

Abstract
Background
Hypertension may be associated with renal cellular injury. Cells in distress release extracellular vesicles (EVs), and their numbers in urine may reflect renal injury. Cellular senescence, an irreversible growth arrest in response to a noxious milieu, is characterized by release of proinflammatory cytokines. We hypothesized that EVs released by senescent nephron cells can be identified in urine of patients with hypertension.
Methods and Results
We recruited patients with essential hypertension (EH) or renovascular hypertension and healthy volunteers (n=14 each). Renal oxygenation was assessed using magnetic resonance imaging and blood samples collected from both renal veins for cytokine‐level measurements. EVs isolated from urine samples were characterized by imaging flow cytometry based on specific markers, including p16 (senescence marker), calyxin (podocytes), urate transporter 1 (proximal tubules), uromodulin (ascending limb of Henle's loop), and prominin‐2 (distal tubules). Overall percentage of urinary p16+ EVs was elevated in EH and renovascular hypertension patients compared with healthy volunteers and correlated inversely with renal function and directly with renal vein cytokine levels. Urinary levels of p16+/urate transporter 1+ were elevated in all hypertensive subjects compared with healthy volunteers, whereas p16+/prominin‐2+ levels were elevated only in EH versus healthy volunteers and p16+/uromodulin+ in renovascular hypertension versus EH.
Conclusions
Levels of p16+ EVs are elevated in urine of hypertensive patients and may reflect increased proximal tubular cellular senescence. In EH, EVs originate also from distal tubules and in renovascular hypertension from Henle's loop. Hence, urinary EVs levels may be useful to identify intrarenal sites of cellular senescence.
Keywords: extracellular vesicles, hypertension, renal artery stenosis, renovascular hypertension, senescence
Subject Categories: Biomarkers, High Blood Pressure, Hypertension, Magnetic Resonance Imaging (MRI), Vascular Disease
Clinical Perspective
What Is New?
Analysis of urinary extracellular vesicles as an index of tissue or organ pathology is a novel, noninvasive, clinically applicable technique.
Our work showed that urinary extracellular vesicles from patients with hypertension express markers of cellular senescence.
What Are the Clinical Implications?
Increased numbers of p16+ extracellular vesicles in the urine of hypertensive patients may reflect cellular processes and serve as an early marker of cellular senescence along the nephron.
Early detection of cellular senescence may help guide disease management and monitor success of therapy.
Further studies are needed to elucidate the mechanisms by which the extent of cellular senescence changes in the different segments of the nephron and its implications for renal outcomes.
Hypertension is often associated with chronic kidney disease and constitutes an independent predictor of chronic kidney disease progression,1 particularly in patients with preexisting renal disease. Glomerular and vascular stretching, compounded by activation of the renin‐angiotensin‐aldosterone system, may cause arteriolosclerosis, inadequate renal blood flow, tubular atrophy, interstitial fibrosis, and glomerular alterations. Renal artery stenosis is a common cause of renovascular hypertension (RVH) and, most frequently, induced by atherosclerosis. A fall in renal blood flow elicits hemodynamic and humoral responses in the poststenotic kidney, including activation of renin‐angiotensin‐aldosterone system and tissue hypoxia, which are associated with cell death and recruitment of inflammatory mechanisms.2
Cellular senescence is an irreversible growth arrest that occurs in response to exposure to potentially oncogenic and other noxious stimuli and has been frequently linked to aging.3 The cyclin‐dependent kinase (Cdk) inhibitor, p16Ink4a (p16), is a key mediator of cellular senescence and aging and has been shown to be upregulated in several diseases involving senescence.4, 5, 6
Senescent cells also develop senescence‐associated secretory phenotype (SASP), a secretome characterized by release of proinflammatory cytokines, including interleukin (IL)‐6, monocyte chemoattractant protein (MCP)‐1, and plasminogen activator inhibitor‐1, which may exert detrimental effects on nearby cells. For example, SASP of senescent endothelial cells may foster vascular calcifications, whereas senescent osteoblasts promote development of osteoporosis and arthritis.3 Cellular senescence has also been implicated in development of renal disease7 and may contribute to development of renal injury in hypertension secondary to barotrauma, activation of renin‐angiotensin‐aldosterone system,8 or other humoral mediators. In addition, renal ischemia distal to renal artery stenosis triggers inflammatory and pro‐oxidant mechanisms and might also lead to cellular senescence and progression to chronic injury.7 However, the identity or tubular location of cells undergoing senescence along the nephron have not been elucidated.
Extracellular vesicles (EVs) are released into body fluids by most cell types, especially under stress conditions,9 and are characterized by their protein, DNA, and RNA content, as well as by cell‐surface markers specific to their cells of origin.10 Previous studies have shown that renal injury, such as that incurred in preeclampsia11 or polycystic kidney disease,12 is reflected in increased numbers of urinary exosomes bearing specific markers. However, whether urinary levels of EVs reflecting renal cellular senescence are elevated in human subjects with hypertension remains unknown.
The aim of the present study was to evaluate and characterize renal cellular senescence‐related EVs in urine of patients with hypertension and relatively preserved renal function, specifically RVH compared with essential hypertension (EH). We tested the hypothesis that the numbers of urinary p16+ EVs would be elevated in hypertensive patients, in association with markers indicating their cellular source along the nephron, which might differ between hypertensive versus ischemic kidney injury.
Methods
The data that support the findings of this study are available from the first author or corresponding author upon reasonable request.
Patient Population
Healthy volunteers (HVs) and patients with EH or RVH (n=14 each) were prospectively enrolled after a written consent was obtained and approval by the Institutional Review Board of the Mayo Clinic. The 3 groups had similar mean age, sex, and body mass index, as shown in Table 1. Inclusion criteria for hypertensive patients included blood pressure ≥140/90 mm Hg, previous diagnosis of hypertension, and/or current use of antihypertensive medications. In patients with RVH, presence of a stenosis was confirmed by Doppler ultrasound (blood velocity, >300 cm/s) and quantitative computed tomography angiographic measurements (>60% vessel occlusion).2 A total of 8 RVH patients had bilateral disease. In these patients, the kidney with the more‐severe stenosis was studied. Exclusion criteria included serum creatinine >2.5 mg/dL, severe hypertension (systolic blood pressure, >180 mm Hg), diabetes mellitus requiring medications, recent cardiovascular event (myocardial infarction, stroke, or congestive heart failure within 6 months), pregnancy, and kidney transplant. HVs enrolled had office blood pressure ≤130/80 mm Hg (arguing against white‐coat hypertension), no history of hypertension or cardiovascular disease, and no antihypertensive or lipid‐lowering drug treatment.13, 14
Table 1.
Demographics of Hypertensive Patients and HVs Included in the Study
| Parameter | HV | EH | RVH |
|---|---|---|---|
| Demographics | |||
| No. of patients | 14 | 14 | 14 |
| Age, y | 72.3 (64–79) | 69.9 (64–78) | 70 (58–79) |
| Sex (female/male) | 8/6 | 7/7 | 9/5 |
| BMI, kg/m2 | 25.3±3.4 | 27.3±4.3 | 29.1±3.95 |
| Renal function | |||
| Creatinine, mg/dL | 0.95 (0.7–1.2) | 1.0 (0.5–1.5) | 1.2 (0.7–1.9) |
| eGFR, mL/min/1.73 m2 | 80.5±10.4 | 72.4±18.2 | 62.8±21.6b |
| Related laboratory measures | |||
| Urine protein, mg/mLa | 96.1 (4–290) | 67 (29–213) | 86.4 (26–255) |
| Mean blood pressure, mm Hg | 87.8±7.9 | 91.3±13.1 | 92.1±9.4 |
| Systolic blood pressure, mm Hg | 120.4±9.3 | 136.6±18.2b | 135.6±20.4b |
| Diastolic blood pressure, mm Hg | 71.4±8.6 | 68.6±13.9 | 70.4±8.1 |
| Total cholesterol, mg/dL | 175.4±30.5 | 178.4±33.7 | 174.8±38.4 |
| Triglycerides, mg/dL | 116.1 (51–207) | 128.9 (85–201) | 127.1 (73–249) |
| HDL, mg/dL | 60.1 (42–88) | 48.7 (35–79) | 52.2 (34–79) |
| LDL, mg/dL | 97.2±28.9 | 103.9±25.2 | 97.1±30.9 |
| Renal oxygenation | |||
| RFH (%R2* >30 1/s) | 9 (1.20–26.25) | 20.5 (4.60–36.05)c | |
| Renal cortical R2* (1/s) | 18.2 (16.45–20.35) | 21.5 (17.3–30.4)c | |
BMI indicates body mass index; eGFR, estimated glomerular filtration rate; EH, essential hypertension; HDL, high‐density lipoprotein; HVs, healthy volunteers; LDL, low‐density lipoprotein; RBF, renal blood flow; RFH, renal fractional hypoxia; RVH, renovascular hypertension.
Spot urine samples were collected in HVs and 24 hours in EH and RVH.
P<0.05 vs HVs.
P<0.05 vs EH.
EH and RVH patients participated in a 3‐day inpatient protocol carried out in the clinical research unit on 2 occasions (3 months apart), as previously described,15 where dietary sodium intake was maintained at 150 mEq/day. EH and RVH patients were all treated with antihypertensive therapy (diuretics, angiotensin‐converting enzyme inhibitors, or angiotensin receptor blockers at usual recommended daily dose).13
On the first study day, measures of clinical and laboratory parameters were collected, including demographics, arterial pressure (average of 3 measurements), serum creatinine, proteinuria, and lipid profile. Estimated glomerular filtration rate (eGFR) was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) formula. Urine was collected over a 24‐hour period.
On day 2, renal oxygenation was assessed by blood oxygen level–dependent magnetic resonance imaging on a 3.0 Tesla system (Twin Speed Signa EXCITE; GE Medical Systems, Waukesha, WI). Intrarenal hypoxia was expressed as R2*, an index of blood and tissue deoxyhemoglobin concentration, and fractional renal hypoxia (which reflects mostly the medulla) as the percentage of kidney voxels with R2* values >30/s.15, 16 R2* was measured in the stenotic kidney of RVH and 1 (randomly selected) kidney of EH and HV.
On the third day of the inpatient protocol, the common femoral vein was cannulated with a 6F sheath and blood samples drawn from the right and left renal veins (RVs) with a 5F pigtail Cobra catheter (Cook, Bloomington, IN) for venous levels of cytokines, neutrophil gelatinase‐associated lipocalin (NGAL) and plasma renin activity.17
Humoral Measurements
Levels of the inflammatory cytokines, soluble vascular cell adhesion molecule‐1, interferon‐γ, IL‐6, and tumor necrosis factor (TNF)‐α, MCP1, soluble E‐selectin, and the anti‐inflammatory cytokine, IL‐10, were measured on samples obtained from the right and left RV of all hypertensive patients by Luminex (Millipore, Burlington, MA).14, 18 In HVs, antecubital blood samples were collected.
NGAL (ng/mL) was tested by ELISA, according to the manufacturer's protocol (Cat# KIT 036; BioPorto Diagnostics, Hellerup, Denmark), in 10 μL of RV, inferior vena cava plasma, and urine samples diluted 1:500. Final volumes of 100 μL were used for assay. Urinary kidney injury molecule‐1 (KIM‐1) levels were evaluated by ELISA using a commercially available kit (Cat# DKM100; R&D Systems, Minneapolis, MN). Urinary NGAL and KIM‐1 were expressed as nanogram/milligram of urinary creatinine.14
Serum creatinine, eGFR, total proteins, low‐density lipoprotein, high‐density lipoprotein, total cholesterol, and triglyceride levels were determined by standard procedures.14 Urine samples were collected for 24 hours in all hypertensive patients, whereas spot urine samples were obtained from HVs. Samples were stored at −80°C until measurement.13
EV Isolation and Characterization
EVs were isolated from whole urine using Total Exosome Isolation reagent (Invitrogen, Waltham, MA), following the manufacturer's guidelines. Urine samples were centrifuged at 2000g for 30 minutes at 4°C to remove cells and debris. Supernatants were mixed with 1 volume of the Total Exosome Isolation reagent and incubated for 1 hour at room temperature. After incubation, samples were centrifuged at 10 000g for 1 hour at 4°C. Pelleted exosomes were resuspended in PBS.
After marking the sample (20 μL) with tag‐it‐violet (TIV), a proliferation and cell‐tracking dye19 (0.5 μL, Cat #425101, dilution 40:1; BioLegend, San Diego, CA), it was vortexed protected from light for 2 hours at 37°C. Once completed, FcR Blocking Reagent human (10 μL, Cat# 130‐059‐901; Miltenyi Biotec, Bergisch Gladbach, Germany) was added and allowed to stand for 10 minutes at room temperature, after which antibodies were added. We added antibodies against the SASP mediator, MCP120 (0.8 μL, Cat#505904, 25:1; BioLegend), senescence marker21 p16INK4a (2 μL, Cat#NB200‐174PE, 10:1; Novus Biologicals, Centennial, CO), urate transporter 1 (URAT‐1) for proximal tubule20 (3 μL, Cat#LS‐C446089‐100; LifeSpan Biosciences, Seattle, WA), the podocyte marker, podocalyxin22 (2 μL, Cat#AF1658; R&D Systems), the distal tubule marker, prominin‐223 (Prom‐2; 2 μL, Cat#MAB2024; R&D Systems), or uromodulin20 (Umod; 0.8 μL, Cat#H00007369‐B01P; Novus Biologicals) that is expressed on the ascending limb of the loop of Henle. After labeling, the sample was vortexed for 1 hour at room temperature.
The FlowSight Imaging Flow Cytometer (Amnis Corporation, Seattle, WA) was used to detect EVs by size and (TIV) fluorescence. Using the Submicron Bead Calibration Kit (Bangs Laboratories, Fishers, IN) at 0.05, 0.1, 0.2, 0.5, and 0.8 μm, the gates for EV size were defined.24 Acquisition gates for EVs are shown in Figure 1A. TIV served to identify EVs, and MCP1, p16, Umod, Podocalyxin, Prom‐2, and URAT‐1 antibodies to identify the source of senescent‐cell–derived (P16+/MCP1±) EVs. Expression of MCP1 on P16+ EVs served to suggest SASP in addition to cell‐cycle arrest propensity. Gates were selected starting from the TIV+ population, after running a sample of unlabeled vesicles and comparing with a single‐stained sample for each antibody. Imaging flow cytometry corroborated the gateways made by observing the running events. At least 50 000 events were acquired for each sample from the gate created in the graph formed by Y=Area TIV and X=Intensity. The percentage of EVs positive for p16, double‐positive for p16 and a nephron marker, or also positive for the SASP marker, MCP1, were calculated out of all TIV EVs.
Figure 1.
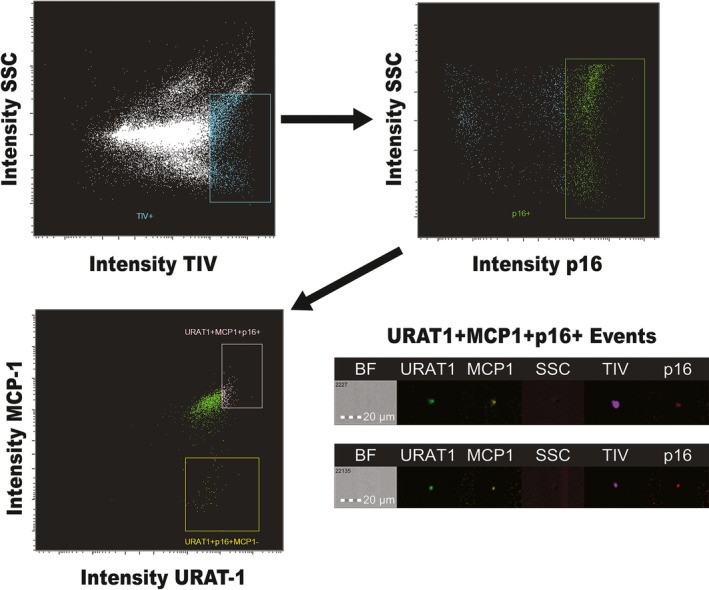
Gating strategy for analysis of extracellular vesicles (EVs). Samples were marked with tag‐it‐violet (TIV) to identify EVs. Anti‐MCP1, p16, uromodulin, podocalyxin, prominin‐2, and URAT‐1 antibodies were added together. Gates were selected starting from the TIV + population, after running a sample of unlabeled vesicles and compared with a single‐stained sample for each antibody. Imaging flow cytometry corroborated the gateways made by observing the running events. BF indicates bright field; MCP1, monocyte chemoattractant protein‐1; SSC, side scatter; URAT‐1, urate transporter 1.
Statistical Analysis
Statistical analysis was performed using the JMP Pro software package (version 13.0; SAS Institute Inc, Cary, NC). Results are expressed as mean±SD for normally distributed data and as median (range) for data not showing normal distribution. Normally distributed data were first compared using ANOVA, with Turkey's post‐hoc test. Non‐normally distributed data were compared using nonparametric tests (Wilcoxon and Kruskal–Wallis) with the Stell–Dawss post‐hoc test. Spearman's correlation and simple linear regression analysis were used for comparisons of EV data, clinical data, and magnetic resonance imaging. Significance was accepted at P<0.05.
Results
Table 1 summarizes the clinical, laboratory, and demographic data of the study participants. No significant differences were found among the groups in age, sex, body mass index, or creatinine levels, although eGFR was lower in RVH compared with HVs. Systolic blood pressure was higher in both groups compared with HVs (both P<0.008), but diastolic and mean blood pressure, cholesterol fractions, and urine protein levels did not differ among groups. Renal tissue oxygenation indices fractional renal hypoxia (%R2* >30/s) and cortical R2* were higher in the stenotic kidney of RVH compared with the EH kidney (P<0.008 and <0.005, respectively).
Markers of Senescence in EV Derived From Different Cells Along the Nephron
No differences were found in overall percentage of urinary EVs out of all events among groups (Table 2). Contrarily, the percentage of p16+ urinary EVs was elevated in EH and RVH in comparison with HV (P<0.002 and <0.04, respectively) and similar between EH and RVH (P=0.2). No significant differences were observed among groups in podocalyxin+ EVs.
Table 2.
Urinary EV Representing Different Nephron Segments in Hypertensive Patients and HVs
| EV Parameter | HV | EH | RVH |
|---|---|---|---|
| Count, TIV+ | 25 389 (10 573–41 903) | 23 689 (11 656–32 585) | 17 639 (11 317–34 723) |
| %, TIV+ | 50.8 (21.4–83.8) | 47.4 (23.3–65.2) | 35.3 (22.6–69.5) |
| %, p16+ | 18.6±7.7 | 27.9±8.4a | 24.6±6.5a |
| Podocalyxin | |||
| %, Podxl+p16+ | 0.035 (0.01–0.07) | 0.046 (0.01–0.13) | 0.039 (0.01–0.11) |
| %, Podxl+p16+MCP1+ | 0.41 (0.03–1.49) | 0.57 (0.02–2.15) | 0.51 (0.03–1.74) |
| URAT1 | |||
| %, URAT1+p16+ | 7.6±5.2 | 31.3±20.6a | 40.2±17.6a |
| %, URAT1+p16+MCP1+ | 15.4±17.3 | 21.7±22.7a | 28.9±14.6a |
| Uromodulin | |||
| %, Umod+p16+ | 7.7 (0.9–16.2) | 15.1 (1.3–36.6) | 36.5 (20.3–43.9)a, b |
| %, Umod+p16+MCP1+ | 2.3 (0.12–10.87) | 6.6 (0.73–22.40) | 9.3 (0.56–28.40)a |
| Prominin2 | |||
| %, Prom2+p16+ | 2.32 (0.32–7.80) | 5.9 (1.1–22.7)a | 3.5 (0.8–7.6) |
| %, Prom2+p16+MCP1+ | 3.0 (0.5–9.3) | 4.6 (1.5–11.4) | 5.6 (1.3–10.6)a |
EH indicates essential hypertension; HVs, healthy volunteers; MCP, monocyte chemoattractant protein; Podxl, podocalyxin; Prom2, prominin‐2; RVH, renovascular hypertension; TIV, tag‐it‐violet; Umod, uromodulin; URAT‐1, urate transporter 1.
P<0.05 vs HV.
P<0.05 vs EH.
Percentages of urinary EVs double positive for the proximal tubule marker, URAT‐1, and for p16, as well as those triple positive for MCP1, were higher in both EH and RVH patients compared with HV (P<0.02 and <0.001; Table 2). EVs positive for both p16 and Umod, a marker of the ascending limb of Henle's loop, were elevated only in RVH urine compared with HVs (P<0.0024) and EH (P<0.012), and the percentage of Umod+/p16+/MCP1+ EVs only in RVH compared with HVs (P<0.037). The percentage of Prom‐2+/p16+ EVs was higher only in EH compared to HVs (P<0.042), whereas the percentage of Prom‐2+/p16+/MCP1+ EVs was higher only in RVH compared with HVs (P<0.021; Table 2).
Relationship of Urinary EVs With Cytokine Levels
Levels of the inflammatory cytokines, soluble vascular cell adhesion molecule‐1, interferon‐γ, IL‐6, TNF‐α, MCP1, and soluble E‐selectin, were elevated in the stenotic kidney RVH‐RV compare with EH‐RV and with systemic HV plasma, whereas anti‐inflammatory cytokine IL‐10 levels were lower (Table 3).
Table 3.
Cytokine Levels and NGAL in Peripheral Blood of HVs and Renal Vein of EH and RVH Patients
| HV | EH | RVH | |
|---|---|---|---|
| NGAL, ng/mL | 9.67 (7.98–14.39) | 13.24 (5.55–28.50) | 15.92 (11.6–25.8) |
| KIM‐1, ng/mL | 0.10 (0.00–0.72) | 0.24 (0.03–0.47) | 0.38 (0.16–0.97) |
| sVCAM‐1, ng/mL | 470.5 (341.4–645.7) | 651.9 (517.8–805.2) | 1060.2 (977.4–1242.3)a, b |
| IF‐γ, pg/mL | 3.7 (2.6–5.4) | 3.8 (3.9–3.9) | 7.4 (6.6–8.2)a, b |
| IL‐6, pg/mL | 1.2 (1.2–2.0) | 1.5 (1.5–2.2) | 4.9 (3.2–6.3)a, b |
| TNF‐α, pg/mL | 3.5 (2.7–4.8) | 3.7 (2.8–4.8) | 5.8 (4.5–7.1)a, b |
| MCP1, pg/mL | 123.1 (97.2–140.8) | 121.2 (93.1–139.7) | 167.3 (146.2–192.4)a, b |
| sE‐selectin, ng/mL | 7.6 (3.6–12.9) | 4.7 (3.9–17.4) | 32.5 (10.9–39.1)a, b |
| IL‐10, pg/mL | 2.8 (2.6–2.8) | 2.4 (1.8–3.8) | 1.4 (0.9–2.1)a, b |
Urinary KIM‐1. EH indicates essential hypertension; HVs, healthy volunteers; IF, interferon; IL, interleukin; KIM‐1, kidney injury molecule‐1; MCP, monocyte chemoattractant protein; NGAL, neutrophil gelatinase‐associated lipocalin; RVH, renovascular hypertension; sE‐selectin, soluble E‐selectin; sVCAM, soluble vascular cell adhesion molecule; TNF, tumor necrosis factor.
P<0.05 vs HVs.
P<0.05 vs EH.
The percentage of p16+ urinary EVs correlated inversely with eGFR (Figure 2A), but directly with levels of soluble vascular cell adhesion molecule‐1 (Figure 2B) and MCP1 (Figure 2C) in the stenotic RV. No correlation was observed with levels of interferon‐γ/TNF‐α, soluble E‐selectin, IL‐6, or IL‐10 levels, with blood pressure or renal hypoxia. Only MCP1 and IL‐6 were also correlated with the number of p16+ EVs when RVH patients were considered alone (R 2=0.35, P=0.027 and R 2=0.41, P=0.013, respectively) and only MCP1 when both hypertensive groups were considered together (R 2=0.14, P=0.0487 and R 2=0.12, P=0.071, respectively).
Figure 2.
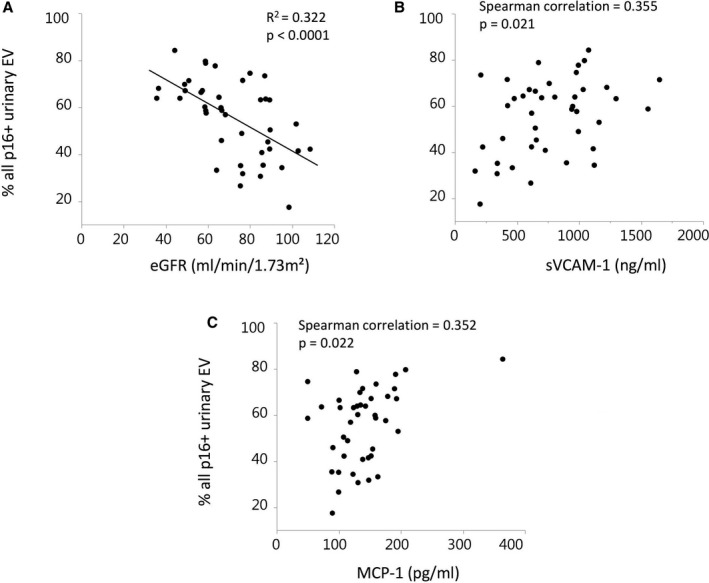
Correlations percentage of p16+ urinary extracellular vesicles (EVs) with renal function and cytokine levels in renal vein (RV) plasma and antecubital blood samples in HVs. The percentage of p16+ EVs correlated inversely with estimated glomerular filtration rate (eGFR; A) and directly with RV levels of soluble vascular cell adhesion molecule (sVCAM)‐1 (B) and monocyte chemoattractant protein (MCP)‐1 (C). HVs indicates healthy volunteers.
Discussion
This study shows that EVs bearing markers of cellular senescence are released from renal cells, detectable in urine, and correlate with renal dysfunction and renal venous plasma levels of cytokines in hypertensive human subjects. Specifically, levels of urinary EVs bearing proximal and distal tubular cell markers are elevated in EH, whereas uromodulin+ EVs from the ascending limb of Henle's loop are elevated only in patients with RVH. This observation suggests that hypertension elicits an increase in release from renal cells of urinary p16+ EVs, which might reflect increased cellular senescence in specific tubular segments.
Senescence is a potent mechanism to curtail malignancies by permanently withdrawing from the cell cycle, but has also been implicated as a driver of aging and age‐related disease.25 Senescent cells may develop SASP, which involves increased expression and release of proinflammatory cytokines, like MCP1 and TNF‐α, that impact nearby cells.25 Therefore, besides the canonical index p16, elevated MCP1 expression may serve as a marker of consolidated cellular senescence.5, 6
In RVH, low perfusion in ischemic kidneys results in development of renal hypoxia26 and damage to proximal and outer medullary tubular cells.27 In turn, development of RVH and an increase in glomerular pressure may potentially cause glomerular and tubulo‐interstitial damage in the contralateral kidney, similar to barotrauma that might be caused by EH.2 For example, early hypertension induced by deoxycorticosterone acetate‐salt feeding elicits in rats proximal and distal tubular atrophy and dilation, which correlates spatially with cellular senescence.8 Conversely, tubular damage might contribute to development of hypertension through alterations in renal sodium reabsorption.28, 29 Therefore, subtle kidney damage in EH and RVH may reflect an increase in senescent cells in specific locations within the kidney30 and, in turn, release of p16+ urinary EVs carrying cellular signatures.
Urinary EVs have been identified in urine20, 31 and characterized by flow cytometry,32 but few markers of cellular senescence are available for detection by flow cytometry. Previous studies have shown that EVs carrying tubular cell signatures can distinguish aging or stone‐forming kidneys.20, 33 The cell‐cycle regulator, p16, is a pivotal mediator and marker of cellular senescence. Whereas cellular p16 is an intracellular protein, many EV membrane proteins are present in a topologically reversed orientation attributed to the unique “inside‐out” topology of EVs and can therefore be identified on their surface.34 Previous studies have shown that EVs obtained using the same kit are mostly between 30 and 100 nm in size and express tumor susceptibility 101 (TSG101) and CD9 proteins that characterize exosomes, supporting their consideration as exosomes.35, 36
In this study, the overall number of urinary EVs was similar among groups, yet the number of p16+ EVs was higher in both hypertensive groups compared with HVs, which might reflect increased renal cellular senescence. The functional significance of this observation is underscored by the finding that the percentage of EVs p16+ found in the urine correlated inversely with eGFR. Hence, renal damage associates with lower renal function and a greater proportion of cells in senescence that, in turn, release EVs carrying the senescence marker, p16.
In concordance with the increased inflammation and senescence in the stenotic kidney, levels of SASP and inflammatory cytokines were elevated in RVH‐RV compared with EH‐RV and with systemic HV plasma, whereas the anti‐inflammatory cytokine, IL‐10, was downregulated, as previously shown.14, 37 Furthermore, p16+ EV levels in hypertensive patients correlated directly with RV levels of soluble vascular cell adhesion molecule‐1 and MCP1. These findings are consistent with the speculation that p16+ EV levels in urine are linked to kidney damage in hypertensive patients and underscore the inflammation. Notably, the lack of correlation of p16+ EVs with other cytokines and SASP mediators might be attributable to marked cytokine release by sources other than senescent cells, such as inflammatory cells. Interestingly, blood pressure levels were not elevated in RVH‐RV compared with EH‐RV, suggesting that inflammatory cytokines may not predominantly regulate blood pressure in RVH.
An increase in podocyte‐specific EVs is viewed as a marker of direct glomerular injury.38 We have also previously shown that RVH patients show elevated levels of urinary podocyte‐derived EVs compared with patients with EH.19 However, no differences were observed among the groups in levels of p16+/podocalyxin+ or p16+/podocalyxin+/MCP‐1+ EVs in this study. These findings argue against significant podocyte senescence or shedding of senescent‐podocyte EVs associated with glomerular damage in these patients with relatively preserved renal function.
Our study suggests that in hypertensive patients, p16+ EVs originate from the proximal convoluted tubule, because they were also positive for URAT‐1. This member of the organic anion transporter family is an anion‐exchanging uptake transporter localized to the apical (brush border) membrane of renal proximal tubular cells.39 The similar increase in p16+/URAT‐1+ urinary EVs in both the EH and RVH groups may link senescence of proximal tubular cells to hypertension. Similarly, release of urinary markers of proximal tubular cells is increased in patients with severe preeclampsia and hypertension.40 Furthermore, URAT‐1+/MCP1+ EVs were also elevated in both EH and RVH, underscoring development of the senescent phenotype and SASP in proximal tubular cells.
An increase in the percentage of p16+ EVs bearing Umod was found only in patients with RVH. Uromodulin (Tamm–Horsfall protein) is exclusively produced in the kidney and is the most abundant protein in normal urine.41 Presumably, the source of these p16+/Umod+ EVs was the ascending limb of Henle's loop. Given the pronounced susceptibility of the renal medulla to hypoxia, this may reflect hypoxic injury in this area of the tubule. Indeed, we found a 2‐fold increase in stenotic‐kidney medullary hypoxia (detected by fractional renal hypoxia) in patients with RVH, whereas cortical hypoxia R2* was less markedly elevated. Yet, the lack of correlation between p16+/uromodulin+ EV levels and renal hypoxia suggests that the relationship between them is complex.
In addition, compared with HVs, we observed elevated levels of p16+/Prom‐2+ urinary EVs in patients with EH, but not in patients with RVH. Prominin‐2 is an integral membrane glycoprotein expressed especially in the distal tubule and shed in urinary EVs.23 Therefore, distal tubular alterations may be more specific to renal damage in patients with EH. Interestingly, urine of patients with RVH contained larger fractions of p16+/prominin‐2+/MCP‐1+ EVs than in EH or HVs, which might suggest some distal tubular cell senescence in these patients. However, we cannot rule out the possibility that in patients with RVH, dilution of the EVs by the stenotic kidney urine reduced our ability to identify distal tubular alterations in the contralateral kidney, which is exposed to RVH.
Limitations
This study was limited by its small size consequent to limited sample availability. Nonsenescent inflammatory cells may also express p16 transiently.42 Yet, coexpression of tubular markers on these EVs is consistent with tubular cell origin, although whether they contribute to, or result from, the pathophysiology of hypertensive kidney injury could not be determined. Urinary EVs are considered to derive from every epithelial cell cells facing the urinary space.43 Nevertheless, URAT‐1 is also expressed (albeit at lower levels) in other organs, podocalyxin in some vascular endothelium cells’ surface, and prominin‐2 in others’ epithelial tissues, and we cannot rule out the presence of some non‐kidney‐derived EVs in urine. Uromodulin is exclusively synthesized in the human kidney, but may also be expressed on the early distal convoluted tubule. Therefore, the cellular origin of p16+ EVs needs to be interpreted cautiously. Future studies including a larger sample size are needed to control for the possibility that medications affect urinary EVs levels. Last, EV characterization may be confounded by elevated levels of proteins in urine,44 but proteinuria was not observed in our patients. Notably, protein levels were measured in 24‐hour urine of patients in the EH and RVH groups, but only a spot sample of urine in HVs.
Conclusions
The present study shows that levels of urinary EVs carrying markers of senescence and SASP are elevated in hypertensive patients compared with age‐matched HVs. Furthermore, the origin of p16+ EVs, and thus cellular senescence, can be attributed to specific tubular segments and was confirmed by their correlation with renal cytokines release. These observations suggest that increased numbers of EVs p16+ in urine of hypertensive patients may reflect cellular processes and serve as an early marker of cellular senescence along the nephron. Additional studies are needed to study the utility of urinary EV signatures as diagnostic or prognostic tools in other forms of kidney disease states.
These observations shed light on the mechanisms implicated in hypertension‐induced renal injury. Our findings may help direct therapy for renal injury using senolytic drugs and highlight a biomarker that may indicate therapeutic success.
Sources of Funding
This study was partly supported by the NIH grant numbers DK100081, DK104273, HL123160, DK120292, and DK102325.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e012584 DOI: 10.1161/JAHA.119.012584.)
Amrutesh S. Puranik is currently located at the Colton Center for Autoimmunity, New York University (New York, NY).
References
- 1. Sternlicht H, Bakris GL. The kidney in hypertension. Med Clin North Am. 2017;101:207–217. [DOI] [PubMed] [Google Scholar]
- 2. Gloviczki ML, Keddis MT, Garovic VD, Friedman H, Herrmann S, McKusick MA, Misra S, Grande JP, Lerman LO, Textor SC. TGF expression and macrophage accumulation in atherosclerotic renal artery stenosis. Clin J Am Soc Nephrol. 2013;8:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khosla S, Farr JN, Kirkland JL. Inhibiting cellular senescence: a new therapeutic paradigm for age‐related osteoporosis. J Clin Endocrinol Metab. 2018;103:1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boquoi A, Arora S, Chen T, Litwin S, Koh J, Enders GH. Reversible cell cycle inhibition and premature aging features imposed by conditional expression of p16Ink4a. Aging Cell. 2015;14:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin J, Tao J, Gu X, Yu Z, Wang R, Zuo G, Li Q, Lv X, Miao D. P16 (INK4a) deletion ameliorated renal tubulointerstitial injury in a stress‐induced premature senescence model of Bmi‐1 deficiency. Sci Rep. 2017;7:7502. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature. 2011;479:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorensen‐Zender I, Rong S, Susnik N, Zender S, Pennekamp P, Melk A, Haller H, Schmitt R. Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am J Physiol Renal Physiol. 2014;306:F907–F915. [DOI] [PubMed] [Google Scholar]
- 8. Westhoff JH, Hilgers KF, Steinbach MP, Hartner A, Klanke B, Amann K, Melk A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension. 2008;52:123–129. [DOI] [PubMed] [Google Scholar]
- 9. van Doormaal FF, Kleinjan A, Di Nisio M, Buller HR, Nieuwland R. Cell‐derived microvesicles and cancer. Neth J Med. 2009;67:266–273. [PubMed] [Google Scholar]
- 10. Zou X, Kwon SH, Jiang K, Ferguson CM, Puranik AS, Zhu X, Lerman LO. Renal scattered tubular‐like cells confer protective effects in the stenotic murine kidney mediated by release of extracellular vesicles. Sci Rep. 2018;8:1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gilani SI, Weissgerber TL, Garovic VD, Jayachandran M. Preeclampsia and extracellular vesicles. Curr Hypertens Rep. 2016;18:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hogan MC, Bakeberg JL, Gainullin VG, Irazabal MV, Harmon AJ, Lieske JC, Charlesworth MC, Johnson KL, Madden BJ, Zenka RM, McCormick DJ, Sundsbak JL, Heyer CM, Torres VE, Harris PC, Ward CJ. Identification of biomarkers for PKD1 using urinary exosomes. J Am Soc Nephrol. 2015;26:1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eirin A, Saad A, Tang H, Herrmann SM, Woollard JR, Lerman A, Textor SC, Lerman LO. Urinary mitochondrial DNA copy number identifies chronic renal injury in hypertensive patients. Hypertension. 2016;68:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eirin A, Gloviczki ML, Tang H, Rule AD, Woollard JR, Lerman A, Textor SC, Lerman LO. Chronic renovascular hypertension is associated with elevated levels of neutrophil gelatinase‐associated lipocalin. Nephrol Dial Transplant. 2012;27:4153–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saad A, Herrmann SM, Crane J, Glockner JF, McKusick MA, Misra S, Eirin A, Ebrahimi B, Lerman LO, Textor SC. Stent revascularization restores cortical blood flow and reverses tissue hypoxia in atherosclerotic renal artery stenosis but fails to reverse inflammatory pathways or glomerular filtration rate. Circ Cardiovasc Interv. 2013;6:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saad A, Crane J, Glockner JF, Herrmann SM, Friedman H, Ebrahimi B, Lerman LO, Textor SC. Human renovascular disease: estimating fractional tissue hypoxia to analyze blood oxygen level‐dependent MR. Radiology. 2013;268:770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gloviczki ML, Glockner JF, Lerman LO, McKusick MA, Misra S, Grande JP, Textor SC. Preserved oxygenation despite reduced blood flow in poststenotic kidneys in human atherosclerotic renal artery stenosis. Hypertension. 2010;55:961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eirin A, Ebrahimi B, Zhang X, Zhu XY, Tang H, Crane JA, Lerman A, Textor SC, Lerman LO. Changes in glomerular filtration rate after renal revascularization correlate with microvascular hemodynamics and inflammation in swine renal artery stenosis. Circ Cardiovasc Interv. 2012;5:720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwon SH, Woollard JR, Saad A, Garovic VD, Zand L, Jordan KL, Textor SC, Lerman LO. Elevated urinary podocyte‐derived extracellular microvesicles in renovascular hypertensive patients. Nephrol Dial Transplant. 2017;32:800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jayachandran M, Lugo G, Heiling H, Miller VM, Rule AD, Lieske JC. Extracellular vesicles in urine of women with but not without kidney stones manifest patterns similar to men: a case control study. Biol Sex Differ. 2015;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hudgins AD, Tazearslan C, Tare A, Zhu Y, Huffman D, Suh Y. Age‐ and tissue‐specific expression of senescence biomarkers in mice. Front Genet. 2018;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kerley RN, McCarthy C. Biomarkers of glomerular dysfunction in pre‐eclampsia—a systematic review. Pregnancy Hypertens. 2018;14:265–272. [DOI] [PubMed] [Google Scholar]
- 23. Jaszai J, Farkas LM, Fargeas CA, Janich P, Haase M, Huttner WB, Corbeil D. Prominin‐2 is a novel marker of distal tubules and collecting ducts of the human and murine kidney. Histochem Cell Biol. 2010;133:527–539. [DOI] [PubMed] [Google Scholar]
- 24. Erdbrugger U, Rudy CK, Etter ME, Dryden KA, Yeager M, Klibanov AL, Lannigan J. Imaging flow cytometry elucidates limitations of microparticle analysis by conventional flow cytometry. Cytometry A. 2014;85:756–770. [DOI] [PubMed] [Google Scholar]
- 25. Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krier JD, Crane JA, Eirin A, Zhu XY, Lerman A, Lerman LO. Hemodynamic determinants of perivascular collateral development in swine renal artery stenosis. Am J Hypertens. 2013;26:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long‐term function. Am J Physiol Renal Physiol. 2001;281:F887–F899. [DOI] [PubMed] [Google Scholar]
- 28. Trepiccione F, Zacchia M, Capasso G. The role of the kidney in salt‐sensitive hypertension. Clin Exp Nephrol. 2012;16:68–72. [DOI] [PubMed] [Google Scholar]
- 29. Gurley SB, Riquier‐Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pomatto MAC, Gai C, Bussolati B, Camussi G. Extracellular vesicles in renal pathophysiology. Front Mol Biosci. 2017;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merchant ML, Rood IM, Deegens JKJ, Klein JB. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol. 2017;13:731–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galbo PM Jr, Ciesielski MJ, Figel S, Maguire O, Qiu J, Wiltsie L, Minderman H, Fenstermaker RA. Circulating CD9+/GFAP+/survivin+ exosomes in malignant glioma patients following survivin vaccination. Oncotarget. 2017;8:114722–114735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turco AE, Lam W, Rule AD, Denic A, Lieske JC, Miller VM, Larson JJ, Kremers WK, Jayachandran M. Specific renal parenchymal‐derived urinary extracellular vesicles identify age‐associated structural changes in living donor kidneys. J Extracell Vesicles. 2016;5:29642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cvjetkovic A, Jang SC, Konecna B, Hoog JL, Sihlbom C, Lasser C, Lotvall J. Detailed analysis of protein topology of extracellular vesicles‐evidence of unconventional membrane protein orientation. Sci Rep. 2016;6:36338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez‐Hernandez J, Forner MJ, Pinto C, Chaves FJ, Cortes R, Redon J. Increased urinary exosomal microRNAs in patients with systemic lupus erythematosus. PLoS One. 2015;10:e0138618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li M, Zeringer E, Barta T, Schageman J, Cheng A, Vlassov AV. Analysis of the RNA content of the exosomes derived from blood serum and urine and its potential as biomarkers. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eirin A, Gloviczki ML, Tang H, Gossl M, Jordan KL, Woollard JR, Lerman A, Grande JP, Textor SC, Lerman LO. Inflammatory and injury signals released from the post‐stenotic human kidney. Eur Heart J. 2013;34:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gilani SI, Anderson UD, Jayachandran M, Weissgerber TL, Zand L, White WM, Milic N, Suarez MLG, Vallapureddy RR, Naav A, Erlandsson L, Lieske JC, Grande JP, Nath KA, Hansson SR, Garovic VD. Urinary extracellular vesicles of podocyte origin and renal injury in preeclampsia. J Am Soc Nephrol. 2017;28:3363–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. [DOI] [PubMed] [Google Scholar]
- 40. Burwick RM, Easter SR, Dawood HY, Yamamoto HS, Fichorova RN, Feinberg BB. Complement activation and kidney injury molecule‐1‐associated proximal tubule injury in severe preeclampsia. Hypertension. 2014;64:833–838. [DOI] [PubMed] [Google Scholar]
- 41. Devuyst O, Olinger E, Rampoldi L. Uromodulin: from physiology to rare and complex kidney disorders. Nat Rev Nephrol. 2017;13:525–544. [DOI] [PubMed] [Google Scholar]
- 42. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin II, Leonova KI, Consiglio CR, Gollnick SO, Chernova OB, Gudkov AV. p16(Ink4a) and senescence‐associated beta‐galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017;9:1867–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Balkom BW, Pisitkun T, Verhaar MC, Knepper MA. Exosomes and the kidney: prospects for diagnosis and therapy of renal diseases. Kidney Int. 2011;80:1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou H, Yuen PS, Pisitkun T, Gonzales PA, Yasuda H, Dear JW, Gross P, Knepper MA, Star RA. Collection, storage, preservation, and normalization of human urinary exosomes for biomarker discovery. Kidney Int. 2006;69:1471–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]