

Abstract
Background
Protein posttranslational modifications by O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) increase with cardiac hypertrophy, yet the functional effects of these changes are incompletely understood. In other organs, O‐GlcNAc promotes adaptation to acute physiological stressors; however, prolonged O‐GlcNAc elevations are believed to be detrimental. We hypothesize that early O‐GlcNAcylation improves cardiac function during initial response to pressure overload hypertrophy, but that sustained elevations during established pathological hypertrophy negatively impact cardiac function by adversely affecting calcium handling proteins.
Methods and Results
Transverse aortic constriction or sham surgeries were performed on littermate controls or cardiac‐specific, inducible O‐GlcNAc transferase knockout (OGTKO) mice to reduce O‐GlcNAc levels. O‐GlcNAc transferase deficiency was induced at different times. To evaluate the initial response to pressure overload, OGTKO was completed preoperatively and mice were followed for 2 weeks post‐surgery. To assess prolonged O‐GlcNAcylation during established hypertrophy, OGTKO was performed starting 18 days after surgery and mice were followed until 6 weeks post‐surgery. In both groups, OGTKO with transverse aortic constriction caused significant left ventricular dysfunction. OGTKO did not affect levels of the calcium handling protein SERCA2a. OGTKO reduced phosphorylation of phospholamban and cardiac troponin I, which would negatively impact cardiac function. O‐GlcNAcylation of protein kinase A catalytic subunit, a kinase for phospholamban, decreased with OGTKO.
Conclusions
O‐GlcNAcylation promotes compensated cardiac function in both early and established pathological hypertrophy. We identified a novel O‐GlcNAcylation of protein kinase A catalytic subunit, which may regulate calcium handling and cardiac function.
Keywords: cardiac failure, cardiac hypertrophy, O‐GlcNAc, OGT, pressure overload, protein kinase A phosphorylation
Subject Categories: Animal Models of Human Disease, Basic Science Research, Cell Signalling/Signal Transduction, Heart Failure, Remodeling
Clinical Perspective
What Is New?
Cardiac specific O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) transferase knockout and the resultant reduction in protein O‐GlcNAcylation negatively impacts cardiac function during both the early hypertrophic response to pressure overload and separately during established pressure overload hypertrophy.
O‐GlcNAc modifies cardiac protein kinase A catalytic subunit, and inhibiting this interaction reduces phosphorylation of proteins targeted by protein kinase A catalytic subunit such as phospholamban and cardiac troponin I.
What Are the Clinical Implications?
Augmenting protein kinase A catalytic subunit O‐GlcNAcylation during pressure overload conditions represents a potential therapeutic target for preventing heart failure.
Posttranslational modification of serine/threonine protein residues by O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) is a dynamic and ubiquitous process that affects protein function and transcriptional events.1, 2, 3, 4, 5, 6 O‐GlcNAc protein modifications occur through a single enzyme for attachment (O‐GlcNAc transferase [OGT]), and a single enzyme for removal (O‐GlcNAcase [OGA]). Several investigative teams have reported increased O‐GlcNAc levels during cardiac hypertrophy and heart failure in both humans and animal models7, 8, 9, 10, 11, 12 However, the functional effects of O‐GlcNAcylation during cardiac hypertrophy still require elucidation.
Prior studies in the heart demonstrate both beneficial and detrimental effects from O‐GlcNAcylation outside of hypertrophy. Acutely augmenting O‐GlcNAc levels pharmacologically during ex vivo or in vivo ischemia/reperfusion or trauma‐hemorrhage improves cardiac functional recovery and cardiomyocyte survival.13, 14, 15, 16, 17, 18, 19, 20 However, chronic O‐GlcNAc elevations cause cardiac dysfunction in mouse models of diabetic cardiomyopathy by mechanisms including negatively affecting calcium handling proteins21, 22, 23, 24 In other organs, the functional effects of O‐GlcNAc are frequently thought to be dichotomous with acute increases promoting cellular adaptation to physiological stressors, while prolonged elevations are believed to contribute to disease pathology (for review see reference 25). Similarly, previous cardiac studies raise the possibility that O‐GlcNAc promotes the successful early adaptation to cardiac stressors, whereas prolonged O‐GlcNAcylation may be detrimental to cardiac function.
Dassanayaka et al11 recently evaluated the effect of O‐GlcNAcylation on the development of pathological cardiomyocyte hypertrophy from transverse aortic constriction (TAC) using inducible, cardiomyocyte‐specific deficient OGT mice11 In their study, OGT deficiency induced before TAC reduced total O‐GlcNAc protein levels but did not alter cardiomyocyte hypertrophy. Interestingly, OGT deficiency significantly impaired cardiac function, although the mechanism was unclear. A limitation of this study is that inducing OGT deficiency before TAC does not separately evaluate the impacts of early O‐GlcNAcylation during the initial response to pressure overload versus potential chronic effects during established hypertrophy.
We hypothesize that early O‐GlcNAcylation improves cardiac function during the initial response to pressure overload, but that sustained elevations during established pathological hypertrophy negatively impact cardiac function by adversely affecting calcium handling proteins. We focused on these proteins based on the noted interactions between prolonged O‐GlcNAcylation, cardiomyopathy, and calcium handling proteins in diabetes21, 22, 23, 24 To test our hypothesis, we performed TAC on the previously described inducible, cardiomyocyte‐specific deficient OGT mice10, 11 and ablated OGT at different times either before or after surgery. To assess early O‐GlcNAcylation in the initial response to pressure overload, OGT deficiency was induced before TAC surgery similar to the previous report11 To evaluate prolonged O‐GlcNAcylation during established pathological hypertrophy, a separate cohort had OGT deficiency induced starting 18 days after TAC surgery, which is beyond the period of rapid hypertrophic growth from pressure overload. Many patients with cardiac hypertrophy secondary to pressure overload lesions, such as aortic stenosis, have established hypertrophy at the time of diagnosis and treatment. Therefore, it is clinically important to assess the functional effect of O‐GlcNAcylation and OGT ablation during isolated established hypertrophy, as was done in the present study.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. All materials used in this study are available commercially from the indicated vendors. The corresponding author reviewed all the results.
Ethics Statement
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication No. 85‐23, revised 1996) and were reviewed and approved by the Office of Animal Care at Seattle Children's Research Institute.
Inducible, Cardiac‐Specific OGT‐Deficient Mice
These mice were previously described10, 11 and kindly supplied by Dr Steven P. Jones (University of Louisville). Experimental mice between the ages of 3 and 5 months were used. After excisional recombination, the inducible, cardiomyocyte‐specific Ogt‐deficient mice are hereafter referred to as OGTKO. The Ogt gene is located on the X chromosome. Prior studies in male mice show that these inducible knockout mice have a low level of mosaic (OGT expressing) cardiomyocytes after excision with OGT mRNA levels from isolated cardiomyocytes at ≈20% of wild‐type values10 To reduce the potential for heterogeneity in mosaic OGT expression from female X‐inactivation, only male mice were used, as was done in prior studies10, 11 Wild‐type mice (control) were Ogt‐floxed/MCM negative littermates. To induce excision recombination, mice were treated with 1 mg of 4‐hydroxytamoxifen (Tam) for 7 days. Tam was prepared as previously described.12 Control mice received Tam in a similar manner. Mice were allowed free access to water and Teklad #7964 chow (Envigo). Teklad ¼‐inch corncob bedding was used in the cages (Envigo). Experiments occurred over 2 years.
Experimental Set‐Up
We hypothesized that early O‐GlcNAcylation improves cardiac function during the initial response to pressure overload but that sustained elevations during established pathological hypertrophy negatively impact cardiac function by adversely affecting calcium handling proteins. Thus, we evaluated the effect of OGT ablation at different times either before or after TAC surgery using 2 separate protocols described below and shown in Figure 1. These experimental protocols were designed prospectively. The primary outcomes were cardiac function determined by echocardiogram and total protein and/or phosphorylation levels of calcium handling proteins assessed by Western blot.
Figure 1.
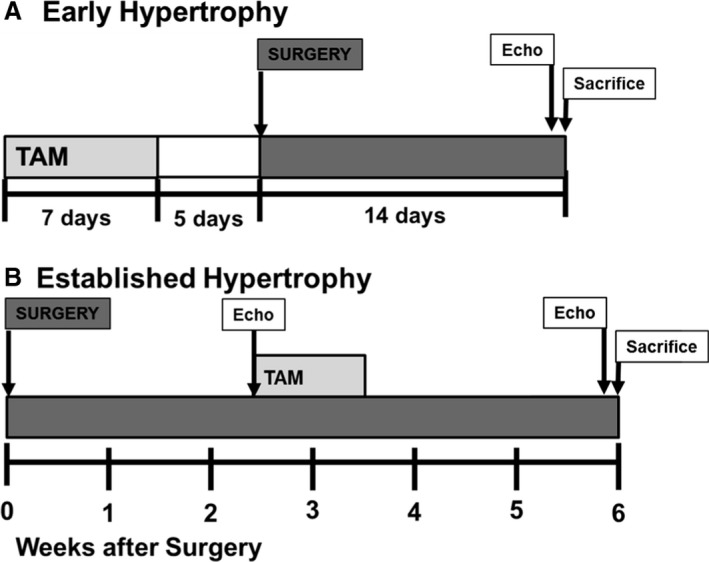
Diagram of the experimental protocol for (A) early hypertrophy and (B) established hypertrophy. Echo indicates echocardiogram; surgery, transverse aortic constriction or sham surgeries; TAM, 4‐hydroxytamoxifen.
To assess the effect of early O‐GlcNAcylation during the initial myocardial response to pressure overload, the first protocol involved Tam injections before surgery. Specifically, mice were injected with Tam for 7 days beginning 12 days preoperatively with a 5‐day washout period before surgery. These mice were then followed for 2 weeks postoperatively, similar to the previous study11 We hereafter refer to these mice as the “early hypertrophy” groups. The early hypertrophy groups include control sham, OGTKO sham, control TAC, and OGTKO TAC.
To assess the effect of prolonged elevations in O‐GlcNAc levels during established hypertrophy, the second protocol involved Tam injections after surgery. Mice underwent TAC or sham surgery and then had Tam injections for 7 days starting at 18 days postoperative. The mice were followed for a total of 6 weeks postoperatively. Since the Tam injections started when the hearts had relatively stable hypertrophy, we hereafter refer to these mice as the “established hypertrophy” groups and they include control sham, OGTKO sham, control TAC, and OGTKO TAC.
A surgical schedule was drafted in the beginning based on the anticipated sample numbers needed for each experimental group. Then, the first age‐ and genotype‐appropriate mice identified in the housing rack for the timeframe of the scheduled surgeries were utilized. No echocardiograms were performed before group assignment nor were any mice excluded after assignment. We were unsure of an effect size of OGTKO on our outcomes of interest; therefore, power calculations were not performed before commencing the experiments. Based on our previous experience and the planned tissue requirements, we initially anticipated needing ≈10 mice per group. However, the histology and immunoprecipitation methods required additional tissue, and supplementary surgeries were scheduled and performed in a similar manner.
TAC Surgery
TAC was used to create pathological pressure overload cardiac hypertrophy. The surgery was performed as previously described in our laboratory with a few changes.12 Briefly, the mice were initially anesthetized with 3% isoflurane in 100% O2 at a flow of 1 L/min and then maintained with 1.5% isoflurane for the duration of the surgery. A midline sternotomy was performed to expose the aorta. For the early hypertrophy groups, the aorta was constricted with a 7‐0 silk suture tied against a 23‐gauge blunt needle. Our initial echocardiograms showed that the 23‐gauge constriction caused decreased cardiac function. For the established hypertrophy groups, we wanted the hearts to have relatively preserved function (ie, be in compensated hypertrophy) immediately before Tam injections. Thus, the aorta was constricted against a 22‐gauge needle to create less narrowing in the established hypertrophy mice. Sham mice (sham) had their aorta mobilized, but no suture tightened. For pain control, all mice received intraperitoneal buprenorphine (0.05–0.1 mg/kg IP) starting intraoperatively and continuing every 8 to 12 hours for 2 days.
Echocardiogram
Echocardiograms were performed as previously described in our laboratory.9, 12, 26 Briefly, mice were initially sedated with 3% isoflurane in O2 at a flow of 1 L/min and placed in a supine position, at which time the isoflurane was reduced to 1.5% administered via a small nose cone. ECG leads were placed for simultaneous ECG monitoring during image acquisition. Echocardiographic images were performed with a Vevo 2100 machine using MS250 or MS550 transducers (VisualSonics, Inc). M‐mode measurements at the midpapillary level of the left ventricle were performed at end‐diastole and end‐systole to determine left ventricular (LV) function via the fractional shortening [(LV end‐diastolic diameter – LV end‐systolic diameter)/LV end‐diastolic diameter ×100] in a parasternal short‐axis mode for at least 3 heart beats. Pulsed Doppler velocity was measured across the aortic constriction site. This velocity serves as a noninvasive estimate of the peak instantaneous pressure gradient across the constriction site and is calculated by the modified Bernoulli equation (Δ pressure gradient=4*velocity2). The echocardiogram reader was blinded to treatment. We previously found that isoflurane anesthesia during echocardiograms increases myocardial total protein O‐GlcNAc levels for several hours after the procedure (W.‐Z. Zhu and A. Olson, unpublished data, 2016). Therefore, the end echocardiograms were performed on the day before euthanization. Established hypertrophy mice had an echocardiogram immediately before starting Tam injections (postoperative day 18).
Histology
Hearts were arrested by perfusion of KB buffer (20 mmol/L KCl, 10 mmol/L KH2PO4, 70 mmol/L K+‐glutamate, 1 mmol/L MgCl2, 25 mmol/L glucose, 20 mmol/L taurine, 0.5 mmol/L EGTA, 10 mmol/L HEPES, 0.1% albumin, and pH adjusted to 7.4 with KOH) and were perfusion fixed with 4% paraformaldehyde at constant pressure. Hearts were fixed an additional 2 days at 4°C with rotation, then processed and embedded in paraffin by standard methods and sectioned at 4 μm. To assess cardiomyocyte hypertrophy, sections were stained with Wheat Germ Agglutinin (catalog number W6748, ThermoFisher Scientific) and imaged on a widefield microscope. DAPI was used to identify nuclei. Cardiomyocyte cross‐sectional areas were measured in cardiomyocytes with a centrally located nuclei using FIJI software27 Fibrosis was measured by staining with Masson's trichrome, and cardiac collagen content was determined as the percentage of collagen in sections of the left ventricle. Cardiac apoptosis was determined by terminal deoxynucleotidyl transferase dUTP nick end‐labeling (TUNEL) positivity. LV sections were subjected to the ApopTag Plus Peroxidase kit (MilliporeSigma, Inc) according to the manufacturer's instructions and costained with DAPI for nucleus identification. TUNEL positivity was calculated by dividing the total number of TUNEL‐positive cells by the number of nuclei.
Western Blots
Immunoblots were performed as previously described on freshly isolated hearts9, 12, 26, 28 Briefly, protein was isolated with a lysis buffer containing 10 mmol/L Tris (pH=7.6), 150 mmol/L NaCl, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, and 1% NP‐40 containing phosphatase inhibitor (ThermoFisher Scientific) plus the OGA inhibitors PUGNAc 20 μmol/L (Toronto Research Chemical) and Thiamet‐G 1 μmol/L (Adooq Bioscience). Fifty micrograms of total protein extract from mouse heart tissue was separated electrophoretically and transferred to the polyvinylidene fluoride membrane. The membranes were probed with the following antibodies: sarco/endoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a, sc‐376235) from Santa Cruz Biotechnology, Inc; phospholamban (Ab85146) from Abcam; phosphorylated phospholamban Ser16 (p‐PLN,07‐052) from MilliporeSigma; cardiac troponin I (cTnI, 4002), phosphorylated cTnI Ser23/24 (4004), protein kinase A catalytic subunit (PKAc, 5842), and phosphorylated PKAc Thr197 (4781) from Cell Signaling Technology, Inc. Western blots were visualized with enhanced chemiluminescence upon exposure to Kodak BioMax Light ML‐1 film or using the chemiDoc‐ItTM imaging system (Analytik Jenna USA LLC). Membranes were stripped by washing for 30 minutes with 100 mmol/L dithiothreitol, 2% (wt/vol) sodium dodecyl sulfate, 62.5 mmol/L Tris·HCl, and pH 6.7, at 70°C, followed by three 10‐minute washes with Tris‐buffered saline for additional antibody analysis. Immunoblots of proteins without a phosphorylated form were normalized to total protein staining in the molecular weight region around the band of interest by Thermo Scientific Pierce Reversible Protein Stain Kit for PVDF Membranes (Thermo Scientific), which are shown in the figures. Phosphorylated proteins were normalized to their appropriate total protein. Immunoblots and protein staining were measured by densitometry analysis using ImageJ software (National Institutes of Health).
The O‐GlcNAc Western blots require additional details from the general immunoblot methods described above. Our unpublished data (W.‐Z. Zhu and A. Olson, 2016) show that isoflurane increases myocardial O‐GlcNAcylation for hours after an exposure. Accordingly, all mice underwent sedated echocardiograms on the day before tissue collection. The protein lysis buffer included the OGA inhibitors PUGNAc and Thiamet‐G (concentrations noted above). Protein O‐GlcNAc levels were determined with the RL‐2 antibody from Abcam. This antibody is raised in mice and the secondary anti‐mouse antibody can react with IgG from the tissue resulting in a nonspecific band at either 55 kDa (from IgG heavy chain) or 25 kDa (from IgG light chain). To allow for unobstructed detection of O‐GlcNAc bands in the 55‐kDa region, we only used a light chain–specific secondary antibody (Peroxidase AffiniPure goat anti‐mouse IgG, light chain specific; Jackson ImmunoResearch Laboratories, Inc) for our immunoblots and excluded the 25‐kDa band during quantification. Total (global) O‐GlcNAc levels were measured by densitometry of the entire Western blot lane. We also measured specific clusters of O‐GlcNAc bands corresponding to molecular weights of (approximately) >150 kDa, 75 to 150 kDa, and <75 kDa. O‐GlcNAc levels were normalized to the protein staining by Thermo Scientific Pierce Reversible Protein Stain Kit for PVDF Membranes (described above) of the entire lane or same molecular weight region as appropriate.
Immunoprecipitations were performed following the instructions detailed in the Pierce Co‐Immunoprecipitatin Kit (#26149, Thermo Scientific). Antibodies phospholamban and PKAc (details above) coupled to agarose beads were used. Twenty milligrams of tissue/immunoprecipitation was homogenized in the kit's immunoprecipitation lysis buffer with Halt protease/phosphatase inhibitor (#78442, ThermoFisher Scientific), Thiamet G 1 μmol/L (Adooq Bioscience), and PUGNAc 20 μmol/L (Toronto Research Chemical). The tissue lysate (1 mg) was precleared for 1 hour at 4°C using the kit's control agarose resin. The precleared lysate was incubated overnight at 4°C with its respective antibody. Immunoblotting was performed as described above. Briefly, the immunoprecipitation complex was separated electrophoretically and transferred to the polyvinylidene fluoride membrane. The membranes were probed with RL‐2 antibody described above. The immunoblots were stripped and reprobed against the antibody target with phospholamban or PKAc for normalization. The immunoblots were visualized as described above.
Statistical Analysis
All reported values are expressed as mean±standard error of the mean. Numerical values for all of the figures are reported in Table S1. For comparisons involving only 2 groups, we initially evaluated the distribution of results from each experimental group for normality using the Shapiro‐Wilk test. We compared normally distributed data using a 2‐way, unpaired t test and non‐normal data using the Mann‐Whitney test.
For comparisons involving all 4 groups, we first evaluated the distribution of results in each group for normality using the Shapiro‐Wilk test. If data were normally distributed, we performed a 2‐factor ANOVA (control or OGTKO and sham or TAC). If the distribution of results in at least 1 experimental group was not normally distributed, then we identified outliers using the Grubbs test (α=0.05). Using this testing, we found that a single outlying value in a group was causing the data to not have a normal distribution. We then performed 2‐factor ANOVAs with and without the outlying value. As outlier exclusion did not change significance for any of the comparisons or appreciably alter the means, we retained the outlying values in our analyses. If the global test for the 2‐factor AVOVA was statistically significant, then we performed follow‐up pairwise comparisons between the groups of interest identified a priori (control sham versus OGTKO sham, control sham versus control TAC, OGTKO sham versus OGTKO TAC, and control TAC versus OGTKO TAC). To limit type II errors for our comparisons of the Western blots containing all 4 experimental groups, which have ≤4 replicates per group (well space limits replicates to 3 or 4 per group), we present P values from the pairwise t tests for comparisons. We also performed a Tukey correction for multiple procedures in this instance and report these P values in the figures when they differ in significance from the t test. Since all of the other experiments did not have limitations on the numbers of replicates per group, we used the Tukey correction alone for multiple comparisons.
Criterion for significance was P<0.05. No statistical comparisons were made between OGTKO sham versus control TAC and control sham and OGTKO TAC. We preformed statistical analysis using GraphPad Prism version 7.03 (GraphPad Software).
Results
Experimental Groups
Table 1 shows the number of mice assigned to each experimental group, deaths, and mice completing the protocol for the early hypertrophy groups. No mortalities occurred during Tam injections. Intraoperative deaths were from arterial hemorrhage or intubation problems. All postoperative deaths occurred within 48 hours of surgery. Mice that died during the protocol were excluded from analysis.
Table 1.
Outcomes of Mice in the Early Hypertrophy Experimental Groups
| Experimental Group | Control Sham | OGTKO Sham | Control TAC | OGTKO TAC |
|---|---|---|---|---|
| Number assigned to group | 15 | 12 | 16 | 17 |
| Intraoperative deaths | 1 | 0 | 2 | 0 |
| Postoperative death <48 h | 0 | 0 | 1 | 0 |
| Number completing protocol | 14 | 12 | 13 | 17 |
The number assigned to group represents the starting population of mice that received the 4‐hydroxytamoxifen injections. Intraoperative deaths were from either arterial hemorrhage or intubation issues. No fatalities occurred >48 hours after surgery. OGTKO indicates O‐linked β‐N‐acetylglucosamine transferase knockout; TAC, transverse aortic constriction.
Table 2 shows the same parameters for the established hypertrophy groups. Intraoperative deaths were also from arterial hemorrhage or intubation issues and all postoperative deaths occurred within 48 hours of the surgery. No deaths occurred during or after the Tam injections. Mice that died during the protocol were excluded from analysis.
Table 2.
Outcomes of Mice in the Established Hypertrophy Groups
| Experimental Group | Control Sham | OGTKO Sham | Control TAC | OGTKO TAC |
|---|---|---|---|---|
| Number assigned to group | 15 | 11 | 24 | 22 |
| Intraoperative death | 1 | 0 | 1 | 1 |
| Postoperative death <48 h | 0 | 0 | 4 | 5 |
| Number completing protocol | 14 | 11 | 19 | 16 |
The number assigned to group represents the starting population of mice that underwent surgery. Intraoperative deaths were either from arterial hemorrhage or intubation issues. No fatalities occurred >48 hours after surgery. OGTKO indicates O‐linked β‐N‐acetylglucosamine transferase knockout; TAC, transverse aortic constriction.
OGT and O‐GlcNAc Levels
We used Western blots to assess OGT deletion and total protein O‐GlcNAc levels. In the early hypertrophy groups, OGT levels significantly decreased with OGTKO in both the sham and TAC groups (Figure 2A). In the control sham versus control TAC groups, OGT levels did not change.
Figure 2.
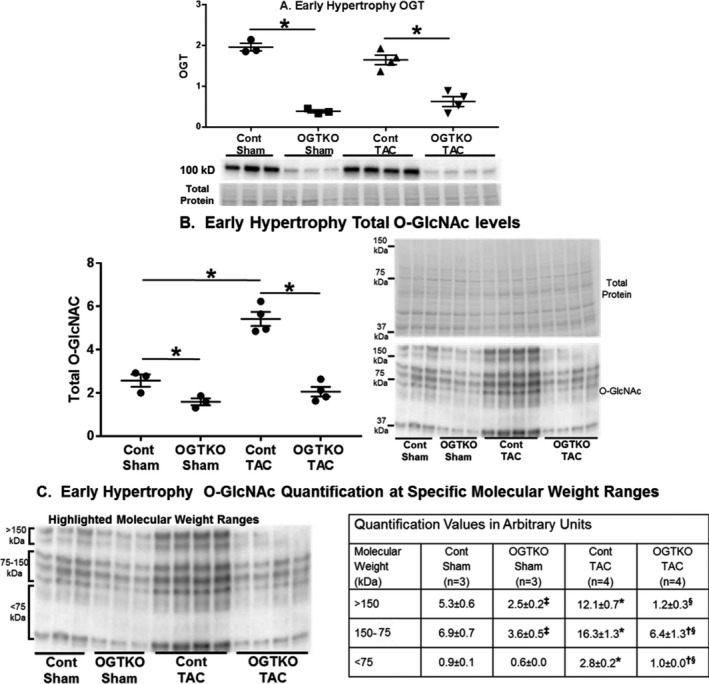
O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) transferase (OGT) and O‐GlcNAc protein levels in early hypertrophy. Immunoblot results are shown for OGT (A), total protein O‐GlcNAc levels (B), and O‐GlcNAc levels at the indicated specific molecular weight ranges (C). Values are arbitrary units reported as mean±standard error of the mean. n=3 control sham, n=3 OGT knockout (OGTKO) sham, n=4 control transverse aortic constriction (TAC), n=4 OGTKO TAC. In (A and B) *P<0.05 between groups indicated with the bar. In (C) *P<0.05 for control sham vs control TAC in same molecular weight range; ‡ P<0.05 for control sham vs OGTKO sham in the same molecular weight range; § P<0.05 for control TAC vs OGTKO TAC at the same molecular weight range; † P<0.05 for OGTKO sham vs OGTKO TAC at the same molecular weight range. For (B) control sham vs OGTKO sham, P value from t test=0.039 and P value from Tukey procedure=0.14. For (C) >150 kDa OGTKO sham vs OGTKO TAC, P value from t test=0.023 and P value from Tukey procedure=0.12. For (C) 150 to 75 kDa control sham vs OGTKO sham, P value from t test=0.022 and P value from Tukey procedure=0.25.
Prior studies show that protein O‐GlcNAc levels do not always uniformly change across the entire range of molecular weight bands8, 9, 29, 30 We measured total (global) O‐GlcNAc levels across all molecular weights. Additionally, we analyzed O‐GlcNAc levels at specific molecular weight ranges corresponding to clusters of bands at approximately >150 kDa, 75 to 150 kDa, and <75 kDa. In the early hypertrophy groups, OGTKO significantly reduced total O‐GlcNAc levels in both the sham and TAC groups compared with their respective controls (Figure 2B). OGTKO produced significant declines in all of the molecular weight ranges with the exception of a trend towards reduced levels at <75 kDa in OGTKO sham versus control sham (P=0.072) (Figure 2C). Compared with control sham, control TAC had significantly higher total O‐GlcNAc levels and at all of the molecular weight ranges (Figures 2B and 2C).
In the established hypertrophy groups, OGTKO significantly diminished OGT levels in both sham and TAC groups (Figures 3A). In control mice, TAC did not affect OGT levels. Total O‐GlcNAc levels declined with OGTKO in both sham and TAC groups compared with their respective controls (Figure 3B). In sham mice, the greatest difference in O‐GlcNAc levels versus OGTKO was at molecular weights >150 kDa and between 75 and 150 kDa (Figure 3C). In TAC hearts, OGTKO significantly reduced O‐GlcNAc levels in all of the molecular weight ranges (Figure 3C). Total protein O‐GlcNAc levels were comparable between control TAC and control sham (Figure 3B). O‐GlcNAc levels were also similar at the various molecular weight ranges (Figure 3C). However, we detected a marginal increase in intensity for the <75 kDa bands (P=0.06) in control TAC versus control sham, justifying a more extensive evaluation with a larger sample number and emphasizing band separation in the lower molecular weights. This analysis showed that O‐GlcNAc levels were significantly higher in control TAC than control sham at protein molecular weights of ≈≤100 kDa (Figure 3D).
Figure 3.
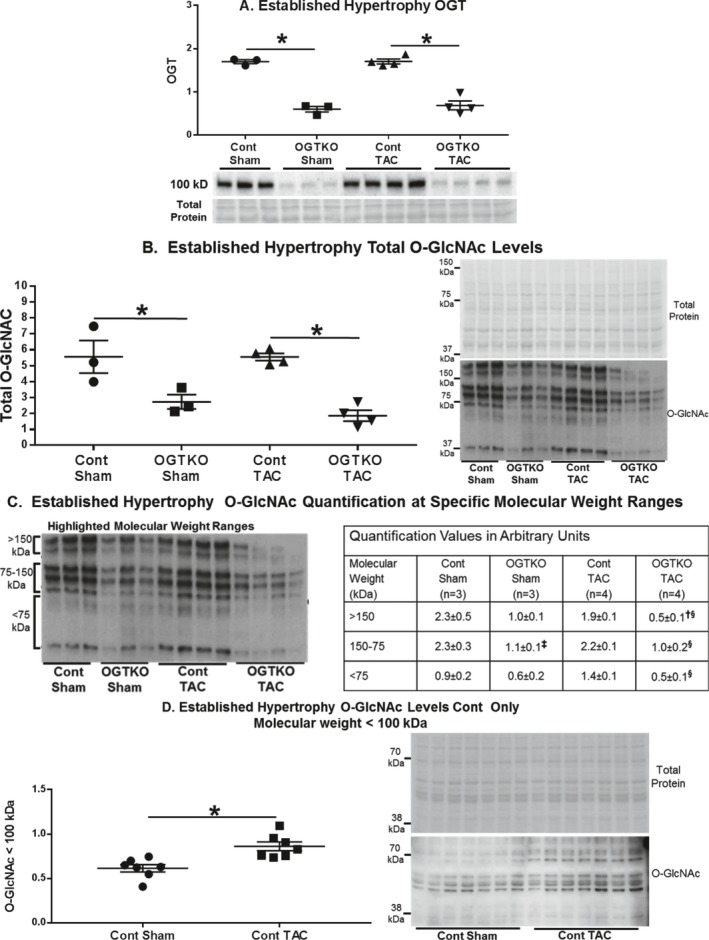
O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) transferase (OGT) and O‐GlcNAc protein levels in established hypertrophy. Immunoblot results are shown for OGT (A), total protein O‐GlcNAc levels (B), O‐GlcNAc levels at the indicated specific molecular weight ranges (C), and O‐GlcNAc protein levels at molecular weights <100 kDa in control (cont) sham and control transverse aortic constriction (TAC) (D). Values are arbitrary units reported as mean±standard error of the mean. For (A through C) n=3 control sham, n=3 O‐GlcNAc transferase knockout (OGTKO) sham, n=4 control TAC, n=4 OGTKO TAC. For D, n=7 control sham and n=7 control TAC. In (A, B, and D) *P<0.05 between groups indicated with the bar. In (C) ‡ P<0.05 for control sham vs OGTKO sham at the same molecular weight range; § P<0.05 for control TAC vs OGTKO TAC at the same molecular weight range; † P<0.05 for OGTKO sham vs OGTKO TAC at the same molecular weight range. For (C) >150 kDa control sham vs OGTKO sham, P value from t test=0.072 and P value from Tukey procedure=0.027. For (C) >150 kDa OGTKO sham vs OGTKO TAC, P value from t test=0.025 and P value from Tukey procedure=0.25.
Echocardiographic Data
For the OGTKO cohort in the early hypertrophy protocol, cardiac measurements were similar from before starting Tam to immediately preoperative (Table 3). Preoperative echocardiographic parameters were also similar between early hypertrophy OGTKO and control mice (Table 3). Additional studies in a separate cohort of early hypertrophy OGTKO mice found that the cardiac measurements did not change during Tam injections (Table 4). End experiment echocardiographic results for the early hypertrophy groups are shown in Table 5. OGTKO sham mice had a mildly lower, but nonsignificant (P=0.098), fractional shortening compared with control sham. To determine the degree of pressure overload from the TAC surgery, we measured the Doppler flow velocity across the TAC site. The velocity was significantly higher in control TAC compared with OGTKO TAC groups. Based on the modified Bernoulli equation, the estimated peak instantaneous pressure gradient would be ≈96 mm Hg in control TAC and 74 mm Hg in OGTKO TAC. Compared with their corresponding sham, TAC hearts had increased posterior wall thickness and decreased systolic function measured by fractional shortening. Similar to the prior report11 OGTKO in TAC hearts caused a further decline in LV systolic function.
Table 3.
Baseline Echocardiogram Measurements in Mice With Early Hypertrophy
| OGTKO | Control | ||
|---|---|---|---|
| Pre‐Tam (n=17) | Pre‐Op (n=25) | Pre‐Op (n=27) | |
| HR, bpm | 454±8 | 453±8 | 441±5 |
| EDD, mm | 4.2±0.1 | 4.4±0.1 | 4.3±0.1 |
| ESD, mm | 3.4±0.1 | 3.4±0.1 | 3.3±0.1 |
| LVPWd, mm | 0.70±0.02 | 0.72±0.02 | 0.72±0.02 |
| FS, % | 20.7±0.6 | 22.6±0.8 | 23.0±0.7 |
| EF, % | 42.3±1.1 | 45.4±1.4 | 46.2±1.1 |
Values are expressed as mean±standard error of the mean. Baseline echocardiograms were performed before surgery. Mice are grouped and evaluated according to their genotypes. O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) mice had echocardiograms immediately before starting 4‐hydroxytamoxifen injections (Pre‐Tam). Preoperative (Pre‐Op) measurements were performed after the 5‐day Tam washout period in both genotypes. Control mice did not have Pre‐Tam echocardiograms. P>0.05 via unpaired t test for all measurements between control Pre‐Op vs OGTKO Pre‐Op. P>0.05 via unpaired t test for all measurements between OGTKO Pre‐Tam vs OGTKO Pre‐Op. In OGTKO mice with echocardiograms at both Pre‐Tam and Pre‐Op (n=17), P>0.05 for all measurements via a paired t test (values not shown separately from the entire cohort). bpm indicates beats per minute; EDD, left ventricular end‐diastolic diameter; EF, left ventricular ejection fraction; ESD, left ventricular end‐systolic diameter; FS, left ventricular fractional shortening; HR, heart rate; LVPWd, left ventricular posterior wall thickness in diastole.
Table 4.
Echocardiogram Measurements Immediately Before and on Day 7 of Tam Injections in a Cohort of OGTKO Mice With Early Hypertrophy
| Pre‐Tam (n=7) | Day 7 Tam (n=7) | |
|---|---|---|
| HR, bpm | 455±14 | 455±17 |
| EDD, mm | 4.5±0.1 | 4.5±0.1 |
| ESD, mm | 3.5±0.0 | 3.5±0.1 |
| LVPWd, mm | 0.71±0.02 | 0.74±0.02 |
| FS, % | 20.8±1.0 | 21.9±1.2 |
| EF, % | 42.3±1.7 | 44.3±2.0 |
Values are expressed as mean±standard error of the mean. A separate cohort of early hypertrophy O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) mice had echocardiograms performed immediately before starting 4‐hydroxytamoxifen (Tam) injections (Pre‐Tam) and on day 7 of Tam injections (Day 7 Tam). P>0.05 by paired t test for all measurements. bpm indicates beats per minute; EDD, left ventricular end‐diastolic diameter; EF, left ventricular ejection fraction; ESD, left ventricular end‐systolic diameter; FS, left ventricular fractional shortening; HR, heart rate; LVPWd, left ventricular posterior wall thickness in diastole.
Table 5.
Echocardiogram Measurements in the Early Hypertrophy Groups at Experimental End
| Sham | TAC | |||
|---|---|---|---|---|
| Control (n=14) | OGTKO (n=12) | Control (n=13) | OGTKO (n=17) | |
| HR, bpm | 469±12 | 494±13 | 470±12 | 523±10* |
| TAC velocity, m/s | N/A | N/A | 4.9±0.1 | 4.3±0.1* |
| EDD, mm | 4.4±0.1 | 4.4±.01 | 4.4±0.01 | 4.7±0.1 |
| ESD, mm | 3.4±0.1 | 3.6±.01 | 3.6±0.1 | 4.1 ±0.2* , † |
| LVPWd, mm | 0.76±0.02 | 0.75±0.02 | 0.98±0.04‡ | 1.02±.04† |
| FS, % | 23.7±1.1 | 19.5±1.1 | 18.8±1.2‡ | 12.2±1.2* , † |
| EF, % | 47.3±1.9 | 40.1±2.0 | 39.4±2.4 | 26.2±2.5* , † |
Values are expressed as mean±standard error of the mean. bpm, beats per minute; EDD, left ventricular end‐diastolic diameter; ESD, left ventricular end‐systolic diameter; FS, left ventricular fractional shortening; HR, heart rate; LVPWd, left ventricular posterior wall thickness in diastole; N/A, not applicable. *P<0.05 control transverse aortic constriction (TAC) vs O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) TAC; † P<0.05 OGTKO sham vs OGTKO TAC; ‡ P<0.05 control sham vs control TAC. For left ventricular ejection fraction (EF) in control sham vs control TAC, P=0.082.
In the established hypertrophy mice, all mice had echocardiograms immediately before starting Tam injections (postoperative day 18) to establish cardiac parameters before OGTKO (Table 6). We wanted TAC mice in these experiments to have relatively preserved systolic function before Tam injections, so the constriction was made against a slightly larger diameter needle as noted in the Methods section. Flow velocity across the aortic constriction site, measured before OGT ablation, was similar in the TAC groups and predicted a peak instantaneous pressure gradient of ≈77 mm Hg in both. Pre‐Tam LV chamber size and systolic function was similar among all of the groups. Both TAC groups had greater LV posterior wall thickness versus their corresponding sham groups. Heart rate was higher in the control TAC compared with the control sham group. These results show that the TAC hearts had relatively compensated cardiac function immediately before the Tam injections. Of note, additional studies in a separate cohort of OGTKO TAC mice showed that there was no acute change in echocardiographic parameters during the Tam injections (Table 7).
Table 6.
Echocardiogram Measurements in the Established Hypertrophy Groups Before Starting 4‐Hydroxytamoxifen (Pre‐Tam) and at Experimental End (End)
| Sham | TAC | |||||||
|---|---|---|---|---|---|---|---|---|
| Pre‐Tam | End | Pre‐Tam | End | |||||
| Control (n=14) | OGTKO (n=11) | Control (n=14) | OGTKO (n=11) | Control (n=19) | OGTKO (n=16) | Control (n=19) | OGTKO (n=16) | |
| HR, bpm | 460±9 | 451±15 | 470±10 | 512±9 | 503±7* | 490±11 | 538±12† | 525±16 |
| TAC velocity, m/s | N/A | N/A | N/A | N/A | 4.4±0.1 | 4.4±0.1 | ND | ND |
| EDD, mm | 4.3±0.0 | 4.4±0.1 | 4.3±0.1 | 4.7±0.1‡ | 4.3±0.1 | 4.1±0.1 | 4.4±0.1 | 5.0±0.1§ |
| ESD, mm | 3.3±0.1 | 3.4±0.1 | 3.2±0.1 | 3.9±0.1‡ | 3.3±0.1 | 3.1±0.1 | 3.4±.01 | 4.5±0.2§ , ∥ |
| LVPWd, mm | 0.81±0.02 | 0.82±0.03 | 0.83±0.03 | 0.87±0.04 | 0.94±0.03* | 0.99±0.02¶ | 0.99±0.03† | 0.93±0.03 |
| FS, % | 22.8±1.1 | 21.9±1.1 | 23.8±0.9 | 17.7±0.7‡ | 22.4±1.0 | 24.2±1.1 | 21.6±1.0 | 10.4±1.0§ , ∥ |
| EF, % | 45.0±2.1 | 44.4±1.9 | 47.6±1.5 | 36.9±1.3‡ | 45.1±1.7 | 48.4±1.9 | 43.8±1.8 | 22.4±2.1§ , ∥ |
Values are expressed as mean±standard error of the mean. For Pre‐Tam heart rate (HR) in O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) sham vs OGTKO transverse aortic constriction (TAC), P=0.073. bpm indicates beats per minute; EDD, left ventricular end‐diastolic diameter; EF, left ventricular ejection fraction; ESD, left ventricular end‐systolic diameter; FS, left ventricular fractional shortening; LVPWd, left ventricular posterior wall thickness in diastole; N/A, not applicable; ND, not done.
*P<0.05 Pre‐Tam control sham vs Pre‐Tam control TAC; † P<0.05 End control sham vs End control TAC; ‡ P<0.05 End control sham vs End OGTKO sham; § P<0.05 End control TAC vs End OGTKO TAC; ∥ P<0.05 End OKGTO sham vs End OGTKO TAC; ¶ P<0.05 Pre‐Tam OGTKO sham vs Pre‐Tam OGTKO TAC.
Table 7.
Echocardiogram Measurements Immediately Before and on Day 7 of 4‐Hydroxytamoxifen (Tam) Injections in a Cohort of Mice With Established Hypertrophy OGTKO TAC
| Pre‐Tam (n=7) | Day 7 Tam (n=7) | |
|---|---|---|
| HR, bpm | 471±30 | 494±12 |
| EDD, mm | 4.4±0.1 | 4.6±0.2 |
| ESD, mm | 3.4±0.1 | 3.7±0.2 |
| LVPWd, mm | 0.89±0.03 | 0.93±0.03 |
| FS, % | 22.6±0.8 | 20.8±1.3 |
| EF, % | 45.9±1.4 | 42.4±2.4 |
| TAC velocity, m/s | 4.1±0.2 | ND |
Values are expressed as mean±standard error of the mean.
A separate cohort of O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) transverse aortic constriction (TAC) mice had echocardiograms performed immediately before starting Tam injections (Pre‐Tam) and on day 7 of Tam injections (Day 7 Tam). P>0.05 via paired t test for all measurements. bpm indicates beats per minute; EDD, left ventricular end‐diastolic diameter; EF, left ventricular ejection fraction; ESD, left ventricular end‐systolic diameter; FS, left ventricular fractional shortening; HR, heart rate; LVPWd, left ventricular posterior wall thickness in diastole; ND, not done.
We subsequently evaluated cardiac measurements in the established hypertrophy groups at experiment end (Table 6). Control sham and control TAC had similar LV chamber sizes and systolic function, but LV posterior wall thickness and heart rate remained increased with TAC. In the sham hearts, OGTKO caused LV chamber dilation and mildly reduced systolic function. OGTKO in TAC hearts caused an even greater degree of LV dilation and reduced systolic function. Overall, these results show that OGT and O‐GlcNAcylation are critical to maintain compensated cardiac function during established pressure overload hypertrophy and also have a similar, but lesser, beneficial effect in sham hearts.
Morphometrics
In the early hypertrophy groups, both TAC groups had increased heart weight/tibial length versus their corresponding sham groups (Table 8). Lung wet weight/tibial length (a marker of pulmonary edema) was greater in the OGTKO TAC compared with the OGTKO sham group. Lung wet weight/tibial weight did not reach significance in the control TAC compared the control sham group, but the large standard error of the mean in the control TAC results suggests that some mice developed pulmonary edema. The combination of pulmonary edema and decreased systolic function in the early hypertrophy OGTKO TAC group suggests the presence of uncompensated cardiac function.
Table 8.
Morphometric Measurements of Heart Weight (Heart) and Lung Weight (Lung) Normalized to TL in the Early Hypertrophy Groups
| Control Sham (n=14) | OGTKO Sham (n=12) | Control TAC (n=13) | OGTKO TAC (n=17) | |
|---|---|---|---|---|
| Heart/TL, mg/mm | 7.1±0.2 | 7.8±0.1 | 10.4±0.3* | 10.6±0.3† |
| Lung/TL, mg/mm | 8.0±0.2 | 8.1±0.1 | 10.2±0.7* | 12.2±1.4† |
Values are expressed as mean±standard error of the mean. All weights are on wet organs. TL indicates tibial length.
*P<0.05 control sham vs control transverse aortic constriction (TAC); † P<0.05 O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) sham vs OGTKO TAC.
With established hypertrophy, both TAC groups had increased heart weight/tibial length ratios versus their corresponding sham (Table 9). Lung weight was similar between control sham and control TAC. Since systolic cardiac function was also similar between these groups at experiment end, the control TAC hearts had compensated cardiac function. The OGTKO TAC hearts had increased lung weight compared with control TAC, as well as OGTKO sham. The pulmonary findings plus the severe reduction in systolic function demonstrate that OGTKO caused functional decompensation during established hypertrophy. Within the sham or TAC groups, OGTKO ablation increased heart weight/tibial length.
Table 9.
Morphometric Measurements of Heart Weight (Heart) and Lung Weight (Lung) Normalized to TL in the Established Hypertrophy Groups
| Control Sham (n=10) | OGTKO Sham (n=7) | Control TAC (n=15) | OGTKO TAC (n=12) | |
|---|---|---|---|---|
| Heart/TL, mg/mm | 7.1±0.1 | 8.7±0.3* | 9.3±0.2† | 11.4±0.9‡ , § |
| Lung/TL, mg/mm | 8.2±0.2 | 9.0±0.4 | 8.7±0.2 | 11.6±0.9‡ , § |
Values are expressed as mean±standard error of the mean. All weights are on wet organs. TL indicates tibial length.
*P<0.05 control sham vs O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) sham; † P<0.05 control sham vs control transverse aortic constriction (TAC); ‡ P<0.05 OGTKO sham vs OGTKO TAC; § P<0.05 control TAC vs OGTKO TAC.
Histology
In the prior study, preoperative OGTKO did not affect the development of cardiomyocyte hypertrophy, measured by cross‐sectional area and cardiac mass, 4 weeks after aortic constriction11 Our studies similarly found that heart mass was unaffected by OGTKO in TAC hearts during early hypertrophy; therefore, we did not repeat cross‐sectional measurements in this group. In the sham hearts, OGTKO increased cardiac mass in the established hypertrophy protocol. Therefore, we measured cross‐sectional area for sham mice in the established hypertrophy groups (Figure 4), but identified no difference between the groups. In established hypertrophy TAC hearts, OGTKO increased heart mass along with the echocardiographic findings of LV chamber dilation. The cardiomyocyte cross‐sectional area was also significantly higher in the OGTKO TAC compared with the control TAC group, showing that the increase in heart mass was secondary to cardiomyocyte hypertrophy (Figure 4). Both TAC groups had increased cardiomyocyte cross‐sectional area compared with their respective controls.
Figure 4.
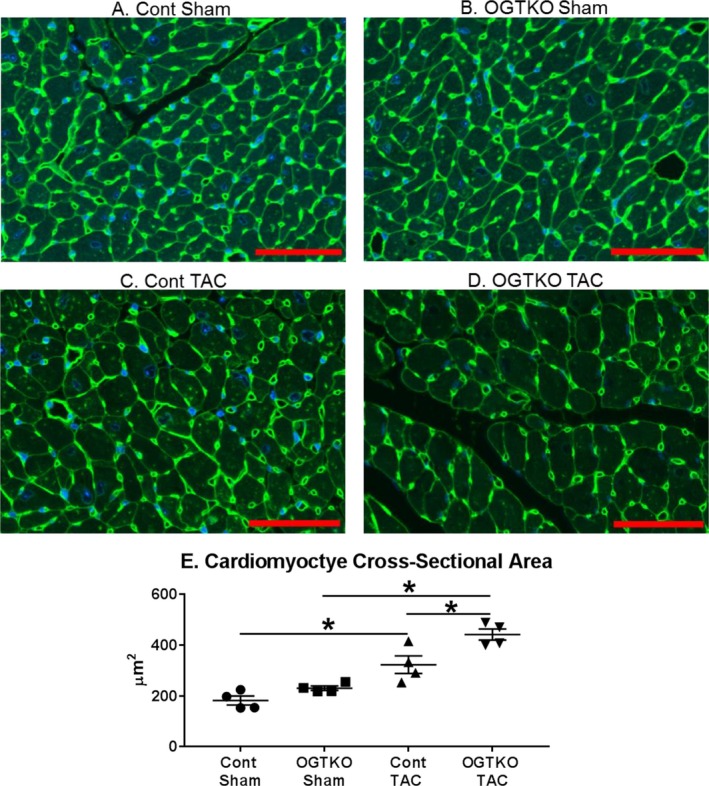
O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) increases cardiomyocyte cross‐sectional area during established hypertrophy in sham and transverse aortic constriction (TAC) hearts. Representative sections from the indicated established hypertrophy groups stained for wheat germ agglutinin and DAPI are shown in (A through D). E, Quantification of the cross‐sectional areas for the experimental groups. Values are expressed as mean±standard error of the mean. n=4 per group. Red bar indicates 50 μm. *P<0.05 between groups indicated with the bar.
Next, we evaluated the established hypertrophy groups for histopathological changes, which impact cardiac function. Previous work showed that OGT ablation before TAC did not affect apoptosis or fibrosis11 OGTKO had a markedly more severe effect on cardiac function with established hypertrophy, so we measured apoptosis and fibrosis in this condition. Although both apoptosis and fibrosis increased with TAC, OGTKO did not cause further changes (Figures 5 and 6).
Figure 5.
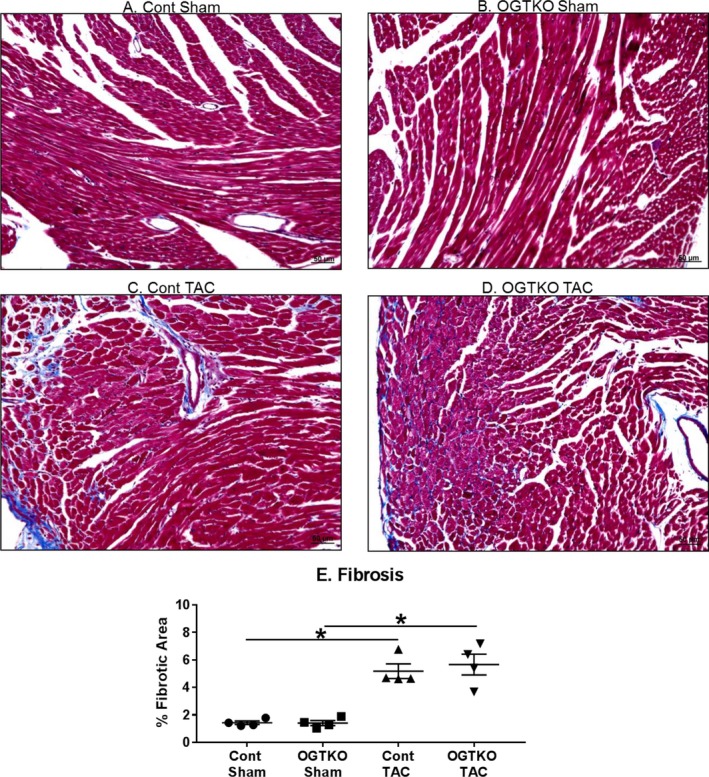
O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) does not alter histological marker of fibrosis by Masson's trichrome staining during established hypertrophy. Representative sections with Masson's trichrome staining from the indicated established hypertrophy groups are shown in (A through D). E, Quantification of the percent fibrotic area for the experimental groups. n=4 per group. Black bar indicates 50 μm. Values are mean±standard error of the mean. *P<0.05 between groups indicated with the bar. TAC indicates transverse aortic constriction.
Figure 6.
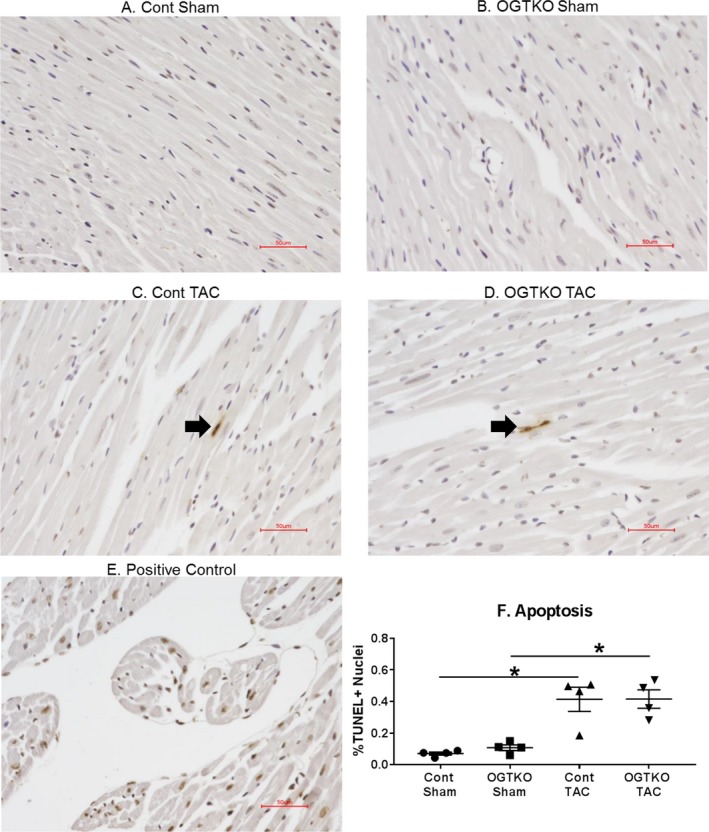
O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) does not alter histological markers of apoptosis during established hypertrophy. Sections of terminal deoxynucleotidyl transferase dUTP nick end‐labeling (TUNEL)–stained hearts from the indicated established hypertrophy groups are shown in (A through D). TUNEL‐positive nuclei in (C and D) are shown by the arrow. Because of the low number of TUNEL‐positive nuclei, these images are not representative of the overall results. E, Positive control myocardium demonstrating multiple TUNEL‐positive nuclei. F, Quantification of the percent TUNEL‐positive nuclei (% TUNEL+ nuclei) for the experimental groups. n=4 per group. Red bar indicates 50 μm. Values are expressed as mean±standard error of the mean. *P<0.05 between groups indicated with the bar. TAC indicates transverse aortic constriction.
Western Blots
Diabetic cardiomyopathy mouse studies indicate that calcium handling proteins are negatively affected by prolonged elevations in O‐GlcNAc level21, 22, 23, 24 SERCA2a transports Ca2+ from cytosol into the sarcoplasmic reticulum. Unless phosphorylated at Ser16, phospholamban inhibits SERCA2a activity, resulting in decreased cardiac function31, 32, 33 In diabetic cardiomyopathy studies, increased O‐GlcNAcylation is associated with decreased phospholamban phosphorylation and/or SERCA2a protein levels21, 22, 23, 24 Accordingly, we evaluated the effect of OGT and O‐GlcNAc status on calcium handling proteins and determined whether the duration of elevated O‐GlcNAcylation (ie, early versus established hypertrophy) differentially regulates these proteins.
In early hypertrophy, SERCA2a protein levels were greater in the TAC groups compared with their corresponding shams; however, OGTKO did not influence levels (Figure 7A). Similarly, in the established hypertrophy groups, SERCA2a protein levels increased with TAC over the corresponding sham without an effect from OGTKO (Figure 7B).
Figure 7.
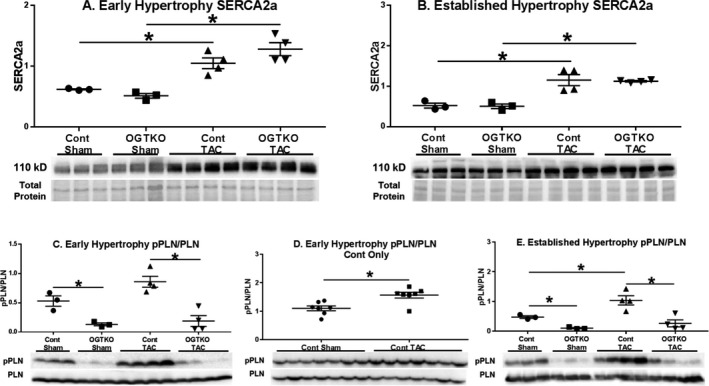
Sarco/endoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a) protein levels and phospholamban phosphorylation (pPLN) in early and established hypertrophy. Immunoblots from early and established hypertrophy for total SERCA2a protein levels (A, B) and phosphorylated phospholamban Ser16 to total phospholamban (PLN) (C through E). For (A through E) n=3 control sham, n=3 O‐linked β‐N‐acetylglucosamine transferase knockout (OGTKO) sham, n=4 control transverse aortic constriction (TAC), n=4 OGTKO TAC. For (D) n=7 control sham, n=7 control TAC. Values are arbitrary units reported as mean±standard error of the mean. *P<0.05 between groups indicated with the bar. For (C) control sham vs OGTKO sham, P value from t test=0.013 and P value from Tukey procedure=0.0522. For (E) control sham vs OGTKO sham, P value from t test=0.0014 and P value from Tukey procedure=0.22.
We next measured phospholamban phosphorylation (Ser16). In early and established hypertrophy, there was an increase in phospholamban phosphorylation in control TAC versus control sham (Figure 7C through 7E). Of note, the P value in the early hypertrophy control TAC group versus the control sham group was 0.053 (Figure 7C) with a low number of replicates per group (n=3 control sham and n=4 control TAC), justifying a repeat immunoblot in control mice with more samples per group to assess significance (Figure 7D). Critically, OGTKO significantly reduced phospholamban phosphorylation in all conditions including sham hearts. Previous studies in diabetic hearts have shown that phospholamban O‐GlcNAcylation inversely affects phosphorylation24 so we assessed phospholamban O‐GlcNAcylation with immunoprecipitation in established hypertrophy. However, no O‐GlcNAc was detected on phospholamban in the control or OGTKO groups after multiple attempts (data not shown).
Because O‐GlcNAc did not directly modify phospholamban, we investigated other factors regulating phospholamban. PKAc phosphorylates phospholamban at Ser1634 Phosphorylation of PKAc at Thr197 increases kinase activity35 Accordingly, we assessed PKAc phosphorylation as a potential mechanism for the changes in phospholamban phosphorylation with OGTKO. PKAc phosphorylation had more intragroup variability than the phospholamban immunoblots. Therefore, the number of samples per group was increased, but this only allowed 2 experimental groups per membrane. PKAc phosphorylation was higher in control sham versus OGTKO sham and control TAC versus OGTKO TAC in both early (Figure 8A and 8B) and established (Figure 9A and 9B) hypertrophy. In control hearts, PKAc phosphorylation was greater in TAC versus sham hearts during early hypertrophy (Figure 8C), but not in established hypertrophy (Figure 9C). These results suggest that OGTKO reduced PKAc activity. A prior study in brain tissue demonstrated that O‐GlcNAcylation of PKAc increases kinase activity36 We determined whether PKAc undergoes O‐GlcNAcylation with immunoprecipitation experiments. O‐GlcNAc levels on PKAc were significantly higher in control TAC versus OGTKO TAC and control sham versus OGTKO sham in both early (Figure 8D) and established hypertrophy (Figure 9D). PKAc O‐GlcNAcylation was also greater in control TAC versus control sham during early hypertrophy. In established hypertrophy, PKAc O‐GlcNAcylation was higher in control TAC compared with control sham; however, this value did not reach significance (P value from t test=0.076, P value from Tukey procedure=0.056). Finally, PKAc is also a kinase for cTnI Ser23/24 phosphorylation37, 38 As an additional assessment of in vivo protein kinase A activity, we measured cTnI Ser23/24 phosphorylation and found that it decreased with OGTKO in all conditions (Figure 10A, 10B, 10D, and 10E). In control hearts, cTnI phosphorylation was higher in sham versus TAC during early hypertrophy (Figure 10C), but not in established hypertrophy (Figure 10F).
Figure 8.
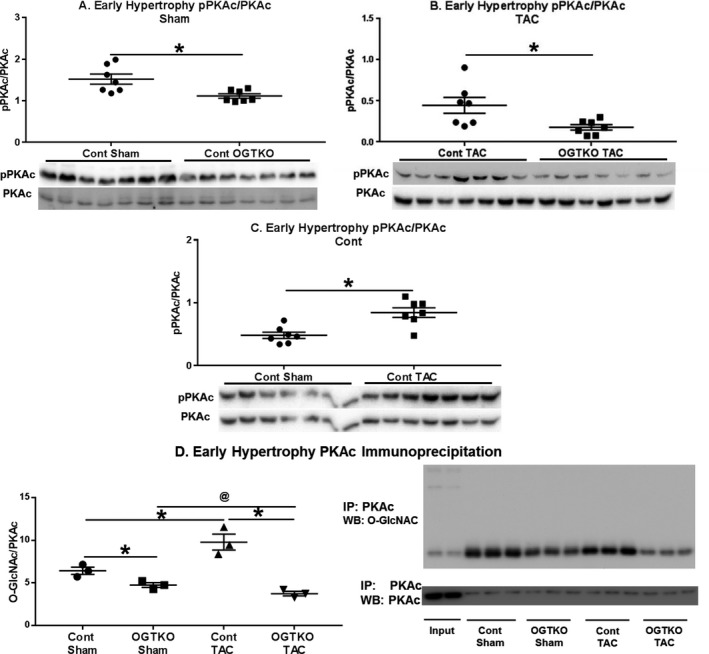
Protein kinase A catalytic subunit (PKAc) phosphorylation (pPKAc) and O‐GlcNAcylation in early hypertrophy. Immunoblots for phosphorylated PKAc at Thr197 normalized to total PKAc in the indicated early hypertrophy groups (A through C). Immunoprecipitation for PKAc with subsequent immunoblotting for O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) and PKAc (for normalization) in early hypertrophy (D). In (D) input lane 1 is the whole cell lysate from control transverse aortic constriction (TAC) and input lane 2 is the whole cell lysate from O‐GlcNAc transferase knockout (OGTKO) TAC. n=7 per group for A–C. n=3 per group in (D). Values are arbitrary units reported as mean±standard error of the mean. *P<0.05 between groups indicated with the bar. @ P value from t test=0.058 and P value from Tukey procedure=0.57 between the groups indicated with the bar. For (D) control sham vs OGTKO sham, P value from t test=0.028 and P value from Tukey procedure=0.20. WB indicates Western blot.
Figure 9.
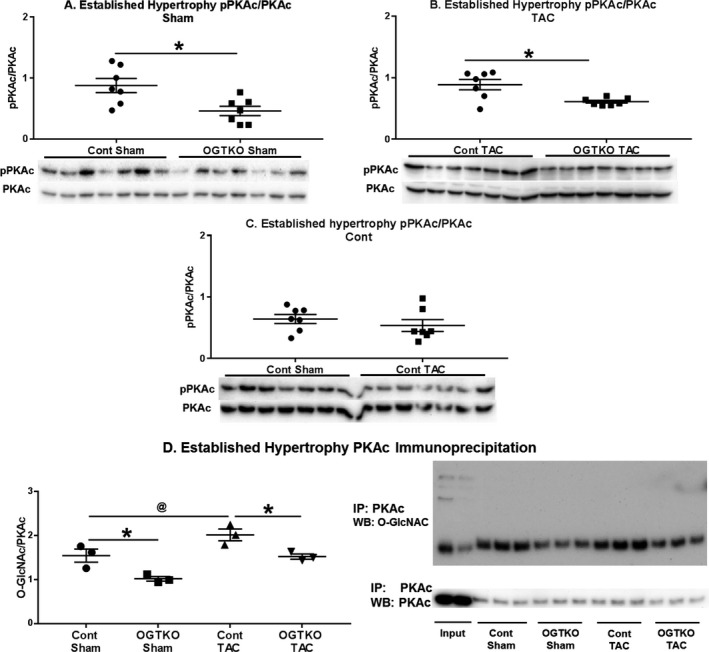
Protein kinase A catalytic subunit (PKAc) phosphorylation (pPKAc) and O‐GlcNAcylation in established hypertrophy. Immunoblots for phosphorylated PKAc at Thr197 normalized to total PKAc in the indicated established hypertrophy groups (A through C). Immunoprecipitation for PKAc with subsequent immunoblotting for O‐linked β‐N‐acetylglucosamine (O‐GlcNAc) and PKAc (for normalization) in established hypertrophy (D). In (D) input lane 1 is the whole cell lysate from control transverse aortic constriction (TAC) and input lane 2 is the whole cell lysate from O‐GlcNAc transferase knockout (OGTKO) TAC. n=7 per group for (A through C). n=3 per group in (D). Values are arbitrary units reported as mean±standard error of the mean. *P<0.05 between groups indicated with the bar. @ P value from t test=0.076 and P value from Tukey procedure=0.056 between the groups indicated with the bar. WB indicates Western blot.
Figure 10.
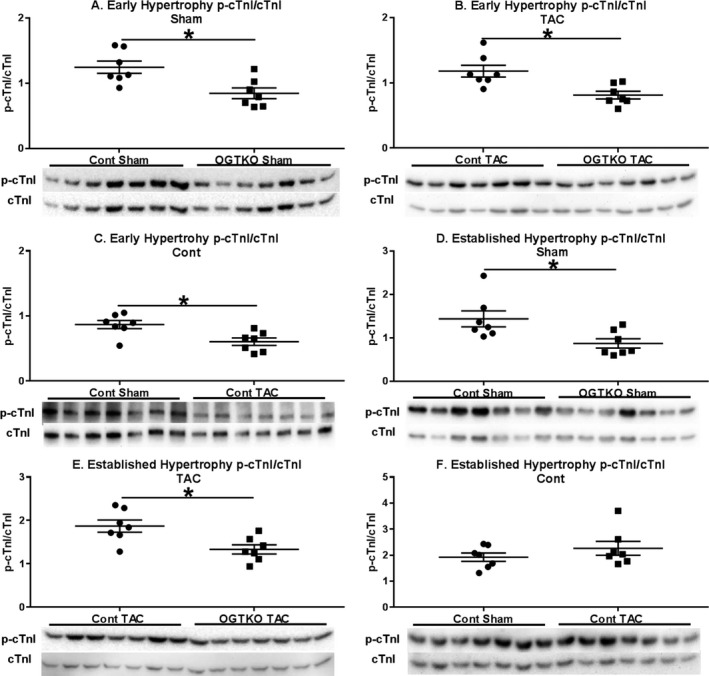
Phosphorylation of cardiac troponin I (p‐cTnI) in early and established hypertrophy. Immunoblots from the indicated early and established hypertrophy groups are show for p‐cTnI Ser23/24 to total cardiac troponin I (cTnI) (A through F). n=7 per group for (A through F). Values are arbitrary units reported as mean±standard error of the mean. *P<0.05 between groups indicated with the bar. OGTKO indicates O‐linked β‐N‐acetylglucosamine transferase knockout; TAC, transverse aortic constriction.
Discussion
O‐GlcNAc levels increase with cardiac hypertrophy, yet the functional impact of this change is incompletely understood. A prior study showed that OGT ablation before TAC reduced cardiac function at 2 and 4 weeks after surgery11 However, the prior work did not determine whether O‐GlcNAc has differential effects during the early response to pressure overload versus potential chronic impacts during established hypertrophy. We tested the hypothesis that early O‐GlcNAcylation improves cardiac function during the initial response to pressure overload, but that sustained elevations during established hypertrophy have an opposite effect on cardiac function by adversely affecting calcium handling proteins. We confirmed that OGTKO and decreased O‐GlcNAcylation reduced cardiac function during early hypertrophy. However, our hypothesis on the effect of prolonged O‐GlcNAcylation during established hypertrophy was incorrect. OGTKO caused severe cardiac dysfunction in established hypertrophy TAC hearts. Further, contrary to our expectations, calcium handling proteins were not adversely affected by O‐GlcNAcylation and phospholamban phosphorylation actually decreased with OGTKO in both the early and established hypertrophy groups. We made a novel finding that cardiac PKAc is O‐GlcNAcylated, which may contribute to the cardiac dysfunction from OGTKO by negatively affecting phosphorylation of phospholamban and cTnI. Overall, our results show that OGT and likely O‐GlcNAcylation benefit cardiac function during both early and established pathological hypertrophy, as well as sham hearts.
O‐GlcNAc Levels During Pressure Overload Hypertrophy
We found an important difference in the O‐GlcNAc profile from TAC in control mice between early and established hypertrophy. While TAC increased total O‐GlcNAc levels in early hypertrophy, the established hypertrophy control TAC mice showed changes only at molecular weights <100 kDa. There was a slight difference in pressure overload applied to these 2 groups. However, this finding suggests that O‐GlcNAcylation is a dynamic process, which still requires an even more thorough evaluation of the molecular mechanisms responsible for regulation. Additionally, the increase in protein O‐GlcNAcylation at <100 kDa in both groups implies that proteins within these molecular weights are important drivers of O‐GlcNAc's effects during pressure overload hypertrophy. In support of this idea, we found that PKAc (molecular weight 42 kDa) is O‐GlcNAcylated in both early and established hypertrophy. Future studies should evaluate additional O‐GlcNAcylated proteins in this molecular weight range, as well as determine the factors regulating O‐GlcNAc specificity.
Cardiac Function
Our early hypertrophy protocol assessed the effects of O‐GlcNAcylation during the initial response to pressure overload. This unique period is characterized by the postoperative recovery, as well as rapid pathological hypertrophic growth. Using the same transgenic mice, it was previously demonstrated that OGT ablation before TAC reduced cardiac function at 2 and 4 weeks postoperative without affecting hypertrophic growth11 The authors did not serially compare cardiac function between 2 and 4 weeks, which would have been useful to determine whether OGT ablation had an ongoing negative effect on cardiac function. The severity of pressure overload in their experiments is unknown, as they did not report the Doppler‐derived pressure gradient at the constriction site and there was no functional comparison to sham‐operated mice. The results of our current study address these unknowns and support that OGTKO deleteriously affects cardiac function during early pathological hypertrophy. The estimated pressure gradient across the constriction site in our early hypertrophy experiments was 96 mm Hg in control TAC and 74 mm Hg in OGTKO TAC, which would clinically be classified as severe stenosis. The pressure gradient was lower in the OGTKO mice for unknown reasons. One possibility is that the suture around the aorta was looser in the OGTKO group, resulting in less constriction. This is unlikely as our previous TAC experiments and the established hypertrophy groups demonstrate that we typically achieve a consistent level of constriction during TAC surgeries12 The pressure gradient through a fixed constriction depends on flow through this area. Therefore, another possible explanation is that the reduced cardiac function in OGTKO TAC hearts lowered cardiac output and, therefore, flows through the constriction site. A similar phenomenon is frequently seen clinically in infants with critical aortic stenosis and depressed cardiac function. However, neither explanation nullifies the finding that OGTKO adversely affected cardiac function in the early hypertrophy mice.
No prior studies evaluated the effect of OGT and O‐GlcNAcylation on isolated established pathological hypertrophy. The established hypertrophy model is important for 2 reasons. First, this model tests whether prolonged O‐GlcNAcylation is maladaptive. Second, clinical therapies are frequently initiated in patients with established hypertrophy. To ensure compensated cardiac function before starting Tam injections, we performed a slightly less severe aortic constriction than in the early hypertrophy groups. The estimated pressure gradient was ≈77 mm Hg in both established hypertrophy TAC groups. Both TAC groups had compensated function before Tam injections, as evidenced by the similar shortening fractions among the TAC and sham groups. In the TAC hearts, OGTKO severely reduced shortening fraction and increased diastolic dilation, which would be considered a dilated cardiomyopathy. Lung wet weight was also increased with OGTKO during TAC, indicating pulmonary congestion and, therefore, decompensated cardiac function. Overall, our results show that OGT and prolonged O‐GlcNAcylation are not maladaptive during established pressure overload hypertrophy.
OGTKO had not been previously reported in sham‐operated hearts. In unoperated mice, both constitutive and inducible cardiomyocyte‐specific OGTKO caused a progressive dilated cardiomyopathy39 In our study, we found a mild negative effect on systolic function from OGTKO in the established hypertrophy sham mice. The totality of these studies shows that OGT and O‐GlcNAc are necessary to maintain cardiac function in nonstressed hearts. However, their importance is clearly amplified during pressure overload hypertrophy.
Calcium Handling Proteins and Mechanisms for Decreased Cardiac Function From OGTKO
Dassanayaka et al11 did not identify a mechanism for the reduced cardiac function with OGTKO before TAC surgery despite a thorough investigation11 OGTKO did not alter fibrosis, apoptosis, capillary density, or expression of metabolic and calcium handling genes. Nkx2‐5 and Acta2 expression increased with OGT deficiency, suggesting cardiomyocyte dedifferentiation, and the investigators speculated that the dedifferentiation made the hearts more sensitive to pressure overload. Further detailing of the mechanistic effects of OGT deficiency and O‐GlcNAcylation during hypertrophy was clearly required.
Based on studies in diabetic cardiomyopathy, we speculated that prolonged O‐GlcNAcylation would detrimentally impact calcium handling proteins. This hypothesis was incorrect as SERCA2a protein levels were unaffected by OGT status, and phospholamban Ser16 phosphorylation actually decreased with OGTKO in all conditions. Our follow‐up experiments, however, identified a potential mechanism for the diminished cardiac function from OGTKO.
We propose that OGTKO is detrimental to cardiac function by reducing PKAc kinase activity with a resultant negative effect of phospholamban and cTnI function. First, multiple findings in our study indicate that OGTKO lowers PKAc kinase activity. OGTKO mice had decreased phosphorylation of the PKAc kinase sites on phospholamban (Ser16) and cTnI (Ser23/24). Additionally, PKAc phosphorylation fell with OGTKO, which would reduce kinase activity35
Potentially most important, OGTKO TAC and OGTKO sham mice both had lower PKAc O‐GlcNAc levels than their corresponding control counterparts. In the brain, PKAc O‐GlcNAcylation increases kinase activity as measured by phosphorylation of the protein kinase A site on the protein tau (Ser214)36 Our results support that PKAc O‐GlcNAcylation similarly promotes kinase activity in the heart. PKAc O‐GlcNAcylation also appears to be part of the normal response to pressure overload hypertrophy as evidenced by the increased PKAc O‐GlcNAcylation in control TAC versus control sham in early hypertrophy with a similar trend in established hypertrophy. To the best of our knowledge, PKAc has not been previously reported to undergo O‐GlcNAcylation in the heart. Of particular interest, PKAc phosphorylation levels dropped with the reduced PKAc O‐GlcNAcylation in the OGTKO groups. Positive and negative crosstalk between these 2 posttranslational modifications is documented on other proteins; thus, further studies are warranted on their interaction in myocardial PKAc2 Overall, our results show that protein kinase A O‐GlcNAcylation is potentially an important mechanism for maintaining cardiac function.
As noted, OGTKO decreased phosphorylation of phospholamban (Ser16) and cTnI (Ser23/24). These changes in phospholamban and cTnI would negatively affect cardiac function40 Lower phospholamban phosphorylation (Ser16) decreases cardiac relaxation by inhibiting SERCA2a transport of Ca2+ from cytosol into the sarcoplasmic reticulum. Inotropy and lusitropy are both diminished with lower cTnI Ser23/24 phosphorylation37, 38 Thus, these mechanisms would decrease cardiac function from OGTKO. Phospholamban and cTnI phosphorylation were similarly altered by OGTKO in all conditions (sham, early hypertrophy, and established hypertrophy), suggesting a fundamental role for OGT and possibly O‐GlcNAcylation in regulating these proteins. Comparing control sham with control TAC, O‐GlcNAc levels correlated with phospholamban phosphorylation but not cTnI phosphorylation. Although our results show regulation of cTnI by O‐GlcNAc, other proteins appear to have a stronger effect during pressure overload.
O‐GlcNAcylation appears to have opposite effects on cardiac function and calcium handling proteins in pressure overload hypertrophy versus diabetic cardiomyopathy. Additional work is required to explain these divergent results. The physiologic stressors are different between these models, which likely explains much of the discrepancy. An additional consideration is that these experimental models employed different methods to alter O‐GlcNAcylation. The diabetic studies lowered O‐GlcNAcylation by increasing OGA levels23 whereas the aortic constriction studies ablated OGT11 It is possible that OGT and OGA may affect different cellular processes beyond O‐GlcNAcylation. For example, OGT is also a protease for the maturation of host cell factor 141 and may also function as a scaffold protein42
It is also notable that the beneficial cardioprotective effects of OGT and O‐GlcNAcylation during ischemia/reperfusion are not operative in pressure overload hypertrophy. In ex vivo or in vivo ischemia, acutely augmenting O‐GlcNAcylation improves cardiomyocyte survival and cardiac function13, 14, 15, 16, 17, 18, 19, 43 Apoptosis and fibrosis were not altered by OGT deletion in the previous study11 For completeness, we measured apoptosis and fibrosis in our established hypertrophy groups and found no difference from OGTKO. Thus, we conclude that cardioprotection is not importantly regulated by OGT and O‐GlcNAc during pathological pressure overload hypertrophy.
Cardiac Hypertrophy
Cardiac hypertrophy was not the primary focus of our experiments, but we had interesting findings that merit follow‐up. In vitro, O‐GlcNAc promotes the development of cardiomyocyte hypertrophy through nuclear factor of activated T‐cell activation.7 Prior work and our results show that OGT and O‐GlcNAcylation are not necessary for the development of in vivo cardiac hypertrophy11 However, our results suggest that OGT and O‐GlcNAcylation may limit in vivo cardiomyocyte hypertrophy. In the established hypertrophy TAC hearts, the cardiomyocyte cross‐sectional area increased with OGTKO, correlating with greater cardiac mass. OGTKO augmented cardiac mass in the sham mice, but we did not identify a change in cardiomyocyte cross‐sectional area. A prior study showed that unoperated constitutive cardiomyocyte‐specific OGTKO had increased hypertrophy (measured by cross‐sectional area) after a longer duration of OGT ablation39 Further studies are necessary to determine whether this effect on limiting cardiomyocyte hypertrophy is directly from O‐GlcNAcylation of proteins involved in cellular growth or secondary to decreased cardiac function. Interestingly, O‐GlcNAc had an opposite effect on cardiac hypertrophy when associated with AMP‐activated protein kinase activation. Gelinas and Mailleux et al44 showed that AMP‐activated protein kinase inhibits cardiac hypertrophy by reducing O‐GlcNAcylation both in vitro and in vivo44 Thus, O‐GlcNAc differentially affects cardiomyocyte growth depending on the model. Further investigations are needed to determine the mechanisms regulating these contrasting effects.
Limitations
As noted, OGT may affect cardiac function through mechanisms outside of O‐GlcNAcylation and we cannot be sure that the findings in our study are solely caused by changes in O‐GlcNAc levels. Additionally, reduced O‐GlcNAcylation likely affects other cellular processes that could diminish cardiac function. Thus, we conclude that altered PKAc activity and phospholamban/cTnI phosphorylation are part of but not necessarily the only, mechanisms causing decreased cardiac function from decreased O‐GlcNAc levels. Third, additional studies are needed to directly assess OGT and O‐GlcNAc's effect on cardiomyocyte calcium handling and PKAc activity in the heart. Finally, we did not include a cre‐positive wild‐type control group. Cardiac‐specific cre activation can cause a transient cardiomyopathy while mice are receiving tamoxifen45; however, we did not find changes in cardiac function during our Tam administration protocol.
Conclusions
OGT and likely O‐GlcNAcylation promote myocardial functional adaptation during both the early response to pressure overload and established pathological hypertrophy. Further, OGT and O‐GlcNAcylation are necessary to maintain cardiac function in sham mice. Mechanistically, OGTKO appeared to reduce PKAc kinase activity, resulting in a detrimental effect on phospholamban and cTnI regulation. The apparent change in PKAc activity with OGTKO correlated to PKAc O‐GlcNAc levels, suggesting that O‐GlcNAcylation regulates myocardial PKAc activity.
Sources of Funding
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number NIH R01HL122546 to Olson. Ledee is funded by the National Institute of Health under award number NIH K01HL120886.
Disclosures
None.
Supporting information
Table S1. Numerical Values for the Figure Graphs
Acknowledgments
The authors thank Miriam Haviland, MSPH, from the Seattle Children's Research Institute Biostatistics, Epidemiology, Econometrics and Programming Core for her statistical assistance.
(J Am Heart Assoc. 2019;8:e011260 DOI: 10.1161/JAHA.118.011260.)
References
- 1. Torres CR, Hart GW. Topography and polypeptide distribution of terminal N‐acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O‐linked GlcNAc. J Biol Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- 2. Butkinaree C, Park K, Hart GW. O‐linked beta‐N‐acetylglucosamine (O‐GlcNAc): extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta. 2010;1800:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O‐GlcNAc cycling in feast or famine. Biochim Biophys Acta. 2010;1800:80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hart GW, Slawson C, Ramirez‐Correa G, Lagerlof O. Cross talk between O‐GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hu P, Shimoji S, Hart GW. Site‐specific interplay between O‐GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett. 2010;584:2526–2538. [DOI] [PubMed] [Google Scholar]
- 6. Zeidan Q, Hart GW. The intersections between O‐GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci. 2010;123:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, Jones SP. O‐GlcNAc signaling is essential for NFAT‐mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol. 2012;302:H2122–H2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lunde IG, Aronsen JM, Kvaloy H, Qvigstad E, Sjaastad I, Tonnessen T, Christensen G, Gronning‐Wang LM, Carlson CR. Cardiac O‐GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics. 2012;44:162–172. [DOI] [PubMed] [Google Scholar]
- 9. Olson AK, Ledee D, Iwamoto K, Kajimoto M, O'Kelly Priddy C, Isern N, Portman MA. C‐Myc induced compensated cardiac hypertrophy increases free fatty acid utilization for the citric acid cycle. J Mol Cell Cardiol. 2013;55:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O‐linked beta‐N‐acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci USA. 2010;107:17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dassanayaka S, Brainard RE, Watson LJ, Long BW, Brittian KR, DeMartino AM, Aird AL, Gumpert AM, Audam TN, Kilfoil PJ, Muthusamy S, Hamid T, Prabhu SD, Jones SP. Cardiomyocyte Ogt limits ventricular dysfunction in mice following pressure overload without affecting hypertrophy. Basic Res Cardiol. 2017;112:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ledee D, Smith L, Bruce M, Kajimoto M, Isern N, Portman MA, Olson AK. c‐Myc alters substrate utilization and O‐GlcNAc protein posttranslational modifications without altering cardiac function during early aortic constriction. PLoS One. 2015;10:e0135262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia‐reperfusion injury via increased protein‐associated O‐GlcNAc. Am J Physiol Cell Physiol. 2007;292:C178–C187. [DOI] [PubMed] [Google Scholar]
- 14. Fulop N, Zhang Z, Marchase RB, Chatham JC. Glucosamine cardioprotection in perfused rat hearts associated with increased O‐linked N‐acetylglucosamine protein modification and altered p38 activation. Am J Physiol Heart Circ Physiol. 2007;292:H2227–H2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, Hart GW, Marban E. Cardioprotection by N‐acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–1182. [DOI] [PubMed] [Google Scholar]
- 16. Liu J, Marchase RB, Chatham JC. Increased O‐GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am J Physiol Heart Circ Physiol. 2007;293:H1391–H1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J, Marchase RB, Chatham JC. Glutamine‐induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O‐GlcNAc levels. J Mol Cell Cardiol. 2007;42:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu J, Pang Y, Chang T, Bounelis P, Chatham JC, Marchase RB. Increased hexosamine biosynthesis and protein O‐GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40:303–312. [DOI] [PubMed] [Google Scholar]
- 19. Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non‐canonical glycosyltransferase modulates post‐hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O‐GlcNAcase in perfused rat hearts by NAG‐thiazolines at the time of reperfusion is cardioprotective in an O‐GlcNAc‐dependent manner. Am J Physiol Heart Circ Physiol. 2010;299:H1715–H1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O‐GlcNAcylation. J Biol Chem. 2003;278:44230–44237. [DOI] [PubMed] [Google Scholar]
- 22. Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez‐Ramirez JA, Banerjee I, Torres‐Gonzalez M, Wang H, Ellrott I, Maya‐Ramos L, Villarreal F, Dillmann WH. Excess protein O‐GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. 2012;303:R689–R699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus‐mediated overexpression of O‐GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96:1006–1013. [DOI] [PubMed] [Google Scholar]
- 24. Yokoe S, Asahi M, Takeda T, Otsu K, Taniguchi N, Miyoshi E, Suzuki K. Inhibition of phospholamban phosphorylation by O‐GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology. 2010;20:1217–1226. [DOI] [PubMed] [Google Scholar]
- 25. Yang X, Qian K. Protein O‐GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Standage SW, Bennion BG, Knowles TO, Ledee DR, Portman MA, McGuire JK, Liles WC, Olson AK. PPARalpha augments heart function and cardiac fatty acid oxidation in early experimental polymicrobial sepsis. Am J Physiol Heart Circ Physiol. 2017;312:H239–H249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ledee D, Portman MA, Kajimoto M, Isern N, Olson AK. Thyroid hormone reverses aging‐induced myocardial fatty acid oxidation defects and improves the response to acutely increased afterload. PLoS One. 2013;8:e65532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Medford HM, Chatham JC, Marsh SA. Chronic ingestion of a Western diet increases O‐linked‐beta‐N‐acetylglucosamine (O‐GlcNAc) protein modification in the rat heart. Life Sci. 2012;90:883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai JY, Grenett MH, Ratcliffe WF, Brewer RA, Nagendran J, Villegas‐Montoya C, Zou C, Zou L, Johnson RL Jr, Dyck JR, Bray MS, Gamble KL, Chatham JC, Young ME. O‐GlcNAcylation, novel post‐translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–44619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kranias EG. Regulation of Ca2+ transport by cyclic 3’,5’‐AMP‐dependent and calcium‐calmodulin‐dependent phosphorylation of cardiac sarcoplasmic reticulum. Biochim Biophys Acta. 1985;844:193–199. [DOI] [PubMed] [Google Scholar]
- 32. Tada M, Inui M, Yamada M, Kadoma M, Kuzuya T, Abe H, Kakiuchi S. Effects of phospholamban phosphorylation catalyzed by adenosine 3’:5’‐monophosphate‐ and calmodulin‐dependent protein kinases on calcium transport ATPase of cardiac sarcoplasmic reticulum. J Mol Cell Cardiol. 1983;15:335–346. [DOI] [PubMed] [Google Scholar]
- 33. Simmerman HK, Jones LR. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev. 1998;78:921–947. [DOI] [PubMed] [Google Scholar]
- 34. Morris GL, Cheng HC, Colyer J, Wang JH. Phospholamban regulation of cardiac sarcoplasmic reticulum (Ca(2+)‐Mg2+)‐ATPase. Mechanism of regulation and site of monoclonal antibody interaction. J Biol Chem. 1991;266:11270–11275. [PubMed] [Google Scholar]
- 35. Cauthron RD, Carter KB, Liauw S, Steinberg RA. Physiological phosphorylation of protein kinase A at Thr‐197 is by a protein kinase A kinase. Mol Cell Biol. 1998;18:1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie S, Jin N, Gu J, Shi J, Sun J, Chu D, Zhang L, Dai CL, Gu JH, Gong CX, Iqbal K, Liu F. O‐GlcNAcylation of protein kinase A catalytic subunits enhances its activity: a mechanism linked to learning and memory deficits in Alzheimer's disease. Aging Cell. 2016;15:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–1065. [DOI] [PubMed] [Google Scholar]
- 39. Watson LJ, Long BW, DeMartino AM, Brittian KR, Readnower RD, Brainard RE, Cummins TD, Annamalai L, Hill BG, Jones SP. Cardiomyocyte Ogt is essential for postnatal viability. Am J Physiol Heart Circ Physiol. 2014;306:H142–H153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta‐adrenergic‐induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol. 2000;278:H769–H779. [DOI] [PubMed] [Google Scholar]
- 41. Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. O‐GlcNAc transferase catalyzes site‐specific proteolysis of HCF‐1. Cell. 2011;144:376–388. [DOI] [PubMed] [Google Scholar]
- 42. Levine ZG, Walker S. The biochemistry of O‐GlcNAc transferase: which functions make it essential in mammalian cells? Annu Rev Biochem. 2016;85:631–657. [DOI] [PubMed] [Google Scholar]
- 43. Laczy B, Marsh SA, Brocks CA, Marchase RB, Chatham JC. Inhibition of O‐GlcNAcase in perfused rat hearts by NAG‐thiazolines at the time of reperfusion is cardioprotective in an O‐GlcNAc dependent manner. Faseb J. 2009;23:H1715–H1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois‐Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Des Rosiers C, Viollet B, Sakamoto K, Balligand JL, Vanoverschelde JL, Beauloye C, Horman S, Bertrand L. AMPK activation counteracts cardiac hypertrophy by reducing O‐GlcNAcylation. Nat Commun. 2018;9:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Willis MS, Holley DW, Wang Z, Chen X, Quintana M, Jensen BC, Tannu M, Parker J, Jeyaraj D, Jain MK, Wolfram JA, Lee HG, Bultman SJ. BRG1 and BRM function antagonistically with c‐MYC in adult cardiomyocytes to regulate conduction and contractility. J Mol Cell Cardiol. 2017;105:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Numerical Values for the Figure Graphs