

Abstract
Background
Sepsis is the overwhelming host response to infection leading to shock and multiple organ dysfunction. Cardiovascular complications greatly increase sepsis‐associated mortality. Although murine models are routinely used for preclinical studies, the benefit of using genetically engineered mice in sepsis is countered by discrepancies between human and mouse sepsis pathophysiology. Therefore, recent guidelines have called for standardization of preclinical methods to document organ dysfunction. We investigated the course of cardiac dysfunction and myocardial load in different mouse models of sepsis to identify the optimal measurements for early systolic and diastolic dysfunction.
Methods and Results
We performed speckle‐tracking echocardiography and assessed blood pressure, plasma inflammatory cytokines, lactate, B‐type natriuretic peptide, and survival in mouse models of endotoxemia or polymicrobial infection (cecal ligation and puncture, [CLP]) of moderate and high severity. We observed that myocardial strain and cardiac output were consistently impaired early in both sepsis models. Suppression of cardiac output was associated with systolic dysfunction in endotoxemia or combined systolic dysfunction and reduced preload in the CLP model. We found that cardiac output at 2 hours post‐CLP is a negative prognostic indicator with high sensitivity and specificity that predicts mortality at 48 hours. Using a known antibiotic (ertapenem) treatment, we confirmed that this approach can document recovery.
Conclusions
We propose a non‐invasive approach for assessment of cardiac function in sepsis and myocardial strain and strain rate as preferable measures for monitoring cardiovascular function in sepsis mouse models. We further show that the magnitude of cardiac output suppression 2 hours post‐CLP can be used to predict mortality.
Keywords: basic science, cardiomyopathy, echocardiography, mouse, sepsis, speckle tracking echocardiography
Subject Categories: Animal Models of Human Disease, Basic Science Research, Biomarkers, Echocardiography
Clinical Perspective
What Is New?
Endotoxemia and cecal‐ligation and puncture exhibit distinct manifestations of septic cardiac dysfunction.
Speckle‐tracking echocardiography may be used in mouse models of sepsis to identify early signs of systolic and diastolic dysfunction even in the context of reduced cardiac load and normal ejection fraction.
Cardiac output measured using echocardiography predicts survival as early as 2 hours after cecal ligation and puncture surgery.
What Are the Clinical Implications?
As endotoxemia and cecal‐ligation and puncture exhibit different characteristics of septic cardiac dysfunction, translational studies should carefully consider how these models may apply to different subpopulations of septic patients.
Use of speckle‐tracking echocardiography is increasingly used to identify cardiac dysfunction in sepsis, and therefore preclinical studies should consider this technology to identify experimental therapies with cardioprotective properties.
Introduction
Sepsis is a leading cause of in‐hospital mortality in the United States and according to the Sepsis‐3 guidelines, is diagnosed using combined evidence of organ dysfunction and clinical suspicion of underlying infection.1 Previous studies have shown that the presence of acute cardiac dysfunction is a negative prognostic indicator in sepsis.2, 3, 4, 5, 6, 7, 8 The manifestations of cardiovascular dysfunction in sepsis are complex and differ between hyperdynamic and hypodynamic sepsis.2, 9 During hyperdynamic sepsis, the reduced peripheral resistance and stimulation of the adrenergic system causes tachycardia, increased cardiac output, and increased surface temperature that result from a decline in afterload. Hypodynamic sepsis results in lower diastolic filling pressure, bradycardia, decreased cardiac output, and ischemic organ damage.10 Despite the burden of sepsis on the healthcare system, no targeted therapies have proven effective at preventing mortality and organ dysfunction,11 and current guidelines emphasize source control, administration of antibiotic therapy, and fluid resuscitation.12
Multiple mouse models of sepsis are frequently used including injection of E coli endotoxin or the cecal ligation and puncture (CLP) polymicrobial peritonitis model.13 Several limitations of the murine models challenge interpretation of the outcomes, which have recently come under scrutiny by the sepsis research community. New guidelines14, 15, 16, 17 emphasize the importance of documenting and resolving acute organ dysfunction in sepsis studies. Our previous studies in animal models of endotoxemic shock and CLP‐induced peritonitis18, 19, 20, 21, 22 identified pharmacological and genetic manipulations that prevent or correct systolic cardiac dysfunction based on ejection fraction (EF) and fractional shortening (FS). Recent advances in rodent cardiac imaging allow for the more detailed and sensitive speckle‐tracking analysis,23 which has been shown in patients to identify subtle changes in cardiac function.7, 8, 24 Thus, we assessed the use of LV‐trace and speckle tracking‐derived parameters in mice that underwent LPS (endotoxin) injection or CLP with and without administration of fluids and antibiotic, and characterized the changes in blood pressure, inflammatory cytokines, lactate, B‐type natriuretic peptide, surface temperature, and survival. Our analyses identified echocardiography‐derived parameters which detect cardiac dysfunction earlier and more reliably than EF, and factors which can be used to predict mortality in CLP sepsis.
Methods
Data Availability Disclosure Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.
Animal Care
Animal protocols were approved by the Temple University Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. Wild‐type (WT) 7‐ to 12‐week old male C57BL/6 mice weighing between 20 and 30 g were purchased from Jackson Laboratory. As our study aimed to identify and compare the cardiovascular manifestations of CLP and LPS‐induced sepsis, and not the effect of biological factors including sex, we restricted our analysis to male mice which have previously been shown to experience cardiac depression to a greater extent compared with female mice.25, 26
CLP and LPS Injection
CLP was performed as previously described.18, 22 Mice were anesthetized with 3% inhaled isoflurane. Under aseptic conditions, a 1 to 2 cm midline laparotomy was performed and exposure of the cecum with adjoining intestine. The cecum was tightly ligated below the ileo‐cecal valve 1 cm from the distal end and was punctured once (CLP1P) or twice (CLP2P) through‐and‐through with a 19‐gauge needle. The length of the ligated cecum was defined as the distance from the distal end of cecum to ligation point, which affects the degree of disease severity. Fecal material was extruded from the punctured cecum, and it was returned to the peritoneal cavity. The peritoneum and the skin were closed with 3 sutures. All mice were resuscitated by injecting subcutaneously 1 mL of pre‐warmed 0.9% saline solution, and for postoperative analgesia the mice received subcutaneously buprenorphine (0.05 mg/kg body weight, ≈100 μL fluid in addition) based upon previous studies detailing the CLP procedure.27 Sham mice underwent procedure to expose the cecum, however the ligation and puncture steps were omitted. To evaluate the effect of antibiotic and fluid treatment, mice received intraperitoneal injection with ertapenem sodium (70 mg/kg) at 2 hours and every 24 hours thereafter for the duration of the protocol, modified from previous studies.28 For each injection, ertapenem was dissolved in normal saline to 7 mg/kg and mice were injected intraperitoneally (I.P.) with 10 μL/g body weight, based on commonly used resuscitation volumes between 5 and 50 μL/g fluid29, 30, 31, 32, 33 and caution against over‐resuscitation.34, 35 Endotoxemic shock was induced by intraperitoneal injection of LPS. E coli. LPS (Sigma) was dissolved in saline to a concentration of 1 mg/mL, and mice were injected I.P. with 5 mg/kg body weight LPS and returned to the cage for monitoring by the indicated analyses.
Measurement of Inflammatory Cytokines, BNP, and Lactate in Plasma
As sepsis has been described as a condition of overactivation of the immune system, we assessed the time‐course of inflammatory cytokine induction after sepsis. We further aimed to associate changes in inflammatory cytokine levels with the cardiac stress marker B‐type natriuretic peptide (BNP) and tissue perfusion marker lactate. We randomized 16 cohorts of C57BL/6 mice to receive sham surgery, CLP with 1 puncture (CLP1P), CLP with 2 punctures (CLP2P), or LPS injection and euthanized them 2, 6, 12, and 24 hours (n=4‐5 per timepoint/group) later for plasma collection. Mice with saline injection alone served as a control for LPS animals. From these cohorts, we collected plasma and assessed circulating levels of interleukin (IL)‐1α, IL‐1β, IL‐6, IL‐10, and tumor necrosis factor‐α (TNF)‐α using the Milliplex MAP Mouse Cytokine kit (MCYTOMAG‐70K‐05) using a 1:4 dilution. Samples were read using the Luminex MAGPIX multiplexing unit. Plasma BNP was measured using the Raybiotech BNP ELISA kit (EIAM‐BNP) following the kit instructions. Plasma L‐Lactate was measured from frozen plasma samples using the Sigma Lactate Assay Kit (MAK064) using the kit protocol.
Echocardiography
We next aimed to assess the timecourse of cardiac dysfunction after CLP surgery, sham surgery, and LPS injection using echocardiography. We therefore randomized 8 cohorts of mice to receive sham surgery, CLP1P, CLP2P, or LPS injection and assessed cardiac function by transthoracic echocardiography using the VisualSonics Vevo 2100 system (VisualSonics, Toronto, ON) at multiple timepoints including baseline, 2, 4, 6, 12, and 24 hours after surgery. These mouse cohorts were separate from those used for inflammatory cytokine analysis. Before echocardiography, hair was removed from the animal's chest wall using Nair for 1 minute. Sedation was induced with 3% isoflurane and maintained at 0.5% isoflurane during the procedure which averaged ≈6 to 10 minutes per mouse during which mice were maintained under a lamp on a heated table at ≈37°C. Parasternal short axis images were taken at the myocardial base using the papillary muscles as an indicator for the position of the probe such that the entire left ventricular wall was visible. Parasternal 3‐chamber long axis (PLAX) images were taken using the position of the right atrium and posterior mitral valve leaflet as landmarks.
Echocardiographic images were processed to perform LV trace analysis of short axis M‐mode images and speckle tracking analysis and automatic tracing of short axis and long axis B‐mode images by 2 independent researchers masked to the outcome and treatment of the animal. Using M‐mode short axis images, we measured EF, FS, end‐diastolic and end‐systolic volumes (EDV, ESV), stroke volume (SV), and cardiac output (CO). VevoStrain software was used to remeasure these same parameters using automatic tracing as well as to detect myocardial strain and strain rate using speckle tracking echocardiography. For this analysis, B‐mode images of 300 frames at >200 frames/s were used. For long‐axis views, global longitudinal strain (GLS) was calculated from anterior and posterior apical, mid, and basal segments (average of 6 left ventricular segments). For short‐axis views, global circumferential strain and global radial strain (GCS, GRS) were calculated from anterior and posterior septal and lateral walls. Left ventricular outflow tract velocity time integral, peak velocity, and cardiac output were also measured using pulsed wave Doppler to assess flow across the aortic valve.29 Diastolic dysfunction was assessed as previously described36, 37 using the reverse peak option to measure reverse GLS and GRS rate from long axis B‐mode traces. We also assessed transmitral inflow velocities by setting the sample volume in the mitral orifice close to the tip of the mitral leaflets. From the pulsed wave Doppler spectral waveforms, we measured the peak early‐ and late‐diastolic transmitral velocities (E and A waves) to obtain the E/A ratio. From the tissue Doppler spectral waveforms, we measured E′ (early‐diastolic myocardial relaxation velocity, A’, and calculated the E’/A’ and E/E′ ratio. This analysis was performed as previously described.37 Representative images are shown using M‐mode short axis traces, and parasternal short axis and parasternal long axis B‐mode traces with the speckle tracking in Figures 2 and 7. In these images, the green area represents the area covered by the myocardial wall movement between systole and diastole and the green lines represent the path of the probe during the wall movement.
Figure 2.
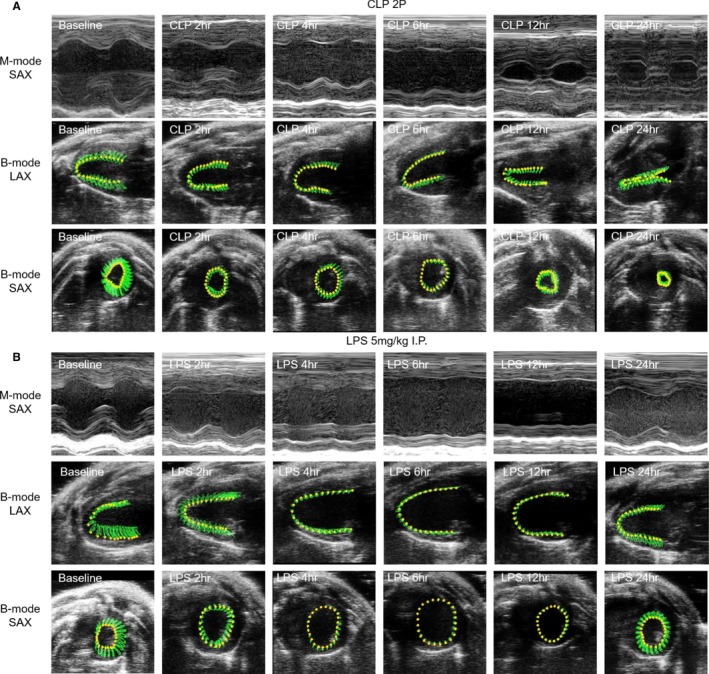
Representative echocardiogram images following CLP surgery or LPS injection. M‐mode parasternal short axis and B‐mode long axis (parasternal long axis ) and parasternal short axis images taken at baseline, 2, 4, 6, 12, and 24 hours after CLP 2 puncture surgery (A) or LPS (5 mg/kg) injection (B). For B‐mode images, the area in green represents the region tracked by the speckle tracking echocardiography throughout a cardiac cycle (contraction and relaxation phases). CLP2P indicates cecal ligation and puncture with 2 punctures; LPS lipopolysaccharide; LAX long axis; SAX short axis.
Figure 7.
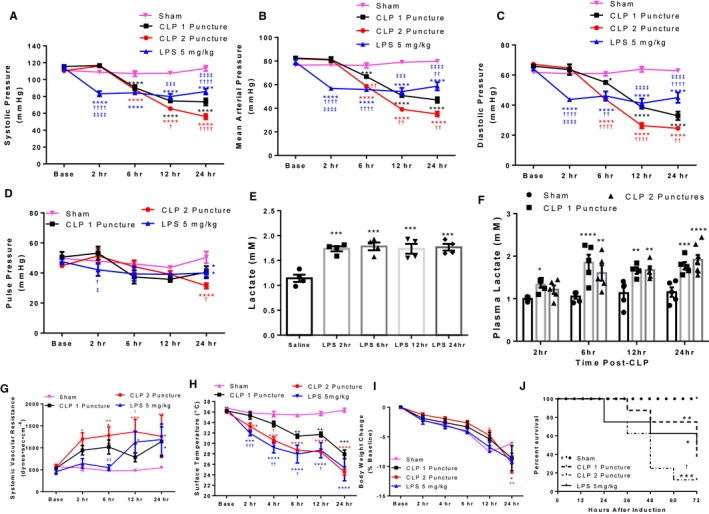
Reduced cardiac output drives hypotension and mortality in CLP and LPS‐induced sepsis. A through D, Non‐invasive tail‐cuff measurement of systolic (A), mean (B), diastolic (C), and pulse (D) pressure. *P<0.05, ***P<0.001, ****P<0.0001 vs Sham, † P<0.05, †† P<0.01, †††† P<0.0001 vs CLP 1 puncture, ‡‡‡ P<0.001, ‡‡‡‡ P<0.0001 vs CLP 2 Puncture by 2‐way ANOVA + LSD multiple momparisons. n=8 sham, n=8 CLP1P, n=8 CLP2P, n=8 LPS. E and F, Plasma lactate in C57BL/6 mice following LPS injection (E) or CLP surgery (F). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs Saline for LPS‐treated animals or sham for CLP animals by 1‐way ANOVA with Tukey multiple comparisons, n=4 saline, n=4 LPS per timepoint; n=5 sham, n=5 CLP1P, and n=5 CLP2P per timepoint. G through J, Systemic vascular resistance (SVR) from automatic tracking derived measurements of cardiac output and mean arterial blood pressure (G), xiphoidal surface temperature (H), and body weight (I). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs Sham, † P<0.05, †† P<0.01, ††† P<0.001 vs CLP 1 puncture, ‡‡ P<0.01 vs CLP 2 puncture by 2‐Way ANOVA + LSD multiple comparisons, n=8 sham, n=8 CLP1P, n=8 CLP2P, n=8 LPS. Kaplan–Meier survival curve (J) assessing percent survival within 72 hours of sepsis induction in mice after LPS injection, CLP1P surgery, CLP2P surgery, or sham surgery. *P<0.05, **P<0.01, ***P<0.001 vs sham, † P<0.05 vs CLP1P by pairwise log‐rank (Mantel‐Cox) test. P=0.007 by overall log‐rank test. n=8 sham, n=8 CLP1P, n=8 CLP2P, n=8 LPS. CLP indicates cecal ligation and puncture; LPS lipopolysaccharide.
Non‐Invasive Blood Pressure Monitoring and Calculation of Systemic Vascular Resistance
Systolic, mean, and diastolic pressure in non‐anesthetized animals was measured non‐invasively using the BP‐2000 tailcuff system. This system uses spectrophotoplethysmographic traces to measure differences in blood flow through the tail during systole and diastole. Prior to randomization, mice were acclimated to the restrainer by taking measurements on 5 different days. Appropriate acclimation to the restrainer is critical to obtaining accurate pressure readings following sepsis induction. During the procedure, animals were placed in the restrainer atop the platform heated to 39°C. Mice were exposed to this environment for at least 10 minutes to allow the tail vasculature to dilate. Thirty measurements were taken at each timepoint and blood pressure curves were assessed to identify appropriate delineation between diastolic and systolic pressures and to ensure reproducibility between measurements. Measurements were averaged to calculate the average blood pressure for each mouse. Systemic vascular resistance (SVR) was calculated from B‐mode measurements of cardiac output and tail‐cuff derived measurements of mean arterial pressure using the equation mean arterial pressure =80×CO×SVR. SVR is represented using the unit dynes×sec×cm−8.
Invasive Hemodynamics
Cardiac function was analyzed by left ventricular pressure‐volume loop measurements performed under Avertin anesthesia. Pressure‐volume (PV) parameters were measured using PVR‐1045 catheter (Millar‐ Houston, TX). The catheter was advanced into the LV through the right common carotid artery and aorta. Data were acquired using the PowerLab system (ADInstruments, Colorado Springs, CO) and analyzed with the Lab‐Chart8 program using the PV‐loop module.
Infrared Thermometer Assessment of Surface Temperature
Xiphoid surface temperature was assessed using the Etekcity Lasergrip infrared as previously described.38 Briefly, hair from the xiphoid surface was removed, and mice were restrained. The thermometer was held ≈3 inches from xiphoid process, and measurements were recorded at 2, 4, 6, 12, and 24 hours after sepsis induction.
Statistical Analysis
Statistical comparisons were generated using GraphPad Prism6 software. Analyses were performed using Students t test, 2‐way ANOVA with LSD multiple comparisons to compare groups within each timepoint, or 1‐way ANOVA with Tukey multiple comparisons. Specific statistical tests used are stated within the figure legends. For scatterplot data, linear regression analysis was performed and the Pearson correlation coefficient (r) and coefficient of determination (r2) were calculated using the GraphPad Prism 6 software regression analysis. Survival analysis was analyzed using both pairwise and overall log‐rank (Mantel‐Cox) test. Statistical significance was determined using α=0.05.
Results
Inflammatory Cytokines and BNP are Elevated Within 2 Hours of Sepsis Induction
Previous studies have shown that inflammation during sepsis is a major component of myocardial depression,39 and that changes in plasma inflammatory cytokine levels differed between sepsis models.40, 41 We therefore hypothesized that induction of inflammatory cytokines may differ between LPS and CLP‐associated sepsis which would be associated with differences in myocardial dysfunction. Beginning 2 hours after CLP surgery, we observed a trend for increased plasma IL‐1α levels (Figure 1A) and significant increases in IL‐1β (Figure 1B), IL‐6 (Figure 1C), and TNF‐α (Figure 1D) which remained elevated at all timepoints. Increased inflammatory cytokine levels were independent of the number of punctures introduced during the CLP procedure. The anti‐inflammatory cytokine IL‐10 was also elevated within 2 hours and increased further 24 hours after CLP surgery (Figure 1E). As inflammation in sepsis has been associated with cardiac stress, we measured plasma BNP by ELISA as a marker of cardiac stress. BNP was elevated >20‐fold at 2, 6, and 24 hours, and was maximally elevated to a 100‐fold increase at 12 hours compared with respective sham groups (Figure 1F).
Figure 1.
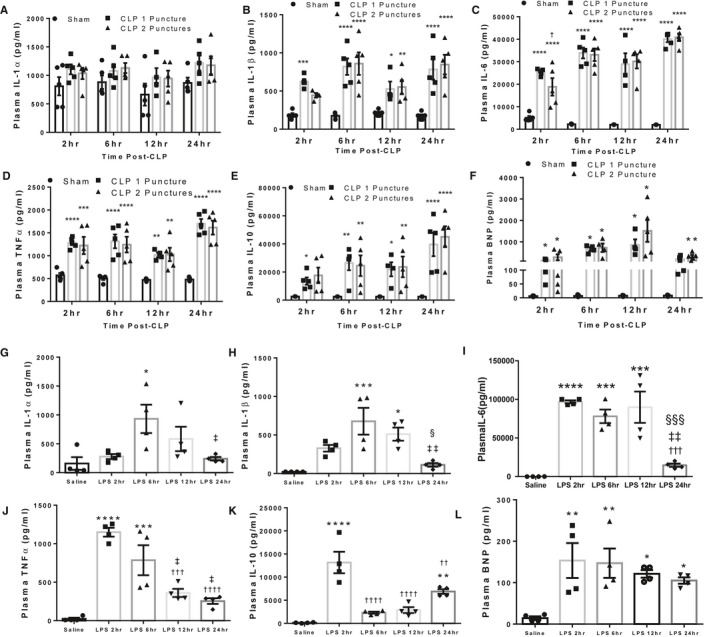
LPS injection and CLP surgery result in rapid release of inflammatory cytokines. A through F, Plasma IL‐1α (A), IL‐1β (B), IL‐6 (C), TNF‐α (D), IL‐10 (E), and B‐type natriuretic peptide (F) levels following sham, CLP 1 puncture, and CLP 2 puncture surgery at 2, 6, 12, and 24 hours after induction. Each timepoint represents a separate cohort of mice. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs Sham, † P<0.5 vs CLP1P by 1‐way ANOVA with Tukey multiple comparisons. n=5 sham, n=5 CLP1P, n=5 CLP2P per timepoint. G‐L: Plasma IL‐1α (G), IL‐1β (H), ΙL‐6 (I), TNF‐α (J), IL‐10 (K), and B‐type natriuretic peptide (L) at 2, 6, 12, and 24 hours post‐LPS injection. Saline mice were injected with equal volume saline collected 6 hours after injection. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs Saline, † P<0.05, †† P<0.01, ††† P<0.001, †††† P<0.0001 vs LPS 2 hours, ‡ P<0.05, ‡‡ P<0.01 vs LPS 6 hours, § P<0.05, §§§ P<0.001 vs 24 hours by 1‐way ANOVA with Tukey multiple comparisons, n=4 saline, n=4 LPS 2 hours, n=4 LPS 6 hours, n=4 LPS 12 hours, n=4 LPS 24 hours. BNP indicates B‐type natriuretic peptide; CLP, cecal ligation and puncture; IL, interleukin; LPS lipopolysaccharide; TNF tumor necrosis factor.
For LPS‐injected mice, IL‐1α increased 6 hours following injection and subsequently returned toward baseline at 12 and 24 hours post‐injection (Figure 1G). The same trend was observed for IL‐1β which was elevated at both 6 and 12 hours (Figure 1H). Conversely, IL‐6 was elevated 1000‐fold 2 hours post‐LPS injection and returned toward normal 24 hours post‐injection (Figure 1I). TNF‐α was increased 45‐fold within 2 hours post‐injection, which progressively declined at 6 and 12 hours (Figure 1J). IL‐10 was elevated 100‐fold after LPS injection and was transiently decreased 6‐ and 12‐hours post‐injection compared with 2 hours (Figure 1K). We found a secondary elevation in IL‐10 levels 24 hours post‐LPS injection (Figure 1K). Plasma BNP levels were elevated ≈8‐fold within 2 hours of LPS injection and remained elevated at all timepoints (Figure 1L).
LPS Injection and CLP Surgery Caused Hypodynamic Sepsis and Impaired Cardiac Function
As we showed that the course of inflammatory cytokine induction differed between CLP and LPS‐induced sepsis, we performed echocardiographic analysis to identify how cardiac function was affected at each timepoint. Cardiac function was assessed serially by echocardiography in 8 cohorts of mice randomized to receive sham, CLP1P, CLP2P, or LPS injection at baseline and at 2, 4, 6, 12, and 24 hours after induction of sepsis (Figure 2). Echocardiograms were analyzed by manual LV‐trace of M‐mode images and using VevoStrain software which provides the same measurements using automatic tracing (Figure 3). We observed that FS and EF were reduced within 4 and 6 hours of CLP2P but not in CLP1P surgery. Beginning 12 hours after CLP surgery, EF and FS increased to baseline levels despite sustained elevation in inflammatory cytokines (Figure 3A and 3B). LPS resulted in a more substantial reduction in FS and EF which remained reduced until 24 hours post‐injection (Figure 3A and 3B) in coordination with the reduction in inflammatory cytokine levels and elevation in IL‐10 (Figure 1). The profound reduction in EF and FS of LPS‐injected mice coincides with trends for expansion of EDV (Figure 3C) and significant expansion of ESV (Figure 3D). Conversely, CLP surgery results in a significant reduction in EDV at all timepoints for CLP2P and at 4 and 12 hours for CLP1P (Figure 3C). Consistently, all mice experienced a significant reduction in SV (Figure 3E) beginning 2 hours after sepsis induction, and in BW normalized CO (Figure 3F) at all timepoints. Conversely, heart rate did not significantly change in any model until 12 and 24 hours when all 3 models had significantly reduced heart rate (Figure 3G). Changes in these parameters were consistent with either LV trace or VevoStrain automatic tracing as shown by significant positive correlation between LV trace‐ and automated tracking‐derived measurements of FS (Figure 3H), CO (Figure 3I), and EDV (Figure 3J).
Figure 3.
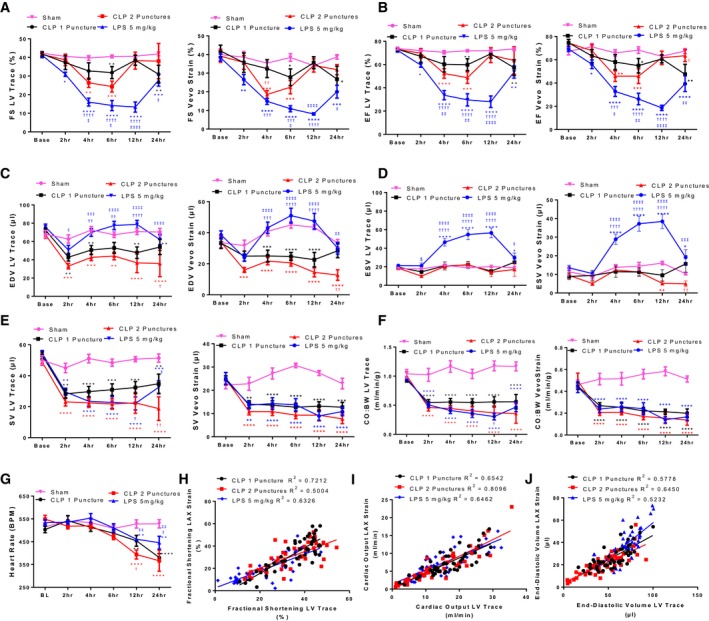
Quantification of cardiac function in sepsis models using speckle tracking echocardiography. A through F, Quantification of classic M‐mode measurements following manual LV‐trace of M‐mode images and automatic tracing of B‐mode parasternal long axis images using VevoStrain software. Fractional shortening (A), ejection fraction (B), end‐diastolic volume (C), end‐systolic volume (D), stroke volume (E), cardiac output normalized to body weight (F), and heart rate (G). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs sham, † P<0.05, †† P<0.01, ††† P<0.001, †††† P<0.0001 vs CLP 1 puncture, ‡ P<0.05, ‡‡ P<0.01, ‡‡‡ P<0.001, ‡‡‡‡ P<0.0001 vs CLP 2 puncture by 2‐way ANOVA + LSD multiple comparisons, n=8 sham, n=8 CLP1P, n=8 CLP2P, n=8 LPS. H through J, Correlation between manual LV Trace and VevoStrain measurements of fractional shortening (H), cardiac output (I), and end‐diastolic volume (J). BL indicates baseline; BPM, beats per minute; CLP, cecal ligation and puncture; CO:BW, cardiac output normalized to body weight; EDV, end‐diastolic volume; EF, ejection fraction; ESV, end‐systolic volume; FS, fractional shortening; LPS, lipopolysaccharide; LV, left ventricle; SV, stroke volume.
Global Radial, Longitudinal, and Circumferential Strain Declined Progressively Following CLP Surgery and LPS Injection
In addition to obtaining M‐mode measures of cardiac function, VevoStrain allows for the quantification of myocardial deformation, which is increasingly being used to assess subtle alterations in cardiac dysfunction in sepsis.42 We hypothesized that myocardial strain would indicate cardiac dysfunction earlier and more consistently compared with standard M‐mode measurements of EF and FS. Using this tool, we measured strain and strain rate in the longitudinal, radial, and circumferential plane at each timepoint following sepsis induction (Figure 4). At baseline, all groups of mice had comparable myocardial strain measurements (GLS ‐22%, GRS 35%, GCS ‐30%). We found significant impairment in all 3 models for GLS (Figure 4A) beginning 2 hours after sepsis induction. GRS began to decline for CLP2P and LPS cohorts beginning at 2 hours post‐induction and for CLP1P cohorts at 4 hours (Figure 4B). GLS and GRS remained decreased at all timepoints except at 12 hours when GRS trended toward decrease for CLP1P and CLP2P mice. No significant difference was observed between CLP1P and CLP2P at any timepoint (Figure 4A and 4B). Conversely, GCS was suppressed for CLP2P at 4 hours, which remained decreased throughout the procedure (Figure 4C). The effect on GCS was severity‐dependent as CLP1P did not deteriorate compared with CLP2P at 4, 6, and 12 hours post‐surgery, however, both groups of mice were similarly impaired 24 hours after surgery (Figure 4C). As with GLS and GRS, GCS was significantly decreased for endotoxemia mice at all timepoints. All 3 strain measurements were reduced most profoundly after LPS injection compared with CLP which supports the profound reduction in EF and FS observed in these mice.
Figure 4.
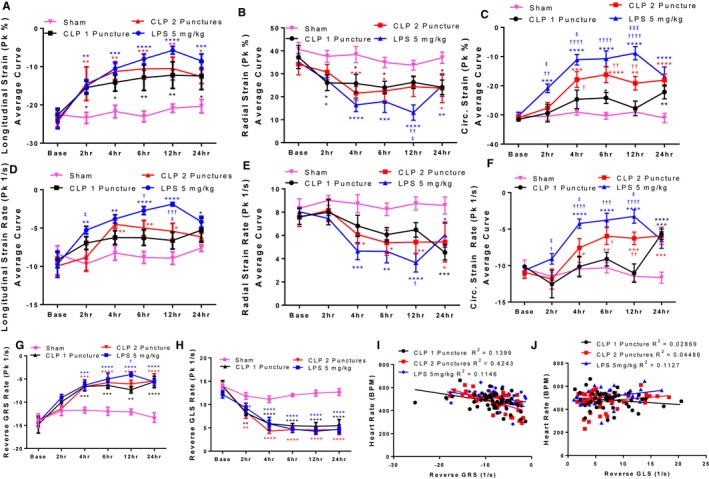
Speckle‐tracking–derived measurements of myocardial deformation. A through F, Speckle‐tracking derived measurements of systolic function. Myocardial strain in the longitudinal (A), radial (B), and circumferential (C) planes. Myocardial strain rate in the longitudinal (D), radial (E), and circumferential (F) planes. G and H, Speckle‐tracking derived measurements of myocardial relaxation. Reverse radial strain rate (G) and reverse longitudinal strain rate (H) in C57BL/6 mice that underwent sham, CLP 1 puncture, CLP 2 punctures, and LPS injection at baseline, 2, 4, 6, 12, and 24 hours after CLP surgery or LPS injection (5 mg/kg). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs sham, † P<0.05, †† P<0.01, ††† P<0.001, †††† P<0.0001 vs CLP 1 puncture, ‡ P<0.05, ‡‡ P<0.01, ‡‡‡ P<0.001 vs CLP 2 puncture by 2‐way ANOVA + LSD multiple comparisons. n=8 sham, n=8 CLP1P, n=8 CLP2P, n=8 LPS. I and J, Association between reverse GRS rate (I), and reverse GLS rate (J) with heart rate. CLP indicates cecal ligation and puncture; GLS, global longitudinal strain; GRS, global radial strain; LPS, lipopolysaccharide; Pk, peak.
In coordination with impairment in myocardial strain, we observed that strain rate which measures the maximal rate of deformation during systole, followed a similar trend compared with the respective strain measurements in the longitudinal (Figure 4D), radial (Figure 4E), and circumferential (Figure 4F) planes. For CLP2P mice, GLS rate was significantly reduced compared with sham mice at 4, 6, and 12 hours post‐surgery (Figure 4D). Trends for reduced GLS rate were observed at all timepoints for CLP1P mice; however, this measurement did not reach statistical significance (Figure 4D). Conversely, GLS rate was significantly impaired at all timepoints following LPS injection (Figure 4D). Similarly, GRS rate was impaired between 4 and 24 hours following CLP2P and at 24 hours following CLP1P (Figure 4E). GRS rate was most profoundly affected by LPS injection and was significantly decreased between 4 and 24 hours (Figure 4E). GCS rate was significantly reduced between 4 and 24 hours for CLP2P mice but only at 24 hours for CLP1P mice (Figure 4F). Significant differences between CLP1P and CLP2P were observed at 4, 6, and 12 hours post‐surgery. Conversely, GCS rate trended to decrease at 2 hours and was significantly reduced beginning 4 hours after LPS injection.
To assess diastolic function, we measured reverse radial (rGRS rate, Figure 4G) and longitudinal (rGLS rate, Figure 4H) strain rate which measures ventricular expansion during early‐diastolic filling, corresponding with the E wave. At baseline, all groups had comparable measurements for rGRS rate and rGLS rate (Figure 4G and 4H). At 2 hours, all 3 models experienced a decrease in both rGRS rate and rGLS rate (Figure 4G and 4H). This decline progressed further 4 hours post‐sepsis induction which remained reduced at this level for CLP1P and CLP2P for subsequent measurements. In comparison, LPS was maximally decreased 12 hours post‐injection (Figure 4G and 4H). As diastolic filling is known to be affected by heart rate,43 we assessed the correlation between heart rate and rGRS rate (Figure 4I) and rGLS rate (Figure 4J). For rGRS rate, a significant association was observed with heart rate suggesting that reduced heart rate in our sepsis models may affect this measurement by reducing myocardial relaxation rate (Figure 4I). For CLP1P and CLP2P, we did not observe a significant association between rGLS and heart rate suggesting that heart rate does not have a significant influence on rGLS rate in these models (Figure 4J).
Invasive Hemodynamics Reveal Reduced CO and Impaired Contractility 12 hours Post‐CLP
As we found significant changes in systolic and diastolic function using speckle tracking echocardiography, we assessed additional measures in a subset of mice using invasive measures of systolic and diastolic function to confirm the presence of cardiac dysfunction in these mice. First, we inserted a PV catheter in the left ventricle 12 hours after sham or CLP2P operation to generate pressure‐volume curves (Figure 5A). During this analysis, 3 out of 7 CLP2P mice were unable to survive anesthesia and catheter insertion and were therefore excluded from the study. We observed a significant suppression of SV (Figure 5B), CO (Figure 5C), and heart rate (Figure 5D) consistent with a hypodynamic sepsis state. We further observed that systolic blood pressure was reduced (Figure 5E) and stroke work was reduced (Figure 5F). This analysis revealed that both dP/dtmax (Figure 5G) and dP/dtmin (Figure 5H) were suppressed after CLP. We also observed a significant reduction in EDV (Figure 5I) consistent with our observation from echocardiographic analysis.
Figure 5.
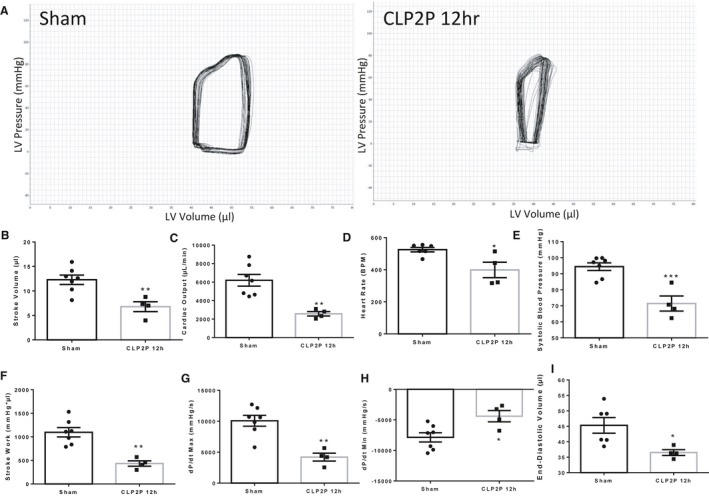
Invasive measurement of cardiac function 12 hour post‐CLP surgery. Ventricular pressure‐volume relationships were assessed invasively via insertion of a catheter into the left ventricle. Representative PV‐loops (A), stroke volume (B), cardiac output (C), heart rate (D), systolic blood pressure (E), stroke work (F), dP/dtmax (G) dP/dtmin (H) and end‐diastolic volume (I) derived from invasive hemodynamics in sham and CLP2P mice. *P<0.05, **P<0.01, ***P<0.001 vs sham by t test, n=7 sham, n=4 CLP2P 12 hours. CLP2P indicates cecal ligation and puncture with 2 punctures; dP/dt, first derivative of left ventricular pressure with respect to time; LV, left ventricle.
Pulse Wave Doppler and Tissue Doppler Reveal Impaired Ejection and Diastolic Filling After CLP
As we observed signs of both systolic and diastolic dysfunction using strain echocardiography and invasive hemodynamics, we used pulse‐wave doppler to assess flow across the aortic valve (Figure 6A), and flow across the mitral valve to assess how CLP2P affects diastolic filling (Figure 6B). We further used tissue doppler to measure mitral annular velocities (Figure 6C). Compared with baseline, CLP2P at 12 hours demonstrated significantly reduced left ventricular outflow tract velocity time integral (Figure 6D) as well as reduced peak velocity (Figure 6E) and CO measured using this approach (Figure 6F). Taken together, these measurements support the presence of systolic dysfunction at this timepoint in coordination with impaired myocardial strain and dP/dtmax. Measurement of flow across the mitral valve and mitral annular velocities revealed impaired diastolic filling. We found reduction in the E/A ratio after CLP2P surgery compared with baseline (Figure 6G) as well as a reduction in the E’/A’ ratio (Figure 6H). Further we found significant impairment in the E/E’ ratio (Figure 6I). Taken together, these results suggest that CLP2P surgery results in an impaired myocardial relaxation pattern of diastolic filling44 consistent with the reduction in rGRS and rGLS as well as reduced dP/dtmin.
Figure 6.
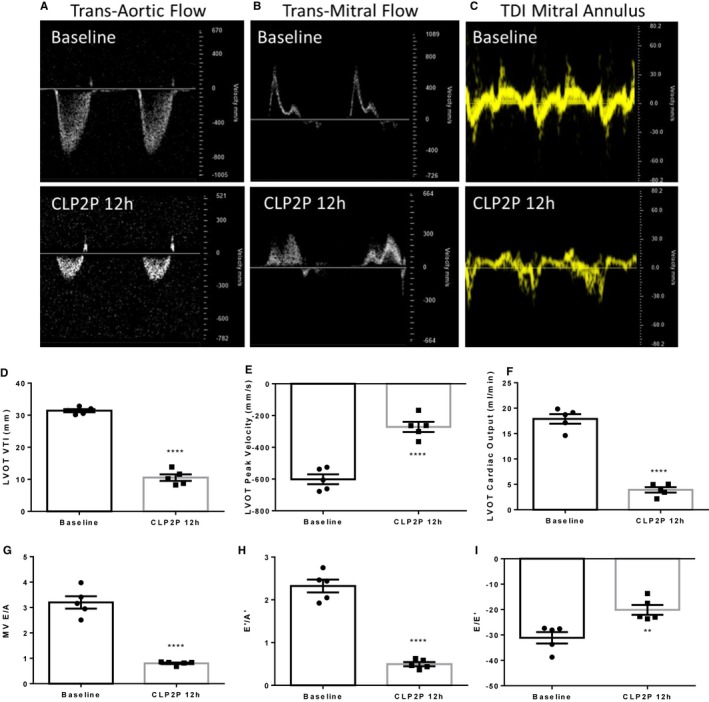
Pulsed‐wave doppler and tissue doppler‐derived measurements of systolic and diastolic function. A through C, Representative images of transaortic flow (A), transmitral flow (B), and mitral annular velocities (C) at baseline and 12 hours after CLP2P surgery. D through F, Measurement of LVOT VTI (D), LVOT Peak Velocity (E), and LVOT cardiac output (F) measured using pulsed‐wave doppler. G through I, Measurement of flow across the mitral valve. Mitral valve E/A ratio (G), E’/A’ ratio (H), and E/E’ ratio (I) at baseline and 12 hours post‐CLP2P. **P<0.01, ****P<0.0001 vs baseline by t test, n=5 mice. CLP2P indicates cecal ligation and puncture with 2 punctures; LVOT left ventricular outflow tract; MV mitral valve; TDI tissue doppler imaging; VTI velocity time integral.
Systolic and Diastolic Blood Pressure Are Reduced Within 2 Hours of LPS Injection and 6 Hours of CLP Surgery
A major component of sepsis disease pathophysiology involves reduced blood pressure and progression to septic shock and tissue hypoperfusion. Reduction in blood pressure reduces the load the heart must pump against and has been proposed as a mechanism underlying normalization in ejection fraction in sepsis45 thereby inversely correlating with cardiac systolic parameters.46 We therefore serially measured blood pressure alongside echocardiogram measurements to have a thorough characterization of the hemodynamic state of our animal models. In all groups, baseline blood pressure was comparable (systolic 116 mm Hg, mean 80 mm Hg, diastolic 68 mm Hg). For animals injected with LPS, systolic (Figure 7A), diastolic (Figure 7B), and mean (Figure 7C) arterial pressure were immediately reduced within 2 hours of injection and remained at this level during subsequent measurements. CLP reduced arterial pressure beginning 6 hours after surgery for both CLP1P and CLP2P, which progressively deteriorated during subsequent measurements (Figure 7A through 7C). CLP1P was significantly increased over CLP2P for all 3 pressures at 12 and 24 hours, and for mean and diastolic pressure at 6 hours. Pulse pressure was not affected, except at 24 hours for CLP2P mice (Figure 7D). We then measured blood lactate levels, which is a marker of shock‐related hypoperfusion which is related to reduced blood pressure and a clinically‐relevant marker of disease severity. Lactate was increased (60%) beginning 2 hours after injection. High plasma lactate levels remained until 24 hours (Figure 7E). Similarly, lactate was increased 2‐fold beginning 6 hours after CLP1P or CLP2P surgery consistent with the reduction in blood pressure at this timepoint (Figure 7F). We then determined the extent to which CO and SVR account for the reduction in mean arterial pressure. Surprisingly, we found that SVR was increased within 2 hours of CLP2P surgery and intermittently increased for CLP1P surgery, whereas LPS injection resulted in a delayed increase in SVR maximizing by 12 and 24 hours (Figure 7G). A significant difference was observed between CLP1P and CLP2P for SVR at 12 hours which was ameliorated 24 hours.
CLP and Endotoxemia Lower Surface Temperature and Body Weight Within 2 Hours
As we observed that reduction in CO was primarily responsible for the decline in blood pressure in our animals, we measured xiphoidal surface temperature. Reduction in surface temperature has previously been used to predict survival in murine sepsis,38 and is responsible for providing the name “cold shock” to hypodynamic sepsis. At baseline, all 3 groups had a surface temperature of 36°C. Surface temperature (Figure 7H) rapidly declined after CLP surgery and LPS injection and progressively deteriorated at all timepoints. Surface temperature was maximally decreased at 24 hours for all septic mice (CLP1P 28°C, CLP2P 25°C, LPS 25°C). In addition to measuring surface temperature, we measured body weight at each timepoint after sepsis induction as previous studies have associated prolonged weight loss with mortality in septic mice.40 All mice including sham mice experienced a decline in body weight likely resulting from surgical stress and reduced food intake (Figure 7I).
Reduced Cardiac Output at 2 Hours After CLP Surgery Differentiates Surviving and Non‐surviving Mice 48 Hours Post‐Sepsis Induction
To assess mortality in our models, we followed the mice at 12 hours intervals for 72 hours after surgery. CLP surgery showed a severity‐dependent mortality rate with 75% of CLP1P and 25% of CLP2P mice surviving until 48 hours, and only 12.5% of CLP2P mice and 37.5% of CLP1P mice surviving until 72 hours (Figure 7J). For mice treated with LPS, mortality began 24 hours after injection despite recovery of cardiac function and suppression of inflammatory cytokines at this timepoint. By 72 hours LPS injection was associated with 50% mortality (Figure 7J).
As it is known that reduced CO in hypodynamic sepsis is a major contributor to progression towards reduced blood pressure and shock,10 we assessed if CO normalized to body weight measured using manual LV Trace of VevoStrain software at early time points may be used to predict mortality within 48 hours post‐CLP induction (Figure 8). From both CLP1P and CLP2P protocols 8 mice survived CLP surgery and 8 mice did not. Separating mice that underwent CLP1P or CLP2P in our analysis, CO:BW predicted survival independent of initial sepsis severity. Decreased VevoStrain CO:BW in non‐surviving mice reached statistical significance at 2, 12, and 24 hours for CLP2P and 12 hours for CLP1P groups (Figure 8A). Conversely, LV Trace CO:BW was significantly reduced in non‐surviving mice at 12 and 24 hours alone for CLP2P and at 2 and 24 hours for CLP1P mice (Figure 8B). As the effect on CO by survival status appeared independent of initial severity, we combined the CLP1P and CLP2P groups to assess the predictive value of reduced CO:BW following CLP for mortality (Figure 8C and 8D). Compared with CLP survivors, non‐survivors had significantly reduced CO:BW beginning at 2 and maximally separating at 24 hours after surgery regardless of whether the measurement was taken using VevoStrain software or manual LV Trace (Figure 8C and 8D). To assess further the validity of reduced CO:BW as a predictor of mortality, we generated receiver operating characteristic (ROC) curves to optimize sensitivity and specificity at each timepoint (Figure 8E and 8F). From this analysis, we proposed cutoff values for CO:BW derived from VevoStrain and manual tracing to differentiate survivors and non‐survivors with greater than 75% sensitivity and specificity (Table). Sensitivity and specificity for differentiating survivors at 2, 4, and 6 hours was greater for VevoStrain derived measurements compared with manual tracing‐derived measurements, however both approaches were able to differentiate survivors and non‐survivors with 100% sensitivity and specificity at 12 and 24 hours after surgery.
Figure 8.
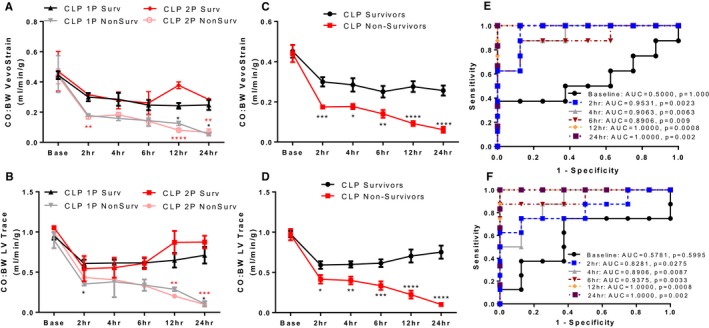
Reduced cardiac output predicts survival after CLP surgery. A and B, B‐mode automatic tracing derived cardiac output normalized to body weight measurements (A) and manual LV Trace derived measurements (B) for mice that survived or did not survive 48 hours post‐CLP1P and CLP2P procedures. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs respective survivors by t test. C and D, Cardiac output normalized measurements for 48‐hour survivors and 48‐hour non‐survivors in CLP1P and CLP2P cohorts combined for automatic tracing (C) and LV‐trace derived (D) measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs survivors by t test. n=8 survivors, n=8 non‐survivors. E and F, receiver operating characteristic analysis to identify optimal values of cardiac output normalized to body weight to differentiate survivors and non‐survivors. Receiver operating characteristic‐curve for VevoStrain (E) and manual trace (F) derived measurements. 1P indicates 1 puncture; 2P, 2 puncture; AUC, area under the curve; BL, baseline; CLP, cecal ligation and puncture; CO:BW, cardiac output normalized to body weight; LV, left ventricle.
Table 1.
Receiver operating characteristic ‐Curve Derived Values of Cardiac Output Normalized to Body Weight to Differentiate Survivors and Non‐Survivors With Optimal Sensitivity and Specificity
| Echo‐Parameter | Timepoint (h, after CLP) | Cutoff Value (mL/min) | Sensitivity | Specificity | Likelihood Ratio |
|---|---|---|---|---|---|
|
Cardiac output Normalized BW VevoStrain |
2 | <0.2279 | 100 | 87.5 | 8 |
| 4 | <0.2047 | 87.5 | 87.5 | 7 | |
| 6 | <0.1790 | 87.5 | 87.5 | 7 | |
| 12 | <0.1569 | 100 | 100 | N/A | |
| 24 | <0.1384 | 100 | 100 | N/A | |
|
Cardiac output Normalized BW LV trace |
2 | <0.4284 | 87.5 | 87.5 | 6 |
| 4 | <0.4777 | 87.5 | 75 | 6 | |
| 6 | <0.4625 | 75 | 87.5 | 7 | |
| 12 | <0.4246 | 100 | 100 | N/A | |
| 24 | <0.3331 | 100 | 100 | N/A |
BW indicates body weight; CLP cecal ligation and puncture; LV left ventricle.
CLP Results in Combined Systolic Dysfunction and Low Preload While LPS Causes Systolic Dysfunction Alone
As CO is regulated by both preload and cardiac contractility, we generated scatterplots to identify the contribution of alterations in EDV and GLS throughout the disease course to changes in CO. We generated scatter plots to identify the correlation between EDV and GLS, EDV and CO, and GLS and CO measurements (Figure 9). There was no correlation between GLS and EDV in either model (Figure 9A and 9B). For the CLP model, a strong correlation between EDV and CO was observed suggesting that preload accounts for ≈76.6% (b=0.3843, P<0.0001) of the variation in CO in CLP2P and 46.1% (b=0.2620, P<0.0001) in CLP1P (Figure 9C), which was not observed for the LPS model (Figure 9D). For both CLP and LPS models, a significant negative correlation between GLS and CO was observed (Figure 9E and 9F), however, the association between impaired GLS and reduced CO was more pronounced for the LPS model (R 2=0.6271, b=‐0.4462, P<0.0001) compared with CLP1P (R 2=0.3796, b=‐0.2596, P<0.0001) or CLP2P (R 2=0.3703, b=‐0.3062, P<0.0001).
Figure 9.
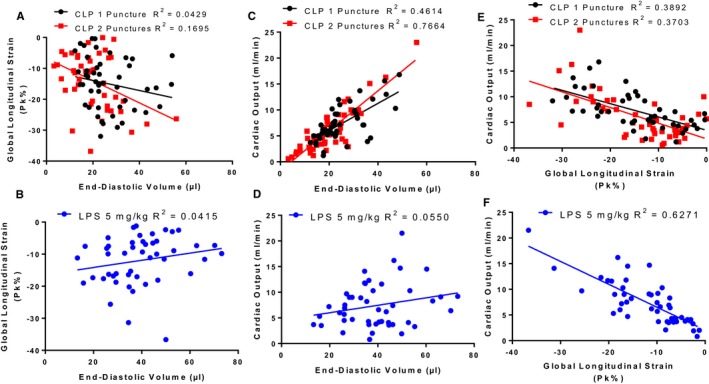
Preload and systolic dysfunction differentially contribute to reduced cardiac output between CLP and LPS sepsis models. A and B, No correlation between end‐diastolic volume and global longitudinal strain for echocardiogram measurements taken throughout the CLP1P, CLP2P, (A) and LPS (B) protocol. C and D, Significant correlation between end‐diastolic volume and cardiac output for CLP1P and CLP2P (C) but not LPS (D) sepsis model. E and F, Significant correlation between global longitudinal strain and cardiac output for both CLP (E) and LPS (F) models. Data points are representative of measurements from 24 mice at baseline, 2, 4, 6, 12, and 24 hours after sepsis induction. n=48 measurements for CLP1P, n=46 measurements for CLP2P, n=47 for LPS measurements 5 mg/kg. CLP indicates cecal ligation and puncture; LPS lipopolysaccharide.
Treatment With Fluids and Antibiotics Restores Diastolic Volume, Increases Cardiac Output, Prevents Hypotension, and Prevents Mortality After CLP
In order to test the validity of our mortality prediction model, we tested if an established treatment would reverse the effects of CLP2P on reduced preload, cardiac output, and eventually mortality. We therefore induced sepsis by CLP2P surgery and performed echocardiographic analysis and blood pressure assessment before the administration of antibiotic treatment (ertapenem 70 mg/kg) at 2 hours dissolved in 10 mL/kg saline (Figure 10A). We then performed subsequent measurements at 4, 6, 12, and 24 hours after surgery. Cardiac function and CO declined similarly between the treated and non‐treated groups before the administration of treatment. FS was partially increased at 4 hours, but not to baseline levels (Figure 10B). The difference in FS between treated and untreated mice was lost as FS began to increase at 6 hours and returned toward normal levels at 12 and 24 hours in untreated mice (Figure 10B). Treatment increased EDV (Figure 10C) beginning at 6 hours and progressively increased further until 24 hours. It also expanded ESV (Figure 10D) at 12 and 24 hours. CO:BW ratio increased at 4 hours and continued to increase further until 24 hours (Figure 10E). Consistent with partial restoration of systolic function, we found that both GLS and GLS rate were partially improved at 4 and 12 hours post‐surgery (Figure 10F and 10G). Likewise, ertapenem treatment partially improved GCS and GCS rate at 6, 12, and 24 hours (Figure 10H and 10I).
Figure 10.
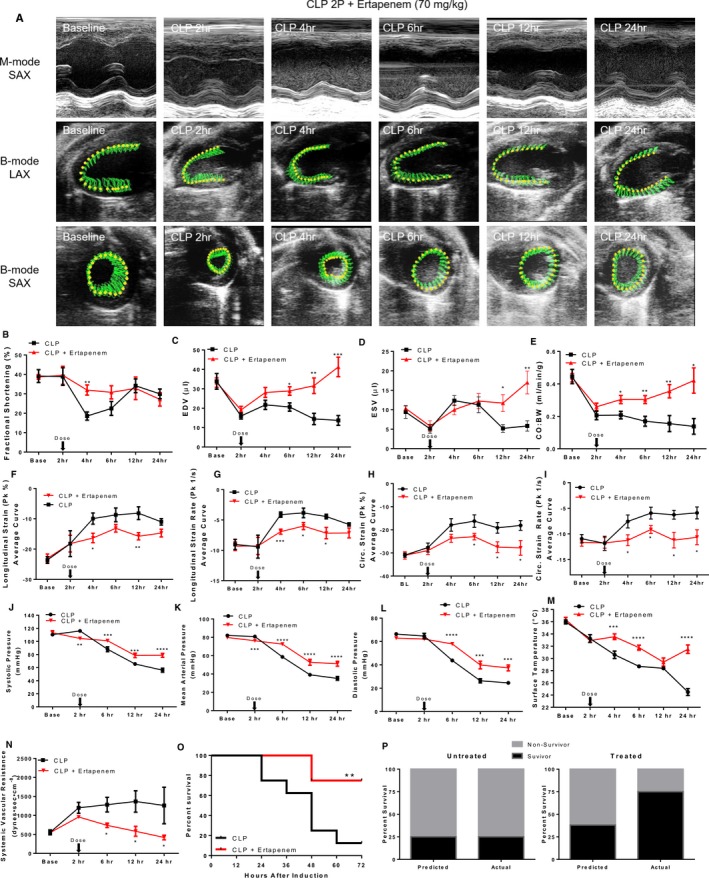
Antibiotic and fluid treatment reverses CLP‐associated suppression of cardiac output and reduces mortality. (A) Representative 2‐dimensional echocardiogram images, fractional shortening (B), end‐diastolic volume (C), end‐systolic volume (D), CO:BW (E) measured by speckle tracking echocardiography, global longitudinal strain (F) and strain rate (G), and global circumferential strain (H) and strain rate (I), systolic (J), mean (K), and diastolic (L) blood pressure measured by tail cuff, xiphoidal surface temperature (M), systemic vascular resistance (SVR) calculated from measurements of cardiac output and mean arterial blood pressure (N), Kaplan–Meier survival (O) in mice that underwent CLP2P and were treated with ertapenem dosed 2 hours post‐CLP. Predicted vs actual 48‐hour mortality for treated and untreated mice using BW:CO measurements less than 0.2279 mL/min per g to differentiate predicted survivors and non‐survivors (P). *P<0.05, **P<0.01. ***P<0.001, ****P<0.0001 vs CLP by t test. n=8 CLP, n=8 CLP + ertapenem. Base indicates baseline; Circ, circumferential; CLP2P, cecal ligation and puncture with 2 punctures; CO:BW cardiac output normalized to body weight; EDV end‐diastolic volume; ESV end‐systolic volume; LAX long axis; Pk peak; SAX short axis.
Although treated mice still experienced a decline in blood pressure, systolic (Figure 10J), mean (Figure 10K), and diastolic (Figure 10L) were significantly increased compared with untreated mice at 6, 12, and 24 hours. In coordination with the delay in hypotension, body surface temperature was significantly increased at 4, 6, and 24 hours (Figure 10M). Body surface temperature was transiently decreased to a similar level with untreated mice at 12 hours (Figure 10M). Interestingly, ertapenem reduced the compensatory induction of SVR observed in untreated mice, and significant reductions in SVR were observed beginning at 6 hours (Figure 10N).
The treated mice had significantly increased 48 hours survival (75% survival, Figure 10O). We used parasternal long axis‐derived measurements of CO:BW ratio to predict mortality and assess the therapeutic potential of the applied treatment. We then applied our receiver operating characteristic derived CO:BW threshold which we showed optimally differentiates surviving and non‐surviving mice at 48 hours. For this assessment, we used measurements taken at 2 hours before treatment was applied. This analysis estimated that 75% of the mice that would not be treated and 67.5% of the mice in the cohort that would be treated had CO:BW ratio below the 0.2279 mL/min per g threshold we established from our receiver operating characteristic analysis. As predicted, the proportion of non‐survivors 48 hours post‐CLP among the mice that were not treated was 75%. The treatment reduced the actual mortality from the 67.5% that was predicted based on CO:BW ratio before application of the treatment to 25% (Figure 10P).
Discussion
The recently published Sepsis3 criteria have emphasized the importance of organ dysfunction in the identification and diagnosis of patients with sepsis.1 As such recent preclinical guidelines emphasize the need to resolve organ dysfunction in preclinical sepsis studies.17 EF and FS derived from LV‐trace measurements are widely used as surrogate markers of cardiac contractility and cardiac function.47 We found that LPS‐ and CLP‐induced sepsis reduced these markers early in the disease course. However, late stage CLP but not LPS treatment resulted in increased EF/FS despite sustained elevation in inflammatory cytokines, and reduced dP/dtmax, left ventricular outflow tract velocity time integral, and myocardial strain. Reductions in preload and reduced blood pressure lower the work developed by the heart for ejection and reduce the stroke volume represented by identical ejection fractions.10 This phenomenon has been described in septic patients, where low diastolic volumes constitutes poor prognosis,48, 49, 50 and is consistent with preclinical data demonstrating impaired cardiac contractility with preserved ejection fraction.26, 29, 51, 52 These studies had focused mainly on later timepoints, while here we show that reductions in preload and afterload occur within 2 and 6 hours post‐CLP, respectively.
As our study and others have demonstrated the limitations of using EF and FS as markers of cardiac dysfunction in the context of reduced cardiac load, we used speckle‐tracking echocardiography to characterize septic cardiac dysfunction by assessing myocardial strain and strain rate changes through the course of different sepsis mouse models. We found that both CLP and LPS injection reduced myocardial strain and strain rate in all 3 planes, which was impaired more consistently and earlier compared with EF and FS throughout the course of disease. Previous literature has shown reduced myocardial strain in the context of preserved EF and FS.51 However, that study focused on later timepoints and did not compare different sepsis disease models. Our study shows the early course of reduced myocardial strain in both endotoxemia and CLP‐induced sepsis, and identified how this parameter changes with respect to myocardial load. Therefore, the present study adds to a growing base of studies demonstrating the benefits of speckle‐tracking echocardiography in assessing septic cardiac dysfunction.
In addition to measuring cardiac systolic function, VevoStrain may be used to assess parameters of myocardial relaxation including rGRS rate and rGLS rate.37 These measurements provide information about the rate of expansion of the left ventricle during early filling and may be used as markers of cardiac diastolic dysfunction. These markers are impaired within 2 to 4 hours of sepsis induction which likely results from the combination of relative hypovolemia and reduced myocardial relaxation as we confirmed with invasive hemodynamics and pulsed‐wave Doppler. These measurements require B‐mode parasternal long axis images of the heart which is faster to obtain compared with the apical 4‐chamber views of the mouse heart that is required to assess transmitral flow. Furthermore, this approach may facilitate future studies aiming to address diastolic dysfunction in sepsis, which is a major manifestation of septic cardiac dysfunction and predictor of mortality.53
We further demonstrate differential manifestations in the hemodynamic state of CLP sepsis and endotoxemia despite being mouse models for the same condition, namely that endotoxemia does not result in preload deficit but rather leads to left ventricular expansion. A larger proportion of the reduction in CO in the LPS model was accounted for by systolic dysfunction. Previous studies of our group discovered experimental treatments that improve systolic function following LPS injection.19, 20, 21 Conversely, most of the reduction in CO in the CLP model is accounted for by reduced preload as well as impaired myocardial strain. Thus, reversal of the preload deficit is a major goal of treatment in the CLP model, as it is in septic patients with a similar presentation.54 In support of CO as a driving factor for hypotension and shock in these models, we show that lower CO can be used as early as 2 hours after CLP surgery to predict mortality at 48 hours. This was confirmed when we used a known treatment with antibiotic and fluid administration. This finding designates CO measurement as one of the tools that allow for early evaluation of the therapeutic potential of an applied treatment. These results are consistent with evidence from septic patients demonstrating an improvement in CO after treatment as a positive prognostic marker,55 and the association of reduced CO measured via continuous hemodynamic signal with non‐surviving septic patients.56
Our animal models share numerous features with human sepsis including activation of inflammatory pathways, reduced cardiac function,2 and elevated BNP.3, 57, 58 CLP of either severity reduced EDV and CO, and increased SVR which has also been described in septic patients.54 Nevertheless, LV dilation, which occurs in the LPS model but not in the CLP model, is also apparent in human sepsis.29, 59 Therefore, the full spectrum of presentations cannot be accounted for fully by either the LPS or CLP model, and the specific question that a researcher aims to address should define the selected mouse model of sepsis. Importantly, human sepsis is classically associated with an initial hyperdynamic phase characterized by high CO and low peripheral resistance.2, 9 This hyperdynamic phase was not observed in any of our animal models despite administering saline post‐surgery. This is consistent with previous evidence showing reductions in diastolic dimension and cardiac output early after CLP with resuscitation.29
Another study has suggested that modifications to the CLP protocol involving vigorous administration of fluids may induce a state of hyperdynamic sepsis in mice, associated with increased heart rate from 407 BPM in sham mice to 524 BPM after CLP.60 This model demonstrated similar progression as our CLP2P model with hypotension initiating within 6 hours of surgery and ≈70% mortality within 48 hours. Progression to a hypodynamic state and reduced preload within the 48 hours period was not observed. That approach included continuous hemodynamic assessment in mice that were constantly anesthetized for 48 hours with ketamine/acepromazine, and therefore the anesthetic regimen of this study differs from our own and other studies. Further, this same protocol resulted in an early hypodynamic state in a subsequent study published by this group.29 Hyperdynamic responses are more consistently observed in other animal models of sepsis including rat and large animals, such as pig and sheep sepsis models.61, 62, 63, 64 However, rats and large animals are not suitable for tissue‐specific gain‐ and loss‐of‐function studies that mice are routinely used for. Further, a proportion of septic patients will enter an initial hyperdynamic state, whereas a significant proportion of patients will rapidly enter hypodynamic sepsis as we observed in our mouse models.46 Therefore, both models of hyperdynamic and hypodynamic sepsis are clinically relevant and may be used for translational studies.
Conclusions
In the present study, we found that myocardial strain and strain rate are reduced early after CLP and LPS‐induced sepsis in coordination with elevation of proinflammatory cytokine levels and plasma BNP. Depression of strain and strain rate occurred even in the context of reduced myocardial preload and reduced blood pressure. Myocardial strain and strain rate measurements may be used as surrogate markers to measure diastolic and systolic function which are impaired within 2 to 4 hours of CLP or LPS injection. Despite being models for the same disease, CLP and endotoxemia models of sepsis have different pathophysiological mechanisms underlying shock which may affect the response to experimental treatment and applicability to different subpopulations of septic patients. Lastly, a reduction in CO measured using either manual LV trace or automatic tracing through VevoStrain strongly predicts survival within 48 hours of surgery in CLP‐induced sepsis, and therefore echocardiography emerges as a useful tool to account for variability in survival between cohorts and to document recovery after treatment.
Limitations
We did not assess the effect of biological factors including sex or age on outcome in our study. It is possible that cardiovascular dysfunction will manifest differently in male and female mice in different sepsis disease models. Specifically, it is known that females in animal studies are resistant to cardiac dysfunction that is characteristic of sepsis.18, 25
For echocardiography, we minimized the level of anesthesia provided to the mice and shortened the duration of each imaging session to minimize the effect of anesthesia. Although all mice including controls were exposed to anesthetic, the effect of isoflurane on heart rate and myocardial contractility cannot be discounted. Further, assessment of myocardial volumes relies on assumptions made by the software about the 3‐dimensional structure of the LV lumen based on dimensions derived from 2‐dimensional images. However, LV‐trace, automatic tracing, and invasive hemodynamics demonstrate the same reduction in ventricular volumes thereby supporting the use of echocardiography to measure preload in murine sepsis.
We characterized and compared multiple measures of cardiac systolic function including EF/FS, dP/dtmax, CO, left ventricular outflow tract velocity time integral, and myocardial strain and strain rate. The ability of the myocardium to eject volume and generate pressure during systole is load‐dependent, and each of these measurements are affected by the changes in both preload and afterload throughout the course of sepsis.10, 65, 66, 67, 68 Although we did not observe a significant association between EDV and GLS in any of our models, other studies have shown that myocardial strain, as with other systolic measures, is preload‐dependent.66, 68 Therefore, the impairment in myocardial strain in our sepsis models likely results from the combined effect of sepsis‐associated myocardial depression and load‐dependent effects on strain.
Sources of Funding
This study was supported by the National Heart Lung and Blood Institute of the National Institutes of Health HL130218 (Dr Drosatos), and P01HL091799 (Dr Koch), and the W.W. Smith Charitable Trust (Dr Drosatos). M. Hoffman was supported by an American Heart Association predoctoral fellowship (18PRE34060115). Dr Kyriazis was supported by the American Heart Association and the Kahn Family Post‐Doctoral Fellowship in Cardiovascular Research (18POST34060150). Dr de Lucia was supported by an American Heart Association postdoctoral fellowship (17POST33660942).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e012260 DOI: 10.1161/JAHA.119.012260.)
References
- 1. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The third international consensus definitions for sepsis and septic shock (sepsis‐3). JAMA. 2016;315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunter JD, Doddi M. Sepsis and the heart. BJA. 2010;104:3–11. [DOI] [PubMed] [Google Scholar]
- 3. Perman SM, Chang AM, Hollander JE, Gaieski DF, Trzeciak S, Birkhahn R, Otero R, Osborn TM, Moretti E, Nguyen HB, Gunnerson KJ, Milzman D, Goyal M, Cairns CB, Ngo L, Rivers EP, Shapiro NI. Relationship between B‐type natriuretic peptide and adverse outcome in patients with clinical evidence of sepsis presenting to the emergency department. Acad Emerg Med. 2011;18:219–222. [DOI] [PubMed] [Google Scholar]
- 4. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. [DOI] [PubMed] [Google Scholar]
- 5. Lin CW, Tang W, Wen F, Chen JJ, Zhen XL, Chen ZG. Diagnostic accuracy of NT‐ProBNP for heart failure with sepsis in patients younger than 18 years. PLoS One. 2016;11:e0147930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai YL, Hu BL, Wen HC, Zhang YL, Zhu JJ. Prognostic value of plasma brain natriuretic peptide value for patients with sepsis: a meta‐analysis. J Crit Care. 2018;48:145–152. [DOI] [PubMed] [Google Scholar]
- 7. Chang WT, Lee WH, Lee WT, Chen PS, Su YR, Liu PY, Liu YW, Tsai WC. Left ventricular global longitudinal strain is independently associated with mortality in septic shock patients. Intensive Care Med. 2015;41:1791–1799. [DOI] [PubMed] [Google Scholar]
- 8. Sanfilippo F, Corredor C, Fletcher N, Tritapepe L, Lorini FL, Arcadipane A, Vieillard‐Baron A, Cecconi M. Left ventricular systolic function evaluated by strain echocardiography and relationship with mortality in patients with severe sepsis or septic shock: a systematic review and meta‐analysis. Crit Care. 2018;22:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gahhos FN, Chiu RCJ, Bethune D, Dion Y, Hinchey EJ, Richards GK. Hemodynamic responses to sepsis: hypodynamic versus hyperdynamic states. J Surg Res. 1981;31:475–481. [DOI] [PubMed] [Google Scholar]
- 10. Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis‐induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20:195–203. [DOI] [PubMed] [Google Scholar]
- 12. Levy MM, Evans LE, Rhodes A. The surviving sepsis campaign bundle: 2018 update. Intensive Care Med. 2018;44:925–928. [DOI] [PubMed] [Google Scholar]
- 13. Lewis AJ, Seymour CW, Rosengart MR. Current murine models of sepsis. Surg Infect. 2016;17:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zingarelli B, Coopersmith CM, Drechsler S, Efron P, Marshall JC, Moldawer L, Wiersinga WJ, Xiao X, Osuchowski MF, Thiemermann C. Part I: minimum quality threshold in preclinical sepsis studies (MQTiPSS) for study design and humane modeling endpoints. Shock. 2019;51:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Libert C, Ayala A, Bauer M, Cavaillon JM, Deutschman C, Frostell C, Knapp S, Kozlov AV, Wang P, Osuchowski MF, Remick DG. Part II: minimum quality threshold in preclinical sepsis studies (MQTiPSS) for types of infections and organ dysfunction endpoints. Shock. 2019;51:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hellman J, Bahrami S, Boros M, Chaudry IH, Fritsch G, Gozdzik W, Inoue S, Radermacher P, Singer M, Osuchowski MF, Huber‐Lang M. Part III: minimum quality threshold in preclinical sepsis studies (MQTiPSS) for fluid resuscitation and antimicrobial therapy endpoints. Shock. 2019;51:33–43. [DOI] [PubMed] [Google Scholar]
- 17. Osuchowski MF, Ayala A, Bahrami S, Bauer M, Boros M, Cavaillon JM, Chaudry IH, Coopersmith CM, Deutschman CS, Drechsler S, Efron P, Frostell C, Fritsch G, Gozdzik W, Hellman J, Huber‐Lang M, Inoue S, Knapp S, Kozlov AV, Libert C, Marshall JC, Moldawer LL, Radermacher P, Redl H, Remick DG, Singer M, Thiemermann C, Wang P, Wiersinga WJ, Xiao X, Zingarelli B. Minimum quality threshold in pre‐clinical sepsis studies (MQTiPSS): an international expert consensus initiative for improvement of animal modeling in sepsis. Shock. 2018;50:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kokkinaki D, Hoffman M, Kalliora C, Kyriazis ID, Maning J, Lucchese AM, Shanmughapriya S, Tomar D, Park JY, Wang H, Yang XF, Madesh M, Lymperopoulos A, Koch WJ, Christofidou‐Solomidou M, Drosatos K. Chemically synthesized secoisolariciresinol diglucoside (LGM2605) improves mitochondrial function in cardiac myocytes and alleviates septic cardiomyopathy. J Mol Cell Cardiol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drosatos K, Drosatos‐Tampakaki Z, Khan R, Homma S, Schulze PC, Zannis VI, Goldberg IJ. Inhibition of c‐Jun‐N‐terminal kinase increases cardiac peroxisome proliferator‐activated receptor alpha expression and fatty acid oxidation and prevents lipopolysaccharide‐induced heart dysfunction. J Biol Chem. 2011;286:36331–36339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, Homma S, Schulze PC, Goldberg IJ. Peroxisome proliferator‐activated receptor‐gamma activation prevents sepsis‐related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, Trent CM, Hu Y, Guo S, Aifantis I, Goldberg IJ. Cardiac myocyte KLF5 regulates Ppara expression and cardiac function. Circ Res. 2016;118:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, Ratner AJ, Madesh M, Drosatos K, Morrow JP. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis‐induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017;2:94248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blessberger H, Binder T. Two dimensional speckle tracking echocardiography: basic principles. Heart. 2010;96:716–722. [DOI] [PubMed] [Google Scholar]
- 24. Potter E, Marwick TH. Assessment of left ventricular function by echocardiography: the case for routinely adding global longitudinal strain to ejection fraction. Imaging. 2018;11:260–274. [DOI] [PubMed] [Google Scholar]
- 25. Chen J, Chiazza F, Collino M, Patel NS, Coldewey SM, Thiemermann C. Gender dimorphism of the cardiac dysfunction in murine sepsis: signalling mechanisms and age‐dependency. PLoS One. 2014;9:e100631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luptak I, Croteau D, Valentine C, Qin F, Siwik D, Remick D, Colucci WS, Hobai I. Myocardial redox hormesis protects the hearts of female mice in sepsis. Shock. 2018. Available at: https://insights.ovid.com/crossref?an=00024382-900000000-97766. Accessed May 15, 2019. [DOI] [PubMed] [Google Scholar]
- 27. Toscano MG, Ganea D, Gamero AM. Cecal ligation puncture procedure. J Vis Exp. 2011;51:e2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wen H. Sepsis induced by cecal ligation and puncture. Methods Mol Biol. 2013;1031:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zanotti Cavazzoni SL, Guglielmi M, Parrillo JE, Walker T, Dellinger RP, Hollenberg SM. Ventricular dilation is associated with improved cardiovascular performance and survival in sepsis. Chest. 2010;138:848–855. [DOI] [PubMed] [Google Scholar]
- 30. Zhou F, Peng ZY, Bishop JV, Cove ME, Singbartl K, Kellum JA. Effects of fluid resuscitation with 0.9% saline versus a balanced electrolyte solution on acute kidney injury in a rat model of sepsis. Crit Care Med. 2014;42:e270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan AI, Coldewey SM, Patel NS, Rogazzo M, Collino M, Yaqoob MM, Radermacher P, Kapoor A, Thiemermann C. Erythropoietin attenuates cardiac dysfunction in experimental sepsis in mice via activation of the β‐common receptor. Dis Models Mech. 2013;6:1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suda K, Kitagawa Y, Ozawa S, Saikawa Y, Ueda M, Ebina M, Yamada S, Hashimoto S, Fukata S, Abraham E, Maruyama I, Kitajima M, Ishizaka A. Anti‐high‐mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J Surg. 2006;30:1755–1762. [DOI] [PubMed] [Google Scholar]
- 33. Fang ZX, Li YF, Zhou XQ, Zhang Z, Zhang JS, Xia HM, Xing GP, Shu WP, Shen L, Yin GQ. Effects of resuscitation with crystalloid fluids on cardiac function in patients with severe sepsis. BMC Infect Dis. 2008;8:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brandt S, Regueira T, Bracht H, Porta F, Djafarzadeh S, Takala J, Gorrasi J, Borotto E, Krejci V, Hiltebrand LB, Bruegger LE, Beldi G, Wilkens L, Lepper PM, Kessler U, Jakob SM. Effect of fluid resuscitation on mortality and organ function in experimental sepsis models. Crit Care. 2009;13:R186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hilton AK, Bellomo R. A critique of fluid bolus resusciation in severe sepsis. Crit Care. 2012;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schnelle M, Catibog N, Zhang M, Nabeebaccus AA, Anderson G, Richards DA, Sawyer G, Zhang X, Toischer K, Hasenfuss G, Monaghan MJ, Shah AM. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J Mol Cell Cardiol. 2018;114:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Lucia C, Wallner M, Eaton DM, Zhao H, Houser SR, Koch WJ. Echocardiographic strain analysis for the early detection of left ventricular systolic/diastolic dysfunction and dyssynchrony in a mouse model of physiological aging. J Gerontol A Biol Sci Med Sci. 2019;74:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laitano O, Van Steenbergen D, Mattingly AJ, Garcia CK, Robinson GP, Murray KO, Clanton TL, Nunamaker EA. Xiphoid surface temperature predicts mortality in a murine model of septic shock. Shock. 2018;50:226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Y, Ge S, Peng Y, Chen X. Inflammation and cardiac dysfunction during sepsis, muscular dystrophy, and myocarditis. Burns Trauma. 2013;1:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Osuchowski MF, Craciun F, Weixelbaumer K, Duffy ER, Remick DG. Sepsis chronically in MARS: systemic cytokine responses are always mixed regardless of the outcome, magnitude or phase of sepsis. J Immunol. 2012;189:4648–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seemann S, Zohles F, Lupp A. Comprehensive comparison of three different animal models for systemic inflammation. J Biomed Sci. 2017;24:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Braga Lima Carvalho Canesso M, Borges IN, de Deus Queiroz Santos TA, Ris TH, de Barros MVL, Nobre V, Nunes MCP. Value of speckle‐tracking echocardiography changes in monitoring myocardial dysfunction during treatment of sepsis: potential prognostic implications. Int J Cardiovasc Imaging. 2019;35:855–859. [DOI] [PubMed] [Google Scholar]
- 43. Yamamoto K, Masuyama T, Tanouchi J, Doi Y, Kondo H, Hori M, Kitabatake A, Kamada T. Effects of heart rate on left ventricular filling dynamics: assessment from simultaneous recordings of pulsed doppler transmitral flow velocity pattern and haemodynamic variables. Cardiovasc Res. 1993;27:935–941. [DOI] [PubMed] [Google Scholar]
- 44. Mottram PM, Marwick TH. Assessment of diastolic function: what the general cardiologist needs to know. Heart. 2005;91:681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Geer LD, Engvall J, Oscarsson A. Strain echocardiography in septic shock—a comparison with systolic and diastolic function parameters, cardiac biomarkers and outcome. Crit Care. 2015;19:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boissier F, Razazi K, Seemann A, Bedet A, Thille AW, de Prost N, Lim P, Brun‐Buisson C, Mekontso Dessap A. Left ventricular systolic dysfunction during septic shock: The role of loading conditions. Intensive Care Med. 2017;43:633–642. [DOI] [PubMed] [Google Scholar]
- 47. Tanaka S. Standard measurement of cardiac function indexes. J Med Ultrasonics. 2006;33:123–127. [DOI] [PubMed] [Google Scholar]
- 48. Jardin F, Fourme T, Page B, Loubieróres Y, Vieillard‐Baron A, Beauchet A, Bourdarias JP. Persistent preload defect in severe sepsis despite fluid loading. Chest. 1999;116:1354–1359. [DOI] [PubMed] [Google Scholar]
- 49. Furian T, Aguiar C, Prado K, Ribeiro RVP, Becker L, Martinelli N, Clausell N, Rohde LE, Biolo A. Ventricular dysfunction and dilation in severe sepsis and septic shock: relation to endothelial function and mortality. J Crit Care. 2012;27:319.e9–15. [DOI] [PubMed] [Google Scholar]
- 50. Dalla K, Hallman C, Bech‐Hanssen O, Haney M, Ricksten SE. Strain echocardiography identifies impaired longitudinal systolic function in patients with septic shock and preserved ejection fraction. Cardiovasc Ultrasound. 2015;13:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haileselassie B, Su E, Pozios I, Nino D, Liu H, Lu DY, Ventoulis I, Fulton WB, Sodhi CP, Hackam D, O'Rourke B, Abraham T. Myocardial oxidative stress correlates with left ventricular dysfunction on strain echocardiography in a rodent model of sepsis. Intensive Care Med Exp. 2017;5:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sakai M, Suzuki T, Tomita K, Yamashita S, Palikhe S, Hattori K, Yoshimura N, Matsuda N, Hattori Y. Diminished responsiveness to dobutamine as an inotrope in mice with cecal ligation and puncture‐induced sepsis: attribution to phosphodiesterase 4 upregulation. Am J Physiol Heart Circ Physiol. 2017;312:H1224–H1237. [DOI] [PubMed] [Google Scholar]
- 53. Landesberg G, Gilon D, Meroz Y, Georgieva M, Levin PD, Goodman S., Avidan A, Beeri R, Weissman C, Jaffe AS, Sprung CL. Diastolic dysfunction and mortality in severe sepsis and septic shock. Eur Heart J. 2012;33:895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schumer W. Pathophysiology and treatment of septic shock. Am J Emerg Med. 1984;2:74–77. [DOI] [PubMed] [Google Scholar]
- 55. Kimmoun A, Ducrocq N, Mory S, Delfosse R, Muller L, Perez P, Fay R, Levy B. Cardiac contractile reserve parameters are related to prognosis in septic shock. Biomed Res Int. 2013;2013:930673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carrara M, Baselli G, Ferrario M. Mortality prediction model of septic shock patients based on routinely recorded data. Comput Math Methods Med. 2015;2015:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rivers EP, McCord J, Otero R, Jacobsen G, Loomba M. Clinical utility of B‐type natriuretic peptide in early severe sepsis and septic shock. J Intensive Care Med. 2007;22:363–373. [DOI] [PubMed] [Google Scholar]
- 58. Post F, Weilemann LS, Messow CM, Sinning C, Munzel T. B‐type natriuretic peptide as a marker for sepsis‐induced myocardial depression in intensive care patients. Crit Care Med. 2008;36:3030–3037. [DOI] [PubMed] [Google Scholar]
- 59. Omar AA, El‐Shahat N, Ramadan MM. Cardiac functions in patients with sepsis and septic shock. Egyptian Heart J. 2012;64:191–196. [Google Scholar]
- 60. Hollenberg SM, Dumasius A, Easington C, Colilia SA, Neumann A, Parrillo JE. Characterization of a hyperdynamic murine model of resuscitated sepsis using echocardiography. Am J Respir Crit Care Med. 2001;164:891–895. [DOI] [PubMed] [Google Scholar]
- 61. Wang P, Ba ZF, Cioffi WG. The pivotal role of adrenomedullin in producing hyperdynamic circulation during the early stage of sepsis. JAMA Surg. 1998;133:1298–1304. [DOI] [PubMed] [Google Scholar]
- 62. Morisaki H, Sibbald W, Martin C, Doig G, Inman K. Hyperdynamic sepsis depresses circulatory compensation to normovolemic anemia in conscious rats. J Appl Physiol. 1996;80:656–664. [DOI] [PubMed] [Google Scholar]
- 63. Giantomasso DD, May CN, Bellomo R. Vital organ blood flow during hyperdynamic sepsis. Chest. 2003;124:1053–1059. [DOI] [PubMed] [Google Scholar]
- 64. Chvojka J, Sykora R, Krouzecky A, Radej J, Varnerova V, Karvunidis T, Hes O, Novak I, Radermacher P, Matejovic M. Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis‐induced septic shock in pigs. Crit Care. 2008;12:R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Konishi T, Nakamura Y, Kato I, Kawai C. Dependence of peak dP/dt and mean ejection rate on load and effect of inotropic agents on the relationship between peak dP/dt and left ventricular developed pressure—assessed in the isolated working rat heart and cardiac muscles. Int J Cardiol. 1992;35:333–341. [DOI] [PubMed] [Google Scholar]
- 66. Burns AT, La Gerche A, D'hooge J, MacIsaac AI, Prior DL. Left ventricular strain and strain rate: characterization of the effect of load in human subjects. Eur J Echocardiogr. 2010;11:283–289. [DOI] [PubMed] [Google Scholar]
- 67. Blaudszun G, Licker MJ, Morel DR. Preload‐adjusted left ventricular dP/dtmax: a sensitive, continuous, load‐independent contractility index. Exp Physiol. 2013;98:1446–1456. [DOI] [PubMed] [Google Scholar]
- 68. Fredholm M, Jorgensen K, Houltz E, Ricksten SE. Load‐dependence of myocardial deformation variables—a clinical strain‐echocardiographic study. Acta Anaesthesiol Scand. 2017;61:1155–1165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare that all supporting data and method descriptions are available within the article or from the corresponding author upon reasonable request.