

Abstract
Background
Branched‐chain amino acid (BCAA) catabolic defect is an emerging metabolic hallmark in failing hearts in human and animal models. The therapeutic impact of targeting BCAA catabolic flux under pathological conditions remains understudied.
Methods and Results
BT2 (3,6‐dichlorobenzo[b]thiophene‐2‐carboxylic acid), a small‐molecule inhibitor of branched‐chain ketoacid dehydrogenase kinase, was used to enhance BCAA catabolism. After 2 weeks of transaortic constriction, mice with significant cardiac dysfunctions were treated with vehicle or BT2. Serial echocardiograms showed continuing pathological deterioration in left ventricle of the vehicle‐treated mice, whereas the BT2‐treated mice showed significantly preserved cardiac function and structure. Moreover, BT2 treatment improved systolic contractility and diastolic mechanics. These therapeutic benefits appeared to be independent of impacts on left ventricle hypertrophy but associated with increased gene expression involved in fatty acid utilization. The BT2 administration showed no signs of apparent toxicity.
Conclusions
Our data provide the first proof‐of‐concept evidence for the therapeutic efficacy of restoring BCAA catabolic flux in hearts with preexisting dysfunctions. The BCAA catabolic pathway represents a novel and potentially efficacious target for treatment of heart failure.
Keywords: amino acids, heart failure, hypertrophy/remodeling, metabolism, therapy
Subject Categories: Basic Science Research, Metabolism, Heart Failure, Remodeling, Pharmacology
1.
Clinical Perspective
What Is New?
This is the first study that demonstrates the therapeutic efficacy of enhancing branched‐chain amino acid catabolism in hearts with preexisting dysfunctions.
What Are the Clinical Implications?
Branched‐chain amino acid catabolism is a promising therapeutic target for the treatment of heart failure.
2.
Despite the significant advances in therapies and prevention, heart failure still remains a major public health problem, affecting an estimated 26 million people worldwide,1, 2 with a 25% to 40% 1‐year mortality rate.3 Thus, there is a clear unmet need for new strategies to treat heart failure and improve the quality of life for patients with systolic dysfunction.
Pathogenesis of heart failure is associated with metabolic remodeling and impaired energetics, often characterized by decreased fatty acid oxidation and increased reliance on glucose use.4, 5 Disturbances in myocardial substrate use and loss of fuel flexibility lead to energy deficiency in diseased hearts and ultimately contribute to systolic and diastolic dysfunctions. Genetic and pharmaceutical modulations of glucose and fatty acid metabolism have been shown to influence the progression of cardiac dysfunction in heart diseases,5, 6, 7 implicating regulating cardiac metabolism as a therapeutic strategy to treat heart failure.
In addition to the current paradigm of lipid and glucose use in healthy and diseased hearts, emerging lines of evidence suggest that branched‐chain amino acid (BCAA) catabolic defect is also a major metabolic hallmark in diseased hearts.8, 9, 10 BCAAs, including leucine, isoleucine, and valine, are essential amino acids for mammals. In addition to de novo protein synthesis, BCAAs also act as important nutrient signal molecules to regulate cellular metabolism and growth.11, 12, 13 The homeostasis of BCAAs is controlled by their catabolic pathway, including a shared first deamination step to branched‐chain α‐ketoacids (BCKAs) by branched‐chain amino‐transferase (BCAT), followed by an irreversible and rate‐limiting catabolic step via BCKDH (branched‐chain α‐ketoacid dehydrogenase) complex. Recent studies show that BCAA catabolism is impaired in diseased hearts, accompanied with the downregulation of key catabolic enzyme expression.8, 10 Furthermore, BCAA catabolic defect is a significant contributor to heart failure progression, at least in part caused by elevated reactive oxygen species and metabolic disturbance.8 Accumulated BCAA in hearts suppresses the glucose metabolism and augments the ischemia‐reperfusion injury.14 These findings highlight the importance of BCAA metabolism in maintaining cardiac function in response to pathological stress.
BCKA dehydrogenase activity is regulated by the phosphorylation status of its regulatory subunit E1α.15, 16 Branched‐chain ketoacid dehydrogenase kinase (BCKDK) phosphorylates E1α to inhibit the BCKA dehydrogenase activity.17 Protein phosphatase 2C in mitochondria (PP2Cm) does the opposite. BCKDK expression is unaffected or increased in diseased hearts in contrast to the other suppressed BCAA catabolic genes, contributing to the BCAA catabolic defect.8, 10 BT2 (3,6‐dichlorobenzo[b]thiophene‐2‐carboxylic acid) is a highly specific small‐molecule BCKDK inhibitor that enhances BCAA catabolic flux by blocking BCKA dehydrogenase phosphorylation.18 Two recent studies reported protective effects of BT2 compound on heart function in pressure‐overload and myocardial infarction mouse models,8, 10 in which the BT2 treatment started from the onset of surgical operation before pathological features emerged. However, in clinical practice, therapeutic intervention usually begins when symptoms of heart dysfunction have developed. Therefore, it is persuasive to ask whether the benefits of BT2 treatment could be achieved in hearts with preexisting pathological changes.
In the present study, we sought to elucidate the therapeutic efficacy of restoring BCAA catabolic flux through pharmacological inhibition of BCKDK in heart failure. Our results showed BCKDK inhibition preserved systolic contractility and diastolic mechanics, and it significantly blunted further pathological deterioration in hearts with preexisting dysfunctions. These findings highlight the therapeutic potential of targeting BCAA catabolism to treat heart failure.
Methods
The authors declare that all supporting data are available within the article. Requests for detailed analytic methods will be addressed by the corresponding author.
An expanded methods section is available in Data S1.
Animals
Wild‐type male C57BL/6N mice were purchased from ENVIGO and housed at 22°C with a 12‐hour light, 12‐hour dark cycle with free access to water and standard chow. Terminal tissue collection was performed on mice under isoflurane anesthesia with additional cervical dislocation. All animal procedures were performed in accordance with the National Institutes of Health guidelines and protocols approved by the University of California at Los Angeles Institutional Animal Care and Use Committee.
Transverse Aortic Constriction
Transverse aortic constriction (TAC) was performed, as described,19 in anesthetized (pentobarbital, 60 mg/kg, IP) and ventilated mice (aged 6–8 weeks) to induce hypertrophy and heart failure. After left anterolateral thoracotomy with blunt dissection through the intercostal muscles, aortic constriction was induced by ligating the transverse aorta around a 27.5‐gauge blunt needle using 6‐0 silk suture. The needle was subsequently removed. Sham‐operated mice underwent a similar surgical procedure without constriction of the aorta. All mice were maintained in the same environment with regular laboratory chow and water ad libitum. At the end of the experiments, animals were euthanized by cervical dislocation, and the hearts and other organs were removed and weighed. Hearts were dissected, and tissues were either immediately immersed into 4% buffered formaldehyde or quickly frozen in liquid nitrogen for further experiments.
Mouse Study With BT2 Treatment
Compound BT2 (3,6‐dichlorobenzo[b]thiophene‐2‐carboxylic acid) was a kind gift from the laboratory of Dr David T. Chuang (University of Texas Southwestern Medical Center, Dallas, TX). BT2 was dissolved in dimethyl sulfoxide and diluted into 5% dimethyl sulfoxide, 10% cremophor EL, and 85% 0.1 mol/L sodium bicarbonate, pH 9.0, buffer for delivery. Vehicle buffer contained the same components without BT2 compound. Administration of vehicle or BT2 was performed, as previously described,18 except that animals (aged 6–8 weeks) were dosed daily by oral gavage at 40 mg/kg per day. C57BL/6N adult male mice (n=21) were subjected to TAC to provoke hypertrophy and contractile dysfunction, and echocardiography was performed to confirm left ventricular (LV) contractility and cardiac dysfunction. At 2 weeks after TAC, 4 mice died. Seventeen survival mice were then intragastrically administered a BCKDK inhibitor BT2 (n=9) or vehicle (n=8) for 6 weeks, and cardiac function was assessed every week. C57BL/6N adult male mice (n=6) were subjected to sham operation, and 2 weeks later, they were randomized to vehicle (n=3) or BT2 (n=3) treatment for an additional 6 weeks. At the end of the experiments (8 weeks after TAC or sham), mice were euthanized by cervical dislocation, and the hearts were collected for further biomolecular analyses.
Neonatal Rat Ventricular Myocyte Culture and BT2 Treatment
Neonatal rat ventricular myocytes (NRVMs) were prepared from 2‐day‐old Sprague Dawley rats by enzymatic digestion with collagenase and pancreatin in 1× ADS buffer at 37°C, as described previously.20 NRVMs were cultured in serum‐free DMEM supplemented with 100 U/mL penicillin/streptomycin and 5% ITS (insulin‐transferrin‐selenium) (w/v). NRVMs were exposed to phenylephrine (100 μmol/L) for 24 hours, and then further treated with BT2 (80 μmol/L, dissolved in dimethyl sulfoxide) or 0.1% dimethyl sulfoxide as control for an additional 24 hours.
Echocardiography
The mice were anesthetized and maintained with 1% to 2% isoflurane in 95% oxygen. Echocardiography was performed with a VisualSonics Vevo 2100 instrument (VisualSonics Inc, Toronto, ON, Canada) equipped with a 30‐MHz linear transducer. A parasternal short‐axis view was used to obtain M‐mode images for analysis of fractional shortening (FS), ejection fraction (EF), and other cardiac parameters.
Speckle‐tracking echocardiography was performed, as described previously.21 Strain analyses were conducted by the same trained investigator (M.C.) on all animals using a speckle‐tracking algorithm provided by VisualSonics (VevoStrain). In brief, suitable B‐mode loops were selected from digitally acquired echocardiographic images based on adequate visualization of the endocardial border and absence of image artifacts. Three consecutive cardiac cycles were selected for analysis, and strain measures were averaged over the obtained cardiac cycles. Each long‐ and short‐axis view of the LV myocardium was divided into 6 standard anatomic segments, and peak strain and strain rate measurements were averaged across all 6 segments, as well as the motion measurements (velocity and displacement). Longitudinal strain analyses were obtained from long‐axis views, whereas circumferential and radial strain analyses were obtained from short‐axis views. For the diastolic myocardial strain analysis, the data presented in this study were the mean global strain rate values at all 3 axes as well. As described in a previous study,22 to quantify the peak strain rate during early LV filling, the “reverse peak” option in the Vevo2100 1.6.0 Image Software was used, and parameters of peak reverse strain rate at different planes (longitudinal, radial, and circumferential) were obtained.
Western Blot Analysis
Proteins from heart tissue were harvested in buffer (50 mmol/L HEPES [pH 7.4], 150 mmol/L NaCl, 1% NP‐40 (nonyl phenoxypolyethoxylethanol‐40), 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L glycerophosphate, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L Na3VO4, 20 mmol/L NaF, 1 mmol/L phenylmethylsulfonyl fluoride, and 1 μg/mL of aprotinin, leupeptin, and pepstatin). Samples were separated onto 4% to 12% Bis‐Tris gels (Invitrogen) and transferred onto a nitrocellulose blot (Amersham). The blot was probed with the indicated primary antibodies. Protein signals were detected using horseradish peroxidase–conjugated secondary antibodies and enhanced chemiluminescence23 Western blotting detection reagents (Pierce). Rabbit polyclonal antisera against the E1α subunit of BCKD complex and phosphorylated E1α antibody, and GAPDH, H3, and BCKDK antibodies, were purchased from Abcam. PP2Cm antibody was generated in the laboratory.
Real‐Time Reverse Transcription–Polymerase Chain Reaction Analysis
Total RNA was extracted from heart tissues using Trizol Reagent (Invitrogen), according to the manufacturer's instructions. Total RNA was reverse transcribed into the first‐strand cDNA using ProtoScript II Reverse Transcriptase (New England BioLabs). Then, cDNA transcripts were quantified by the CFX Real‐Time PCR System (BioRad). 18S ribosomal RNAs were used for normalization, except where indicated otherwise. Polymerase chain reaction primer information is available upon request.
Cardiac BCAA and BCKA Assay
Mice were subjected to TAC for 2 weeks to provoke dysfunction and then intragastrically administered a BCKDK inhibitor BT2 or vehicle for an additional 6 weeks. At the end of the experiment, mice were fasted for 6 hours before tissue collection. Heart tissues were collected, and LVs were separated and frozen in liquid nitrogen. Frozen tissue was weighed and homogenized in a 3‐fold volume of PBS using a Bead‐Bug homogenizer. Tissue lysate (50 μL) was precleared of protein by addition of an equal volume of methanol, followed by 2 rounds of centrifugation to precipitate protein. The supernatant was lyophilized and resuspended in 30 μL distilled water. For BCAA measurement, standards (l‐valine, l‐leucine, l‐isoleucine, and l‐allo‐isoleucine) were prepared from 10 mmol/L stocks, and serial pooled dilutions were made (10–2000 μmol/L). A total of 2 μL of standard or 2 μL of sample was diluted in 50 μL butanolic HCL and heated to 60℃ for 20 minutes, and then speed‐vac was used to obtain dryness. The samples were resuspended in 200 μL of (50:50) distilled water/acetonitrile containing 0.1% formic acid. BCAA concentrations in samples and standard were determined by liquid chromatography–tandem mass spectrometry in a 3200 QTRAP mass spectrometer coupled to a Shimadzu Prominence liquid chromatographic instrument. An Agilent C18XDB 5 µm packing column (50×4.6 mm) was used for chromatography with the following conditions: buffer A, distilled water+0.1% formic acid; buffer B, acetonitrile+0.1% formic acid; flow rate, 1.0 mL/min. BCAA concentrations in vehicle‐ or BT2‐treated heart samples were determined on the basis of the standard curve. BCKA measurements were performed, as described elsewhere.24 Briefly, the perchloric acid heart supernatants were derivatized by o‐phenylenediamine, extracted with ethyl acetate, and dried down in glass tubes in an unheated vacuum centrifuge. After drying, the ketoacids were reconstituted in 200 mmol/L ammonium acetate, pH 6.8, and analyzed using a Shimadzu ultrafast liquid chromatography 20ADXR liquid chromatographic system in line with an AB‐Sciex 3200 QTRAP mass spectrometer. Both instruments used in this analysis were housed in the University of Texas Southwestern Medical Center Core Facility. BCAA/BCKA concentrations were measured and normalized to the weight of tissue.
Statistical Analysis
For 2‐group comparison, statistical analyses were performed with Student's t test. For repeated measurements over time, as shown in longitudinal echocardiographic analysis, 2‐way ANOVA, followed by Bonferroni's post hoc test, was performed to evaluate the differences between groups over time or at each time point. For multiple group comparison, differences were analyzed by 1‐ or 2‐way ANOVA with Bonferroni's method. All data were presented as mean±SEM or mean±SD, and were analyzed using GraphPad Prism 7 software (GraphPad Software). P<0.05 was considered as statistically significant.
Results
Establishment of a Mouse Model With Preexisting Cardiac Dysfunction for Subsequent Pharmacological Treatment
Often, patients present to medical attention at the time of symptom development. As such, preventing the further deterioration of existing pathological conditions is the major objective of therapeutic intervention. To assess the therapeutic impacts of restoring BCAA catabolism on the progression of heart failure, pressure overload to LV was initiated by TAC for 2 weeks, followed by treating the animals with BT2 (a small‐molecule inhibitor of BCKDK) or vehicle via oral gavage for additional 6 weeks (Figure 1A and 1B). Before the BT2 treatment, echocardiography detected robust hypertrophic growth with dramatically increased LV mass and ventricular dilatation and significant declines in systolic function in hearts with aortic stricture (Figure 1C, D, E, F). Mice were then randomly assigned to receive BT2 (40 mg/kg per day) or vehicle treatment. As expected, BT2 treatment significantly reduced BCKDK‐mediated BCKDE1α (branched‐chain ketoacid dehydrogenase subunit E1α) phosphorylation without significantly affecting BCKDK protein expression (Figure 1G and 1H). Cardiac intratissue concentrations of BCAAs and BCKAs were significantly reduced (Figure 1I and 1J), consistent with the enhanced BCAA catabolic activities.
Figure 1.
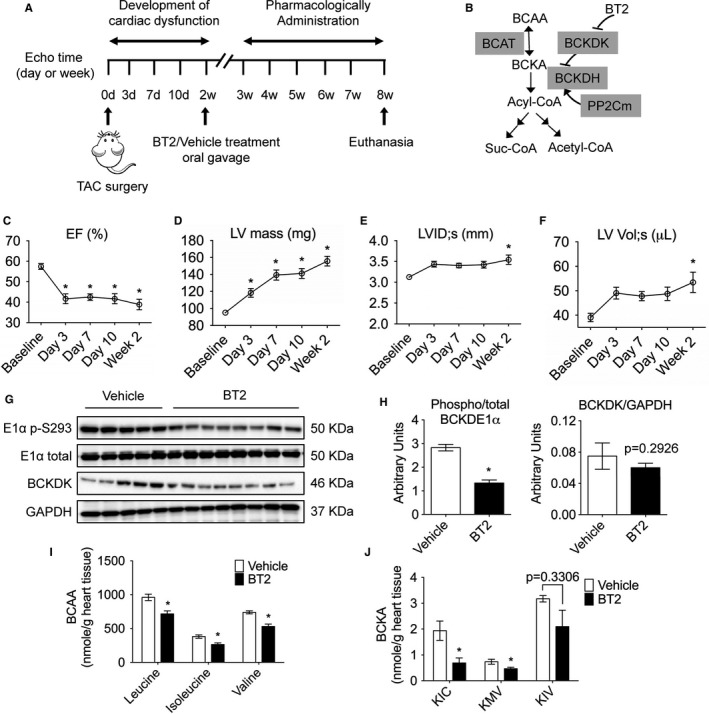
Establishment of a mouse model with preexisting cardiac dysfunction for subsequent pharmacological treatment. A, Schematic timeline and experimental design. C57BL/6N adult male mice (n=21) were subjected to transverse aortic constriction (TAC) to provoke hypertrophy and contractile dysfunction, and echocardiography was performed to confirm left ventricular (LV) contractility and cardiac dysfunction. Four mice died before drug treatment. Mice were then intragastrically administered with BT2 (n=9) or vehicle (n=8) for 6 weeks, and cardiac function was assessed every week. B, A schematic illustration of key enzymes and regulators involved in branched‐chain amino acid (BCAA) catabolism. C through F, Echocardiographic parameters in the first 2 weeks after TAC. Average values of LV ejection fraction (EF; C), LV mass (D), LV internal diameter at systole (LVID;s; E), and LV volume at systole (LV Vol;s; F) obtained from echocardiography are shown. G through J, Analyses of hearts at 8 weeks after TAC procedure (n=5 in vehicle group, n=7 in BT2 group). G, Expression of total, phosphorylated branched‐chain ketoacid dehydrogenase (BCKD) subunit E1α, and BCKD kinase (BCKDK) in mouse heart was determined by Western blotting. H, The relative phosphorylated E1α level and BCKDK level were calculated. Cardiac BCAA (I) and branched‐chain α‐ketoacid (BCKA; J) abundances were measured, as described in Methods. For data of Figure 1 C and 1 F, comparison of indicated time points vs baseline was analyzed by 1‐way ANOVA with Bonferroni's test. For Figure 1 H and 1 J, differences were evaluated by Student's t test. Data were presented as mean±SEM. BCAT indicates branched‐chain amino‐transferase; BCKDH, branched‐chain ketoacid dehydrogenase; KIC, α‐ketoisocaproic; KMV, α‐keto‐β‐methylvaleric; KIV, α‐ketoisovaleric; Suc‐CoA; Succinyl‐CoA. *P<0.05.
BCKDK Inhibitory Treatment Alleviated Pressure‐Overload Induced Systolic Dysfunction Without Affecting Hypertrophy
To assess responses in individual mouse, we measured FS and EF at each time point and calculated ∆FS and ∆EF of each mouse using parameters at 2 weeks after TAC (time right before drug treatment) as baseline (defined as 0); then, the average ∆FS and ∆EF at each time point were plotted. Through this approach, minus ∆FS or ∆EF indicated worsened systolic function, whereas plus values indicated improvement (Figure 2A and 2B). After 2 weeks of treatment (4 weeks after TAC), BT2‐treated mouse hearts started to show an improvement of systolic performance, as indicated by positive values of ∆FS and ∆EF. Six weeks of BT2 treatment led to well‐preserved cardiac functions, whereas progressive deterioration of cardiac dysfunction was observed in vehicle‐treated animals, as indicated by the continuous reduction of FS and EF during the treatment period (Figure 2A and 2B, Figure S1A and S1B). Similar analyses of structural measurements showed preserved chamber structure in BT2‐treated mouse hearts, as seen in approximately unchanged ∆LV internal dimensions (Figure 2C, Figure S1C) and ∆LV volume (Figure 2D, Figure S1D). In contrast, vehicle‐treated animals developed significant chamber dilatation, as indicated by continuous increases of LVID and LV volume during the treatment period. BT2 treatment did not affect cardiac hypertrophy, indicated by heart weights (Figure 2E), hypertrophic marker expression (Figure 2F), and LV posterior wall thickness (Table S1). Heart rate was not affected by BT2 treatment (Table S2). Together, these data demonstrate that BCKDK inhibitory treatment is capable of attenuating existing systolic dysfunction.
Figure 2.
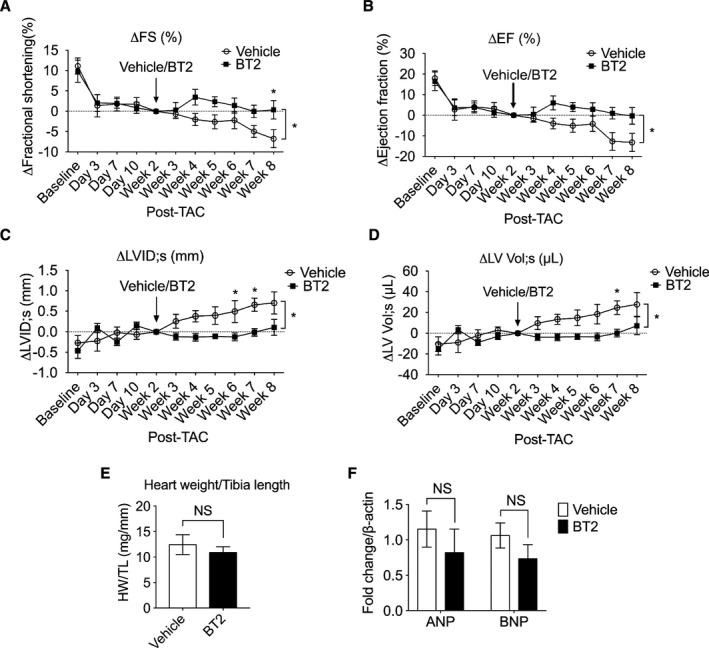
Inhibition of branched‐chain α‐ketoacid dehydrogenase kinase (BCKDK) by BT2 preserved systolic function in hearts with preexisting dysfunctions. Mice were subjected to transverse aortic constriction (TAC) procedure for 2 weeks and then were randomized to be treated with BT2 (n=9) or vehicle (n=8) for an additional 6 weeks. Relative changes of left ventricular (LV) fractional shortening (∆FS; A), ejection fraction (∆EF; B), LV internal diameters (∆LVID;s; C), and LV volume (∆LV Vol;s; D) were presented at indicated time points. E, Heart weight/tibia length (HW/TL) ratios at 8 weeks after TAC procedure were presented (n=5 in vehicle group, n=7 in BT2 group). F, Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) expression was quantified by real‐time quantitative reverse transcription–polymerase chain reaction (n=5 in vehicle group, n=7 in BT2 group). For A through D, 2‐way ANOVA, followed by Bonferroni's post hoc test, was performed to evaluate the differences between groups over the treatment period (from week 3 to week 8). For E and F, Student's t test was used for 2‐group comparison. Data were presented as mean±SEM. NS indicates not significant. *P<0.05.
BCKDK Inhibitory Treatment Preserved Myocardial Contractile Function
Conventional echocardiographic measures, such as EF and FS, lack sensitivity for capturing subtle changes in LV ventricular performance and fail to detect early myocardial dysfunction.25 Recent studies have reported that mechanical indexes derived from strain analysis provide more accurate and sensitive measurement of myocardial contractile function.25, 26, 27 To fully describe the impact of BT2 treatment on murine myocardium contractile function in the setting of heart failure, we took advantage of a 2‐dimensional speckle‐tracking echocardiography technique to measure myocardial mechanics, including deformation (strain and strain rate) and wall motion (velocity and displacement). Consistent with the conventional echocardiographic measurements, BT2 treatment significantly improved myocardial strain (Figure 3A and 3B) and strain rate (Figure 3C and 3D) in both longitudinal and radial axes. Furthermore, BT2 treatment increased systolic velocity and displacement in both longitudinal and radial planes (Figure 3E, F, G, H), indicating that BT2 significantly improved myocardial wall motion. Together, these data suggest that BT2 improves myocardial mechanics to preserve cardiac contractile function under pathological stress.
Figure 3.
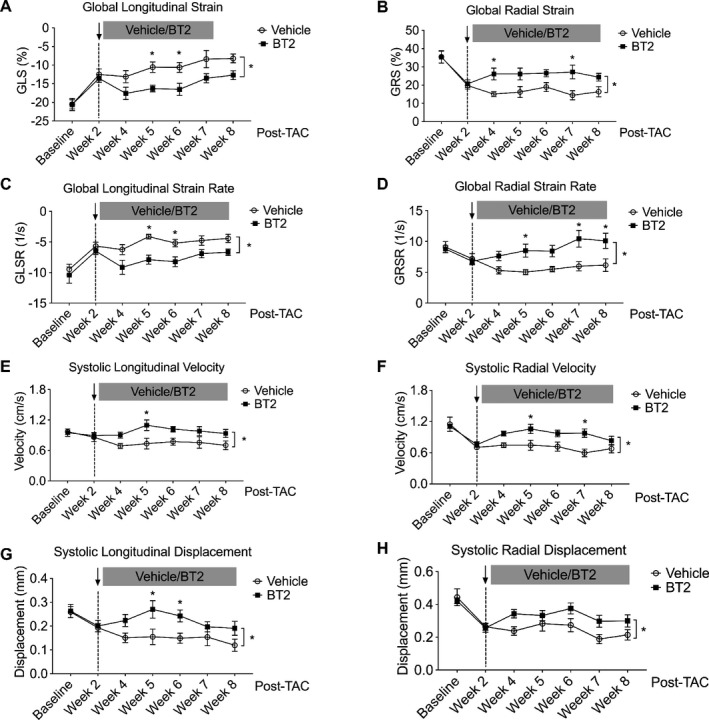
Branched‐chain α‐ketoacid dehydrogenase kinase (BCKDK) inhibitory treatment improved myocardium contractility and wall motion. Mice were subjected to transverse aortic constriction (TAC) procedure for 2 weeks and then were randomized to be treated with BT2 (n=9) or vehicle (n=8) for an additional 6 weeks. Relative global longitudinal strain (GLS; A), global radial strain (GRS; B), and respective global strain rate (GLSR and GRSR; C and D) for both groups at indicated time points were presented. Means of systolic velocity (E and F) and displacement (G and H) at longitudinal and radial planes were presented. Two‐way ANOVA, followed by Bonferroni's post hoc test, was performed to evaluate the differences between groups over the treatment period (from week 4 to week 8). Data were presented as mean±SEM. *P<0.05.
BT2. Treatment Improved Diastolic LV Mechanics
Several previous investigations suggest that early diastolic strain rate can serve as a novel marker of elevated LV filling pressure28, 29, 30 and contributes to the evaluation of diastolic function22 and myocardial viability.31, 32, 33 We assessed the effects of BT2 treatment on diastolic myocardial mechanics in mouse hearts by measuring peak strain rate during early LV filling through 2‐dimensional speckle‐tracking echocardiography. Compared with vehicle, BT2‐treated mouse hearts displayed a higher longitudinal diastolic strain rate by 2 weeks of treatment (4 weeks after TAC), and a similar trend was observed in the subsequent 4 weeks (Figure 4A). At radial plane, BT2 treatment prevented the reduction of radial strain rate over the observation period (from week 4 to week 8) (Figure 4B). Furthermore, BT2 treatment preserved the diastolic velocity of wall motion (Figure 4C and 4D). Together, these data imply that BT2 preserves diastolic mechanics to blunt deteriorated myocardial performance under pathological stress.
Figure 4.
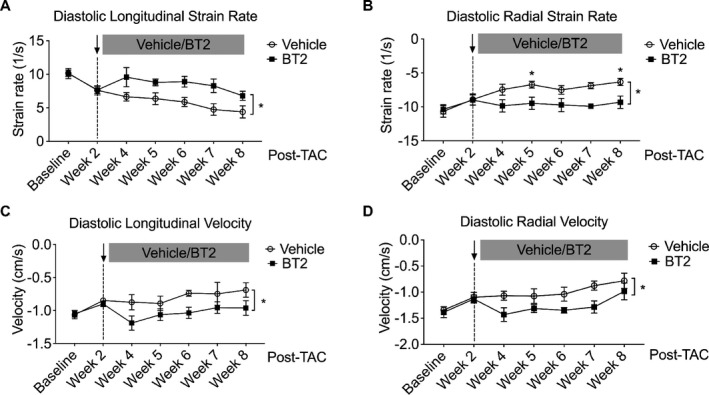
Branched‐chain α‐ketoacid dehydrogenase kinase (BCKDK) inhibitory treatment improved diastolic left ventricular mechanics. Mice were subjected to transverse aortic constriction (TAC) procedure for 2 weeks and then were randomized to be treated with BT2 (n=9) or vehicle (n=8) for an additional 6 weeks. Relative diastolic longitudinal strain rate (A) and diastolic radial strain rate (B) for both groups at indicated time points were presented. Diastolic longitudinal velocity (C) and radial velocity (D) were presented. Two‐way ANOVA, followed by Bonferroni's post hoc test, was performed to evaluate the differences between groups over the treatment period (from week 4 to week 8). Data are presented as mean±SEM. *P<0.05.
BT2. Treatment Altered the Expression of Genes Involved in Substrate Metabolism
Although the direct contribution of BCAA catabolism to myocardial energetics is limited,4, 34, 35 disruptive BCAA catabolism suppresses glucose oxidation, promotes fatty acid oxidation,14 and affects global metabolism.8, 10 To investigate the potential mechanisms underlying the therapeutic benefits of BT2, we assessed the expression of genes involved in major metabolic processes in mouse hearts. We also examined the direct impacts of BT2 on these genes in cultured primary NRVMs treated with or without phenylephrine (Figure S2). The mitochondrial mass seemed unchanged because mitochondrial DNA amount remained unaffected by BT2 treatment in both hearts and NRVMs (Figure 5A and 5B). Interestingly, although the expression of genes involved in glucose transport was unaffected, BT2 treatment significantly increased PDK4 (pyruvate dehydrogenase kinase 4) expression in cardiomyocytes and a similar trend was observed in the hearts (Figure 5C and 5D), which may indicate a decreased glucose oxidation via PDK4‐inhibited pyruvate dehydrogenase complex. Moreover, mRNA level of CD36 (cluster of differentiation 36), a key gene for fatty acid uptake, was increased by BT2 in cardiomyocytes and hearts (Figure 5E and 5F). In addition, expression of genes involved in fatty acid β‐oxidation (Acox1, Acadl, and Acadm) also showed a trend of increase in BT2‐treated mouse hearts and NRVMs (Figure 5E and 5F). Collectively, these gene expression data indicate that BCKDK inhibitory treatment may lead to reduced glucose oxidation and increased fatty acid use in hearts.
Figure 5.
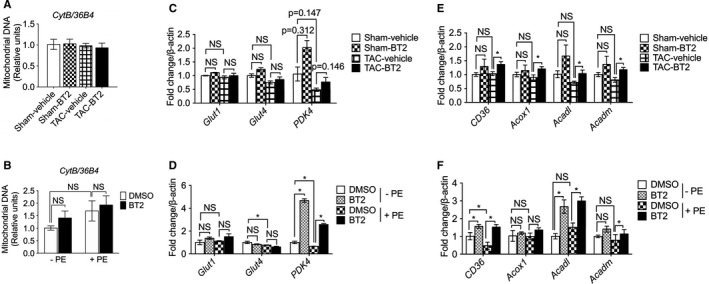
BT2 treatment altered the expression of genes involved in substrate metabolism. A, C, and E, Hearts were collected after 6 weeks of treatment with either vehicle or BT2 (8 weeks after surgery). B, D, and F, Neonatal rat ventricular myocytes (NRVMs) were treated with vehicle or phenylephrine (100 μmol/L) for 24 hours, followed by treatments of BT2 (80 μmol/L, dissolved in dimethyl sulfoxide [DMSO]) or 0.1% DMSO as control for an additional 24 hours. Mitochondrial DNA amounts in mouse hearts (A) and NRVMs (B) were analyzed by quantitative polymerase chain reaction (PCR) using primers specific for the mitochondrial cytochrome b (CytB) gene and normalized to genomic DNA by amplification of the large ribosomal protein p0 (36B4) nuclear gene. mRNA expression of genes involved in glucose transportation (Glut1 and Glut4) and oxidation (PDK4) in mouse hearts (C) and NRVMs (D) was determined by quantitative PCR. mRNA expression of genes involved in fatty acid transport (CD36) and β‐oxidation (Acox1, Acadl, and Acadm) in mouse hearts (E) and NRVMs (F) was analyzed by quantitative PCR. For animal studies: n=3 for sham‐vehicle group, n=3 for sham‐BT2 group, n=5 for transverse aortic constriction (TAC)–vehicle group, n=7 for TAC‐BT2 group. For NRVM experiment: n=3 for each group. Two‐way ANOVA, followed by Bonferroni's post hoc test, was performed for multiple comparison. Data were presented as mean±SD. NS indicates not significant. *P<0.05.
BT2. Treatment Showed No Signs of Apparent Toxicity
Drug toxicity is a major concern in clinic. To evaluate whether BT2 treatment resulted in toxic effects, animals were subjected to sham operation and 2 weeks later randomized to receive vehicle or BT2 compound for an additional 6 weeks. Echocardiographic measurements showed that BT2 treatment did not affect the systolic function and myocardial structure as no significant changes were detected in EF and LVID between BT2 and vehicle groups (Figure 6A, B, C). Furthermore, body weight and organ weight were not affected by BT2 treatment (Figure 6D and 6E). BT2 treatment reduced BCKDE1α (branched‐chain ketoacid dehydrogenase subunit E1α) phosphorylation (Figure 6F). Together, these data indicate that BT2 treatment in the current study does not cause apparent toxicity in mice.
Figure 6.
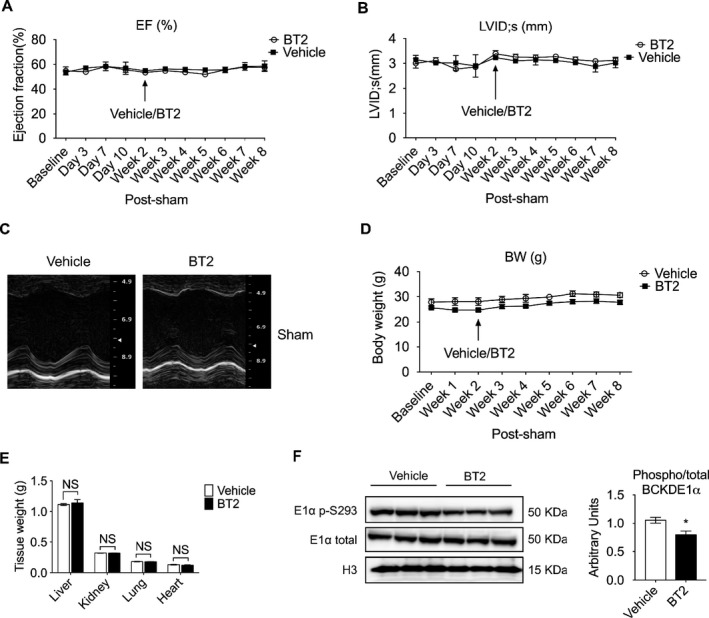
BT2 treatment showed no signs of apparent toxicity. Mice were subjected to sham procedure. Two weeks later, animals were randomized into 2 groups to be treated with either BT2 (n=3) or vehicle (n=3). Mean left ventricular ejection fraction (EF; A) and left ventricular internal diameter at systole (LVID;s; B) were obtained from echocardiography. C, Representative M‐mode echocardiographs of mouse hearts after sham procedure, treated with either vehicle or BT2. D, Body weight (BW) from both groups. E, Tissue weight of mice at 8 weeks after sham procedure. F, Expression of total and phosphorylated branched‐chain ketoacid dehydrogenase subunit E1α (BCKDE1α) in mouse heart at 8 weeks after sham procedure was determined by Western blot (left). The relative phosphorylated E1α level was calculated (right). For A through C, statistical analysis was performed using 2‐way ANOVA, followed by Bonferroni's post hoc test. Student's t test was performed for E and F. Data were presented as mean±SEM. NS indicates not significant. *P<0.05.
Discussion
The current study explored the therapeutic value of targeting the BCAA catabolic flux to treat heart failure. After the establishment of cardiac dysfunction by pressure overload, animals were subjected to BT2 treatment to enhance BCAA catabolic flux. The results showed that BT2 compound was capable of preserving systolic function and alleviating structural remodeling in mouse hearts with preexisting dysfunction. These therapeutic benefits were associated with significantly improved myocardial contractility and diastolic deformation. Moreover, gene expression analysis indicated that enhanced BCAA catabolic flux might improve fatty acid oxidation in the dysfunctional hearts. Finally, BT2 treatment did not affect the body weight and cardiac function in sham‐operated mice, suggesting the lack of apparent toxicity of BT2 treatment.
Emerging studies report impaired BCAA catabolic activity in dysfunctional hearts.8, 9, 10, 36 Previous studies investigated roles of BCAA catabolism in heart failure progression by administrating BT2 at the onset of pathological stresses induced by myocardial infarction or pressure overload.8, 10 In the present study, BT2 is administrated after cardiac dysfunction and pathological remodeling have been established. This design provides a virtual approach to imitate the situation in clinic in which patients usually seek medical care after symptoms of cardiac dysfunction develop. The clear beneficial effects of administrating BT2 in mouse with preexisting cardiac dysfunction provide the first proof‐of‐concept evidence for the therapeutic effects of restoring BCAA catabolic flux. Therefore, the BCAA catabolic pathway represents a novel and potentially efficacious target for the treatment of heart failure.
The accurate assessment of myocardial function is of great significance for the diagnosis and management of cardiac dysfunction.37, 38 Compared with the conventional parameters for evaluating cardiac function, strain analysis using 2‐dimensional speckle‐tracking echocardiography provides a tool with more sensitivity and accuracy as it directly detects the intrinsic mechanical property of myocardium.25, 27, 29 Because of the complex cardiac geometry, myocardium conducts multidirectional motion and distortion to perform proper contraction and relaxation to generate sufficient cardiac output.39, 40 Cardiac performance is partially based on the intrinsic ability of myocardial fibers to conduct these physical movements. Thus, myocardial mechanical impairment can result in cardiac dysfunction.41, 42 In the present study, BT2 treatment improved systolic deformation and wall motion in dysfunctional hearts. And similar effects were also observed in diastolic strain measurements. These findings suggest that BT2 could preserve myocardial mechanical function in the setting of heart failure and ultimately improve LV pump performance, indicating a mechanism underlying the therapeutic benefits from targeting BCAA catabolic flux.
Fatty acid oxidation is the preferred fuel source in the healthy myocardium, accounting for ∼70% of the ATP generated in the mitochondria. In failing hearts, fatty acid use is substantially decreased.43 Although whether the deficiency of fatty acid oxidation contributes to heart failure remains controversial, several lines of evidence suggest that enhancing fatty acid oxidation by peroxisome proliferator‐activated receptor α activation44, 45, 46 or ACC2 (acetyl‐CoA carboxylase 2) deletion47 is beneficial in heart failure. Our gene expression data indicate that BT2 treatment may increase fatty acid use and decrease glucose oxidation. An early study showed that inhibition of glucose metabolism, resulting from increased fatty acid oxidation, led to preserved cardiac function and energetics in stressed heart.47 Therefore, it is tempting to speculate that the BT2 treatment acts through optimization of cardiac substrate use to attenuate the deterioration of heart dysfunction. Additional studies on myocardial metabolism, including protein levels, enzyme activities, substrate use analysis, and metabolic fluxes, are needed to provide more supporting evidence. On the other hand, BT2 may exert its beneficial effects on heart by alleviating the oxidative stress via reducing BCKA or reducing the mTORC1 (mammalian target of rapamycin complex 1) activity by lowering BCAA.8, 10 The molecular mechanisms underlying the cardioprotective effects observed in these studies remain to be fully investigated.
Despite the limited contribution of BCAA metabolism to the energy supply in heart tissue,4, 34, 35 increasing lines of evidence suggest that a defect in BCAA catabolic pathway is another metabolic hallmark of cardiac hypertrophy and failure.9, 48 The promise of targeting BCAA metabolism for heart failure therapy has been highlighted in previous investigations.8, 10 The current study clearly demonstrates the therapeutic effect of BCKDK inhibition by showing BT2 is capable of preserving myocardial contractility and mechanical function in hearts with preexisting dysfunctions. Thus, targeting BCAA catabolic flux provides a novel and promising therapeutic strategy for the treatment of heart failure.
Sources of Funding
This work was supported by National Natural Science Foundation of China (NSFC81570717 and 81522011), National Institutes of Health (HL140116, HL108186, HL103205, HL098954, HL080111, and DK62306), American Heart Association Postdoctoral Fellowship Award (Gao), the Laubisch Fund (University of California, Los Angeles), the Welch Foundation (I‐1286), and Science and Technology Commission of Shanghai Municipality (13ZR1423300 and 16JC1404400). Chen was supported by scholarship from the China Scholarship Council.
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Table S1. Left ventricular posterior wall thickness of sham or TAC mice treated with vehicle or BT2.
Table S2. Heart rate of sham or TAC mice treated with vehicle or BT2.
Figure S1. Absolute values of echocardiographic parameters before and after BT2 treatments in pressure‐overloaded mouse hearts. Average left ventricular fractional shortening (FS) (A) and ejection fraction (EF) (B) for both groups were presented at indicated time points. Structural measurements of LV internal diameters (LVID;s) (C) and LV volume (LV Vol;s) (D) were presented at indicated time points. Two‐way ANOVA followed by Bonferroni's post‐hoc test was performed to evaluate the differences between groups over the treatment period (from week 3 to week 8). *P<0.05. Data was presented as mean±SEM.
Figure S2. BT2 treatment led to a decrease of BCKDE1α phosphorylation in NRVM. (A) Schematic timeline of primary cardiomyocytes study. NRVM was pretreated with PE (100 μmol/L) for 24 h, and then further exposed to DMSO or BT2 (80 μmol/L) for additional 24 h. Cells were collected and proceeded for further measurements. (B) At day 3, NRVM were collected and expression of total, phosphorylated BCKD subunit E1α, and GAPDH protein was determined by Western blotting. PE indicates phenylephrine; NRVM, neonatal rat ventricular myocytes.
Acknowledgments
The authors thank Ms Haiying Pu at Departments of Anesthesiology, Medicine, and Physiology, University of California, for technical assistance.
(J Am Heart Assoc. 2019;8:e011625 DOI: 10.1161/JAHA.118.011625.)
References
- 1. Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CSP, Sato N, Shah AN, Gheorghiade M. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. 2014;63:1123–1133. [DOI] [PubMed] [Google Scholar]
- 2. Rajadurai J, Tse HF, Wang CH, Yang NI, Zhou J, Sim D. Understanding the epidemiology of heart failure to improve management practices: an Asia‐pacific perspective. J Card Fail. 2017;23:327–339. [DOI] [PubMed] [Google Scholar]
- 3. Ezekowitz JA, Kaul P, Bakal JA, Quan H, McAlister FA. Trends in heart failure care: has the incident diagnosis of heart failure shifted from the hospital to the emergency department and outpatient clinics? Eur J Heart Fail. 2011;13:142–147. [DOI] [PubMed] [Google Scholar]
- 4. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–448. [DOI] [PubMed] [Google Scholar]
- 6. Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation. 2004;110:894–896. [DOI] [PubMed] [Google Scholar]
- 7. Lee L, Horowitz J, Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur Heart J. 2004;25:634–641. [DOI] [PubMed] [Google Scholar]
- 8. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. Catabolic defect of branched‐chain amino acids promotes heart failure. Circulation. 2016;133:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 2010;3:420–430. [DOI] [PubMed] [Google Scholar]
- 10. Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, Lee Y, Li C, Zhang L, Lian K, Gao E, Cheng H, Tao L. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;311:H1160–H1169. [DOI] [PubMed] [Google Scholar]
- 11. Zhang S, Zeng X, Ren M, Mao X, Qiao S. Novel metabolic and physiological functions of branched chain amino acids: a review. J Anim Sci Biotechnol. 2017;8:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoon M‐S. The emerging role of branched‐chain amino acids in insulin resistance and metabolism. Nutrients. 2016;8:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. D'Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F, Caliaro F, Corsetti G, Bottinelli R, Carruba MO, Valerio A, Nisoli E. Branched‐chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle‐aged mice. Cell Metab. 2010;12:362–372. [DOI] [PubMed] [Google Scholar]
- 14. Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, Zhou B, Cao Y, Ritterhoff J, Gu H, Raftery D, Sun H, Tian R. Defective branched‐chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia‐reperfusion injury. Cell Metab.2017;25:374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu G, Sun H, She P, Youn JY, Warburton S, Ping P, Vondriska TM, Cai H, Lynch CJ, Wang Y. Protein phosphatase 2cm is a critical regulator of branched‐chain amino acid catabolism in mice and cultured cells. J Clin Invest. 2009;119:1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burrage LC, Nagamani SC, Campeau PM, Lee BH. Branched‐chain amino acid metabolism: from rare Mendelian diseases to more common disorders. Hum Mol Genet. 2014;23:R1–R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tso SC, Gui WJ, Wu CY, Chuang JL, Qi X, Skvora KJ, Dork K, Wallace AL, Morlock LK, Lee BH, Hutson SM, Strom SC, Williams NS, Tambar UK, Wynn RM, Chuang DT. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched‐chain alpha‐ketoacid dehydrogenase kinase. J Biol Chem. 2014;289:20583–20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee JH, Gao C, Peng G, Greer C, Ren S, Wang Y, Xiao X. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circ Res. 2011;109:1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ota A, Zhang J, Ping P, Han J, Wang Y. Specific regulation of noncanonical p38α activation by hsp90‐cdc37 chaperone complex in cardiomyocyte. Circ Res. 2010;106:1404–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhan A, Sirker A, Zhang J, Protti A, Catibog N, Driver W, Botnar R, Monaghan MJ, Shah AM. High‐frequency speckle tracking echocardiography in the assessment of left ventricular function and remodeling after murine myocardial infarction. Am J Physiol Heart Circ Physiol. 2014;306:H1371–H1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schnelle M, Catibog N, Zhang M, Nabeebaccus AA, Anderson G, Richards DA, Sawyer G, Zhang X, Toischer K, Hasenfuss G, Monaghan MJ, Shah AM. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J Mol Cell Cardiol. 2017;114:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hernández‐Alvarez MI, Díaz‐Ramos A, Berdasco M, Cobb J, Planet E, Cooper D, Pazderska A, Wanic K, O'Hanlon D, Gomez A. Early‐onset and classical forms of type 2 diabetes show impaired expression of genes involved in muscle branched‐chain amino acids metabolism. Sci Rep. 2017;7:13850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olson KC, Chen G, Lynch CJ. Quantification of branched‐chain keto acids in tissue by ultra fast liquid chromatography–mass spectrometry. Anal Biochem. 2013;439:116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bauer M, Cheng S, Jain M, Ngoy S, Theodoropoulos C, Trujillo A, Lin FC, Liao R. Echocardiographic speckle‐tracking based strain imaging for rapid cardiovascular phenotyping in mice. Circ Res. 2011;108:908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ternacle J, Berry M, Alonso E, Kloeckner M, Couetil JP, Rande JL, Gueret P, Monin JL, Lim P. Incremental value of global longitudinal strain for predicting early outcome after cardiac surgery. Eur Heart J Cardiovasc Imaging. 2013;14:77–84. [DOI] [PubMed] [Google Scholar]
- 27. Adamo L, Perry A, Novak E, Makan M, Lindman BR, Mann DL. Abnormal global longitudinal strain predicts future deterioration of left ventricular function in heart failure patients with a recovered left ventricular ejection fraction. Circ Heart Fail. 2017;10:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang J, Khoury DS, Thohan V, Torre‐Amione G, Nagueh SF. Global diastolic strain rate for the assessment of left ventricular relaxation and filling pressures. Circulation. 2007;115:1376–1383. [DOI] [PubMed] [Google Scholar]
- 29. Ersboll M, Andersen MJ, Valeur N, Mogensen UM, Fakhri Y, Thune JJ, Moller JE, Hassager C, Sogaard P, Kober L. Early diastolic strain rate in relation to systolic and diastolic function and prognosis in acute myocardial infarction: a two‐dimensional speckle‐tracking study. Eur Heart J. 2014;35:648–656. [DOI] [PubMed] [Google Scholar]
- 30. Flachskampf FA, Biering‐Sorensen T, Solomon SD, Duvernoy O, Bjerner T, Smiseth OA. Cardiac imaging to evaluate left ventricular diastolic function. JACC Cardiovasc Imaging. 2015;8:1071–1093. [DOI] [PubMed] [Google Scholar]
- 31. Park TH, Nagueh SF, Khoury DS, Kopelen HA, Akrivakis S, Nasser K, Ren G, Frangogiannis NG. Impact of myocardial structure and function postinfarction on diastolic strain measurements: implications for assessment of myocardial viability. Am J Physiol Heart Circ Physiol. 2006;290:H724–H731. [DOI] [PubMed] [Google Scholar]
- 32. Park SM, Miyazaki C, Prasad A, Bruce CJ, Chandrasekaran K, Rihal C, Bell MR, Oh JK. Feasibility of prediction of myocardial viability with doppler tissue imaging following percutaneous coronary intervention for ST elevation anterior myocardial infarction. J Am Soc Echocardiogr. 2009;22:183–189. [DOI] [PubMed] [Google Scholar]
- 33. Hoffmann R, Altiok E, Nowak B, Kuhl H, Kaiser HJ, Buell U, Hanrath P. Strain rate analysis allows detection of differences in diastolic function between viable and nonviable myocardial segments. J Am Soc Echocardiogr. 2005;18:330–335. [DOI] [PubMed] [Google Scholar]
- 34. Brand K. Metabolism of 2‐oxoacid analogues of leucine, valine and phenylalanine by heart muscle, brain and kidney of the rat. Biochim Biophys Acta. 1981;677:126–132. [DOI] [PubMed] [Google Scholar]
- 35. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turer AT, Stevens RD, Bain JR, Muehlbauer MJ, van der Westhuizen J, Mathew JP, Schwinn DA, Glower DD, Newgard CB, Podgoreanu MV. Metabolomic profiling reveals distinct patterns of myocardial substrate use in humans with coronary artery disease or left ventricular dysfunction during surgical ischemia/reperfusion. Circulation. 2009;119:1736–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC); European Association for Cardio‐Thoracic Surgery (EACTS) , Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Barón‐Esquivias G, Baumgartner H, Borger MA, Carrel TP, De Bonis M. Guidelines on the management of valvular heart disease (version 2012) the joint task force on the management of valvular heart disease of the European Society of Cardiology (ESC) and the European Association for Cardio‐Thoracic Surgery (EACTS). Eur Heart J. 2012;33:2451–2496.22922415 [Google Scholar]
- 38. McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez‐Sanchez MA. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur J Heart Fail. 2012;14:803–869. [DOI] [PubMed] [Google Scholar]
- 39. Cottrell C, Kirkpatrick JN. Echocardiographic strain imaging and its use in the clinical setting. Expert Rev Cardiovasc Ther. 2010;8:93–102. [DOI] [PubMed] [Google Scholar]
- 40. Sengupta PP, Korinek J, Belohlavek M, Narula J, Vannan MA, Jahangir A, Khandheria BK. Left ventricular structure and function: basic science for cardiac imaging. J Am Coll Cardiol. 2006;48:1988–2001. [DOI] [PubMed] [Google Scholar]
- 41. MacIver DH. The relative impact of circumferential and longitudinal shortening on left ventricular ejection fraction and stroke volume. Exp Clin Cardiol. 2012;17:5. [PMC free article] [PubMed] [Google Scholar]
- 42. MacIver DH, Adeniran I, Zhang H. Left ventricular ejection fraction is determined by both global myocardial strain and wall thickness. IJC Heart Vasc. 2015;7:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia FA. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid x receptor‐α in pacing‐induced heart failure. Circulation. 2002;106:606–612. [DOI] [PubMed] [Google Scholar]
- 44. Brigadeau F, Gelé P, Wibaux M, Marquié C, Martin‐Nizard F, Torpier G, Fruchart J‐C, Staels B, Duriez P, Lacroix D. The PPARα activator fenofibrate slows down the progression of the left ventricular dysfunction in porcine tachycardia‐induced cardiomyopathy. J Cardiovasc Pharmacol. 2007;49:408–415. [DOI] [PubMed] [Google Scholar]
- 45. Wayman NS, Hattori Y, McDonald MC, Mota‐Filipe H, Cuzzocrea S, Pisano B, Chatterjee PK, Thiemermann C. Ligands of the peroxisome proliferator‐activated receptors (PPAR‐γ and PPAR‐α) reduce myocardial infarct size. FASEB J. 2002;16:1027–1040. [DOI] [PubMed] [Google Scholar]
- 46. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kolwicz SC Jr, Olson DP, Marney LC, Garcia‐Menendez L, Synovec RE, Tian R. Cardiac‐specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure‐overload hypertrophy. Circ Res. 2012;111:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, Dungan J, Newby LK, Hauser ER, Ginsburg GS, Newgard CB, Kraus WE. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet. 2010;3:207–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. Left ventricular posterior wall thickness of sham or TAC mice treated with vehicle or BT2.
Table S2. Heart rate of sham or TAC mice treated with vehicle or BT2.
Figure S1. Absolute values of echocardiographic parameters before and after BT2 treatments in pressure‐overloaded mouse hearts. Average left ventricular fractional shortening (FS) (A) and ejection fraction (EF) (B) for both groups were presented at indicated time points. Structural measurements of LV internal diameters (LVID;s) (C) and LV volume (LV Vol;s) (D) were presented at indicated time points. Two‐way ANOVA followed by Bonferroni's post‐hoc test was performed to evaluate the differences between groups over the treatment period (from week 3 to week 8). *P<0.05. Data was presented as mean±SEM.
Figure S2. BT2 treatment led to a decrease of BCKDE1α phosphorylation in NRVM. (A) Schematic timeline of primary cardiomyocytes study. NRVM was pretreated with PE (100 μmol/L) for 24 h, and then further exposed to DMSO or BT2 (80 μmol/L) for additional 24 h. Cells were collected and proceeded for further measurements. (B) At day 3, NRVM were collected and expression of total, phosphorylated BCKD subunit E1α, and GAPDH protein was determined by Western blotting. PE indicates phenylephrine; NRVM, neonatal rat ventricular myocytes.