

Abstract
Background
We previously reported that vascular smooth muscle cells (VSMCs) from spontaneously hypertensive rats (SHRs) show the increased expression of complement 3 (C3) and the synthetic phenotype. We targeted the SHR C3 gene (C3 knockout [C3KO] SHRs) by the zinc finger gene editing method. In the current study, we investigated the mechanisms underlying the increased expression of C3 and the role of endogenous C3 in the synthetic phenotype of SHR VSMCs in comparison to cells from Wistar‐Kyoto (WKY) rats and C3KO SHRs.
Methods and Results
Nonmuscle myosin heavy chain staining of aortas from SHRs at 1 day after birth was stronger in comparison to WKY rats and C3KO SHRs. DNA synthesis in VSMCs from SHRs was significantly higher in comparison to WKY rats and C3KO SHRs. Immunohistochemical staining of renin and liver X receptor α in VSMCs from SHRs was stronger in comparison to WKY rats and C3KO SHRs. The expression of renin, Krüppel‐like factor 5, and liver X receptor α proteins in VSMCs from SHRs was significantly higher in comparison to WKY rats and C3KO SHRs. The expression of synthetic phenotype markers osteopontin, matrix gla, and l‐caldesmon, growth factors transforming growth factor‐β1 and platelet‐derived growth factor‐A, transcription factors Krüppel‐like factor 5 and liver X receptor α, and angiotensinogen mRNAs in VSMCs from SHRs was significantly higher in comparison to WKY rats and C3KO SHRs. The expression of miR‐145 mRNA in VSMCs from SHRs was suppressed in comparison to cells from WKY rats. miR‐145 inhibitor significantly increased the expression of C3 in VSMCs from WKY rats, but not in cells from SHRs.
Conclusions
These findings indicate that the increased C3 with the suppression of miR‐145 induces the synthetic phenotype through Krüppel‐like factor 5 and the activation of the renin‐angiotensin system through liver X receptor α in VSMCs from SHRs.
Keywords: complement 3, gene editing, miR‐145, spontaneously hypertensive rat, vascular smooth muscle
Subject Categories: Smooth Muscle Proliferation and Differentiation, Hypertension
Clinical Perspective
What Is New?
Using complement 3 (C3) knockout spontaneously hypertensive rats, established by zinc finger nuclease gene editing technology, we investigated the mechanisms underlying the increased expression of C3 in vascular smooth muscle cells from spontaneously hypertensive rats.
We found that the increased expression of C3 is involved in the synthetic phenotype and the exaggerated growth of vascular smooth muscle cells from spontaneously hypertensive rats.
We found that the mechanisms underlying the increases in the expression of C3 are associated with the suppression of miR‐145.
What Are the Clinical Implications?
Vascular smooth muscle cells in spontaneously hypertensive rats constitutively and increasingly express C3, which is independent of the immune system.
The increased expression of C3 may contribute to the activation of renin‐angiotensin system in vascular smooth muscle cells, which contributes to the pathogenesis of hypertension.
Introduction
Essential hypertension is eventually associated with cardiovascular complications, such as stroke, hypertensive heart disease, and renal sclerosis. Spontaneously hypertensive rats (SHRs) are widely used as a genetic model of essential hypertension to investigate the pathogenesis of essential hypertension. Previous studies have demonstrated that SHRs show the exaggerated growth of mesenchymal cells, such as vascular smooth muscle cells (VSMCs) and renal mesangial cells in comparison to normotensive Wistar‐Kyoto (WKY) rats.1, 2, 3 SHRs have been reported to show enhanced DNA synthesis and hypertrophy in cardiovascular organs from as early as the day of birth.4, 5 We also found that VSMCs from prehypertensive SHRs showed hyperproliferation in comparison to VSMCs from WKY rats.2 These findings indicate that the exaggerated growth of VSMCs from SHRs is not only caused by hypertension, but also by intrinsic abnormalities. We have demonstrated that SHR‐derived VSMCs generate angiotensin II, with increases in angiotensin II–generating enzymes, cathepsin D, and angiotensin‐converting enzyme, which was associated with a phenotypic change from contractile to synthetic in VSMCs.2, 6, 7 To find the intrinsic factors inducing the synthetic phenotype of VSMCs from SHRs, we performed a microarray analysis of aortic smooth muscle; and we found that the transcript encoding complement 3 (C3) is only expressed in VSMCs from SHRs, and not in cells from WKY rats. We then verified that C3 contributes to the synthetic phenotype and exaggerated growth of VSMCs and mesangial cells from SHRs; inhibition of C3a and C3a receptor in SHR‐derived VSMCs and MCs leads to the transition from the synthetic phenotype to the contractile phenotype.3, 8, 9 Moreover, we demonstrated that C3a stimulates the promoter activity of Krüppel‐like factor 5 (KLF5), which induces the synthetic phenotype of VSMCs.10 In addition, we recently demonstrated that unilateral ureteral obstruction model mice show increased blood pressure and strong staining of C3 and renin in the degenerated nephrotubulus through the epithelial‐mesenchymal transition (EMT) in the kidney. Intrarenal angiotensin II levels were markedly higher in the unilateral ureteral obstruction kidney. However, the EMT phenomenon and the activation of the intrarenal renin‐angiotensin system (RAS) were abolished in C3 knockout (C3KO) mice, indicating that C3 induces the EMT, maintains the synthetic phenotype of mesenchymal cells, and activates the intrarenal RASs.11 To verify the contribution of C3 in the pathogenesis of hypertension in SHRs, we recently established C3KO SHRs using zinc finger nuclease (ZFN) gene editing technology and found that the salt‐sensitive hypertension with intrarenal activation of the RAS by the EMT phenomenon shown in SHRs was abolished in C3KO SHRs.12 Thus, we have clarified that the increased C3 expression induces salt‐sensitive hypertension with the activation of the RAS through the EMT phenomenon and dedifferentiation of mesenchymal tissues in SHRs.
In the current study, we investigated mechanisms underlying the increased expression of C3 and the contribution of endogenous C3 in the synthetic phenotype and the exaggerated growth in VSMCs from SHRs using VSMCs from C3KO SHRs established by the ZFN gene editing technology.
Methods
Ethics and Animals
Our research followed the guidelines of the ethics committee of the Nihon University School of Medicine (Tokyo, Japan; No. 11‐034) and the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (publication No. 85‐23, 1996). We also received official acknowledgement of the generation of C3KO SHR/Izm by ZFN methods from the Disease Model Cooperative Research Association. C3KO SHR/Izm (SHR‐C3emKyo) are preserved in the National Bio Resource Project for the Rat in Japan (http://www.anim.med.kyoto-u.ac.jp/nbr).
Generation of C3KO SHRs
The generation of ZFN mutants was performed with reference to a previously published article.13 ZFN constructs targeting bases 1803 to 1841 (National Center for Biotechnology Information reference sequence: NM_016994) of C3 (target sequence: cagggggcccgagtgggctagtggctgtggacaagggg) were designed, assembled, and validated by Sigma‐Aldrich (Tokyo, Japan). Briefly, transcribed mRNA encoding the C3 ZFNs was diluted and injected into the pronucleus of SHR/Izm embryos. One hundred embryos were injected and transplanted to pseudopregnant SHR/Izm females. At 10 days of age, DNA was extracted from ear tissue and screened for ZFN‐induced mutations using primers flanking the target sequence (forward primer: 5′‐ACTCTTCCCTGTCTTGCGTC‐3′; and reverse primer: 5′‐AATAGAGGCCACCAATGCAC‐3′) and polyacrylamide gel electrophoresis. Positive mutant pups were identified, and polymerase chain reaction (PCR) products from the two pups were cloned using the PCR2.1‐TOPO kit (Invitrogen, Carlsbad, CA), according to the user guide. Sequencing of the mutant revealed a 9‐base frameshift deletion of bases 1815 to 1824 (ggctagtgg). The F4 generation of homozygote C3KO SHRs was used for the practical experiments.
Immunohistochemical Staining of Phenotype Markers in the Aorta
Aortas were removed from WKY rats, SHRs, and C3KO SHRs at 1 day, 8 weeks, and 20 weeks of age; and the endothelium was completely denudated. The specimens were incubated with smooth muscle heavy chain 1 (SM1) and nonmuscle myosin heavy chain (SMemb) monoclonal antibodies (Yamasa, Tokyo, Japan) for 1 hour at room temperature. They were then washed with phosphate‐buffered saline (PBS), and incubated with secondary antibody for 30 minutes. Next, they were washed with PBS, incubated with horseradish peroxidase–conjugated antibiotin labeling solution (ABC kit; Vector) for 30 minutes, then developed with 3,3′‐diaminobenzidine, and counterstained with hematoxylin.
Culture of VSMCs
VSMCs were obtained from the aortas of 3‐week‐old male WKY rats, SHRs, and C3KO SHRs by the explant method, as described previously.14 The explants of aorta were from 3 rats. They were seeded and grown in Dulbecco's Modified Eagle Medium(DMEM) with 10% fetal bovine serum (FBS; Gibco Life Technologies), penicillin (100 U/mL), and streptomycin (100 mg/mL). The cells achieved confluence after 7 to 10 days, at which time we confirmed the hill‐and‐valley pattern characteristic of smooth muscle cells in culture. The cultures were uniformly composed of VSMCs from WKY rats, SHRs, and C3KO SHRs that were positively stained for smooth muscle–specific α‐actin. They were passaged by trypsinization with 0.05% trypsin (Gibco) in Ca2+‐ and Mg2+‐free Dulbecco's PBS and incubated in 75‐cm2 tissue culture flasks at a density of 105 cells/mL. Experiments were performed after 3 to 5 passages.
Immunocytochemistry
After serum starvation with 0.5% FBS for 24 hours, VSMCs were cultured in 4‐well chamber slides; fixed with 4% paraformaldehyde; permeabilized with 0.5% Triton X‐100; blocked in 10% goat serum and 1% BSA; incubated with rabbit polyclonal antibody against α‐smooth muscle actin (1:100; Abcam), rabbit polyclonal antibody against C3 α chain (1:100; Abcam), rabbit polyclonal antibody against renin (1:100; Proteintech Group, Rosemont, IL), and rabbit polyclonal antibody against liver X receptor α (LXRα; 1:100; Abcam) at 4°C overnight, followed by Alexa Fluor 594 antirabbit secondary antibody (Thermo Fisher Scientific); and mounted in Prolong Gold with 4’,6‐diamidino‐2‐phenylindole.
Cell Contractility of VSMCs
Cell contractility was evaluated using the collagen gel contraction assay (Cell Contraction Assay CBA‐201; Cell Biolabs, San Diego, CA), according to the product manual. Briefly, VSMCs were suspended (3.0×106 cells/mL) in medium, and the collagen gel working solution/cell mixture (4:1) was dispensed (0.5 mL/well) into 24‐well plates and incubated for 1 hour at 37°C. One milliliter of culture medium was added to each well. After incubation for 2 days, the collagen gels were released and photographed, and their diameter was measured after releasing for 20 and 30 minutes. Cell contractility was expressed in terms of the decrease in gel diameter in comparison to the initial diameter.
DNA Synthesis of VSMCs
DNA synthesis in VSMCs was assessed by 5‐bromo‐2‐deoxyuridine labeling and detection with an ELISA kit (Amersham Biosciences, Buckinghamshire, UK). Briefly, VSMCs were subcultured in 96‐well plates (5×103 cells/well) and starved of 0.5% FBS for 24 hours, then incubated with 0.5%, 2%, and 5% FBS for 24 hours. Next, 5‐bromo‐2‐deoxyuridine labeling reagent was added, and the cells were cultured for a further 2 hours. Cellular viability was quantified as the absorbance at 450 nm, measured by the microplate manager software program (version 5.2; Bio‐Rad Laboratories, Tokyo, Japan).
Western Blotting of Aorta Specimens and VSMCs
For Western blotting of aorta specimens, aortas were removed from WKY rats, SHRs, and C3KO SHRs at 3 weeks of age; then, the endothelium was removed using a surgical knife, and the specimens were homogenized using a Polytron PT10 Homogenizer (Kinematica AG, Luzern, Switzerland). For Western blotting of VSMCs, after serum starvation for 24 hours, cell protein extracts were obtained by lysing whole cells in radioimmunoprecipitation assay buffer (08714‐04; Nacalai Tesque, Kyoto, Japan) and the protein concentration was quantified with a Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). Equal amounts of protein were mixed with LDS Sample Buffer (Thermo Fisher Scientific) and Sample Reducing Agent (Thermo Fisher Scientific), heated for 10 minutes at 70°C, and loaded in NuPAGE 4% to 12% Bis‐Tris gel (Invitrogen). The separated proteins were electrophoretically transferred onto a polyvinylidene difluoride membrane. The membranes were blocked with blocking buffer. The membranes were then incubated overnight with primary antibodies: C3 α chain (1:100; Abcam, Cambridge, UK), h‐caldesmon (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), renin (1:200; Proteintech Group), KLF‐5 (1:500; Abcam), and LXRα (1:4000; Abcam) at 4°C. Then, they were washed and incubated with horseradish peroxidase–conjugated goat antirabbit (Jackson ImmunoResearch, West Grove, PA) (used for C3, renin, KLF5, and LXRα) and antimouse IgG (Jackson ImmunoResearch) (used for h‐caldesmon). Finally, the membranes were treated with chemiluminescent reagent and images were acquired using a cooled charge‐coupled device camera (LAS‐3000 Mini; Fujifilm, Tokyo, Japan). Each protein was quantified and normalized by β‐actin using the Image J software program (National Institutes of Health, Bethesda, MD). The C3 protein expression in the aorta tissue of 3‐week old male WKY rats, SHRs, and C3KO SHRs was evaluated using the same method.
Real‐Time PCR Relative Quantitation of VSMCs
After serum starvation for 24 hours, RNA was extracted using an RNeasy mini kit (Qiagen, Tokyo, Japan), treated with RNase‐free DNase I (Qiagen), and reverse transcribed to cDNA with PrimeScript RT Master Mix (Takara, Japan). Quantitative PCR (qPCR) relative quantitation analysis was performed with Probe qPCR Mix (Takara Bio, Shiga, Japan) using the Step‐One Plus Real‐Time PCR System (Applied Biosystems). The TaqMan probes used in the qPCR were as follows: osteopontin (Rn00681031_m1), matrix gla (Rn00563463_m1), l‐caldesmon (Rn00565719_m1), transforming growth factor (TGF)‐β1 (Rn00572010_m1), platelet‐derived growth factor (PDGF)‐A (Rn00709363_m1), KLF5 (Rn00821442_g1), LXRα (Rn00581185_ml), angiotensinogen (Rn00593114_m1), h‐caldesmon (sense, GGAGGAGGCGAAGGCTAGG; antisense, CTCTCTCCGCTCCCTTCTCC; probe, [6FAM]CCTTTGCTTCCTGCCTCTCACTCCTTTGC[TAM]), and 18S ribosomal RNA (4352930). The miR‐145 (has‐miR‐145, 002278) and miR‐143 (has‐miR‐143, 000466) expression levels were measured by microRNA quantitative real‐time analysis using TaqMan MicroRNA assays (Thermo Fisher Scientific), according to the method of a previous publication.15 Approximately 10 ng of total RNA was used for microRNA‐specific reverse transcription, which was performed using the TaqMan MicroRNA reverse transcription kit (Thermo Fisher Scientific); qPCR was performed with the TaqMan microRNA assay kit using the Step‐One Plus Real‐Time PCR system (Thermo Fisher Scientific). All thermal cycling conditions were performed according to the manufacturer's instructions. The relative expression was determined by the relative standard curve method and normalized to the expression of 18S ribosomal RNA.
Suppression of miR‐143 and miR‐145 With Inhibitors
VSMCs (105 cells/cm2) were transfected with miR‐145 inhibitor or miR‐143 inhibitor (Thermo Fisher Scientific) in 6‐well plates using 100 nmol/L lipofectamine (Introgen, Austin, TX). Two days later, total RNA was extracted and real‐time qPCR was performed.
Statistical Analysis
Values are reported as mean±SEM. Comparisons between 3 groups were analyzed by using 1‐ or 2‐way ANOVA, followed by the Bonferroni post hoc test. One‐way repeated‐measure ANOVA was used to determine the statistical difference at different time points. Statistical differences within groups or between 2 groups were assessed by using the Student t test. P<0.05 was considered to indicate statistical significance.
Results
Expression of C3 α Chain
The abundance of C3 α‐chain protein in aortas from SHRs was significantly higher in comparison to aortas (P=0.012) from WKY rats, which was significantly (P<0.01) lower in aortas from C3KO SHRs (Figure 1A). Staining of C3 α chain was stronger in VSMCs from SHRs than VSMCs from WKY rats and C3KO SHRs (Figure 1B). The present results demonstrate that the expression of C3 in the aortas and VSMCs from C3KO SHRs was much lower in comparison to SHRs, indicating that ZFN gene editing technology could target the C3 gene from SHRs.
Figure 1.
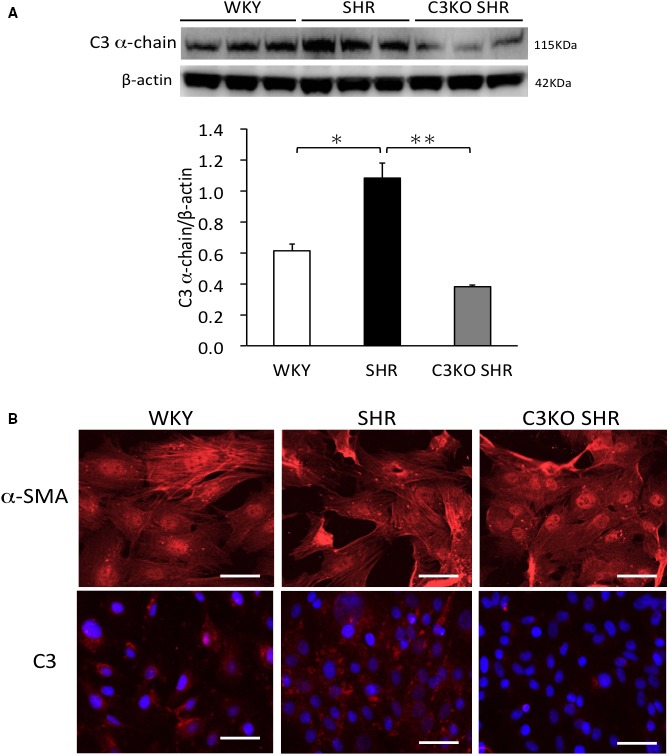
A, Western blotting of complement 3 (C3) α chain in aortas. Aortas were removed from 3‐week‐old Wistar‐Kyoto (WKY) rats, spontaneously hypertensive rats (SHRs), and C3 knockout (C3KO) SHRs, and then the endothelium was removed using surgical knife and homogenized. (One‐way ANOVA was performed, followed by Bonferroni post hoc test.) Data indicate the mean±SEM (n=3). *P<0.05, **P<0.01 between indicated columns. B, Immunofluorescence of α‐smooth muscle–specific actin (α‐SMA) and C3 α chain in vascular smooth muscle cells (VSMCs). VSMCs were acquired from the aortas of 3‐week‐old WKY rats, SHRs, and C3KO SHRs by the explant method. Bar=50 μm.
Expression of SM1 and SMemb in the Aorta
Staining of SM1 (a contractile phenotype marker) in aortas from WKY rats was stronger in comparison to aortas from SHRs at 1 day after birth. SM1 staining of aortas obtained at 8 and 20 weeks of age from WKY rats, SHRs, and C3KO SHRs did not differ to a statistically significant extent (Figure 2A). Staining of SMemb (a synthetic phenotype marker) in aortas obtained from SHRs at 1 day and 8 weeks was stronger in comparison to that in aortas obtained from WKY rats, which was weaker in aortas from C3KO SHRs. SMemb staining of aortas obtained at 20 weeks of age from WKY rats, SHRs, and C3KO SHRs did not differ to a statistically significant extent (Figure 2B).
Figure 2.
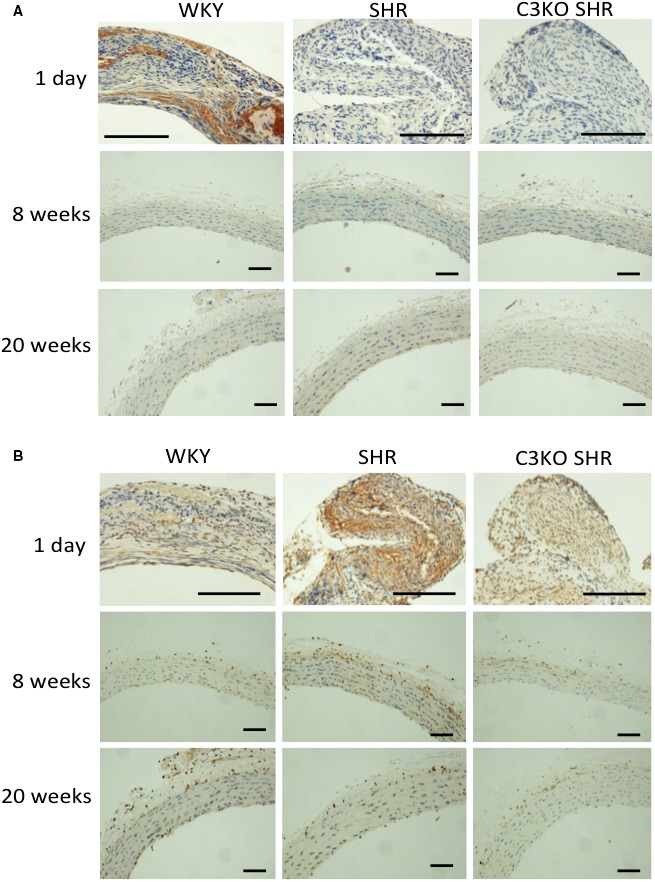
Immunohistochemical staining of smooth muscle heavy chain 1 (SM1) (A) and nonmuscle myosin heavy chain (SMemb) (B) in aortas. The aortas were removed from 1‐day‐old, 8‐week‐old, and 20‐week‐old Wistar‐Kyoto (WKY) rats, spontaneously hypertensive rats (SHRs), and complement 3 knockout (C3KO) SHRs; and the endothelium was completely denudated (n=3). Aorta sections were stained with SM1 and SMemb monoclonal antibodies. Bar=25 μm.
Contractility of VSMCs
Gels incubated with VSMCs from WKY rats and C3KO SHRs showed significant shrinkage in gel diameter, which did not show significant shrinkage in gel diameter in cells from SHRs. This result indicates that the VSMCs from WKY rats and C3KO SHRs have strong contractility, causing collagen gel significant shrinkage, but the VSMCs from SHRs did not have such ability, indicating C3 may inhibit the contractility of VSMCs from SHRs (Figure 3A).
Figure 3.
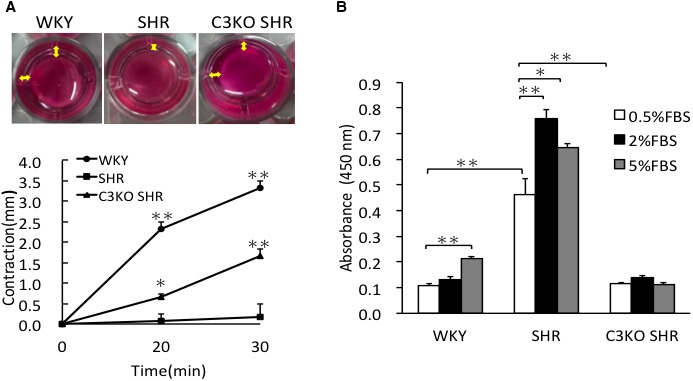
A, Cell contractility of vascular smooth muscle cells (VSMCs). Cell contractility was evaluated using collagen gel contraction assay. VSMCs from 3‐week‐old Wistar‐Kyoto (WKY) rats, spontaneously hypertensive rats (SHRs), and complement 3 knockout (C3KO) SHRs were incubated in collagen gel for 2 days; the change in collagen gel diameter was measured after releasing the gel for 20 and 30 minutes. The representative photographs of collagen gel contraction of VSMCs after being released 30 minutes; the yellow doubled‐headed arrow indicates the contracted diameter of the gel (top). The contraction ability was expressed as the decrease in gel diameter in comparison to the initial diameter (bottom). One‐way repeated‐measure ANOVA was used to determine the statistical difference at different time points. Data are the mean±SEM (n=4). *P<0.05, **P<0.01 vs 0 minutes. B, DNA synthesis of VSMCs. The incorporation of 5‐bromo‐2‐deoxyuridine by VSMCs from 3‐week‐old WKY rats, SHRs, and C3KO SHRs under basal condition; they were incubated with 0.5% fetal bovine serum (FBS). The DNA synthesis response to serum was evaluated by 5‐bromo‐2‐deoxyuridine with the incorporation of 0.5%, 2%, and 5% FBS. Significance was determined using 2‐way ANOVA, followed by Bonferroni post hoc test. Data are the mean±SEM (n=6). *P<0.05, **P<0.01 between indicated columns.
DNA Synthesis of VSMCs
DNA synthesis, evaluated by the incorporation of 5‐bromo‐2‐deoxyuridine in VSMCs from SHRs, was significantly (P<0.01) higher in comparison to VSMCs from WKY rats and C3KO SHRs under basal conditions (0.5% FBS). DNA synthesis in VSMCs from SHRs in response to 2% serum significantly (P<0.01) increased, which did not increase in VSMCs from WKY rats and C3KO SHRs. DNA synthesis in VSMCs from SHRs in response to 5% serum significantly (P=0.037) increased, which did not increase in C3KO SHRs (Figure 3B).
Immunocytochemistry for Renin and LXRα in VSMCs
In immunocytochemistry, staining of renin in VSMCs from SHRs was stronger than that in VSMCs from WKY rats, which was abolished in VSMCs from C3KO SHRs. Staining of LXRα as a transcription factor to promote the renin gene was also stronger in VSMCs from SHRs than in those from WKY rats and C3KO SHRs, especially localized in the nucleus of VSMCs (Figure 4A).
Figure 4.
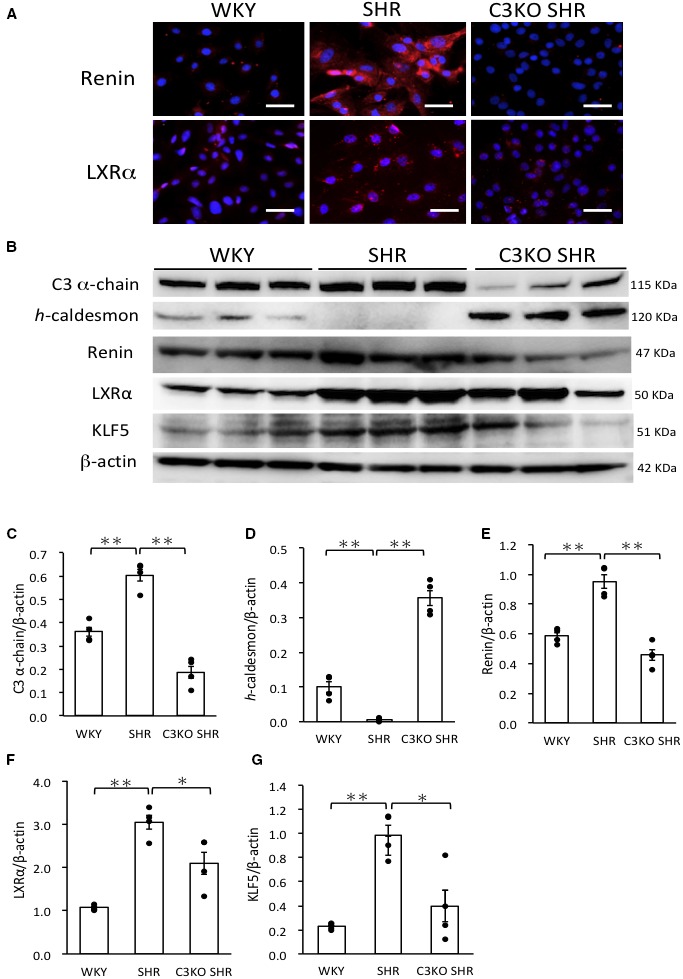
A, Immunocytochemical staining of renin and liver X receptor α (LXRα) in vascular smooth muscle cells (VSMCs). VSMCs were obtained from the aortas of 3‐week‐old male Wistar‐Kyoto (WKY) rats, spontaneously hypertensive rats (SHRs), and complement 3 knockout (C3KO) SHRs by the explant method. VSMCs were stained for renin and LXRα. Bar=50 μm. B, Western blotting was performed in VSMC whole‐cell lysates for C3, h‐caldesmon, renin, LXRα, and Krüppel‐like factor 5 (KLF5). C through G, Results were normalized by β‐actin using the Image J software program. (One‐way ANOVA was performed, followed by Bonferroni post hoc test.) Data are the mean±SEM (n=4). *P<0.05, **P<0.01 between indicated columns.
Western Blotting of VSMCs
The abundance of h‐caldesmon in VSMCs from SHRs was significantly (P<0.01) lower in comparison to VSMCs from WKY rats, which was significantly (P<0.01) higher in VSMCs from C3KO SHRs (Figure 4B 4D). The abundance of C3 α‐chain protein, renin, LXRα and KLF5 proteins in VSMCs from SHRs was significantly (P<0.01) higher in comparison to VSMCs from WKY rats, which was significantly (C3 α chain, P<0.01; renin, P<0.01; LXRα, P=0.035; KLF5, P=0.016) lower in VSMCs from C3KO SHRs (Figure 4B, 4C, and 4E through 4G).
Expression of Phenotype Marker, Growth Factor, and Transcription Factor mRNAs in VSMCs
The abundance of osteopontin, matrix gla, and l‐caldesmon mRNAs (synthetic phenotype markers) and PDGF‐A, KLF5, and angiotensinogen mRNAs in VSMCs from SHRs was significantly (P<0.01) higher in comparison to VSMCs from WKY rats, which was significantly (P<0.01) lower in VSMCs from C3KO SHRs (Figure 5A through 5C, 5E, 5F, and 5H). The abundance of TGF‐β and LXRα mRNAs in VSMCs from SHRs was significantly (P<0.01) higher in comparison to VSMCs from C3KO SHRs (Figure 5D and 5G). The abundance of h‐caldesmon mRNA (a contractile phenotype marker) in VSMCs from SHRs was significantly (P<0.01) lower in comparison to VSMCs from WKY rats, which was significantly (P<0.01) higher in VSMCs from C3KO SHRs (Figure 5I).
Figure 5.
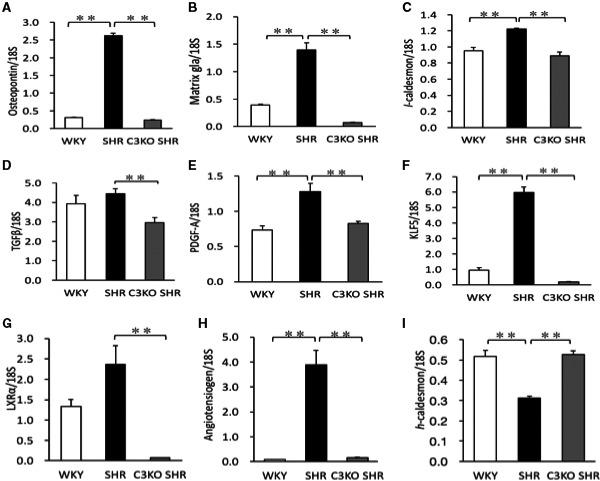
The expression of mRNAs in vascular smooth muscle cells (VSMCs). VSMCs were obtained from the aortas of 3‐week‐old male Wistar‐Kyoto (WKY) rats, spontaneously hypertensive rats (SHRs), and complement 3 knockout (C3KO) SHRs by the explant method. The expression of osteopontin (A), matrix gla (B), l‐caldesmon (C), transforming growth factor (TGF)‐β1 (D), platelet‐derived growth factor (PDGF)‐A (E), Krüppel‐like factor 5 (KLF5) (F), liver X receptor α (LXRα) (G), angiotensinogen (H), and h‐caldesmon (I) mRNAs in VSMCs. Total RNA from VSMCs was reverse transcribed into cDNA. Real‐time quantitative PCR was performed, analyzed with standard curves, and normalized to 18S ribosomal RNA. (One‐way ANOVA was performed, followed by Bonferroni post hoc test.) Data are the mean±SEM (n=6). **P<0.01 between indicated columns.
Expression of miR‐143 and miR‐145 and Effects of the microRNA Inhibitors on Expression of C3 and KLF5 in VSMCs
The abundance of miR‐145 mRNA in VSMCs from WKY rats was significantly (P<0.01) higher in comparison to VSMCs from SHRs (Figure 6A). The abundance of miR‐143 mRNA in VSMCs from the 3 strains did not differ to a statistically significant extent (Figure 6B). We examined the effects of miR‐145 inhibitor on the expression of C3 and KLF5 mRNAs in VSMCs. The miR‐145 inhibitor significantly (P<0.01) increased the abundance of C3 and KLF‐5 mRNAs in VSMCs from WKY rats (Figure 6C and 6E). In VSMCs from SHRs, the inhibition of miR‐145 did not increase the C3 expression under the same condition, but it significantly (P=0.023) increased the KLF5 expression (Figure 6D and 6F). In VSMCs from C3KO SHRs, the inhibition of miR‐145 also significantly (P<0.01) increased the KLF5 mRNA expression (Figure 6G). Inhibition of miR‐143 did not affect the expression of C3 or KLF5 mRNAs in VSMCs from WKY rats, SHRs, or C3KO SHRs (Figure 6C through 6G).
Figure 6.
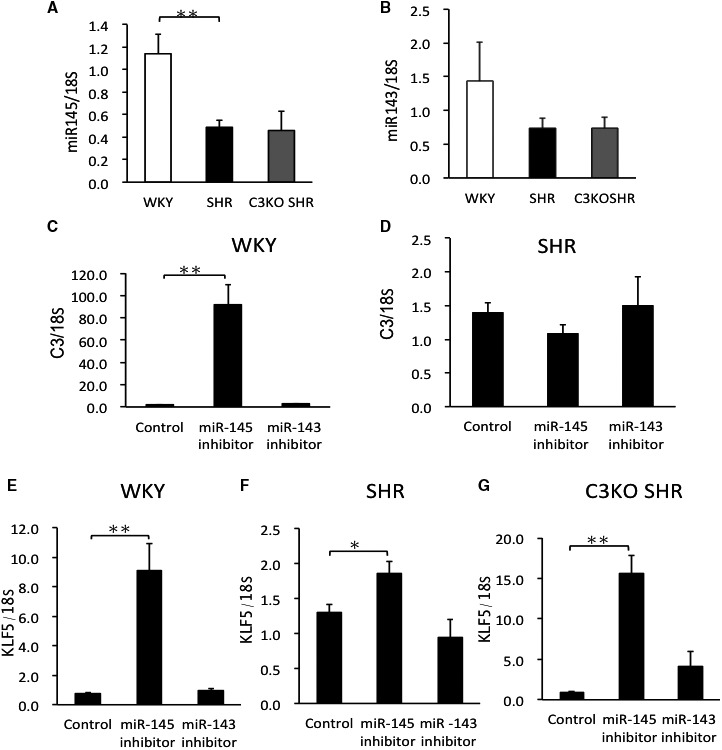
The expression of miR‐145 (A) and miR‐143 (B) in vascular smooth muscle cells (VSMCs). VSMCs were obtained from the aortas of 3‐week‐old male Wistar‐Kyoto (WKY) rats, spontaneously hypertensive rats (SHRs), and complement 3 knockout (C3KO) SHRs by the explant method. The effects of miR‐143 and miR‐145 inhibitors on the expression C3 mRNA in VSMCs from WKY rats (C) and SHRs (D). The effects of miR‐143 and miR‐145 inhibitors on the expression of Krüppel‐like factor 5 (KLF5) mRNA in VSMCs from WKY rats (E), SHRs (F), and C3KO SHRs (G). VSMCs (105 cells/cm2) were transfected with miR‐145 inhibitor or miR‐143 inhibitor using lipofectamine. Two days later, total RNA was extracted, and real‐time PCR was performed. Significance was determined using 1‐way ANOVA, followed by Bonferroni post hoc test. Comparisons between 2 groups were assessed by using Student t test. Data are the mean±SEM (n=4–6). *P<0.05, **P<0.01 between indicated columns.
Discussion
C3 has been known to be increased in atherosclerotic lesions and renal diseases.16, 17 C3 is not only critical for eliciting the complement system, but it also exerts several other biological functions. The complement cascade can be activated by the classic, alternative, or lectin pathways. Once C3 is activated, C3 is cleaved into C3a and C3b (C3a as anaphylatoxin). C3 and C3a, which are known to be vasoactive molecules, are enormously increased by the formation of immunocomplexes, induce the migration of blood cells, and increase the permeability of vessels.18, 19 Our previous studies demonstrated that C3a promotes the synthetic phenotype and increases the production of angiotensinogen and angiotensin II by synthetic organelles, such as Golgi, mitochondria, and endoplasmic reticulum, which produce a series of proteolytic enzymes, including cadherin D and angiotensin‐converting enzyme.6, 7, 20 In the present study, at 1 day after birth in vivo, staining of SM1 (a contractile phenotype marker) in aortas from WKY rats was stronger in comparison to that in aortas from SHRs. In contrast, at 1 day after birth in vivo, staining of SMemb (a synthetic phenotype marker) in aortas from SHRs was stronger in comparison to aortas from WKY rats, which showed weaker staining in aortas from C3KO SHRs. These results indicate that aorta from SHRs is synthetic phenotype at birth already, which is dependent on the increases in endogenous C3 in SHRs.
In addition, DNA synthesis in response to serum was higher in VSMCs from SHRs than that in VSMCs from WKY rats or C3KO SHRs. The VSMCs from WKY rats and C3KO SHRs showed strong contractility, but VSMCs from SHRs showed weak contractility, indicating that C3 suppresses the contractility of VSMCs from SHRs, which depends on an increase in endogenous C3. The expression of osteopontin, matrix gla, l‐caldesmon, and mRNAs (synthetic phenotype markers) in VSMCs from SHRs was higher than that in VSMCs from WKY rats, which was lower in VSMCs from C3KO SHRs. Moreover, the expression of SM1, SM2, and h‐caldesmon (contractile phenotype markers) was lower in VSMCs from SHRs. These findings suggest that VSMCs of SHRs showed a synthetic phenotype caused by increases in endogenous C3.
The change from the contractile phenotype to the synthetic phenotype in VSMCs occurs as a result of the dedifferentiation of VSMCs.21, 22 It has been reported that C3 participates in cell differentiation and proliferation.23, 24, 25 C3a maintains the dedifferentiation of mesenchymal stem cells through extracellular signal‐regulated kinase 1/2.26 These reports suggest that C3 induces the differentiation process of mesenchymal cells. Our previous study demonstrated that C3 activates KLF5 promoter through the extracellular signal‐regulated kinase signaling pathway to induce the synthetic phenotype of VSMCs.10 KLF5 is upregulated in activated VSMCs in atherosclerosis in association with the synthetic phenotype modulation of VSMCs.27, 28, 29 Moreover, it has been reported that angiotensin II induces the expression of KLF5, which, in turn, activates the expression of TGF‐β1 and PDGF‐A to stimulate the growth of VSMCs.29, 30 TGF‐β1 is known to be a growth inhibitor for almost cells; however, it is known that TGF‐β1 is changed to a growth stimulator in mesenchymal cells that have changed to the synthetic phenotype.31 In the present study, the expression of TGF‐β1 and PDGF‐A mRNAs was significantly elevated in VSMCs from SHRs in comparison to cells from WKY rats or C3KO SHRs, indicating that the increases in the expression of TGF‐β1 and PDGF‐A mRNA are associated with the higher expression of C3 in VSMCs from SHRs. We previously demonstrated that antisense oligonucleotides (ODN) to pre‐pro‐C3 significantly inhibited the expression of TGF‐β1, PDGF‐A chain, and basic fibroblast growth factor mRNAs in VSMCs from SHRs. We have shown that levels of angiotensin II–generating enzymes cathepsin D and angiotensin‐converting enzyme are increased with the synthetic phenotype.7, 32 We demonstrated that the endogenous production of angiotensin II is associated with increased levels of growth factors in VSMCs from SHRs. These previous findings and the findings of the present study on VSMCs from SHRs and C3KO SHRs indicate that C3 changes VSMCs from SHRs to the synthetic phenotype and then induces exaggerated growth, with increases in angiotensin II–related growth factors, such as TGF‐β1 and PDGF‐A chain.33 The increases in growth factors may induce the exaggerated growth of VSMCs from SHRs, depending on the increased expression of C3.
In the present study, VSMCs from SHRs showed higher expression levels of angiotensinogen and renin in comparison to VSMCs from WKY rats and C3KO SHRs. KLF5 in VSMCs from SHRs was elevated with the activation of RAS in comparison to VSMCs from WKY rats and C3KO SHRs, indicating that C3 induces the dedifferentiation of VSMCs through KLF5 and RAS activation in VSMCs from SHRs.
LXRα, which has been known to regulate the expression of renin by binding cAMP‐response element binding protein in the renin promoter, acts as a cAMP‐responsive nuclear modulator of renin.34 Juxtaglomerular (JG) cells display the highest of LXRα expression level in the mouse kidney cortex.35 LXRα also stimulates cellular proliferation through c‐myc, which may lead to JG cell hyperplasia.34 These findings indicate that LXRα plays an important role in the expression of renin in cardiovascular and renal diseases. Our previous study found C3a induces the high expression of LXRα and the nuclear localization of LXRα.11, 12 Our present results also showed the higher expression of LXRα with nuclear localization in VSMCs from SHRs. This indicates that C3 may regulate the expression of renin through LXRα, whether the dedifferentiation of VSMCs resembles JG cells or not. JG cells account for 0.1% of all kidney cells, and little is known about JG cells because they are difficult to maintain in cell culture. Once JG cells (renin‐producing cells) were thought to be derived from smooth muscle cells through metaplastic transformation.35 However, it also was reported that renin‐producing cells can produce smooth muscle cells, and the smooth muscle cells derived from renin‐producing cells can undergo metaplasia to renin‐producing cells when required.36, 37 We showed that synthetic and contractile VSMCs, which represent different stages of differentiation, can transit between each other. Synthetic VSMCs express high levels of renin and display similar characteristics to JG cells (renin‐producing cells). Thus, it is possible that there is an intimate relationship between synthetic VSMCs and JG cells. C3 plays an important role in the formation, maintenance, and hyperplasia of synthetic VSMCs.
Recently, microRNAs have been established as master regulators of gene expression by the sequence‐specific binding with target genes to inhibit their expression by either mRNA degradation or translational repression. The circulating microRNAs have been known to exert major cellular processes, including cell cycle differentiation and metabolism, to induce cardiovascular diseases in the specific patterns.38 It has been reported that miR‐145 and miR‐143 regulate smooth muscle cell fate decisions, promote differentiation, and repress the proliferation of VSMCs by converging on serum response factor (SRF)‐dependent coactivators and corepressors.39, 40, 41 KLF5 also acts as a target gene of miR‐145, leading to the phenotypic modulation of VSMCs through its downstream signaling molecule, myocardin. PDGF‐A stimulation suppresses the expression of miR‐145, increases the expression of KLF5, and causes the dedifferentiation of VSMCs.42 Our present study showed that the expression of miR‐145 in VSMCs from SHRs was lower than that in VSMCs from WKY rats. miR‐145 inhibitor significantly increased the expression of C3 and KLF‐5 mRNAs in VSMCs from WKY rats. In VSMCs from SHRs, the inhibition of miR‐145 did not increase the expression of C3 under the same conditions, but it did significantly increase the expression of KLF5. This indicated that although the inhibition of miR‐145 was involved in the C3‐induced increase in KLF5, it also increased the KLF5 expression independently of C3. In VSMCs from C3KO SHRs, the inhibition of miR‐145 also significantly increased the expression of KLF5 mRNA. On the other hand, the expression of miR‐143 in VSMCs from SHRs did not differ from that in VSMCs from WKY rats and C3KO SHRs, and it did not affect the expression of C3 or KLF5 or the phenotype of VSMCs. These results indicate miR‐145 can regulate VSMCs through the suppression of the C3‐ and KLF5‐induced synthetic phenotype. miR‐145 may have the potential to suppress the expression of C3. All of these findings indicate that C3 promotes the expression of KLF5, leading to the dedifferentiation of VSMCs; the acquisition of synthetic phenotype modulation can further promote dedifferentiation through a type of positive feedback regulation through the activation of RAS‐ and miR‐145–related pathways.
In this study, we investigated the contribution of the increased expression of C3 in the exaggerated growth of VSMCs in comparisons of growth and phenotype of VSMCs from WKY rats, SHRs, and C3KO SHRs. From these in vitro experiments, assessment of the exaggerated growth of VSMCs in vivo could not be clarified completely as one limitation of this study is to investigate the role of C3 in the pathogenesis of hypertension in vivo.
In conclusion, the increased expression of C3 with the suppression of miR‐145 induced KLF5, which changes VSMCs from the contractile phenotype to the synthetic phenotype in SHRs. The increased level of C3 stimulates LXRα, which induces RAS, TGF‐β1, and PDGF‐A chain in VSMCs from SHRs. Thus, the increased C3 eventually contributes growth of VSMCs and pathogenesis of hypertension in SHRs (Figure 7).
Figure 7.
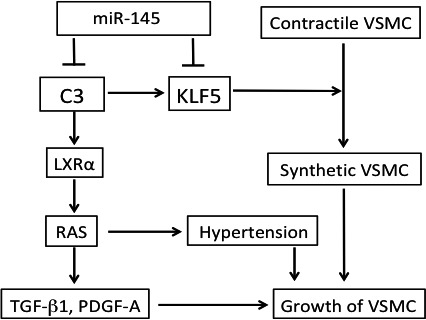
A summary diagram. The increased expression of complement 3 (C3) with the suppression of miR‐145 induces the expression of Krüppel‐like factor 5 (KLF5), which changes vascular smooth muscle cells (VSMCs) in spontaneously hypertensive rats (SHRs) from the contractile phenotype to the synthetic phenotype. The increased C3 stimulates liver X receptor α (LXRα), which induces renin‐angiotensin system (RAS), transforming growth factor (TGF)‐β1, and platelet‐derived growth factor (PDGF)‐A chain in VSMCs from SHRs. Thus, the increased C3 eventually contributes to the growth of VSMCs and pathogenesis of hypertension in SHRs.
Sources of Funding
This study was supported by financial grants from the MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (S1411018) and by a financial grant from Grant‐in‐Aid for Scientific Research from MEXT (Ministry of Education, Culture, Sports, Science and Technology, Japan) (15K09300).
Disclosures
None.
Acknowledgments
We thank Akiko Tsunemi and Mayumi Katakawa for their technical support.
(J Am Heart Assoc. 2019;8:e012327. DOI: 10.1161/JAHA.119.012327.)
Contributor Information
Noboru Fukuda, Email: fukuda.noboru@nihon-u.ac.jp.
Masanori Abe, Email: fukuda.noboru@nihon-u.ac.jp.
References
- 1. Sen S, Tarazi RC, Khairallah PA, Bumpus FM. Cardiac hypertrophy in spontaneously hypertensive rats. Circ Res. 1974;35:775–781. [DOI] [PubMed] [Google Scholar]
- 2. Hu WY, Fukuda N, Kanmatsuse K. Growth characteristics, angiotensin II generation, and microarray‐determined gene expression in vascular smooth muscle cells from young spontaneously hypertensive rats. J Hypertens. 2002;20:1323–1333. [DOI] [PubMed] [Google Scholar]
- 3. Ikeda K, Fukuda N, Ueno T, Endo M, Kobayashi N, Soma M, Matsumoto K. Role of complement 3a in the growth of mesangial cells from stroke‐prone spontaneously hypertensive rats. Clin Exp Hypertens. 2014;36:58–63. [DOI] [PubMed] [Google Scholar]
- 4. Gray SD. Spontaneous hypertension in the neonatal rat: a review. Clin Exp Hypertens A. 1984;6:755–781. [DOI] [PubMed] [Google Scholar]
- 5. Walter SV, Hamet P. Enhanced DNA synthesis in heart and kidney of newborn spontaneously hypertensive rats. Hypertension. 1986;8:520–525. [DOI] [PubMed] [Google Scholar]
- 6. Fukuda N, Satoh C, Hu WY, Soma M, Kubo A, Kishioka H, Watanabe Y, Izumi Y, Kanmatsuse K. Production of angiotensin II by homogeneous cultures of vascular smooth muscle cells from spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 1999;19:1210–1217. [DOI] [PubMed] [Google Scholar]
- 7. Hu WY, Fukuda N, Satoh C, Jian T, Kubo A, Nakayama M, Kishioka H, Kanmatsuse K. Phenotypic modulation by fibronectin enhances the angiotensin II‐generating system in cultured vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:1500–1505. [DOI] [PubMed] [Google Scholar]
- 8. Lin ZH, Fukuda N, Jin XQ, Yao EH, Ueno T, Endo M, Saito S, Matsumoto K, Mugishima H. Complement 3 is involved in the synthetic phenotype and exaggerated growth of vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 2004;44:42–47. [DOI] [PubMed] [Google Scholar]
- 9. Wan J, Fukuda N, Endo M, Tahira Y, Yao E, Matsuda H, Ueno T, Matsumoto K. Complement 3 is involved in changing the phenotype of human glomerular mesangial cells. J Cell Physiol. 2007;213:495–501. [DOI] [PubMed] [Google Scholar]
- 10. Yao EH, Fukuda N, Ueno T, Tsunemi A, Endo M, Matsumoto K. Complement 3 activates KLF5 gene in vascular smooth muscle cells. Biochem Biophys Res Commun. 2008;367:468–473. [DOI] [PubMed] [Google Scholar]
- 11. Zhou X, Fukuda N, Matsuda H, Endo M, Wang X, Saito K, Ueno T, Matsumoto T, Matsumoto K, Soma M, Kobayashi N, Nishiyama A. Complement 3 activates the renal renin‐angiotensin system by induction of epithelial‐to‐mesenchymal transition of the nephrotubulus in mice. Am J Physiol Renal Physiol. 2013;305:F957–F967. [DOI] [PubMed] [Google Scholar]
- 12. Negishi E, Fukuda N, Otsuki T, Katakawa M, Komatsu K, Chen L, Tanaka S, Kobayashi H, Hatanaka Y, Ueno T, Endo M, Mashimo T, Nishiyama A, Abe M. Involvement of complement 3 in the salt‐sensitive hypertension by activation of renal renin‐angiotensin system in spontaneously hypertensive rats. Am J Physiol Renal Physiol. 2018;315:F1747–F1758. [DOI] [PubMed] [Google Scholar]
- 13. Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene‐specific mutated rats using zinc‐finger nucleases. Methods Mol Biol. 2010;597:211–225. [DOI] [PubMed] [Google Scholar]
- 14. Ross R. The smooth muscle cell II: growth of smooth muscle in the culture and formation of elastic fibers. J Cell Biol. 1971;50:172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Youssef Y, White NM, Grigull J, Krizova A, Samy C, Mejia‐Guerrero S, Evans A, Jewett M, Yousef GM. MiRNA profiling in kidney cancer subtypes: accurate molecular classification and correlation with cytogenetic and mRNA data identifies unique and shared biological pathways. Eur Urol. 2011;59:721–730. [DOI] [PubMed] [Google Scholar]
- 16. Seifert PS, Kazatchkine MD. The complement system in atherosclerosis. Atherosclerosis. 1988;73:91–104. [DOI] [PubMed] [Google Scholar]
- 17. Welch TR. The complement system in renal diseases. Nephron. 2001;88:199–204. [DOI] [PubMed] [Google Scholar]
- 18. Li W, Tada T, Miwa T, Okada N, Ito J, Okada H, Tateyama H, Eimoto T. mRNA expression of complement components and regulators in rat arterial smooth muscle cells. Microbiol Immunol. 1999;43:585–593. [DOI] [PubMed] [Google Scholar]
- 19. Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. [DOI] [PubMed] [Google Scholar]
- 20. Han Y, Fukuda N, Ueno T, Endo M, Ikeda K, Xueli Z, Matsumoto T, Soma M, Matsumoto K. Role of complement 3a in the synthetic phenotype and angiotensin II‐production in vascular smooth muscle cells from spontaneously hypertensive rats. Am J Hypertens. 2012;25:284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Louis SF, Zahradka P. Vascular smooth muscle cell motility: from migration to invasion. Exp Clin Cardiol. 2010;15:e75–e85. [PMC free article] [PubMed] [Google Scholar]
- 22. Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45:A25–A32. [DOI] [PubMed] [Google Scholar]
- 23. Del Rio‐Tsonis K, Tsonis PA, Zarkadis IK, Tsagas AG, Lambris JD. Expression of the third component of complement, C3, in regenerating limb blastema cells of urodeles. J Immunol. 1998;161:6819–6824. [PubMed] [Google Scholar]
- 24. Usami M, Mitsunaga K, Miyajima A, Sunouchi M, Doi O. Complement component C3 functions as an embryotrophic factor in early postimplantation rat embryos. Int J Dev Biol. 2010;54:1277–1285. [DOI] [PubMed] [Google Scholar]
- 25. Hsieh CC, Chou HS. The role of complement component 3 (C3) in differentiation of myeloid‐derived suppressor cells. Blood. 2013;121:1760–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003;17:1003–1014. [DOI] [PubMed] [Google Scholar]
- 27. Nagai R, Suzuki T, Aizawa K, Shindo T, Manabe I. Significance of the transcription factor KLF5 in cardiovascular remodeling. J Thromb Haemost. 2005;3:1569–1576. [DOI] [PubMed] [Google Scholar]
- 28. Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai‐Kowase K, Moriyama N, Imai Y, Kawakami H, Nishimatsu H, Ishikawa T, Suzuki T, Morita H, Maemura K, Sata M, Hirata Y, Komukai M, Kagechika H, Kadowaki T, Kurabayashi M, Nagai R. Krüppel‐like zinc‐finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–863. [DOI] [PubMed] [Google Scholar]
- 29. Kawai‐Kowase K, Kurabayashi M, Hoshino Y, Ohyama Y, Nagai R. Transcriptional activation of the zinc finger transcription factor BTEB2 gene by Egr‐1 through mitogen‐activated protein kinase pathways in vascular smooth muscle cells. Circ Res. 1999;85:787–795. [DOI] [PubMed] [Google Scholar]
- 30. Fujiu K, Manabe I, Ishihara A, Oishi Y, Iwata H, Nishimura G, Shindo T, Maemura K, Kagechika H, Shudo K, Nagai R. Synthetic retinoid Am 80 suppresses smooth muscle phenotypic modulation and in‐stent neointima formation by inhibiting KLF5. Circ Res. 2005;97:1132–1141. [DOI] [PubMed] [Google Scholar]
- 31. Fukuda N, Hu W‐Y, Kubo A, Endoh M, Kishioka H, Satoh C, Soma M, Izumi Y, Kanmatsuse K. Abnormal regulation of TGF‐β receptors on vascular smooth muscle cells from spontaneously hypertensive rats by angiotensin II. Hypertension. 1998;31:672–677. [DOI] [PubMed] [Google Scholar]
- 32. Fukuda N, Hu WY, Satoh C, Nakayama M, Kishioka H, Kubo A, Kanmatsuse K. Contribution of synthetic phenotype on the enhanced angiotensin II‐generating system in vascular smooth muscle cells from spontaneously hypertensive rats. J Hypertens. 1999;17:1099–1107. [DOI] [PubMed] [Google Scholar]
- 33. Satoh C, Fukuda N, Hu WY, Nakayama M, Kishioka H, Kanmatsuse K. Role of endogenous angiotensin II in the increased expression of growth factors in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2001;37:108–118. [DOI] [PubMed] [Google Scholar]
- 34. Tamura K, Chen YE, Horiuchi M, Chen Q, Daviet L, Yang Z, Lopez‐Ilasaca M, Mu H, Pratt RE, Dzau VJ. LXRα functions as a cAMP‐responsive transcriptional regulator of gene expression. Proc Natl Acad Sci USA. 2000;97:8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morello F, de Boer RA, Steffensen KR, Gnecchi M, Chisholm JW, Boomsma F, Anderson LM, Lawn RM, Gustafsson JK, Lopez‐Ilasaca M, Pratt RE, Dzau VJ. Liver X receptors alpha and beta regulate renin expression in vivo . J Clin Invest. 2005;115:1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA. Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol. 2001;281:F345–F356. [DOI] [PubMed] [Google Scholar]
- 37. Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell. 2004;6:719–728. [DOI] [PubMed] [Google Scholar]
- 38. Wronska A, Kurkowska‐Jastrzebska I, Santulli G. Application of microRNAs in diagnosis and treatment of cardiovascular disease. Acta Physiol. 2015;213:60–83. [DOI] [PubMed] [Google Scholar]
- 39. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR‐145 and miR‐143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel‐Duby R, Olson EN. MicroRNAs miR‐143 and miR‐145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rangrez AY, Massy ZA, Metzinger‐Le Meuth V, Metzinger L. miR‐143 and miR‐145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet. 2011;4:197–205. [DOI] [PubMed] [Google Scholar]
- 42. Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA‐145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]