

Abstract
Background
Turner syndrome (TS) is the most common sex chromosome abnormality in women and is associated with increased morbidity and mortality. We describe long‐term outcomes in a large cohort of patients with TS.
Methods and Results
Retrospective review of patients with TS followed at Mayo Clinic Rochester from 1950 to 2017 was performed. Clinical, imaging, surgical, and genetic data were analyzed. Survival analysis was performed with the Kaplan–Meier method using age‐ and sex‐matched Olmsted County residents as the reference group. The study cohort comprised 317 patients with TS. Average age at diagnosis was 9 (range, 2–12) years, genetic testing was performed in 202 (64%), and pure monosomy X was present in 75 (37%). Congenital heart disease occurred in 131 (41%), with the most frequent lesions being bicuspid aortic valve (n=102, 32%) and coarctation of the aorta (n=43, 14%). Ascending aortic dilation was common, with mean aortic root size index 2 cm/m2, and aortic dissection occurred in 6 (2%) patients. The average follow‐up was 11 (range, 2–26) years, yielding 3898 patient‐years, and during this period 46 (14%) patients died; mean age at the time of death was 53±17 years. Patients with TS had reduced survival compared with the control group (82% versus 94% at 30 years; P<0.001), and the leading causes of death were cardiovascular disease, liver disease, and malignancy.
Conclusions
Patients with TS have reduced survival compared with age‐matched controls, and cardiovascular disease is the major cause of death. Further studies are required to determine if targeted cardiovascular risk factor modification will result in improved survival in this population.
Keywords: aortic disease, congenital heart disease, Turner syndrome
Subject Categories: Genetics, Echocardiography
Clinical Perspective
What Is New?
In a large cohort with long follow up, Turner syndrome patients have reduced survival compared with age‐ and sex‐matched controls.
Cardiovascular diseases are a major cause of death in Turner syndrome.
What Are the Clinical Implications?
Turner syndrome patients are at elevated risk of a cardiovascular event, and close attention should be paid in this population to primary and secondary prevention of cardiovascular disease.
Introduction
Turner syndrome (TS) is a sex chromosome disorder characterized by partial or complete loss of an X chromosome. It is the most common sex chromosome abnormality in women,1 with a prevalence of ≈1 in 2000 live births.2 The syndrome was initially described separately by Dr Otto Ullrich in 1930,3 and by Dr Henry Turner in 1938.4 Since the initial description more than 8 decades ago, studies have reported comorbidities such as congenital heart disease, aortic disease, renal and genitourinary anomalies, and autoimmune disease in patients with TS.1, 2, 5, 6, 7 Available data suggest reduced survival in patients with TS, but some of the data are based on studies with limited follow‐up.1, 2 We present a 68‐year single‐center retrospective review of children and adults with TS, investigating overall mortality and long‐term outcomes.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. This is a retrospective study of patients with TS followed at Mayo Clinic Rochester from January 1, 1950, to December 31, 2017. Medical records were reviewed, and all patients with a diagnosis of Turner syndrome contained in the diagnosis list were identified. Patients with TS and a prior echocardiogram were enrolled in the study. The Mayo Clinic Institutional Review Board approved this study and waived informed consent for patients who provided research authorization.
Hard copies of medical records (1950–1994) and electronic health records (1994–2017) were reviewed by 2 of the investigators (C.A. and M.F). Clinical, imaging, surgical, and genetic data were collected and analyzed. The status of each patient (alive or dead) as of December 2017 was verified using the electronic health records and the Social Security Death Index.
Categorical variables were reported as percentages, and continuous variables were reported as mean±standard deviation or median (interquartile range) for skewed data. We selected a reference group (reference group A) of Olmsted County residents. The study cohort and the reference group were matched 1:1 by year of birth and sex. To validate the sampling used for the reference group, 2 additional reference groups (reference groups B and C) were also selected from Olmsted County residents. Similar to reference group A, reference groups B and C were matched 1:1 to the cases by year of birth and sex. Survival was assessed using the Kaplan–Meier method and compared between study cohort and reference groups using a log‐rank test. The time of first encounter at the Mayo Clinic was used as “time zero” or the beginning of the “at‐risk” period for the study participants and their respective matched subjects from the reference group. We performed all statistical analyses with JMP software (version 13.0; SAS Institute Inc, Cary, NC), and P<0.05 was considered statistically significant.
Results
Baseline Characteristics
A total of 317 patients with TS were seen at the Mayo Clinic Rochester from 1950 to 2017. The average age at the time of diagnosis of TS was 9 (range, 2–21) years, and diagnosis was based on genetic testing in 202 (64%) and physical examination in 115 (36%) patients. Of the 202 patients with genetic testing, 75 (37%) had a pure monosomy X, 101 (50%) had mosaic TS, and 26 (13%) had a more complex chromosome anomaly with chromosome X loss. Y chromosome material was present in 30 (15%) patients. The age at first encounter at the Mayo Clinic was 22±18 years, and 57 (18%) patients presented in the early era (January 1950 to December 1980) while 260 (82%) patients presented in the late era (January 1981 to December 2017). Congenital heart disease was present in 131 (41%), and the most common congenital heart disease diagnoses were bicuspid aortic valve (n=102, 32%) and coarctation of the aorta (n=43, 14%). Table 1 shows the baseline clinical data of the cohort.
Table 1.
Baseline Characteristics (N=317)
| Age at the beginning of study, y | 22±18 |
| Height, cm | 143±16 |
| Weight, kg | 57±11 |
| Body surface area, m2 | 1.49±0.41 |
| Body mass index, kg/m2 | 26±8 |
| Comorbidities | |
| Hypertension | 98 (31%) |
| Hyperlipidemia | 79 (25%) |
| Coronary artery disease | 19 (6%) |
| Current or prior smoker | 26 (8%) |
| Diabetes mellitus | 37 (12%) |
| Sleep apnea | 27 (9%) |
| Prior stroke | 11 (4%) |
| Hypothyroidism | 104 (33%) |
| Congenital heart disease | |
| Bicuspid aortic valve | 102 (32%) |
| Coarctation of aorta | 43 (14%) |
| Persistent left superior vena cava | 24 (8%) |
| Anomalous pulmonary vein | 18 (6%) |
| Others | 22 (7%) |
| Medications | |
| Diuretics | 36 (11%) |
| Beta and/or calcium channel blockers | 54 (17%) |
| RAAS antagonist | 48 (15%) |
| Warfarin | 15 (5%) |
| Direct oral anticoagulants | 1 (3%) |
| Aspirin | 39 (12%) |
| Statin | 37 (12%) |
| Levothyroxine | 72 (23%) |
| Estrogen | 181 (57%) |
| Growth hormone | 88 (28%) |
RAAS indicates renin‐angiotensin‐aldosterone system.
Table 2 shows echocardiographic data of the cohort. The mean aortic root dimension was 28±6 mm, mean aortic size index of the aortic root was 2.0±0.6 cm/m2, and 44 (14%) patients had an aortic root Turner‐specific Z score >2. The mean mid–ascending aorta dimension was 28±8 mm, mean aortic size index of the mid–ascending aorta was 1.9±0.6 cm/m2, and 59 (19%) patients had a mid–ascending aorta Turner‐specific Z score >2. There were 36 patients (11%) with cross‐sectional imaging (cardiac computed tomography scan or cardiac magnetic resonance imaging scan) performed within 12 months from the time of echocardiogram. There was a modest correlation between the dimensions obtain by echocardiography and cross‐sectional imaging for aortic root (r=0.73, P=0.002) and mid–ascending aorta (r=0.79; P<0.001).
Table 2.
Echocardiography
| Left ventricle | |
| Left ventricular ejection fraction, % | 62±7 |
| Left ventricular end‐diastolic dimension, mm | 43±7 |
| Left ventricular end‐systolic dimension, mm | 29±4 |
| Left ventricular mass index, g/m2 | 78±24 |
| Aortic valve annulus | |
| Aortic valve annulus, mm | 19±3 |
| Aortic valve annulus, Z score | 0.4 (−0.3 to 1.1) |
| Aortic root/sinus of Valsalva | |
| Aortic root/sinus of Valsalva, mm | 28±6 |
| Aortic root/sinus of Valsalva, Z score | 0.8 (−0.2 to −1.6) |
| Aortic root/sinus of Valsalva, Z score >2 | 44 (14%) |
| Aortic size index, cm/m2 | 2.0±0.6 |
| Aortic size index, ≥2.5 cm/m2 | 38 (12%) |
| Mid–ascending aorta | |
| Mid–ascending aorta, mm | 28±8 |
| Mid–ascending aorta, Z score | 1.1 (−0.1 to 2.0) |
| Mid–ascending aorta, Z score >2 | 59 (19%) |
| Aortic size index, cm/m2 | 1.9±0.6 |
| Aortic size index, ≥2.5 cm/m2 | 37 (12%) |
Mortality and Cardiovascular Events
There were 46 (14%) deaths, and the mean age at the time of death was 53±17 years. The causes of death were cardiac (n=10, 22%), malignancy (n=5, 11%), gastrointestinal bleeding (n=4, 9%; ages 41, 47, 53, and 62 years), cirrhosis (n=2, 4%; ages 49 and 71 years), end‐stage renal failure (n=1, 2%; age 49 years), and unknown (n=24, 52%). The 10 cardiac deaths were attributable to postoperative death after cardiovascular surgery (n=5; ages 17, 31, 37, 42, and 70 years), end‐stage heart failure (n=2; ages 63 and 72 years), tamponade after diagnostic coronary angiogram (n=1; age 63 years), aortic dissection at an outside hospital (n=1; age 72 years), and sudden cardiac death (n=1; age 33 years). Deaths after cardiovascular surgery included a 17‐year‐old undergoing third sternotomy for pulmonary valve replacement, a 31‐year‐old with suspected spontaneous coronary artery dissection and attempted coronary artery bypass grafting, a 37‐year‐old undergoing aortic arch and subclavian artery repair, a 42‐year‐old with subacute ascending aortic dissection undergoing aortic root replacement, and a 70‐year‐old presenting for aortic valve replacement and coronary artery bypass grafting.
There were 4 cases of type A dissection, with an average age of 50 years. Three of the patients underwent emergent surgery at the Mayo Clinic, and 2 of these 3 patients survived to hospital dismissal. The maximum aortic dimension at the time of dissection ranged from 47 to 66 mm. The fourth case of type A dissection occurred at another hospital, and the patient died during the hospitalization. There were 2 cases of type B dissection, average age 30 years, and both patients had successful delayed surgical repair at the Mayo Clinic. The 5 deaths attributable to malignancy resulted from lung cancer in 2 patients (ages 77 and 56 years), mesothelioma in 1 patient (age 57 years), leukemia in 1 patient (age 40 years), and osteosarcoma in 1 patient (age 52 years).
The 317 patients were followed for 11 (2–26) years, yielding a total of 3898 patient‐years of follow‐up. Patients with TS had significantly lower survival compared with reference group A of year of birth– and sex‐matched Olmsted County residents (82% versus 94%; P<0.001 at 30 years; Figure 1). The 30‐year survival in reference groups B and C was 94% and 95%, respectively, and did not differ from 94% survival in reference group A (P=0.314).
Figure 1.
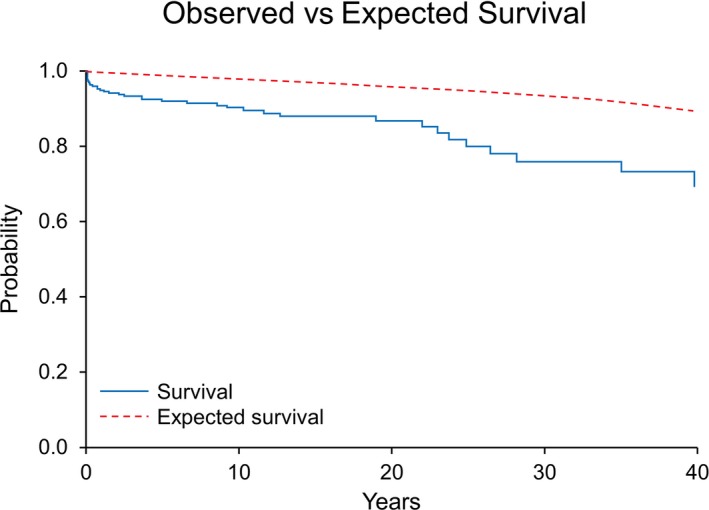
Kaplan–Meier curve comparing survival between the patients with Turner syndrome (blue) vs Olmsted County residents (red).
There was no difference in survival between patients with TS with and without congenital heart disease, patients whose diagnosis was made by genetic testing versus physical examination, and patients with or without bicuspid aortic valve (Figure 2). Similarly, there was no survival difference in patients who presented in the early era versus late era, or in patients with monosomy versus other genetic mutations (data not shown).
Figure 2.
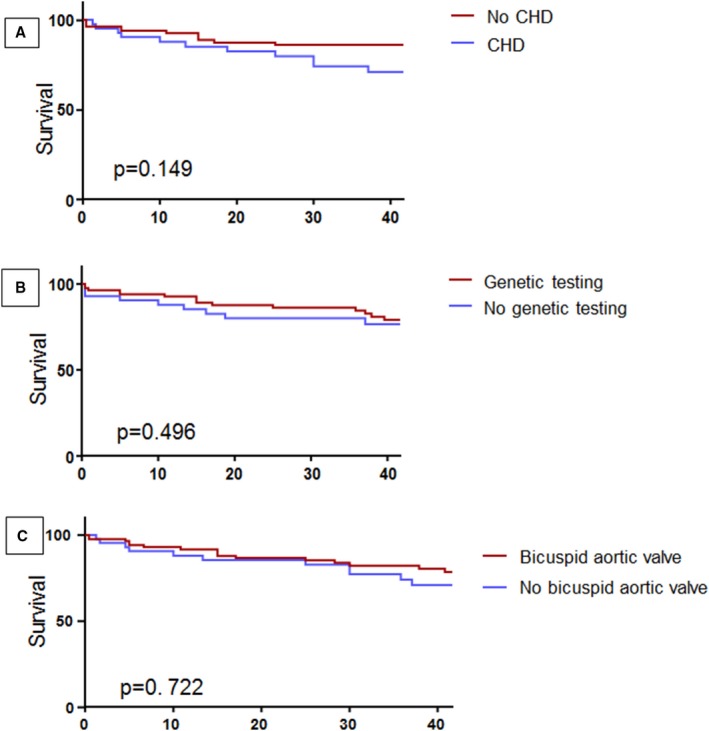
Kaplan–Meier curve comparing survival between. A, Patient with congenital heart disease (CHD) (blue) vs patients without CHD (red); 30‐year survival was 79% in CHD patients vs 83% in patients without CHD. B, Patients with genetic testing (red) vs patients without genetic testing (blue); 30‐year survival was 84% in patients who had genetic testing vs 77% in patients without genetic testing. C, Patients with bicuspid aortic valve (red) vs patients without bicuspid aortic valve (blue); 30‐year survival was 83% in patients with bicuspid aortic valve vs 82% in patients without bicuspid aortic valve.
Discussion
Our study demonstrates that patients with TS have 3‐fold increased mortality over age‐matched controls, which is similar to previously published cohort studies.1, 2, 5 While reduced survival has been reported in patients with 45,X monosomy,1, 2 in our study there was no survival reduction in this group. This may be related to the number of patients with genetic testing available for analysis. Alternately, it may represent greater recognition of anticipated sequelae and thereby improving care of this patient population.
Cardiovascular disease, both congenital and acquired, is common in TS,6 and has been recognized as a principle cause of increased morbidity and mortality in this population.1, 2 Patients in our study had many risk factors for acquired heart disease, including hypertension, hyperlipidemia, diabetes mellitus, and body mass index >25 in 50%. Of the 10 confirmed cardiac deaths, at least 4 can be attributed to acquired heart disease. Congenital heart disease was present in close to half of the cohort, with bicuspid aortic valve and coarctation of the aorta the most frequently observed anomalies. The reported prevalence of congenital heart disease in TS varies on the basis of spectrum of disease studied and type of investigation employed and has been estimated at 22% to 70%.8 Similar to prior studies, bicuspid aortic valve, coarctation of the aorta, anomalous pulmonary venous drainage, and persistent left superior vena cava were the most common congenital heart diseases noted in our cohort.8, 9 As with acquired heart disease, congenital heart disease has been associated with increased mortality.1 Despite its prevalence in our cohort, patients with congenital heart disease did not have higher mortality than those without congenital heart disease. This may represent improving care of our patients with adult congenital heart disease, such that many life‐threatening conditions are able to be anticipated and addressed before they result in mortality.
Aortic dissection remains a serious concern in patients with TS, largely due to the young age and small aortic dimension at which it occurs.10 Incidence of dissection in our cohort was 1.9%, with average age of 40 years at the time of dissection. A Danish registry study estimated a similar overall incidence of 1.4%, with a median age of 35 years compared with 77 years in the general population.11 Studies of mortality in these patients have demonstrated that, of all disorders of the circulatory system, aortic dissection is the most morbid, with a standardized mortality ratio of 23.6.1 Prior literature review of 88 cases found a mortality rate from aortic dissection of 58%.12 Thus, while the absolute number of cases of aortic dissection in TS is small, this complication tends to occur at a much younger age compared with the general population. Dissections can occur in any portion of the aorta, though ascending aortic dissection is most common and carries the highest risk of death.9 Additionally, because dissection of the ascending aorta in TS has been noted to occur at smaller aortic dimensions than otherwise expected in the general population, aortic size index (aortic dimension indexed to body surface area) has been suggested as a method to identify patients with pathologic aortic dilation.13 An aortic size index of 2 cm/m2 represents the 95th percentile for healthy female control subjects, while an aortic size index of 2.5 cm/m2 correlates with highest risk of aortic dissection.13 In our study, the average aortic size index of the aortic root was 2 cm/m2, emphasizing the prevalence of aortic dilation in this population. More recently, Turner‐specific Z scores have also been devised, allowing for further risk stratification.14 These Z scores are preferred for the assessment of aortic dissection risk in children under the age of 15, where a healthy girl with TS and no aortic pathology may have an aortic size index >2.5 cm/m2.10, 15 The authors have also had some experience using Turner‐specific Z score to assess the risk of an adult with extremely short stature.
The etiology of aortic dissection in TS is likely multifactorial, related to structural and hemodynamic factors as well as inherent abnormalities of the aortic tissue. Most patients with TS with aortic dissection have comorbid conditions that are established risk factors for dissection. A literature review of patients with TS with aortic dissection revealed that 89% of cases occurred in patients with hypertension, congenital heart disease (primarily bicuspid aortic valve and/or coarctation of the aorta), or both conditions.12 There are, however, a small number of patients with TS who have aortic dissection without an identifiable cause, and TS itself has been identified as an independent risk factor for aortic dilation and dissection.16 A study of vascular function in adults with TS demonstrates a generalized arteriopathy with greater intima media thickness and enlargement of conduit arteries over controls.17 Estrogen deficiency may play a role in arteriopathy, with increased intima media thickness also noted in 46,XX women with primary amenorrhea; however, arterial enlargement appears unique to patients with TS.17 Magnetic resonance imaging in TS supports these findings, with statistically significant associations between TS and enlargement of the aortic branch vessels.18 Notably, vasculopathy has also been appreciated in children with TS,19 before estrogen deficiency or adult comorbidities would be expected. Thus, while the molecular signaling pathways for arteriopathy in TS are not well understood, these patients appear to have both inherent and acquired risk factors for aortic dissection.
After cardiovascular events, liver disease was the second‐leading cause of death in our population, with 4 deaths from gastrointestinal bleeding and 2 deaths from cirrhosis, at an average age of 54 years. Hepatic disease in TS most commonly manifests as an asymptomatic rise in liver function tests, and prevalence increases with age.10 While in many cases liver injury does not progress to cirrhosis, there is a 6‐fold increased risk of cirrhosis in patients with TS compared with the general population.10 The pathophysiology of liver disease in this population is variable, though nonalcoholic fatty liver disease is felt to play an important role.20 This is likely driven in part by coincident central obesity and metabolic syndrome, which also occur more frequently in the TS population.21 Additionally, there is some evidence that congenital vascular abnormalities result in liver dysfunction, with correlations noted between patients with aortic coarctation and cerebral aneurysm and those with severe microvascular liver disease.22 Because of increased risk of severe liver disease in this population, it is recommended that patients with TS follow healthy eating patterns, avoid elevated body mass index, and have annual liver function testing to screen for asymptomatic disease.10
Malignancy was the third‐leading cause of death in our study. Overall risk of malignancy in TS is felt to be slightly higher than in the general population, with standardized incidence ratio up to 1.34 for solid tumors.23 Multiple studies have demonstrated increased prevalence of central nervous system tumors and melanoma, as well as decreased prevalence of breast cancer attributable to reduced estrogen exposure.23, 24 Of the 5 patients in our study dying of malignancy, none of them had a cancer demonstrated to occur at higher rates in the TS population.24 It is our conclusion that these deaths were likely incidental, rather than TS related. The risk of gonadoblastoma in patients with Y chromosome material has been reported with varying prevalence, and the true risk to patients is uncertain.24, 25 There were 30 patients in our study who had Y chromosome material present, and no episodes of gonadoblastoma were identified. Nevertheless, recommendations are to perform gonadectomy for individuals with Y chromosome fragments.10
Limitations
This is a single‐center retrospective study, and as a result it is subject to the inherent bias associated with this type of study design. Cause of death was not available for all patients, limiting conclusions regarding mode of death in the population. The presence of congenital heart disease was determined by echocardiographic imaging and surgical findings, and it is possible that mild congenital heart disease may have been missed by screening transthoracic echocardiogram. Echocardiographic measurements of aortic size were collected from reports, though images were reviewed and measurements verified where possible. The prevalence of coronary artery disease is likely underestimated, as routine assessment of coronary artery disease was not performed.
Conclusions
Patients with TS have multiorgan involvement resulting in multiple medical comorbidities. The overall survival is reduced in this population, and cardiovascular disease is the leading cause of mortality. The current study provides important outcomes data, albeit retrospective data, to guide counseling of patients with TS. Further studies are required to determine if targeted cardiovascular risk factor modification will result in improved survival in this population.
Sources of Funding
Dr Egbe is supported by the National Institutes of Health, K23 HL141448‐01.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e011501 DOI: 10.1161/JAHA.118.011501.)
References
- 1. Schoemaker MJ, Swerdlow AJ, Higgins CD, Wright AF, Jacobs PA; United Kingdom Clinical Cytogenetics G . Mortality in women with Turner syndrome in Great Britain: a national cohort study. J Clin Endocrinol Metab. 2008;93:4735–4742. [DOI] [PubMed] [Google Scholar]
- 2. Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab. 2006;91:3897–3902. [DOI] [PubMed] [Google Scholar]
- 3. Kinderheilk Z. Uber typische Kombinationsbilder multipler Abartungen [Over typical combination pictures of multiple modifications]. Eur J Pediatr. 1930;49:271. [Google Scholar]
- 4. Turner HH. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology. 1938;23:566–574. [PubMed] [Google Scholar]
- 5. Price WH, Clayton JF, Collyer S, De Mey R, Wilson J. Mortality ratios, life expectancy, and causes of death in patients with Turner's syndrome. J Epidemiol Community Health. 1986;40:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gravholt CH, Juul S, Naeraa RW, Hansen J. Morbidity in Turner syndrome. J Clin Epidemiol. 1998;51:147–158. [DOI] [PubMed] [Google Scholar]
- 7. Levitsky LL, Luria AH, Hayes FJ, Lin AE. Turner syndrome: update on biology and management across the life span. Curr Opin Endocrinol Diabetes Obes. 2015;22:65–72. [DOI] [PubMed] [Google Scholar]
- 8. Mortensen KH, Andersen NH, Gravholt CH. Cardiovascular phenotype in Turner syndrome—integrating cardiology, genetics, and endocrinology. Endocr Rev. 2012;33:677–714. [DOI] [PubMed] [Google Scholar]
- 9. Wong SC, Cheung M, Zacharin M. Aortic dilatation and dissection in Turner syndrome: what we know, what we are unclear about and what we should do in clinical practice? Int J Adolesc Med Health. 2014;26:469–488. [DOI] [PubMed] [Google Scholar]
- 10. Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, Lin AE, Mauras N, Quigley CA, Rubin K, Sandberg DE, Sas TCJ, Silberbach M, Soderstrom‐Anttila V, Stochholm K, van Alfen‐van derVelden JA, Woelfle J, Backeljauw PF; International Turner Syndrome Consensus Group . Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177:G1–G70. [DOI] [PubMed] [Google Scholar]
- 11. Gravholt CH, Landin‐Wilhelmsen K, Stochholm K, Hjerrild BE, Ledet T, Djurhuus CB, Sylven L, Baandrup U, Kristensen BO, Christiansen JS. Clinical and epidemiological description of aortic dissection in Turner's syndrome. Cardiol Young. 2006;16:430–436. [DOI] [PubMed] [Google Scholar]
- 12. Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. J Med Genet. 2007;44:745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation. 2007;116:1663–1670. [DOI] [PubMed] [Google Scholar]
- 14. Quezada E, Lapidus J, Shaughnessy R, Chen Z, Silberbach M. Aortic dimensions in Turner syndrome. Am J Med Genet A. 2015;167A:2527–2532. [DOI] [PubMed] [Google Scholar]
- 15. Silberbach M, Roos‐Hesselink JW, Andersen NH, Braverman AC, Brown N, Collins RT, De Backer J, Eagle KA, Hiratzka LF, Johnson WH Jr, Kadian‐Dodov D, Lopez L, Mortensen KH, Prakash SK, Ratchford EV, Saidi A, van Hagen I, Young LT. Cardiovascular health in Turner syndrome: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2018;11:e000048. [DOI] [PubMed] [Google Scholar]
- 16. Lopez L, Arheart KL, Colan SD, Stein NS, Lopez‐Mitnik G, Lin AE, Reller MD, Ventura R, Silberbach M. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics. 2008;121:e1622–e1627. [DOI] [PubMed] [Google Scholar]
- 17. Ostberg JE, Donald AE, Halcox JP, Storry C, McCarthy C, Conway GS. Vasculopathy in Turner syndrome: arterial dilatation and intimal thickening without endothelial dysfunction. J Clin Endocrinol Metab. 2005;90:5161–5166. [DOI] [PubMed] [Google Scholar]
- 18. Mortensen KH, Hjerrild BE, Andersen NH, Sorensen KE, Horlyck A, Pederson EM, Lundorf E, Christiansen JS, Gravholt CH. Abnormalities of the major intrathoracic arteries in Turner syndrome as revealed by magnetic resonance imaging. Cardiol Young. 2010;20:191–200. [DOI] [PubMed] [Google Scholar]
- 19. Lawson SA, Urbina EM, Gutmark‐Little I, Khoury PR, Gao Z, Backeljauw PF. Vasculopathy in the young Turner syndrome population. J Clin Endocrinol Metab. 2014;99:E2039–E2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roulot D. Liver involvement in Turner syndrome. Liver Int. 2013;33:24–30. [DOI] [PubMed] [Google Scholar]
- 21. El‐Mansoury M, Berntorp K, Bryman I, Hanson C, Innala E, Karlsson A, Landin‐Wilhelmsen K. Elevated liver enzymes in Turner syndrome during a 5‐year follow‐up study. Clin Endocrinol (Oxf). 2008;68:485–490. [DOI] [PubMed] [Google Scholar]
- 22. Roulot D, Degott C, Chazouilleres O, Oberti F, Cales P, Carbonell N, Benferhat S, Bresson‐Hadni S, Valla D. Vascular involvement of the liver in Turner's syndrome. Hepatology. 2004;39:239–247. [DOI] [PubMed] [Google Scholar]
- 23. Ji J, Zoller B, Sundquist J, Sundquist K. Risk of solid tumors and hematological malignancy in persons with Turner and Klinefelter syndromes: a national cohort study. Int J Cancer. 2016;139:754–758. [DOI] [PubMed] [Google Scholar]
- 24. Schoemaker MJ, Swerdlow AJ, Higgins CD, Wright AF, Jacobs PA. Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. Lancet Oncol. 2008;9:239–246. [DOI] [PubMed] [Google Scholar]
- 25. Gravholt CH, Fedder J, Naeraa RW, Muller J. Occurrence of gonadoblastoma in females with Turner syndrome and Y chromosome material: a population study. J Clin Endocrinol Metab. 2000;85:3199–3202. [DOI] [PubMed] [Google Scholar]