [DOI] [PubMed] [Google Scholar]
- 97. Du XJ, Cole TJ, Tenis N, Gao XM, Kontgen F, Kemp BE, Heierhorst J. Impaired cardiac contractility response to hemodynamic stress in S100A1‐deficient mice. Mol Cell Biol. 2002;22:2821–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PM, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004;114:1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Davis J, Maillet M, Miano JM, Molkentin JD. Lost in transgenesis: a user's guide for genetically manipulating the mouse in cardiac research. Circ Res. 2012;111:761–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ladenson PW, Sherman SI, Baughman KL, Ray PE, Feldman AM. Reversible alterations in myocardial gene expression in a young man with dilated cardiomyopathy and hypothyroidism. Proc Natl Acad Sci USA. 1992;89:5251–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)‐ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. [DOI] [PubMed] [Google Scholar]
- 102. Feldman AM, Weinberg EO, Ray PE, Lorell BH. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ Res. 1993;73:184–192. [DOI] [PubMed] [Google Scholar]
- 103. Arai M, Matsui H, Periasamy M. Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ Res. 1994;74:555–564. [DOI] [PubMed] [Google Scholar]
- 104. Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left‐ventricular function in aortic‐banded rats in transition to heart failure. Proc Natl Acad Sci USA. 2000;97:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. del Monte F, Hajjar RJ, Harding SE. Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ Res. 2001;88:E66–E67. [DOI] [PubMed] [Google Scholar]
- 106. Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, Nakajima‐Takenaka C, Tsuji T, Konishi N, del Monte F, Hajjar RJ, Takaki M. Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Physiol Heart Circ Physiol. 2007;292:H1204–H1207. [DOI] [PubMed] [Google Scholar]
- 107. Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, Hadri L, Yoneyama R, Hoshino K, Takewa Y, Sakata S, Peluso R, Zsebo K, Gwathmey JK, Tardif JC, Tanguay JF, Hajjar RJ. Reversal of cardiac dysfunction after long‐term expression of SERCA2a by gene transfer in a pre‐clinical model of heart failure. J Am Coll Cardiol. 2008;51:1112–1119. [DOI] [PubMed] [Google Scholar]
- 108. Carter PJ, Samulski RJ. Adeno‐associated viral vectors as gene delivery vehicles. Int J Mol Med. 2000;6:17–27. [DOI] [PubMed] [Google Scholar]
- 109. Greenberg B, Yaroshinsky A, Zsebo KM, Butler J, Felker GM, Voors AA, Rudy JJ, Wagner K, Hajjar RJ. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up‐regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail. 2014;2:84–92. [DOI] [PubMed] [Google Scholar]
- 110. MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. [DOI] [PubMed] [Google Scholar]
- 111. Feldman AM, Ray PE, Silan CM, Mercer JA, Minobe W, Bristow MR. Selective gene expression in failing human heart. Quantification of steady‐state levels of messenger RNA in endomyocardial biopsies using the polymerase chain reaction. Circulation. 1991;83:1866–1872. [DOI] [PubMed] [Google Scholar]
- 112. Williams RE, Kass DA, Kawagoe Y, Pak P, Tunin RS, Shah R, Hwang A, Feldman AM. Endomyocardial gene expression during development of pacing tachycardia‐induced heart failure in the dog. Circ Res. 1994;75:615–623. [DOI] [PubMed] [Google Scholar]
- 113. McTiernan CF, Frye CS, Lemster BH, Kinder EA, Ogletree‐Hughes ML, Moravec CS, Feldman AM. The human phospholamban gene: structure and expression. J Mol Cell Cardiol. 1999;31:679–692. [DOI] [PubMed] [Google Scholar]
- 114. McTiernan CF, Lemster BH, Frye C, Brooks S, Combes A, Feldman AM. Interleukin‐1 beta inhibits phospholamban gene expression in cultured cardiomyocytes. Circ Res. 1997;81:493–503. [DOI] [PubMed] [Google Scholar]
- 115. Gupta RC, Mishra S, Rastogi S, Imai M, Habib O, Sabbah HN. Cardiac SR‐coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am J Physiol Heart Circ Physiol. 2003;285:H2373–H2381. [DOI] [PubMed] [Google Scholar]
- 116. Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli‐Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N. Increased expression of cardiac phosphatases in patients with end‐stage heart failure. J Mol Cell Cardiol. 1997;29:265–272. [DOI] [PubMed] [Google Scholar]
- 118. Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli‐Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–766. [DOI] [PubMed] [Google Scholar]
- 119. Chen G, Zhou X, Florea S, Qian J, Cai W, Zhang Z, Fan GC, Lorenz J, Hajjar RJ, Kranias EG. Expression of active protein phosphatase 1 inhibitor‐1 attenuates chronic beta‐agonist‐induced cardiac apoptosis. Basic Res Cardiol. 2010;105:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, Mitton B, Pathak A, Robbins J, Hajjar RJ, Jones K, Kranias EG. Inducible expression of active protein phosphatase‐1 inhibitor‐1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res. 2009;104:1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schwab DM, Tilemann L, Bauer R, Heckmann M, Jungmann A, Wagner M, Burgis J, Vettel C, Katus HA, El‐Armouche A, Muller OJ. AAV‐9 mediated phosphatase‐1 inhibitor‐1 overexpression improves cardiac contractility in unchallenged mice but is deleterious in pressure‐overload. Gene Ther. 2018;25:13–19. [DOI] [PubMed] [Google Scholar]
- 122. Wittkopper K, Fabritz L, Neef S, Ort KR, Grefe C, Unsold B, Kirchhof P, Maier LS, Hasenfuss G, Dobrev D, Eschenhagen T, El‐Armouche A. Constitutively active phosphatase inhibitor‐1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest. 2010;120:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Fish KM, Ladage D, Kawase Y, Karakikes I, Jeong D, Ly H, Ishikawa K, Hadri L, Tilemann L, Muller‐Ehmsen J, Samulski RJ, Kranias EG, Hajjar RJ. AAV9.I‐1c delivered via direct coronary infusion in a porcine model of heart failure improves contractility and mitigates adverse remodeling. Circ Heart Fail. 2013;6:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ishikawa K, Fish KM, Tilemann L, Rapti K, Aguero J, Santos‐Gallego CG, Lee A, Karakikes I, Xie C, Akar FG, Shimada YJ, Gwathmey JK, Asokan A, McPhee S, Samulski J, Samulski RJ, Sigg DC, Weber T, Kranias EG, Hajjar RJ. Cardiac I‐1c overexpression with reengineered AAV improves cardiac function in swine ischemic heart failure. Mol Ther. 2014;22:2038–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wittkopper K, Dobrev D, Eschenhagen T, El‐Armouche A. Phosphatase‐1 inhibitor‐1 in physiological and pathological beta‐adrenoceptor signalling. Cardiovasc Res. 2011;91:392–401. [DOI] [PubMed] [Google Scholar]
- 126. Overman EL, Rivier JE, Moeser AJ. CRF induces intestinal epithelial barrier injury via the release of mast cell proteases and TNF‐alpha. PLoS One. 2012;7:e39935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Stirrat CG, Venkatasubramanian S, Pawade T, Mitchell AJ, Shah AS, Lang NN, Newby DE. Cardiovascular effects of urocortin 2 and urocortin 3 in patients with chronic heart failure. Br J Clin Pharmacol. 2016;82:974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Venkatasubramanian S, Newby DE, Lang NN. Urocortins in heart failure. Biochem Pharmacol. 2010;80:289–296. [DOI] [PubMed] [Google Scholar]
- 129. Gheorghiade M, Greene SJ, Ponikowski P, Maggioni AP, Korewicki J, Macarie C, Metra M, Grzybowski J, Bubenek‐Turconi SI, Radziszewski W, Olson A, Bueno OF, Ghosh A, Deckelbaum LI, Li LY, Patel AR, Koester A, Konstam MA. Haemodynamic effects, safety, and pharmacokinetics of human stresscopin in heart failure with reduced ejection fraction. Eur J Heart Fail. 2013;15:679–689. [DOI] [PubMed] [Google Scholar]
- 130. Chan WY, Frampton CM, Crozier IG, Troughton RW, Richards AM. Urocortin‐2 infusion in acute decompensated heart failure: findings from the UNICORN study (urocortin‐2 in the treatment of acute heart failure as an adjunct over conventional therapy). JACC Heart Fail. 2013;1:433–441. [DOI] [PubMed] [Google Scholar]
- 131. Giamouridis D, Gao MH, Lai NC, Tan Z, Kim YC, Guo T, Miyanohara A, Blankesteijn WM, Biessen E, Hammond HK. Effects of urocortin 2 versus urocortin 3 gene transfer on left ventricular function and glucose disposal. JACC Basic Transl Sci. 2018;3:249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Giamouridis D, Gao MH, Lai NC, Tan Z, Kim YC, Guo T, Miyanohara A, Blankesteijn MW, Biessen EAL, Hammond HK. Urocortin 3 gene transfer increases function of the failing murine heart. Hum Gene Ther. 2019;30:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Behl C. Breaking BAG: the co‐chaperone BAG3 in health and disease. Trends Pharmacol Sci. 2016;37:672–688. [DOI] [PubMed] [Google Scholar]
- 134. Myers VD, McClung JM, Wang J, Tahrir FG, Gupta MK, Gordon J, Kontos CH, Khalili K, Cheung JY, Feldman AM. The multifunctional protein BAG3: a novel therapeutic target in cardiovascular disease. JACC Basic Transl Sci. 2018;3:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Feldman AM, Gordon J, Wang J, Song J, Zhang XQ, Myers VD, Tilley DG, Gao E, Hoffman NE, Tomar D, Madesh M, Rabinowitz J, Koch WJ, Su F, Khalili K, Cheung JY. BAG3 regulates contractility and Ca(2+) homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016;92:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Su F, Myers VD, Knezevic T, Wang J, Gao E, Madesh M, Tahrir FG, Gupta MK, Gordon J, Rabinowitz J, Ramsey FV, Tilley DG, Khalili K, Cheung JY, Feldman AM. Bcl‐2‐associated athanogene 3 protects the heart from ischemia/reperfusion injury. JCI Insight. 2016;1:e90931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tahrir FG, Knezevic T, Gupta MK, Gordon J, Cheung JY, Feldman AM, Khalili K. Evidence for the role of BAG3 in mitochondrial quality control in cardiomyocytes. J Cell Physiol. 2017;232:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, Lao DH, Dalton ND, Gu Y, Wang C, Wang M, Liang Y, Lange S, Ouyang K, Peterson KL, Evans SM, Chen J. Loss‐of‐function mutations in co‐chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Knezevic T, Myers VD, Gordon J, Tilley DG, Sharp TE III, Wang J, Khalili K, Cheung JY, Feldman AM. BAG3: a new player in the heart failure paradigm. Heart Fail Rev. 2015;20:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Myers VD, Tomar D, Madesh M, Wang J, Song J, Zhang XQ, Gupta MK, Tahrir FG, Gordon J, McClung JM, Kontos CD, Khalili K, Cheung JY, Feldman AM. Haplo‐insufficiency of Bcl2‐associated athanogene 3 in mice results in progressive left ventricular dysfunction, beta‐adrenergic insensitivity, and increased apoptosis. J Cell Physiol. 2018;233:6319–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Feldman AM, Begay RL, Knezevic T, Myers VD, Slavov DB, Zhu W, Gowan K, Graw SL, Jones KL, Tilley DG, Coleman RC, Walinsky P, Cheung JY, Mestroni L, Khalili K, Taylor MR. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J Cell Physiol. 2014;229:1697–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Toro R, Perez‐Serra A, Campuzano O, Moncayo‐Arlandi J, Allegue C, Iglesias A, Mangas A, Brugada R. Familial dilated cardiomyopathy caused by a novel frameshift in the BAG3 gene. PLoS One. 2016;11:e0158730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dominguez F, Cuenca S, Bilinska Z, Toro R, Villard E, Barriales‐Villa R, Ochoa JP, Asselbergs F, Sammani A, Franaszczyk M, Akhtar M, Coronado‐Albi MJ, Rangel‐Sousa D, Rodriguez‐Palomares JF, Jimenez‐Jaimez J, Garcia‐Pinilla JM, Ripoll‐Vera T, Mogollon‐Jimenez MV, Fontalba‐Romero A, Garcia‐Medina D, Palomino‐Doza J, de Gonzalo‐Calvo D, Cicerchia M, Salazar‐Mendiguchia J, Salas C, Pankuweit S, Hey TM, Mogensen J, Barton PJ, Charron P, Elliott P, Garcia‐Pavia P; European Genetic Cardiomyopathies Initiative Investigators . Dilated cardiomyopathy due to BLC2‐associated athanogene 3 (BAG3) mutations. J Am Coll Cardiol. 2018;72:2471–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Myers VD, Gerhard GS, McNamara DM, Tomar D, Madesh M, Kaniper S, Ramsey FV, Fisher SG, Ingersoll RG, Kasch‐Semenza L, Wang J, Hanley‐Yanez K, Lemster B, Schwisow JA, Ambardekar AV, Degann SH, Bristow MR, Sheppard R, Alexis JD, Tilley DG, Kontos CD, McClung JM, Taylor AL, Yancy CW, Khalili K, Seidman JG, Seidman CE, McTiernan CF, Cheung JY, Feldman AM. Association of variants in BAG3 with cardiomyopathy outcomes in African American individuals. JAMA Cardiol. 2018;3:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Knezevic T, Myers VD, Su F, Wang J, Song J, Zhang XQ, Gao E, Gao G, Muniswamy M, Gupta MK, Gordon J, Weiner KN, Rabinowitz J, Ramsey FV, Tilley DG, Khalili K, Cheung JY, Feldman AM. Adeno‐associated virus serotype 9–driven expression of BAG3 improves left ventricular function in murine hearts with left ventricular dysfunction secondary to a myocardial infarction. JACC Basic Transl Sci. 2016;1:647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]