

Abstract
In the course of infecting their hosts, pathogenic bacteria secrete numerous effectors, namely, bacterial proteins that pervert host cell biology. Many Gram-negative bacteria, including context-dependent human pathogens, use a type IV secretion system (T4SS) to translocate effectors directly into the cytosol of host cells. Various type IV secreted effectors (T4SEs) have been experimentally validated to play crucial roles in virulence by manipulating host cell gene expression and other processes. Consequently, the identification of novel effector proteins is an important step in increasing our understanding of host–pathogen interactions and bacterial pathogenesis. Here, we train and compare six machine learning models, namely, Naïve Bayes (NB), K-nearest neighbor (KNN), logistic regression (LR), random forest (RF), support vector machines (SVMs) and multilayer perceptron (MLP), for the identification of T4SEs using 10 types of selected features and 5-fold cross-validation. Our study shows that: (1) including different but complementary features generally enhance the predictive performance of T4SEs; (2) ensemble models, obtained by integrating individual single-feature models, exhibit a significantly improved predictive performance and (3) the ‘majority voting strategy’ led to a more stable and accurate classification performance when applied to predicting an ensemble learning model with distinct single features. We further developed a new method to effectively predict T4SEs, Bastion4 (Bacterial secretion effector predictor for T4SS), and we show our ensemble classifier clearly outperforms two recent prediction tools. In summary, we developed a state-of-the-art T4SE predictor by conducting a comprehensive performance evaluation of different machine learning algorithms along with a detailed analysis of single- and multi-feature selections.
Keywords: type IV secreted effector, bioinformatics, sequence analysis, comprehensive performance evaluation, machine learning, feature analysis
Introduction
Pathogenic bacteria are microorganisms that cause infections. During this process, bacteria invade a host organism where they multiply, producing and secreting effector proteins. Such effector proteins fulfill a range of functions critical for the virulence of the pathogen, that is the degree of damage that the bacterium causes to the host. In most cases, effector proteins are directly injected into host cells via dedicated secretion systems, enabling them to modulate or manipulate a wide range of cellular processes, including actin dynamics (e.g. Beps secreted by Bartonella spp.) [1–3], phagocytosis (e.g. various effectors of Yersinia and Salmonella enterica) [4, 5], endocytic trafficking (e.g. effectors of Legionellapneumophila) [6–8], apoptosis (e.g. Shigella effectors IpgD and OspG) [9, 10], immune response (YopJ from Yersinia enterocolitica) [4, 11] and secretion (e.g. Escherichia coli effector EspG) [12].
Currently, Gram-negative bacterial secretion systems are classified into six types (I-VI) [13]. Among them, type III and type IV secretion systems (T3SS and T4SS, respectively) and their associated effectors (T3SEs and T4SEs, respectively) have been widely studied, as they are critical for virulence of various human pathogens. For example, S. enterica, Yersinia pestis and Pseudomonas syringae use type III secretion systems [14], while Brucella spp., Bartonella spp., Helicobacterpylori and L. pneumophila use T4SSs [15]. Despite their clinical significance, a fundamental biological question remains: How does a given secretion system recognize a given effector protein as a substrate, which it must bind and secrete? These secretion systems are highly selective nanomachines, and do not inadvertently secrete non-effector proteins. Clearly, some element or elements of effector protein sequence and/or structure must dictate recognition by the cognate secretion system, but there is an outstanding need for an integrative understanding what these recognition elements are and how they determine substrate protein secretion. While specific wet-lab experimental studies can answer underlying questions for individual effector proteins, bioinformatics-based tools are needed to address the matter more efficiently and comprehensively.
Recently, machine learning algorithms were introduced to predict T4SEs [16–18]. For instance, Burstein et al. [16] developed a machine learning model for differentiating T4SEs from non-effectors in L. pneumophila. Their model used seven types of features including ‘taxonomic distribution among bacteria and metazoa’, ‘sequence similarity to known effectors’ and ‘homology to known eukaryotic proteins’, which the authors concluded from their analysis were the three best representative features [16]. To examine the classification performance of different algorithms, they used support vector machine (SVM), multilayer perceptron (MLP), Naïve Bayes (NB), Bayesian networks (BNs) and a Voting Algorithm, the latter of which was based on the former four classifiers. The study successfully predicted and experimentally verified 40 novel T4SEs from Legionella. In another recent work, Zou et al. [17] developed an SVM-based classifier called T4EffPred using four distinct feature types, including amino acid composition (AAC) and position-specific scoring matrix (PSSM), as well as feature combinations. T4EffPred could distinguish IVA and IVB effectors, which are the two main subtypes of T4SEs [17]; it has also been successfully applied to perform genome-wide predictions of effectors in the bacterium Bartonella henselae, where ∼50 putative T4SEs were found. In a third study, Wang et al. [18] presented a T4SE inter-species cross-prediction tool based on C-terminal features, such as AACs, motifs, secondary structures (SSs) and solvent accessibility (SA). The tool comprises three computational models that were trained using SVM-based machine learning (T4SEpre_psAac, trained using position-specific, sequence-based AACs; T4SEpre_bpbAac, trained using AACs based on bi-profile Bayes feature extraction combined with SVM; T4SEpre_Joint, trained using position-specific AACs, SSs and SA). When applied to the genome of H. pylori, 25 candidate T4SEs were identified by the authors. Also based on C-terminal Signals, Zou et al. [19] analyzed the performance of C-terminal sequence features such as AAC and position-specific amino acid composition (PSAac). They used multiple machine learning algorithms to train models of T4SEs with a majority vote strategy. Based on their findings, an SVM predictor of type IV-B effectors trained with PSAac and AAC was developed and validated through a genome-scale prediction in Coxiella burnetii. Our previous work [20] comprehensively reviewed the currently available bioinformatics approaches for T4SE prediction, and offered an assessment of these approaches in terms of software utilities and prediction performance. A recent review from Zeng et al. [21] further discussed and highlighted some potential improvements of the prediction performance after benchmarking the available identification tools of secreted effector proteins in bacteria. The schematic figures in such article give a bird’s-eye view of computational toolkits in the field of secreted effector predictions.
While previous work has demonstrated that machine learning approaches can successfully predict effector proteins, the features or combinations of features that are most appropriate for efficient T4SE prediction have not been systematically assessed. Here, we used 10 types of features and 6 different machine learning algorithms to train predictors with 390 T4SS effectors and 1112 non-effectors. We first compared the 10 types of features with their combinations on multiple performance assessments and found that, while combinations of features in a single model do not yield statistically significant improvements, the ensemble of multiple individual models trained with different single features significantly improved the overall performance. Our direct comparison of six representative models, namely, NB, K-nearest neighbor (KNN), logistic regression (LR), random forest (RF), SVM and MLP, shows that RF and SVM outperformed all others in terms of predictive and computational performance. In addition, the ensemble model that integrated all six machine learning methods further improved the prediction performance. With this valuable knowledge, we developed Bastion4, an online T4SE predictor that operates as an ensemble classifier based on six machine learning models, each of which consists of individual models trained with various types of selected features. Our subsequent analysis presented here shows that Bastion4 outperforms T4Effpred and T4SEpred based on independent tests. Bastion4 is available at http://bastion4.erc.monash.edu/.
Materials and methods
The Bastion4 methodology development (Figure 1) involved five major stages: Data set Curation, Feature Extraction, Feature Selection, Model Training and Validation and Prediction. Each of these major stages is described in the following sections.
Figure 1.

Overview of the proposed methodology for predicting T4SEs. First, a large number of protein sequences are collected, forming the input data set. Then, 10 types of features are extracted that characterize those proteins in different ways. Using the mRMR/Gain Ratio technique, a subset of features is selected to optimize the following model training. Next, the performance of trained models is evaluated by a 5-fold cross-validation test and an independent test. Finally, by applying a voting mechanism to various models, an ensemble classifier is formed, which separates the input into putative effectors and non-effectors.
Data sets collection
The input data set consisted of two parts: the training data set and the independent data set. We constructed the training data set by extracting known T4SEs from independent data sets described in the literature. Specifically, 347 T4SE sequences were extracted from the T4SEpre data set constructed by Wang et al. [18]. The pathogen B.henselae has two subtypes of T4SS (IVA and IVB), and 340 effectors including 30 IVA proteins and 310 IVB proteins were acquired from Zou et al. [17]. Finally, we added 120 proteins identified by Burstein et al. [16]. For the negative training set, we chose the entire set of 1132 non-effectors in Zou et al. [17]. After forming the preliminary data set, CD-HIT [22] was used to remove highly homologous sequences (defined as having 60% sequence identity) to reduce sequence redundancy, which may otherwise lead to a potential bias in the trained models. The final training data set contained 390 positive and 1112 negative sequences.
To evaluate the model performance in comparison with existing T4SE prediction tools, we generated an independent data set containing both positive and negative samples. For the former, 43 positive samples were acquired from the UniProt Database [23] and Meyer et al. [24], while for the latter, we used 150 samples from the data set of Vibrio parahaemolyticus serotype O3: K6 (strain RIMD 2210633) [25]. After removal of duplicate samples, which appear in our training set and the data sets used by the existing T4SE predictors, we obtained a final independent data set made up of 30 positive and 150 negative samples.
Feature extraction
The variety of features used in this work can be categorized into three main types: local sequence encoding, global sequence encoding and structural descriptor encoding. Extracted from the type-specific information available for any given protein, each feature is represented by a number of encoding vectors.
Local sequence encoding
Feature associated with local sequence encoding refers to distinguishable patterns in the protein sequence.
(1) Amino acid composition
AAC is represented as a 20-dimensional feature vector, in which each element characterizes the frequency of an amino acid type in the whole protein sequence [26].
Each element in this feature vector was calculated according to the following formula:
where is the number of occurrences of amino acid in the whole protein sequence, and is the length of the sequence. Finally, represents thei-th element in the feature vector, which indicates the frequency of the amino acid in the protein sequence.
(2) Dipeptide composition
A protein’s dipeptide composition (DPC) is encoded in a 400-dimensional feature vector {}, which represents the frequencies of all 400 possible amino acid pairs in the protein sequence. Each element is obtained using the following formula:
where denotes the number of occurrences of the -th amino acid pair [17], and refers to the total number of dipeptides in the whole sequence.
(3) Composition of -spaced amino acid pairs
As a widely used feature type in sequence analysis [27, 28], the composition of -spaced amino acid pairs (CKSAAPs) is in effect a generalization of the DPC. Two amino acids form a -spaced amino acid pair if they have amino acids in-between them. In this sense, amino acid pairs in the DPC can be viewed as -spaced amino acid pairs in the CKSAAP. For CKSAAP, all pairs with space ≤ are considered. Thus, CKSAAP outputs a -dimensional feature vector for a given protein sequence. We use k = 5, and, consequently, a 2400-dimensional vector is constructed.
(4) Property composition
The property composition (PPT) [29] maps amino acids to three distinct amino acid alphabets, namely, the classical amino acid alphabet, the amino acid property alphabet and the hydrophobic/hydrophilic alphabet. Each amino acid corresponds to a certain property class. When an amino acid fits to multiple property classes, it was categorized into the most specific (smallest) class. For each property class, di- and tripeptides were measured in terms of frequency. Moreover, only the features that occur more than one time in both positive and negative data sets were selected to avoid over-fitting. Consequently, a 72-dimensional feature vector is formed for each protein sequence.
Global sequence encoding
PSSMs have proved beneficial for incorporating evolutionary information in machine learning methods [17, 30–35]. Here, we generated PSSM profiles by running PSI-BLAST against the nonredundant database of NCBI with parameters and . There are two types of methods for exploiting patterns from PSSM profiles, which are explained below.
(5) PSSM profiles with auto covariance transformation
A PSSM is a matrix, where is the length of the corresponding protein sequence. The (i, j)-th element of the matrix denotes the probability of amino acid j to appear at the i-th position of the protein sequence. The PSSM encoding converts the PSSM profile into a matrix by summing up all rows of the same amino acid residue [34], thereby forming a 400-dimensional vector as part of the input for model training.
Based on the original matrix, the PSSM_AC encoding uses the auto covariance (AC) transformation to further measure the correlation between two properties [17, 36] by using the following formula:
where j refers to one of the 20 amino acids, L denotes the length of the whole protein sequence, denotes the PSSM score of amino acid j at position i and is the average score for amino acid j along the whole sequence:
Consequently, the number of AC components amounts to , where lg runs from , with . Here, we set as previously used in Zou et al. [17], yielding a 200-dimensional feature vector.
(6) Smoothed PSSM encoding
A transformation of the standard PSSM profile, the Smoothed PSSM (PSSM_SMTH) encoding, has been previously used to predict RNA-binding sites of proteins [37] and drug-binding residues [38]. Assuming the size of a smoothing window is and represents the th row vector of the PSSM, each row vector of the PSSM_SMTH can be constructed by summation of the current row vector and the following row vectors (Figure 2):
Figure 2.

Example of a PSSM_SMTH profile using a smoothing window of size 7. The PSSM profile shows the evolutionary information extracted from the PSSM file, which is generated by PSI-BLAST. When the size of the smoothed window is set to 7, the values of the 7th row in the PSSM_SMTH profile are equal to the sum of the corresponding values from the 7th row to the 13th row of the PSSM file.
For this method, we use values of ranging from 1 to 10. Therefore, 10 PSSM_SMTH profiles are obtained. For each PSSM_SMTH profile, rows corresponding to the first 50 amino acids starting from the protein’s N-terminus are considered to form a vector with dimension 50 × 20 = 1000. As a result, a 10 000-dimensional vector is constructed.
To extract the PSSM_AC encoding and the 10 PSSM_SMTH encodings, we used the POSSUM server, which is a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles [39].
Structural descriptor encoding
Protein structural information has been widely used to improve the prediction performance in a number of bioinformatics applications [40–45], but has not been comprehensively analyzed for the prediction of T4SEs. In our machine learning framework described here, we extract SS, SA and natively disordered region information for T4SE sequences and use them as features for model training.
(7) Predicted SS
Protein SS is a widely used attribute in bioinformatics predictors. As the SS is known for only a relatively small number of proteins, we instead predicted protein SSs from amino acid sequences using SSpro [46]. Specifically, for each residue of the query sequence, SSpro predicts one of three types of SS: alpha-helix, beta-strand or coil. Here, we represent these types of predicted SS by using a 3-bit encoding and encode the first 50 residues of the queried sequence, thereby forming a vector of length 3 × 50 = 150.
(8) Predicted SA
SA is another important feature for prediction. The SSpro program can be used to predict SA from protein sequence data. For each residue in a sequence SSpro predicts, it being in one of the two possible states ‘exposed’ or ‘buried’. Therefore, we use a 2-bit encoding to represent predicted SA and encode the first 50 residues of the queried sequence, forming a vector of length 2 × 50 = 100.
(9) Predicted natively disordered region
Disordered (DISO) protein regions lack fixed tertiary structure, being either fully or partially unfolded [47]. Contrary to initial concerns that these regions were functionally ‘useless’, recent studies indicate that such regions are commonly involved in many biological functions [47]. Here, we predict the native disorder information using DISOPRED2 [48], which provides a quantitative real-valued score ranging from 0 to 1, which represents the probability of a residue being disordered. For this structural descriptor, we used seven different sizes of smoothing windows as previously suggested [49] () to encode the first 50 residues of the queried sequence, resulting in a feature vector of length 7 × 50 = 350.
Feature normalization
After feature extraction, we found that some features have values ranging between 0 and 0.01, while others have values ranging from 1 to 1000. However, features that can frequently assume larger numeric values are also more likely to have a larger impact on the prediction as compared with features with ranges of smaller numeric values. Thus, to improve the prediction accuracy and avoid having a particular feature dominating the prediction because of it assuming larger numerical values, we normalize values of different features so that all values fall into the same numeric interval [50].
Here, we use the following formula to normalize all feature values to the numeric interval [0, 1]:
where denote the original value, the minimum value and the maximum value in the feature vector, respectively, and denotes the output value of after scaling. If the numbers in a feature vector are equal to each other, i.e. , we assign the value 0.
Feature selection
Feature selection plays an important role in machine learning. Biological data sets are usually characterized by a large number of initial features, making it a formidable task to deal with oversized feature sets; some of the typical problems include slow algorithm speed and a low predictor performance. Thus, the objective of feature selection is 3-fold: improving the prediction performance of the predictors, providing faster and more cost-effective predictors and providing a better understanding of the underlying process that generated the data [51].
Gain ratio
The gain ratio algorithm is a powerful method based on information theory [52]. In this binary classification problem, we assume the probability of a positive sample to be and the probability of a negative sample to be . The entropy of the classification can be denoted as:
where denotes the positive class label. The conditional entropy of the feature can be calculated as follows:
where denotes the total number of features. Therefore, we can express the formula of gain ratio as:
mRMR
The mRMR algorithm is based on mutual information [53]. It was originally proposed by Peng et al. [53] and can be downloaded from http://penglab.janelia.org/proj/mRMR/. The mRMR algorithm has been widely used in a number of feature-selection tasks in many research areas [54–59], including protease cleavage sites prediction, acetylation site prediction and other posttranslational modification site predictions.
Model training
Naive Bayes
NB is a commonly used statistical classifier that is generally adopted to calculate the conditional probability without assuming any dependence between features. It has been successfully applied in many disciplines of science, and performs consistently well even when considering relatively few attributes [60]. NB operates based on the Bayes’ theorem:
where C represents the binary class variable, and F denotes the input feature vector of the classifier.
K-nearest neighbor
KNN is a simple but powerful classification method, which predicts a new candidate by evaluating the distance functions to k nearest known neighbors. It has been successfully used in many bioinformatics endeavors such as the prediction of protein function [61], protein subcellular localization [62] and membrane protein architecture [63]. According to the KNN algorithm, a new instance is classified by a majority vote of its top KNNs. The instance is then assigned to the most common class among the top KNNs.
The choice of parameter k is important, and has a direct effect in the performance and outcome of a KNN classifier. In this work, k was optimized so as to minimize the classification error for values , where is the number of features used during model training.
Logistic regression
As a widely used algorithm [64, 65], LR results from a linear regression using the following equation:
where refers to the expected probability of dependent variables, and are constants.
As the values of LR range from 0 to 1, it is a useful technique for handling classification problems, especially in situations where only the probability of occurrence of the response is concerned.
Random forest
The RF algorithm is a classification algorithm developed by Leo Breiman [66] using an ensemble of classification trees. It has been widely used and implemented as the RF package in R [67]. RF is one of the most powerful algorithms in machine learning [68]. In RF, two key parameters are the number of the trees, , and the number of features selected randomly, .
Here, we selected , and optimized the parameter over the set of integers between 1 and to minimize the classification error. Here, is the total number of features.
Support vector machine
SVM is a powerful machine learning algorithm and is commonly used to deal with binary classification problems. SVM has been widely applied to solve many classification and regression problems in bioinformatics and computational biology [17, 18, 26, 27] and, particularly, SVM with a Gaussian radial basis kernel is widely used for nonlinear classification problems. There are two parameters that affect the performance of the nonlinear SVM model: Cost (C), which controls the cost of misclassification during data training, and , which is the free parameter of the Gaussian radial basis function.
In this study, we adopt the radial basis kernel for SVM model training by using the e1071 package [69] in R language. We use the grid search method to identify the optimal parameters and . Accordingly, our number of grid points is 13 × 13 = 169. Based on the training data, the SVM is optimized by finding the optimal values for and that minimize the classification error by performing 10-fold cross-validation.
Neural networks
A neural network is a nonlinear statistical classifier that is able to detect complex relationships between dependent and independent variables [70]. One type of neural network is called MLP. An MLP is characterized by multiple layers, that is there can be one or more nonlinear layers (hidden layers) between the input and the output layers. An increase in the number of hidden layers facilitates neural network models to solve increasingly nonlinear problems.
Using RSNNS [71], an R implementation of SNNS [72], we train an MLP classifier with two hidden layers. The numbers of nodes in the first and second hidden layers are set to 64 and 32, respectively, while the maximum number of iterations to learn is set to 1000.
Randomized 5-fold cross-validation test
Cross-validation is a common method for estimating the performance of a classification model. In this study, the benchmark data set is randomly partitioned into five equal-sized subsets, and tests are repeated five times. For each cross-validation test, one subset is used as testing data, while the remaining four subsets form the training set are used to train the classifier. Hence, each subset is used once for testing and four times for training. The five numerical results obtained from these tests are averaged to obtain a single value that represents the performance of the classification model.
Independent test
In this study, we compare the performance of our models with three previously published classifiers: T4Effpred [17] and two variant models of T4SEpred (i.e. T4SEpred_bpbAac and T4SEpred_psAac) [18]. As noted earlier, we constructed an independent test data set, which is completely different from the training data sets of these three models. Performance comparison is conducted on this independent data set.
Performance assessment
Six performance measures, namely, Sensitivity (SN), Specificity (SP), Precision (PRE), Accuracy (ACC), F-value and Matthew’s correlation coefficient (MCC) [73], are used to evaluate the overall predictive performance of classification models. These measures are defined as:
where represent the numbers of true positives, true negatives, false positives and false negatives, respectively.
Additionally, the receiver operating characteristic (ROC) curve, which is a plot of the true-positive rate versus the false-positive rate, is depicted to visually measure the comprehensive performance of different classifiers. The area under the curve (AUC) is also provided in each of the ROC plots, to quantify the respective performance.
Results and discussion
Sequence analysis
We analyzed the amino acid occurrences (including those overrepresented and underrepresented) on each position of T4SS effectors. We examined the first 50 N-terminal and 50 C-terminal positions of sequences of T4SEs [18, 20], non-effectors and control proteins with the pLogo program [74], and studied the differences among the three groups of proteins with respect to their amino acid preferences (Figure 3).
Figure 3.

Position-specific amino acid sequence profiles of T4SEs and non-effectors for N- and C-terminal 50 positions. Images were generated by pLogo. The vertical axis denotes the log-odds binomial probability, while the horizontal one represents the N-terminal position number. The red horizontal bars on the images denote the statistical significant thresholds (P = 0.05) following a Bonferroni correction. (A), (B) and (C) illustrate sequence logo representations for T4SEs, non-effectors and control effectors (i.e. cytoplasmic proteins), respectively.
For the N-terminus, remarkable consensus was found in T4SE sequences, while amino acid residues tended to be more disordered in non-T4SE and control sequences. Specifically, the N-terminal sequences of T4SEs showed a significant overrepresentation of lysine and asparagine residues, with glycine and alanine largely absent. Likewise, the C-terminal sequences showed an enrichment for glutamate residues at positions 35–42 for the T4SEs (i.e. in residues located at −16 to −8 positions relative to the C-terminus). There was no significant motif pattern in the C-terminal sequences of non-T4SEs or the control sequences. Such characteristic features distinguish T4SEs from non-effectors, and are useful for explaining protein features that might be captured in machine learning models. Previous work on several specific T4SEs has shown that the C-terminal segment of the proteins incorporates at least part of the signal for engagement by the T4SS [75].
As shown in Table 1, L. pneumophila has 291 T4SEs, thereby accounting for the largest proportion (74.6%) of T4SEs. To address whether this biases the outcomes of putative signal sequence motifs, we analyzed sequences from L. pneumophila and C.burnetii, respectively (Figure 4). The enrichment of glutamate (‘E’) residues is clear in sequences from L. pneumophila. While sequences from C. burnetii commonly have glutamic residues, these have a much reduced preference. Biologically, this could indicate two distinct targeting signals, with the one composed of glutamic residues being the predominant form in species, such as L. pneumophila, but with this glutamic acid-rich signal used by fewer of the T4SEs in species like C. burnetii. Computationally, this finding reveals that there is no common motif in T4SS effectors across multiple species, which further supports the need to look at many features to develop globally effective machine learning models.
Table 1.
The components of various species in T4SEs
| Species | Number |
|---|---|
| Agrobacterium rhizogenes | 4 |
| Agrobacterium tumefaciens str. C58 | 2 |
| Agrobacterium tumefaciens | 4 |
| Anaplasma marginale str. St. Maries | 3 |
| Anaplasma phagocytophilum HZ | 2 |
| Bartonella grahamii as4aup | 1 |
| Bartonella henselae str. Houston-1 | 5 |
| Bordetella pertussis Tohama I | 4 |
| Brucella melitensis biovar Abortus 2308 | 6 |
| Brucella melitensis bv. 1 str. 16M | 2 |
| Coxiella burnetii CbuG_Q212 | 1 |
| Coxiella burnetii CbuK_Q154 | 3 |
| Coxiella burnetii Dugway 5J108-111 | 7 |
| Coxiella burnetii RSA 331 | 15 |
| Coxiella burnetii RSA 493 | 34 |
| Ehrlichia chaffeensis str. Arkansas | 1 |
| Helicobacter pylori 26695 | 1 |
| Legionella pneumophila subsp. pneumophila str. Philadelphia 1 | 35 |
| Ochrobactrum anthropi ATCC 49188 | 1 |
| Legionella pneumophila subsp. pneumophila (strain Philadelphia 1/ATCC 33152/DSM 7513) | 256 |
| Unknown | 3 |
| Total | 390 |
The top two species with the largest numbers of known T4SEs are highlighted in bold.
Figure 4.

Position-specific amino acid sequence profiles of L. pneumophila effectors and C. burnetii effectors for both N- and C-terminal 50 positions.
Performance evaluation using randomized 5-fold cross-validation tests
For each of the 10 feature encodings, all six classifiers were trained and validated to predict T4SEs based on a randomized 5-fold cross-validation test. As negative samples, 390 protein sequences were randomly selected from the non-type IV effector data set, to generate a balanced training data set with a 1:1 ratio of positive to negative samples. All experiments were repeated five times. The results are documented in Table 2, Figures 5 and 6, and discussed below.
Table 2.
The performance of various classifiers based on the 5-fold cross-validation tests
| Feature | Method | PRE | SN | SP | F-score | ACC | MCC |
|---|---|---|---|---|---|---|---|
| AAC | RF | 0.836 ± 0.009 | 0.825 ± 0.005 | 0.839 ± 0.012 | 0.829 ± 0.006 | 0.831 ± 0.007 | 0.663 ± 0.014 |
| SVM | 0.856 ± 0.007 | 0.845 ± 0.011 | 0.859 ± 0.009 | 0.849 ± 0.007 | 0.851 ± 0.007 | 0.703 ± 0.014 | |
| LR | 0.816 ± 0.006 | 0.834 ± 0.005 | 0.813 ± 0.007 | 0.824 ± 0.003 | 0.823 ± 0.004 | 0.647 ± 0.009 | |
| NB | 0.792 ± 0.005 | 0.837 ± 0.004 | 0.782 ± 0.005 | 0.813 ± 0.004 | 0.809 ± 0.003 | 0.619 ± 0.007 | |
| KNN | 0.827 ± 0.005 | 0.838 ± 0.009 | 0.826 ± 0.006 | 0.831 ± 0.005 | 0.831 ± 0.003 | 0.664 ± 0.008 | |
| MLP | 0.864 ± 0.010 | 0.727 ± 0.008 | 0.886 ± 0.011 | 0.788 ± 0.007 | 0.805 ± 0.007 | 0.620 ± 0.013 | |
| PPT | RF | 0.816 ± 0.006 | 0.816 ± 0.014 | 0.817 ± 0.005 | 0.815 ± 0.010 | 0.816 ± 0.008 | 0.633 ± 0.017 |
| SVM | 0.818 ± 0.009 | 0.828 ± 0.007 | 0.817 ± 0.011 | 0.822 ± 0.005 | 0.822 ± 0.005 | 0.645 ± 0.010 | |
| LR | 0.803 ± 0.007 | 0.788 ± 0.003 | 0.808 ± 0.008 | 0.794 ± 0.004 | 0.797 ± 0.004 | 0.596 ± 0.008 | |
| NB | 0.715 ± 0.006 | 0.348 ± 0.003 | 0.860 ± 0.004 | 0.464 ± 0.004 | 0.603 ± 0.002 | 0.243 ± 0.007 | |
| KNN | 0.808 ± 0.008 | 0.745 ± 0.008 | 0.824 ± 0.008 | 0.773 ± 0.010 | 0.783 ± 0.009 | 0.570 ± 0.016 | |
| MLP | 0.843 ± 0.016 | 0.689 ± 0.035 | 0.872 ± 0.019 | 0.755 ± 0.020 | 0.779 ± 0.014 | 0.571 ± 0.027 | |
| DPC | RF | 0.811 ± 0.015 | 0.810 ± 0.006 | 0.812 ± 0.017 | 0.809 ± 0.010 | 0.810 ± 0.011 | 0.621 ± 0.023 |
| SVM | 0.837 ± 0.007 | 0.805 ± 0.010 | 0.844 ± 0.010 | 0.819 ± 0.005 | 0.823 ± 0.004 | 0.648 ± 0.007 | |
| LR | 0.812 ± 0.003 | 0.839 ± 0.005 | 0.806 ± 0.002 | 0.824 ± 0.003 | 0.822 ± 0.002 | 0.645 ± 0.005 | |
| NB | 0.793 ± 0.002 | 0.840 ± 0.003 | 0.782 ± 0.004 | 0.815 ± 0.002 | 0.811 ± 0.003 | 0.623 ± 0.006 | |
| KNN | 0.797 ± 0.004 | 0.820 ± 0.006 | 0.793 ± 0.003 | 0.807 ± 0.005 | 0.806 ± 0.003 | 0.612 ± 0.006 | |
| MLP | 0.813 ± 0.015 | 0.681 ± 0.012 | 0.843 ± 0.014 | 0.739 ± 0.013 | 0.761 ± 0.012 | 0.531 ± 0.023 | |
| CKSAAP | RF | 0.840 ± 0.004 | 0.813 ± 0.009 | 0.846 ± 0.005 | 0.825 ± 0.003 | 0.829 ± 0.002 | 0.659 ± 0.006 |
| SVM | 0.877 ± 0.005 | 0.726 ± 0.009 | 0.900 ± 0.006 | 0.793 ± 0.004 | 0.812 ± 0.002 | 0.635 ± 0.005 | |
| LR | 0.737 ± 0.009 | 0.742 ± 0.012 | 0.736 ± 0.012 | 0.738 ± 0.009 | 0.738 ± 0.009 | 0.477 ± 0.018 | |
| NB | 0.819 ± 0.003 | 0.831 ± 0.004 | 0.817 ± 0.004 | 0.824 ± 0.003 | 0.823 ± 0.003 | 0.648 ± 0.006 | |
| KNN | 0.763 ± 0.008 | 0.860 ± 0.007 | 0.732 ± 0.010 | 0.808 ± 0.006 | 0.796 ± 0.006 | 0.598 ± 0.011 | |
| MLP | 0.831 ± 0.008 | 0.733 ± 0.006 | 0.852 ± 0.007 | 0.779 ± 0.005 | 0.792 ± 0.005 | 0.589 ± 0.010 | |
| PSSM | RF | 0.909 ± 0.004 | 0.900 ± 0.005 | 0.911 ± 0.003 | 0.904 ± 0.004 | 0.905 ± 0.003 | 0.811 ± 0.007 |
| SVM | 0.933 ± 0.001 | 0.861 ± 0.008 | 0.939 ± 0.003 | 0.895 ± 0.004 | 0.900 ± 0.003 | 0.803 ± 0.006 | |
| LR | 0.808 ± 0.007 | 0.851 ± 0.016 | 0.797 ± 0.011 | 0.828 ± 0.008 | 0.824 ± 0.006 | 0.649 ± 0.012 | |
| NB | 0.888 ± 0.004 | 0.887 ± 0.003 | 0.889 ± 0.003 | 0.887 ± 0.004 | 0.888 ± 0.003 | 0.776 ± 0.006 | |
| KNN | 0.899 ± 0.003 | 0.911 ± 0.003 | 0.898 ± 0.003 | 0.904 ± 0.003 | 0.904 ± 0.003 | 0.809 ± 0.005 | |
| MLP | 0.935 ± 0.013 | 0.859 ± 0.010 | 0.943 ± 0.010 | 0.895 ± 0.009 | 0.902 ± 0.008 | 0.806 ± 0.016 | |
| PSSM_AC | RF | 0.906 ± 0.006 | 0.771 ± 0.009 | 0.921 ± 0.005 | 0.832 ± 0.007 | 0.846 ± 0.006 | 0.699 ± 0.012 |
| SVM | 0.897 ± 0.012 | 0.765 ± 0.022 | 0.914 ± 0.012 | 0.825 ± 0.015 | 0.839 ± 0.012 | 0.686 ± 0.022 | |
| LR | 0.720 ± 0.011 | 0.757 ± 0.012 | 0.705 ± 0.015 | 0.736 ± 0.008 | 0.730 ± 0.008 | 0.463 ± 0.017 | |
| NB | 0.610 ± 0.001 | 0.867 ± 0.003 | 0.447 ± 0.003 | 0.715 ± 0.002 | 0.656 ± 0.002 | 0.346 ± 0.006 | |
| KNN | 0.833 ± 0.004 | 0.816 ± 0.004 | 0.836 ± 0.004 | 0.823 ± 0.002 | 0.825 ± 0.002 | 0.652 ± 0.006 | |
| MLP | 0.896 ± 0.021 | 0.690 ± 0.009 | 0.921 ± 0.018 | 0.777 ± 0.007 | 0.805 ± 0.007 | 0.628 ± 0.018 | |
| PSSM_SMTH | RF | 0.859 ± 0.006 | 0.825 ± 0.007 | 0.865 ± 0.006 | 0.840 ± 0.005 | 0.844 ± 0.005 | 0.691 ± 0.011 |
| SVM | 0.873 ± 0.007 | 0.790 ± 0.014 | 0.886 ± 0.004 | 0.828 ± 0.010 | 0.837 ± 0.008 | 0.679 ± 0.017 | |
| LR | 0.733 ± 0.017 | 0.734 ± 0.014 | 0.730 ± 0.026 | 0.732 ± 0.008 | 0.732 ± 0.011 | 0.466 ± 0.020 | |
| NB | 0.658 ± 0.003 | 0.870 ± 0.002 | 0.548 ± 0.006 | 0.748 ± 0.001 | 0.708 ± 0.002 | 0.441 ± 0.006 | |
| KNN | 0.804 ± 0.004 | 0.784 ± 0.005 | 0.809 ± 0.007 | 0.793 ± 0.003 | 0.796 ± 0.005 | 0.594 ± 0.010 | |
| MLP | 0.886 ± 0.016 | 0.756 ± 0.022 | 0.909 ± 0.013 | 0.815 ± 0.018 | 0.835 ± 0.016 | 0.675 ± 0.030 | |
| DISO | RF | 0.714 ± 0.011 | 0.733 ± 0.015 | 0.708 ± 0.011 | 0.722 ± 0.012 | 0.719 ± 0.011 | 0.441 ± 0.022 |
| SVM | 0.736 ± 0.016 | 0.726 ± 0.020 | 0.739 ± 0.020 | 0.728 ± 0.015 | 0.732 ± 0.014 | 0.466 ± 0.027 | |
| LR | 0.604 ± 0.008 | 0.607 ± 0.018 | 0.602 ± 0.020 | 0.603 ± 0.009 | 0.603 ± 0.007 | 0.209 ± 0.016 | |
| NB | 0.631 ± 0.026 | 0.657 ± 0.033 | 0.625 ± 0.009 | 0.637 ± 0.033 | 0.640 ± 0.016 | 0.283 ± 0.037 | |
| KNN | 0.695 ± 0.005 | 0.746 ± 0.008 | 0.674 ± 0.010 | 0.718 ± 0.004 | 0.709 ± 0.004 | 0.422 ± 0.006 | |
| MLP | 0.733 ± 0.016 | 0.570 ± 0.032 | 0.791 ± 0.016 | 0.639 ± 0.022 | 0.680 ± 0.014 | 0.371 ± 0.028 | |
| SA | RF | 0.611 ± 0.005 | 0.642 ± 0.010 | 0.590 ± 0.005 | 0.623 ± 0.008 | 0.613 ± 0.006 | 0.232 ± 0.013 |
| SVM | 0.604 ± 0.010 | 0.606 ± 0.022 | 0.600 ± 0.022 | 0.601 ± 0.013 | 0.600 ± 0.010 | 0.206 ± 0.018 | |
| LR | 0.585 ± 0.014 | 0.591 ± 0.015 | 0.581 ± 0.016 | 0.585 ± 0.012 | 0.583 ± 0.012 | 0.172 ± 0.026 | |
| NB | 0.543 ± 0.006 | 0.911 ± 0.011 | 0.207 ± 0.007 | 0.672 ± 0.006 | 0.560 ± 0.007 | 0.179 ± 0.015 | |
| KNN | 0.633 ± 0.014 | 0.498 ± 0.007 | 0.711 ± 0.019 | 0.555 ± 0.008 | 0.603 ± 0.010 | 0.214 ± 0.020 | |
| MLP | 0.576 ± 0.019 | 0.449 ± 0.036 | 0.671 ± 0.017 | 0.502 ± 0.030 | 0.560 ± 0.014 | 0.123 ± 0.032 | |
| SS | RF | 0.560 ± 0.022 | 0.535 ± 0.030 | 0.579 ± 0.016 | 0.544 ± 0.025 | 0.555 ± 0.022 | 0.115 ± 0.046 |
| SVM | 0.562 ± 0.021 | 0.463 ± 0.043 | 0.634 ± 0.023 | 0.492 ± 0.034 | 0.540 ± 0.021 | 0.102 ± 0.037 | |
| LR | 0.536 ± 0.017 | 0.542 ± 0.022 | 0.531 ± 0.018 | 0.537 ± 0.019 | 0.536 ± 0.018 | 0.073 ± 0.037 | |
| NB | 0.543 ± 0.007 | 0.673 ± 0.018 | 0.432 ± 0.010 | 0.597 ± 0.012 | 0.555 ± 0.007 | 0.111 ± 0.015 | |
| KNN | 0.530 ± 0.017 | 0.493 ± 0.018 | 0.564 ± 0.020 | 0.505 ± 0.017 | 0.524 ± 0.016 | 0.057 ± 0.032 | |
| MLP | 0.535 ± 0.024 | 0.361 ± 0.029 | 0.688 ± 0.032 | 0.428 ± 0.025 | 0.525 ± 0.018 | 0.052 ± 0.037 | |
| Group 1 | RF | 0.835 ± 0.004 | 0.825 ± 0.003 | 0.838 ± 0.005 | 0.829 ± 0.002 | 0.831 ± 0.003 | 0.663 ± 0.006 |
| SVM | 0.850 ± 0.008 | 0.833 ± 0.004 | 0.854 ± 0.010 | 0.840 ± 0.004 | 0.842 ± 0.005 | 0.687 ± 0.011 | |
| LR | 0.828 ± 0.009 | 0.829 ± 0.017 | 0.829 ± 0.011 | 0.827 ± 0.010 | 0.828 ± 0.009 | 0.658 ± 0.018 | |
| NB | 0.831 ± 0.001 | 0.820 ± 0.004 | 0.835 ± 0.003 | 0.824 ± 0.002 | 0.826 ± 0.003 | 0.654 ± 0.005 | |
| KNN | 0.805 ± 0.002 | 0.849 ± 0.004 | 0.796 ± 0.003 | 0.825 ± 0.002 | 0.821 ± 0.001 | 0.645 ± 0.003 | |
| MLP | 0.882 ± 0.005 | 0.743 ± 0.017 | 0.902 ± 0.007 | 0.805 ± 0.009 | 0.821 ± 0.007 | 0.652 ± 0.013 | |
| Group 2 | RF | 0.930 ± 0.003 | 0.865 ± 0.004 | 0.935 ± 0.003 | 0.895 ± 0.003 | 0.899 ± 0.003 | 0.802 ± 0.006 |
| SVM | 0.938 ± 0.003 | 0.856 ± 0.012 | 0.945 ± 0.003 | 0.895 ± 0.007 | 0.900 ± 0.005 | 0.804 ± 0.010 | |
| LR | 0.827 ± 0.012 | 0.852 ± 0.009 | 0.822 ± 0.013 | 0.838 ± 0.007 | 0.836 ± 0.006 | 0.674 ± 0.012 | |
| NB | 0.679 ± 0.003 | 0.881 ± 0.002 | 0.584 ± 0.006 | 0.765 ± 0.001 | 0.731 ± 0.002 | 0.487 ± 0.006 | |
| KNN | 0.905 ± 0.006 | 0.894 ± 0.004 | 0.907 ± 0.007 | 0.899 ± 0.005 | 0.900 ± 0.004 | 0.800 ± 0.008 | |
| MLP | 0.964 ± 0.007 | 0.789 ± 0.052 | 0.972 ± 0.007 | 0.864 ± 0.033 | 0.879 ± 0.026 | 0.775 ± 0.044 | |
| Group 3 | RF | 0.730 ± 0.012 | 0.737 ± 0.014 | 0.728 ± 0.015 | 0.731 ± 0.011 | 0.730 ± 0.010 | 0.465 ± 0.019 |
| SVM | 0.742 ± 0.011 | 0.736 ± 0.017 | 0.744 ± 0.014 | 0.737 ± 0.013 | 0.738 ± 0.012 | 0.481 ± 0.020 | |
| LR | 0.622 ± 0.006 | 0.629 ± 0.017 | 0.617 ± 0.009 | 0.623 ± 0.010 | 0.621 ± 0.007 | 0.246 ± 0.016 | |
| NB | 0.582 ± 0.010 | 0.829 ± 0.016 | 0.393 ± 0.014 | 0.679 ± 0.010 | 0.611 ± 0.010 | 0.250 ± 0.028 | |
| KNN | 0.718 ± 0.006 | 0.703 ± 0.009 | 0.725 ± 0.010 | 0.708 ± 0.006 | 0.712 ± 0.006 | 0.427 ± 0.011 | |
| MLP | 0.684 ± 0.009 | 0.445 ± 0.021 | 0.794 ± 0.014 | 0.536 ± 0.015 | 0.619 ± 0.007 | 0.255 ± 0.014 | |
| All features | RF | 0.912 ± 0.005 | 0.860 ± 0.006 | 0.919 ± 0.004 | 0.885 ± 0.005 | 0.889 ± 0.005 | 0.779 ± 0.008 |
| SVM | 0.931 ± 0.004 | 0.864 ± 0.009 | 0.937 ± 0.003 | 0.896 ± 0.007 | 0.900 ± 0.006 | 0.803 ± 0.010 | |
| LR | 0.887 ± 0.006 | 0.873 ± 0.010 | 0.890 ± 0.006 | 0.878 ± 0.007 | 0.880 ± 0.006 | 0.762 ± 0.012 | |
| NB | 0.809 ± 0.005 | 0.885 ± 0.003 | 0.792 ± 0.007 | 0.844 ± 0.002 | 0.838 ± 0.003 | 0.680 ± 0.007 | |
| KNN | 0.900 ± 0.006 | 0.887 ± 0.006 | 0.904 ± 0.006 | 0.893 ± 0.003 | 0.894 ± 0.002 | 0.790 ± 0.005 | |
| MLP | 0.943 ± 0.009 | 0.715 ± 0.039 | 0.956 ± 0.007 | 0.806 ± 0.027 | 0.833 ± 0.017 | 0.692 ± 0.026 |
Note: The values were expressed as mean ± standard error. Except for AAC (20 D) and PPT (72 D), all the feature vectors were 200-dimensional, and their selection was performed using GainRatio. Group 1 denotes the AAC group (AAC, DPC, CKSAAP and PPT); Group 2 denotes the PSSM group (PSSM, PSSM_AC and PSSM_SMTH); Group 3 denotes the structure group (SA, SS and DISO), while all features include all the 10 feature types and are used as a whole group. For each group, individual models were trained with the corresponding group and then integrated as an ensemble model using the majority vote scheme. For each performance measure, the best performance value across different machine learning methods within a feature group is highlighted in bold for clarification. These highlights also apply to Tables 3, 4 and 6.
Figure 5.
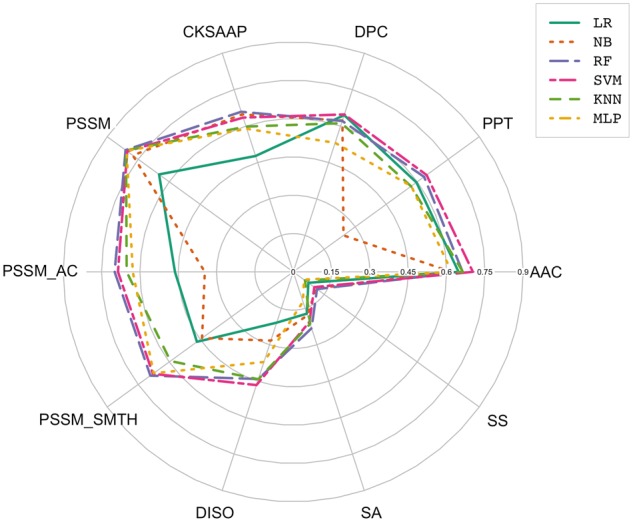
Prediction performance of different machine learning models trained with various feature encodings in terms of MCC on the 5-fold cross-validation test.
Figure 6.

ROC curves of RF, SVM, NB, KNN, LR and MLP predictors of T4SEs with different feature encodings. Group 1 denotes the AAC group (AAC, DPC, CKSAAP and PPT); Group 2 denotes the PSSM group (PSSM, PSSM_AC and PSSM_SMTH); Group 3 denotes the structure group (SA, SS and DISO), while all features include all the 10 feature types and are used as a whole group. For each group, individual models were trained with the corresponding group and then integrated as an ensemble model using the majority vote scheme.
Performance evaluation of various classifiers
For most of the feature encodings, RF and SVM predictors clearly outperformed the other classifiers in terms of ACC, F-score and MCC (Table 2, Figures 5 and 6). This observation is consistent with and supports the conclusion drawn by Fernández-Delgado et al. [68], who found that RF and SVM are most likely the best classifiers among all compared 17 machine learning algorithms based on 112 different data sets. Among all the classifiers corresponding to various feature encoding methods, RF classifiers achieved the highest F-score (0.905) and MCC (0.811) when PSSM was used for training.
To make a fair performance comparison of a variety of different classifiers, the trade-off between SN and SP was taken into consideration. The difference between SN and SP for RF models, in most cases, is lower than for other models. This implies that the RF classifier provides a better trade-off between SN and SP, and achieves a more comprehensive and stable performance on the prediction of T4SEs. As an ensemble classifier, RF can even fit training data that suffers heavily from noisy, high-dimensional and highly correlated features without over-fitting [76].
To evaluate the computational efficiency of various classifiers, we compared the computational time for each classifier, using 200-dimensional PSSM features (selected by GainRatio) for model training. The total computational time for each classifier included parameter tuning time (Tuning time) and randomized 5-fold cross-validation time (CV time). As can be seen in Figure 7A, SVM and MLP were most time-consuming among all methods in terms of the total computational process, which consists of parameter tuning and model training. Parameter tuning for SVM was computationally costliest (Figure 7C), highlighting difficulties associated with optimizing parameters for SVM models. In contrast, training MLP model (without performing parameter optimization in advance, which is another extremely complex task)-associated cross-validations are most time-consuming. Finally, when compared with SVM, RF achieved a better trade-off with remarkably less tuning time and only slightly longer CV time (Figure 7B and C).
Figure 7.

(A) Computational time of various classifiers when using the PSSM feature for training (after applying feature selection to form a 200-dimensional vector using GainRatio). Parameter tuning time and CV time are counted into the overall computational time for the classifiers. For classifiers without parameter optimization (LR, NB and MLP), the tuning time is 0. (B) and (C) represent the proportions of parameter tuning time and CV time of RF and SVM, respectively.
Performance evaluation of various feature encoding schemes
Among all feature encoding schemes, the most powerful one is PSSM (Table 2), achieving the highest AUC values for five of six classifiers when compared with other feature encodings (Figure 6). The local sequence encoding and global sequence encoding (except for PSSM) achieved similar performances, while the structural descriptor encoding showed a poor performance (Figure 6). CKSAAP performed worse than DPC for most classifiers (Table 2 and Figure 6): a possible explanation is that DPC might recognize the most valuable patterns in protein sequences, while CKSAAP may introduce redundant and noisy information that reduce the performance of T4SE prediction.
We explored the contribution of all features and three distinctive groups of them (AAC group, PSSM group and structure group) in two ways: feature ensemble and feature combination. For feature ensemble, we trained single-feature models and then integrated these as an ensemble model. For feature combination, features were first combined into a vector to train a model.
As shown, for each machine learning method, models trained based on all features and the three distinctive groups using feature ensemble (Figure 6 and Table 2) outperformed those trained using feature combination (Supplementary Figure S1 and Supplementary Table S1). When compared with single feature-based models, feature ensemble models achieved more stable performance across various machine learning methods (Figure 6 and Table 2).
Performance evaluation of feature selection methods
To remove redundant features and properly characterize feature importance, we conducted feature selection experiments (Figure 8). For different feature encodings, models trained using GainRatio-selected features (such as the top 50, 100, 150, 200, 250, 300 and 350 features) generally resulted in a comparable or, in some cases, better performance compared with models trained using all original features (Figure 8A). This finding indicates that the most discriminative features from the original set could be extracted to form a subset that preserved the original semantics of the variables. Owing to the removal of noisy features, a selected feature set is also likely to be better modeled and interpreted by machine learning methods [77]. It is also advantageous to use selected feature sets, which can help significantly reduce the computational time during model training. This is especially so for feature encodings with a large number of features (such as PSSM_SMTH). By using the mRMR feature selection, we obtained similar results as with GainRatio (Figure 8B). It is noteworthy that mRMR failed to recognize an informative feature set for PSSM_SMTH encoding, leading to a decreased performance after feature selection as compared with the full original feature set. A side-by-side performance comparison of GainRatio and mRMR revealed that, overall, GainRatio achieved a more stable performance (Figure 8C).
Figure 8.

Feature selection by using GainRatio and mRMR methods. The error bars indicate the SDs of MCC values over five different randomized 5-fold cross-validation tests. (A) Performance of various feature encodings with different numbers of top features selected by GainRatio; (B) performance of various feature encodings with different numbers of top features selected by mRMR; (C) side-by-side performance comparison of various feature encodings with different numbers of top features selected by GainRatio versus mRMR.
Performance comparison of models trained using individual feature types versus feature combinations
Although previous studies have used a combination of features to train prediction models [17, 18, 28], our experiments indicate that simply combining features did not help in further enhancing the model performance. Classifiers trained with different combinations of feature types did not show improved performances, compared with the model trained using PSSM feature encoding only (Figure 9). There are possible reasons for this. As the PSSM features dominate others for T4SE prediction [17] (also refer to Figure 6, Table 2), the performance of a feature combination model could largely depend on the proportion of PSSM features among the combined features. Other features, when directly combined with the PSSM features, may not contribute to the performance improvement and/or could even result in a decreased performance.
Figure 9.

Performance comparison of models trained using single features versus combined features, based on 5-fold cross-validation using the training data set. Combine1 denotes the composition of PSSM and PSSM_AC; Combine2 denotes the composition of PSSM, PSSM_AC and AAC; Combine3 denotes the composition of PSSM, PSSM_AC, AAC and DPC.
These observations support training single-feature models and subsequently assembling them into ensemble models, instead of merging all features into a vector to train a model.
A majority voting strategy based on ensemble learning models further improves the prediction performance
We first assessed the performance of various classifiers (single-feature encoding-based models and ensemble models) using RF by performing independent tests. All experiments were conducted five times. Each time, 30 negative randomly chosen samples were used to form the balanced independent data set along with the positive samples. The performance results are shown in Table 3 and Figure 10. The predictive performance of models trained by single-feature encodings showed a highly consistent trend with respect to the performance evaluation based on 5-fold cross-validation, further confirming the effectiveness of local and global sequence encodings (Table 3 and Figure 10).
Table 3.
The performance of various classifiers based on the independent tests
| Model a | Voting b | PRE | SN | SP | F-value | ACC | MCC |
|---|---|---|---|---|---|---|---|
| 1. AAC | – | 0.826 ± 0.044 | 1.000 ± 0.000 | 0.787 ± 0.069 | 0.904 ± 0.027 | 0.893 ± 0.035 | 0.806 ± 0.057 |
| 2. PPT | – | 0.787 ± 0.057 | 0.967 ± 0.000 | 0.733 ± 0.091 | 0.867 ± 0.035 | 0.850 ± 0.046 | 0.721 ± 0.077 |
| 3. DPC | – | 0.791 ± 0.039 | 0.900 ± 0.000 | 0.760 ± 0.055 | 0.842 ± 0.022 | 0.830 ± 0.027 | 0.667 ± 0.050 |
| 4. CKSAAP | – | 0.839 ± 0.014 | 0.933 ± 0.000 | 0.820 ± 0.018 | 0.883 ± 0.008 | 0.877 ± 0.009 | 0.758 ± 0.017 |
| 5. PSSM | – | 0.821 ± 0.033 | 1.000 ± 0.000 | 0.780 ± 0.051 | 0.901 ± 0.020 | 0.890 ± 0.025 | 0.800 ± 0.042 |
| 6. PSSM_AC | – | 0.882 ± 0.049 | 0.833 ± 0.000 | 0.887 ± 0.051 | 0.857 ± 0.023 | 0.860 ± 0.025 | 0.722 ± 0.053 |
| 7. PSSM_SMTH | – | 0.811 ± 0.080 | 0.800 ± 0.000 | 0.807 ± 0.095 | 0.804 ± 0.039 | 0.803 ± 0.048 | 0.609 ± 0.097 |
| 8. DISO | – | 0.778 ± 0.061 | 0.800 ± 0.000 | 0.767 ± 0.082 | 0.788 ± 0.032 | 0.783 ± 0.041 | 0.568 ± 0.080 |
| 9. SA | – | 0.645 ± 0.059 | 0.667 ± 0.000 | 0.627 ± 0.095 | 0.655 ± 0.030 | 0.647 ± 0.048 | 0.294 ± 0.095 |
| 10. SS | – | 0.665 ± 0.065 | 0.700 ± 0.000 | 0.640 ± 0.092 | 0.681 ± 0.032 | 0.670 ± 0.046 | 0.342 ± 0.093 |
| {1, 2, 3, 4} | 3-in-4 | 0.854 ± 0.025 | 0.967 ± 0.000 | 0.833 ± 0.033 | 0.906 ± 0.014 | 0.900 ± 0.017 | 0.807 ± 0.030 |
| {5, 6, 7} | 2-in-3 | 0.880 ± 0.048 | 0.867 ± 0.000 | 0.880 ± 0.051 | 0.873 ± 0.023 | 0.873 ± 0.025 | 0.748 ± 0.052 |
| {8, 9, 10} | 2-in-3 | 0.788 ± 0.092 | 0.800 ± 0.000 | 0.773 ± 0.123 | 0.792 ± 0.047 | 0.787 ± 0.062 | 0.576 ± 0.121 |
| {1, 2, 3, 4, 5, 6, 7} | 4-in-7 | 0.854 ± 0.030 | 0.967 ± 0.000 | 0.833 ± 0.041 | 0.907 ± 0.017 | 0.900 ± 0.020 | 0.808 ± 0.036 |
| {1, 2, 3, 4, 8, 9, 10} | 4-in-7 | 0.850 ± 0.045 | 0.967 ± 0.000 | 0.827 ± 0.060 | 0.904 ± 0.025 | 0.897 ± 0.030 | 0.802 ± 0.054 |
| {5, 6, 7, 8, 9, 10} | 4-in-6 | 0.903 ± 0.058 | 0.900 ± 0.000 | 0.900 ± 0.067 | 0.901 ± 0.029 | 0.900 ± 0.033 | 0.801 ± 0.066 |
| {1, 2, 3, 4, 5, 6, 7, 8} | 5-in-8 | 0.918 ± 0.025 | 0.967 ± 0.000 | 0.913 ± 0.030 | 0.942 ± 0.014 | 0.940 ± 0.015 | 0.882 ± 0.028 |
| {1-10} | 6-in-10 | 0.908 ± 0.045 | 0.967 ± 0.000 | 0.900 ± 0.053 | 0.936 ± 0.024 | 0.933 ± 0.026 | 0.869 ± 0.050 |
| {1, 3, 5, 6, 8, 10} | 4-in-6 | 0.922 ± 0.042 | 1.000 ± 0.000 | 0.913 ± 0.051 | 0.959 ± 0.023 | 0.957 ± 0.025 | 0.918 ± 0.046 |
Note: aEach term in this column refers to a single encoding-based model or an ensemble model of different single encoding-based models (e.g. 1. AAC means the model trained with AAC encoding features, while {5, 6, 7} stands for the ensemble model of number 5, 6 and 7 classifiers).
bThe majority voting scheme was used for voting in ensemble models.
Figure 10.

The ROC curve of both single encoding-based models and ensemble models based on the independent test.
Ensemble models based on selections of single-feature-encoding models were assessed in combination with majority voting, to determine whether this could further improve the predictive performance. Table 3 reports only on a few representative ensemble models selected after comprehensively examining the behaviors of all possible combinations of single-feature models. Several important conclusions were drawn from these results. First, ensemble models achieved a better and more robust performance as compared with single encoding-based models. In particular, the majority voting scheme {1, 3, 5, 6, 8, 10} achieved the overall best performance, with a maximum accuracy of 95.7%, an F-score of 0.959 and an MCC value of 0.918 (Table 3). Second, combinations of similar-feature-group encoding-based models did not lead to visible performance improvement. This has been observed in the case of ensemble classifiers {1, 2, 3} (AAC feature group), {5, 6, 7} (PSSM feature group) and {8, 9, 10} (structural feature group). On the other hand, ensembles of models that were trained on different feature groups resulted in clear performance improvements, e.g. ensemble classifiers {1, 2, 3, 4, 5, 6, 7}, {1, 2, 3, 4, 8, 9, 10}, {5, 6, 7, 8, 9, 10} and {1–10}. This is in agreement with the result in [17] by exploring the vote of various feature-based models (including two sequence-based models and two PSSM-based models). The ensemble classifier {1, 2, 3, 4, 5, 6, 7, 8} is an excellent example portraying the advantages of ensemble learning. Comparing it with the ensemble classifier {1, 2, 3, 4, 5, 6, 7} showed that the DISO feature-based model still contributes to an improved performance of the ensemble classifier, while it only gives a moderate performance when used as a single-feature model.
For each of machine learning methods (i.e. SVM, KNN, NB, LR and MLP), we trained an ensemble model by integrating eight top single-feature-based models (i.e. AAC, PPT, DPC, CKSAAP, PSSM, PSSM_AC, PSSM_SMTH and DISO). By further integrating ensemble models with the majority vote scheme, we studied the prediction performance of these single machine learning-based models and their ensemble models using the independent test. As shown in Table 4, the RF- and SVM-based models outperformed other method-based models, while the ensemble model of these two models ({a, b}) further improved the prediction performance. The ensemble model integrating all six method-based models ({a, b, c, d, e, f}) achieved the best performance in terms of F-value, ACC and MCC, consistent with the observations reported in [19]. Based on these findings, we constructed Bastion4 with a default setting: all six machine learning methods were integrated, and for each of them the eight top single-feature-based models were generated for assembling.
Table 4.
The performance of various machine learning methods based on the independent tests
| Method a | Voting b | PRE | SN | SP | F-value | ACC | MCC |
|---|---|---|---|---|---|---|---|
| a. RF | – | 0.918 ± 0.025 | 0.967 ± 0.000 | 0.913 ± 0.030 | 0.942 ± 0.014 | 0.940 ± 0.015 | 0.882 ± 0.028 |
| b. SVM | – | 0.940 ± 0.014 | 0.933 ± 0.000 | 0.940 ± 0.015 | 0.936 ± 0.007 | 0.937 ± 0.007 | 0.873 ± 0.015 |
| c. KNN | – | 0.880 ± 0.055 | 1.000 ± 0.000 | 0.860 ± 0.072 | 0.935 ± 0.031 | 0.930 ± 0.036 | 0.870 ± 0.064 |
| d. NB | – | 0.834 ± 0.033 | 0.933 ± 0.000 | 0.813 ± 0.045 | 0.881 ± 0.019 | 0.873 ± 0.022 | 0.753 ± 0.041 |
| e. LR | – | 0.875 ± 0.056 | 1.000 ± 0.000 | 0.853 ± 0.069 | 0.932 ± 0.031 | 0.927 ± 0.035 | 0.864 ± 0.063 |
| f. MLP | – | 0.906 ± 0.004 | 0.960 ± 0.043 | 0.900 ± 0.000 | 0.932 ± 0.023 | 0.930 ± 0.022 | 0.862 ± 0.046 |
| {a, b} | 2-in-2 | 0.966 ± 0.024 | 0.933 ± 0.000 | 0.967 ± 0.024 | 0.949 ± 0.011 | 0.950 ± 0.012 | 0.901 ± 0.024 |
| {a, b, c} | 2-in-3 | 0.918 ± 0.025 | 0.967 ± 0.000 | 0.913 ± 0.030 | 0.942 ± 0.014 | 0.940 ± 0.015 | 0.882 ± 0.028 |
| {a, b, c, d} | 3-in-4 | 0.918 ± 0.025 | 0.967 ± 0.000 | 0.913 ± 0.030 | 0.942 ± 0.014 | 0.940 ± 0.015 | 0.882 ± 0.028 |
| {a, b, c, d, e} | 3-in-5 | 0.907 ± 0.028 | 0.967 ± 0.000 | 0.900 ± 0.033 | 0.936 ± 0.015 | 0.933 ± 0.017 | 0.869 ± 0.031 |
| {a, b, c, d, e, f} | 4-in-6 | 0.942 ± 0.025 | 0.967 ± 0.000 | 0.940 ± 0.028 | 0.954 ± 0.013 | 0.953 ± 0.014 | 0.907 ± 0.027 |
Note: aEach term in this column refers to a single method-based model or an ensemble model that integrates different single method-based models (e.g. ‘a. RF’ means the model is trained based on the RF method, while ‘{a, b, c}’ stands for the ensemble model that integrates a, b and c models).
bThe majority voting scheme is used for voting in ensemble models.
Performance evaluation of specific training data sets
To investigate whether the diversity of positive samples affects the performance of the predictors, we trained another two predictors using a part of the training data set. In more detail, from the positive samples in the training data set, 291 Legionella samples were chosen to construct a new balanced independent data set together with randomly selected negative samples from V.parahaemolyticus serotype O3: K6. The remaining 99 positive samples and an equal number of negative samples from the original training set were used to form a new training data set. Based on this new data set, we used eight feature types (i.e. AAC, PPT, DPC, CKSAAP, PSSM, PSSM_AC, PSSM_SMTH and DISO) to train individual models and aggregated their outputs to form an ensemble model for each of the six machine learning methods. We further integrated all these single method-based models with the majority vote scheme to construct a new predictor (labeled ‘Predictor_without_Legionella’). The new independent data set (containing all 291 Legionella samples as positives) was used to analyze the predictive performance. We applied the same procedures to construct a predictor (labeled ‘Predictor_without_Coxiella’) and analyzed its performance on the new independent data set (containing all 60 Coxiella samples as positives). In addition, eight single models trained using the full training data set were assembled as a reference predictor (labelled ‘Predictor_with_Full_Dataset’). The overall performance of the three predictors was assessed based on their respective independent test data sets and is listed in Table 5.
Table 5.
Performance comparison between Predictor_without_Coxiella, Predictor_without_Legionella and Predictor_with_Full_Dataset based on the independent test
| Classifier | PRE | SN | SP | F-value | ACC | MCC |
|---|---|---|---|---|---|---|
| Predictor_with_Full_Dataset | 0.942 | 0.967 | 0.940 | 0.954 | 0.953 | 0.907 |
| Predictor_without_Coxiella | 1.000 | 0.733 | 1.000 | 0.846 | 0.867 | 0.761 |
| Predictor_without_Legionella | 0.841 | 0.691 | 0.869 | 0.758 | 0.780 | 0.569 |
The Predictor_with_Full_Dataset outperformed the Predictor_without_Coxiella and the Predictor_without_Legionella in terms of F-value, ACC and MCC (Table 5). These results indicate that the increase of samples diversity can improve the performance of predictors. Owing to limited training data, the Predictor_without_Legionella failed to achieve a competitive performance. Note that the high SN value of the Predictor_without_Coxiella suggests that it is well able to identify the Coxiella T4SEs even without using such data set for model training. This hints at an underlying similarity between Legionella, Coxiella and other T4SEs. Here, we used an unsupervised learning approach to investigate the potential similarity further. We encoded all T4SEs using PSSM encoding, partitioned them into three groups using the k-means clustering algorithm [78] and then performed dimension-reduction using t-SNE [79] to map to the 2D space for better visualization (Figure 11A). From Figure 11B–D, we can see that each of the identified three clusters is a mixture of Legionella, Coxiella and other T4SEs. This supports the idea that, because of their similarity, these types of T4SEs are not separable. The observation that Legionella samples dominate all three clusters can be attributed to their abundance in the original three classes of positive samples (Figure 11E).
Figure 11.

(A) Representation of the positive samples from Coxiella, Legionella and other T4SEs. The representation of each sample (which constituted a 400-dimensional space generated by the PSSM encoding scheme) was reduced to two dimensions by using t-SNE. Samples were clustered into three groups using K-means algorithm, and these three clusters were indicated by colors. (B–D) Detailed components of the three clusters. Each cluster contains samples from all three T4SE classes, namely, Coxiella, Legionella and others. (E) Detailed numbers and proportions of original three classes of samples.
While there are similarities between Legionella’s, Coxiella’s and other T4SEs, it is noteworthy that the performance of Predictor_without_Legionella was less than that of the Predictor_without_Coxiella. To explore why this is so, we used Clustal Omega [80] to do multiple sequence alignment on the T4SE data set. Based on the alignment results, a phylogenetic tree of T4SS effectors was generated (Supplementary Figure S2) using iTOL [81]. From inspection of Supplementary Figure S2, we found that T4SEs in Legionella, Coxiella and other species were often mixed, while some T4SEs in Legionella clustered alone (marked in light green). This finding indicated that some T4SEs in Legionella differ from those in other species, shedding light on why Predictor_without_Legionella could not distinguish some of T4SEs in Legionella species.
Performance comparison with existing tools
There are currently two tools [17, 18] available for T4SE prediction. Three classifiers (T4SEpred_bpbAac, T4SEpred_psAac and T4SEpre_Joint) were implemented in Wang et al. [18], while a second tool (T4Effpred) with multiple options was developed in Zou et al. [17]. Accordingly, we compared their performance on the independent test data set (Table 6).
Table 6.
Performance comparison between our ensemble classifier and other existing predictors based on the independent test
| Classifier | PRE | SN | SP | F-value | ACC | MCC |
| Bastion4 | 0.942 ± 0.025 | 0.967 ± 0.000 | 0.940 ± 0.028 | 0.954 ± 0.013 | 0.953 ± 0.014 | 0.907 ± 0.027 |
| T4Effpred | 0.940 ± 0.020 | 0.833 ± 0.000 | 0.947 ± 0.018 | 0.883 ± 0.009 | 0.890 ± 0.009 | 0.785 ± 0.020 |
| T4SEpred_bpbAac | 0.959 ± 0.060 | 0.433 ± 0.000 | 0.980 ± 0.030 | 0.597 ± 0.012 | 0.707 ± 0.015 | 0.495 ± 0.046 |
| T4SEpred_psAac | 0.983 ± 0.037 | 0.367 ± 0.000 | 0.993 ± 0.015 | 0.534 ± 0.006 | 0.680 ± 0.007 | 0.462 ± 0.026 |
All options of T4Effpred were explored but, for the sake of brevity, Table 6 only presents the best-performing model from among different T4Effpred variant models [17]: an ensemble model based on a 3-in-4 voting scheme. For the same reason, only the results of T4SEpred_bpbAac and T4SEpred_psAac are listed in Table 6. In Bastion4, default settings were used to construct the predictor. As can be seen from Table 6, Bastion4 achieved an overall accuracy of 95.3% with an F-value of 0.954 and an MCC of 0.907. This is the best performance among all compared predictors. T4Effpred achieved the second-best performance, also using an ensemble classifier based on multiple feature encodings. Moreover, we observed that both T4SEpred_bpbAac and T4SEpred_psAac performed poorly on the independent test data set.
The poorer performance of T4SEpred_bpbAac and T4SEpred_psAac is intriguing, especially considering important features of T4SE proteins that might be biologically important. The implementation of the two predictors did not extract features from the PSSM profiles, which are regarded as the primary features [18], and these have proved to be powerful for T4SE prediction both in our current work and in the work by Zou et al. [17]. Coupled with this, in T4SEpred_bpbAac and T4Sepred_psAac, only the 50 C-terminal amino acids, rather than whole protein sequences, were used to extract features [18]. As also shown in this study, pronounced sequence signals are present at the C-terminus of L. pneumophila effectors, but are not universal and diagnostic of all T4Ses. Our results presented here demonstrate that the complete sequences contain important information that is relevant for accurate T4SE prediction and, presumably, for their recognition by the T4SS.
Genome-wide prediction of T4SEs in Klebsiella pneumoniae
Klebsiella pneumoniae is emerging as a devastating pathogen of humans [82]. The T4SS of this pathogen has only been recently noted [83, 84], and effector proteins and T4SEs have not been identified to our knowledge. We took this opportunity to predict T4SEs with Bastion4 using our default settings, and to identify these on physical genome maps of three clinically relevant strains: K.penumoniae AJ218, B5055 and MGH 78578. Studies with other bacteria have identified the physical location of genomic regions encoding T4SEs [85–87], and the genes encoding certain T4SEs were found to be clustered within specific genomic regions with an observed bias in G + C content, leading to models, whereby T4SE genes are acquired by lateral gene transfer between different bacterial species [16, 88, 89].
Circular maps [90] of extant genome sequence data were generated (Figure 12) to graphically depict the relationships between genome properties and the distribution of predicted effectors in these genomes [91]. The G + C content of the tentative T4SEs in each of the three genomes is significantly lower than expected from the overall G + C contents (Table 7), all with significant P-values according to the Welch’s t-test. The Venn diagram in Figure 12D illustrates the distributions of both predicted strain-specific and common effectors across these three bacterial genomes. While they share some common effectors (four common effectors shared across the three strains), AJ218, B5055 and MGH 78578 had 42, 33 and 33 strain-specific effectors possibly because of relatively recent horizontal gene transfer events [89]. This is consistent with our knowledge that genes encoding effector proteins are often shared via lateral gene transfer from other species. In the K.penumoniae B5055 genome, there is a cluster of predicted T4SEs genes that sit spatially in the nearby genes encoding the components of the T4SS nanomachine. Taken together, the genome-wide predictions of T4SEs provide a basis to explore their genome-level distributions, and to build a compendium of novel putative T4SEs that can be characterized by genetic and biochemical experiments.
Figure 12.

(A–C) Circular maps of representation of the genomes of K. pneumoniae AJ218, B5055 and MGH 78578 strains and (D) Venn diagram of the distributions of predicted effectors in these three bacterial genomes. In the (A–C), the tracks from the outside to the inside represent: (1) locations of predicted T4SEs; (2) forward coding DNA sequence (CDS); (3) reverse CDS; (4) GC content (yellow denotes that G + C content is higher than the average; purple denotes that G + C content is lower than the average); and (5) GC skew [(GC)/(G + C)].
Table 7.
Statistical analysis of the G + C contents between the putative T4SEs and non-T4SEs in the K. pneumoniae strain AJ218, B5055 and MGH 78578 genomes
| Strain type | Mean of G + C content (%) |
P-value by Welch’s t-test | |
|---|---|---|---|
| Putative T4SEs | Non-T4SEs | ||
| AJ218 | 43.33 | 57.73 | 3.755e-16 |
| B5055 | 44.99 | 57.55 | 3.51e-11 |
| MGH 78578 | 45.45 | 57.99 | 4.314e-10 |
Note: For each species, the G + C content (%) of each sequence of putative T4SEs was calculated to form a percentage set. Similarly, the G + C content (% of each non-T4SE sequence was calculated to form another percentage set. Note that the percentage set of non-T4SE sequences was significantly larger than that of the putative T4SEs percentage set. Based on the two sets, the mean values of the G + C content (%) of both putative T4SEs and non-T4SEs were calculated. The Welch’s t-test was performed and P-value calculated to assess the statistical significance.
Availability of online Web servers
As an implementation of the methodology presented here, we developed Bastion4, an online Web server for characterizing protein sequences of interest. Bastion4 is freely accessible at http://bastion4.erc.monash.edu/. The Bastion4 Web server was programmed using the Perl CGI and J2EE framework, and configured on the cloud computing facility provided by the Monash University e-Research Centre. Users can submit multiple protein sequences in raw or FASTA format to the online Web server. The computational time of the Bastion4 server to process a submitted sequence depends not only on the length of the submitted sequence but also considerably on the choice of the selected models.
Conclusion
Identifying effector proteins is necessary to understand host–pathogen interactions and bacterial pathogenesis. Here, we have systematically assessed the use and performance of different protein sequence and protein structure-related features and their combinations along with various machine learning approaches for T4SE prediction. Our main findings are (1) of the six machine learning classifiers (NB, KNN, LR, RF, SVM and MLP), RF and SVM performed best according to the performance measures ACC, F-value and MCC based on 5-fold cross-validation, while RF achieved a good trade-off between the predictive performance and computational time; (2) of the 10 different features, PSSM achieved the highest performance values for all classifiers, emphasizing the importance of global sequence encoding with PSSMs; (3) ensemble models performed better than single-feature-based models; (4) when applied to the predictions of an ensemble model, the diversity in the selected features resulted in a more stable and accurate classification performance. These findings led to the development of Bastion4, a tool that reflects the state of the art in effector prediction for T4SEs. Together with the compendium of predicted tentative T4SEs of the three bacterial genomes, we anticipate Bastion4 to be extensively used for T4SE prediction and, in conjunction with comparative genomics of bacterial pathogens, to improve our understanding of host–pathogen interactions.
Supplementary Material
Biographies
Jiawei Wang is currently a PhD candidate in the Biomedicine Discovery Institute and the Department of Microbiology at Monash University, Australia. He received his bachelor degree in software engineering from Tongji University and his master degree in computer science from Peking University, China. His research interests are computational biology, bioinformatics, machine learning and data mining.
Bingjiao Yang received his master degree at the National Engineering Research Center for Equipment and Technology of Cold Strip Rolling, College of Mechanical Engineering from Yanshan University, China. His research interests are bioinformatics, machine learning and data mining.
Yi An is currently a master student in the College of Information Engineering, Northwest A&F University, China. During her stay as a visiting student at the Biomedicine Discovery Institute and Department of Microbiology at Monash University, she undertook a bioinformatics project focused on computational analysis of bacterial secreted effector proteins. Her research interests include bioinformatics, data mining and Web-based information systems.
Tatiana Marquez-Lago is an Associate Professor in the Department of Genetics, University of Alabama at Birmingham (UAB) School of Medicine, USA. She is additionally affiliated with the UAB Comprehensive Cancer Center and the Informatics Institute. Her research interests include systems biology and biomedicine, gene expression and bioengineering, big data informatics, multiscale modeling and simulations. Her interdisciplinary laboratory studies stochastic gene expression, chromatin organization and microbiota/microbiome interactions in complex diseases.
André Leier is currently an Assistant Professor in the Department of Genetics and the Informatics Institute, University of Alabama at Birmingham (UAB) School of Medicine, USA. He is also an associate scientist in the UAB Comprehensive Cancer Center. He received his PhD in Computer Science (Dr rer. nat.), University of Dortmund, Germany. He conducted postdoctoral research at Memorial University of Newfoundland, Canada, the University of Queensland, Australia and ETH Zürich, Switzerland. His research interests are in Biomedical Informatics and Computational and Systems Biomedicine.
Jonathan Wilksch received his PhD degree in 2012 from The University of Melbourne, Australia. He is a Research Fellow in the Department of Microbiology and Immunology at the University of Melbourne, Australia. His research background and current interests include the mechanisms of bacterial pathogenesis, biofilm formation, gene regulation and host–pathogen interactions.
Qingyang Hong received his bachelor degree from Central South University, China, and his master degree in computer science from the University of Melbourne, Australia. His research interests are machine learning and data mining.
Yang Zhang received his PhD degree in Computer Science and Engineering in 2015 from Northwestern Polytechnical University, China. He is a Professor and Vice-Dean in the College of Information Engineering, Northwest A&F University, China. His research interests are big data analytics, machine learning and data mining.
Morihiro Hayashida received his PhD degree in Informatics in 2005 from Kyoto University, Japan. He is an assistant professor in Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan. His research interests include functional analysis of proteins and development of computational methods.
Tatsuya Akutsu received his Dr Eng. degree in Information Engineering in 1989 from University of Tokyo, Japan. Since 2001, he has been a professor in Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan. His research interests include bioinformatics and discrete algorithms.
Geoffrey I. Webb received his PhD degree in 1987 from La Trobe University, Australia. He is a professor in the Faculty of Information Technology and director of the Monash Centre for Data Science at Monash University. His research interests include machine learning, data mining, computational biology and user modeling.
Richard A. Strugnell undertook his PhD training at Monash University and Postdoctoral research at University of Birmingham and the Wellcome Research Laboratories in the UK, and at Monash University in Australia. He is currently Pro Vice-Chancellor (Graduate and International Research) and Professor in the Department of Microbiology and Immunology, Faculty of Medicine Dentistry and Health Sciences, the University of Melbourne. His laboratory is interested in how bacteria cause disease and what interventions can be made to stop this happening.
Jiangning Song is a senior research fellow and group leader in the Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology, Monash University, Australia. He is affiliated with the Monash Centre for Data Science, Monash University. He is also an Associate Investigator at the ARC Centre of Excellence in Advanced Molecular Imaging, Monash University. His research interests include bioinformatics, systems biology, machine learning, functional genomics and enzyme engineering.
Trevor Lithgow received his PhD degree in 1992 from La Trobe University, Australia. He is an ARC Australian Laureate Fellow in the Biomedicine Discovery Institute and the Department of Microbiology at Monash University, Australia. His research interests particularly focus on molecular biology, cellular microbiology and bioinformatics. His laboratory develops and deploys multidisciplinary approaches to identify new protein transport machines in bacteria, understand the assembly of protein transport machines and dissect the effects of anti-microbial peptides on anti-biotic resistant ‘superbugs’.
Key Points
In this work, we systematically train and compare six commonly used machine learning models for accurate and efficient identification of T4SEs using 10 different types of selected features.
Our study shows that (1) including different but complementary features generally enhance the predictive performance of T4SEs; (2) ensemble models obtained by integrating individual single-feature models exhibit a significantly improved predictive performance. The majority voting strategy enables the ensemble models to achieve the most stable and accurate classification performance.
We propose and built a new ensemble model, Bastion4, to further improve the performance in predicting effector proteins of the T4SS. Independent tests demonstrate that the ensemble models outperform all current predictors of types IV secretion systems. We make Bastion4 publicly accessible at http://bastion4.erc.monash.edu/.
Genome-wide prediction of T4SEs provides important insights into the distribution of T4SEs in three bacterial pathogens. We provide a valuable compendium of novel T4SEs that can be further validated by genetic and biochemical experiments.
Funding
This work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC) (grant number 1092262), the Australian Research Council (ARC) (grant numbers LP110200333 and DP120104460) and the National Institute of Allergy and Infectious Diseases of the National Institute of Health (grant number R01 AI111965). G.I.W. is a recipient of the Discovery Outstanding Research Award (DORA) of the ARC. T.L. is an ARC Australian Laureate Fellow (grant number FL130100038).
References
- 1. Eicher SC, Christoph D.. Bartonella entry mechanisms into mammalian host cells. Cell Microbiol 2012;14(8):1166–73. 10.1111/j.1462-5822.2012.01806.x [DOI] [PubMed] [Google Scholar]
- 2. Rhomberg TA, Truttmann MC, Guye P.. A translocated protein of Bartonella hensela: interferes with endocytic uptake of individual bacteria and triggers uptake of large bacterial aggregates via the invasome. Cell Microbiol 2009;11(6):927–45. 10.1111/j.1462-5822.2009.01302.x [DOI] [PubMed] [Google Scholar]
- 3. Truttmann MC, Rhomberg TA, Dehio C.. Combined action of the type IV secretion effector proteins BepC and BepF promotes invasome formation of Bartonella henselae on endothelial and epithelial cells. Cell Microbiol 2011;13(2):284–99. 10.1111/j.1462-5822.2010.01535.x [DOI] [PubMed] [Google Scholar]
- 4. Navarro L, Alto NM, Dixon JE.. Functions of the Yersinia effector proteins in inhibiting host immune responses. Curr Opin Microbiol 2005;8(1):21–7. 10.1016/j.mib.2004.12.014 [DOI] [PubMed] [Google Scholar]
- 5. Mcghie EJ, Brawn LC, Hume PJ, et al. Salmonella takes control: effector-driven manipulation of the host. Curr Opin Microbiol 2009;12(1):117–24. 10.1016/j.mib.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O’Brien KM, Lindsay EL, Starai VJ.. The Legionella pneumophila effector protein, LegC7, alters yeast endosomal trafficking. PLoS One 2015;10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ku B, Lee KH, Park WS, et al. VipD of Legionella pneumophila targets activated Rab5 and Rab22 to interfere with endosomal trafficking in macrophages. PLoS Pathog 2012;8(12):e1003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hubber A, Roy CR.. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol 2010;26(1):261–83. 10.1146/annurev-cellbio-100109-104034 [DOI] [PubMed] [Google Scholar]
- 9. Clark CS, Maurelli AT.. Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells. Infect Immun 2007;75(5):2531–9. 10.1128/IAI.01866-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ashida H, Kim M, Sasakawa C.. Manipulation of the host cell death pathway by Shigella. Cell Microbiol 2014;16(12):1757–66. 10.1111/cmi.12367 [DOI] [PubMed] [Google Scholar]
- 11. Trosky JE, Liverman AD, Orth K.. Yersinia outer proteins: Yops. Cell Microbiol 2008;10(3):557–65. 10.1111/j.1462-5822.2007.01109.x [DOI] [PubMed] [Google Scholar]
- 12. Dong N, Zhu Y, Lu Q, et al. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 2012;150(5):1029–41. 10.1016/j.cell.2012.06.050 [DOI] [PubMed] [Google Scholar]
- 13. Green ER, Mecsas J.. Bacterial secretion systems: an overview. Microbiol Spectr 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gophna U, Ron EZ, Dan G.. Bacterial type III secretion systems are ancient and evolved by multiple horizontal-transfer events. Gene 2003;312:151–63. 10.1016/S0378-1119(03)00612-7 [DOI] [PubMed] [Google Scholar]
- 15. Burns DL. Type IV transporters of pathogenic bacteria. Curr Opin Microbiol 2003;6(1):29–34. 10.1016/S1369-5274(02)00006-1 [DOI] [PubMed] [Google Scholar]
- 16. Burstein D, Zusman T, Degtyar E, et al. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog 2009;5(7):6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zou L, Nan C, Hu F.. Accurate prediction of bacterial type IV secreted effectors using amino acid composition and PSSM profiles. Bioinformatics 2013;29(24):3135–42. 10.1093/bioinformatics/btt554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Wei X, Bao H, et al. Prediction of bacterial type IV secreted effectors by C-terminal features. BMC Genomics 2014;15(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zou L, Chen K. Computational prediction of bacterial type IV-B effectors using C-terminal signals and machine learning algorithms. In: 2016 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Chiang Mai, Thailand, 2016. IEEE, pp. 1–5.
- 20. An Y, Wang J, Li C, et al. Comprehensive assessment and performance improvement of effector protein predictors for bacterial secretion systems III, IV and VI. Brief Bioinform 2016, doi:10.1093/bib/bbw100. [DOI] [PubMed] [Google Scholar]
- 21. Zeng C, Zou L.. An account of in silico identification tools of secreted effector proteins in bacteria and future challenges. Brief Bioinform 2017, doi:10.1093/bib/bbx078. [DOI] [PubMed] [Google Scholar]
- 22. Huang Y, Niu B, Gao Y, et al. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26(5):680–2. 10.1093/bioinformatics/btq003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. UniProt Consortium. The universal protein resource (uniprot). Nucleic Acids Res 2010;38:D142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyer DF, Noroy C, Moumène A, et al. Searching algorithm for type IV secretion system effectors 1.0: a tool for predicting type IV effectors and exploring their genomic context. Nucleic Acids Res 2013;41:9218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Makino K, Oshima K, Kurokawa K, et al. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 2003;361(9359):743–9. [DOI] [PubMed] [Google Scholar]
- 26. Li K, Xu C, Jian H, et al. Prediction and identification of the effectors of heterotrimeric G proteins in rice (Oryza sativa L.). Brief Bioinform 2017;18:270–8. [DOI] [PubMed] [Google Scholar]
- 27. Wang XB, Wu LY, Wang YC, et al. Prediction of palmitoylation sites using the composition of k-spaced amino acid pairs. Protein Eng Des Sel 2009;22(11):707–12. 10.1093/protein/gzp055 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z, Zhou Y, Song J, et al. hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties. Biochim Biophys Acta 2013;1834(8):1461–7. 10.1016/j.bbapap.2013.04.006 [DOI] [PubMed] [Google Scholar]
- 29. Grynberg M, Godzik A.. Sequence-based prediction of type III secreted proteins. PLoS Pathog 2009;5:e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Delorenzi M, Speed T.. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 2002;18(4):617–25. 10.1093/bioinformatics/18.4.617 [DOI] [PubMed] [Google Scholar]
- 31. Kaur H, Raghava GPS.. Prediction of alpha-turns in proteins using PSI-BLAST profiles and secondary structure information. Proteins Struct Func Bioinform 2004;55(1):83–90. 10.1002/prot.10569 [DOI] [PubMed] [Google Scholar]
- 32. Kaur H, Raghava SPG.. A neural network method for prediction of beta-turn types in proteins using evolutionary information. Bioinformatics 2004;20(16):2751–8. 10.1093/bioinformatics/bth322 [DOI] [PubMed] [Google Scholar]
- 33. Xie D, Li A, Wang M, et al. LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res 2005;33:105–10. 10.1093/nar/gki359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu T, Zheng X, Wang J.. Prediction of protein structural class for low-similarity sequences using support vector machine and PSI-BLAST profile. Biochimie 2010;92(10):1330–4. 10.1016/j.biochi.2010.06.013 [DOI] [PubMed] [Google Scholar]
- 35. Chen SA, Ou YY, Lee TY, et al. Prediction of transporter targets using efficient RBF networks with PSSM profiles and biochemical properties. Bioinformatics 2011;27(15):2062–7. 10.1093/bioinformatics/btr340 [DOI] [PubMed] [Google Scholar]
- 36. Dong Q, Zhou S, Guan J.. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics 2009;25(20):2655–62. 10.1093/bioinformatics/btp500 [DOI] [PubMed] [Google Scholar]
- 37. Cheng-Wei C, Emily Chia-Yu S, Jenn-Kang H, et al. Predicting RNA-binding sites of proteins using support vector machines and evolutionary information. BMC Bioinformatics 2008;12:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J, Zhang Y, Qin W, et al. Using the improved position specific scoring matrix and ensemble learning method to predict drug-binding residues from protein sequences. Nat Sci 2012;04(05):304. [Google Scholar]
- 39. Wang J, Yang B, Revote J, et al. POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017;33:2756–8. 10.1093/bioinformatics/btx302 [DOI] [PubMed] [Google Scholar]
- 40. Chen K, Kurgan L.. PFRES: protein fold classification by using evolutionary information and predicted secondary structure. Bioinformatics 2007;23(21):2843–50. 10.1093/bioinformatics/btm475 [DOI] [PubMed] [Google Scholar]
- 41. Gnad F, Ren S, Cox J, et al. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol 2007;8(11):561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song J, Yuan Z, Tan H, et al. Predicting disulfide connectivity from protein sequence using multiple sequence feature vectors and secondary structure. Bioinformatics 2007;23(23):3147–54. 10.1093/bioinformatics/btm505 [DOI] [PubMed] [Google Scholar]
- 43. Li T, Du P, Xu N, Uversky VN.. Identifying human kinase-specific protein phosphorylation sites by integrating heterogeneous information from various sources. PLoS One 2010;5(11):419–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mizianty MJ, Stach W, Chen K, et al. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010;26(18):i489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song J, Tan H, Shen H, et al. Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 2010;26(6):752–60. 10.1093/bioinformatics/btq043 [DOI] [PubMed] [Google Scholar]
- 46. Magnan CN, Baldi P.. SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning, and structural similarity. Bioinformatics 2014;30(18):2592–7. 10.1093/bioinformatics/btu352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dunker AK, Obradovic Z.. The protein trinity-linking function and disorder. Nat Biotechnol 2001;19(9):805–6. 10.1038/nbt0901-805 [DOI] [PubMed] [Google Scholar]
- 48. Ward JJ, Sodhi JS, Mcguffin LJ, et al. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004;337(3):635–45. 10.1016/j.jmb.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 49. Radivojac P, Vacic V, Haynes C, et al. Identification, analysis, and prediction of protein ubiquitination sites. Proteins Struct Funct Bioinform 2010;78(2):365–80. 10.1002/prot.22555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aksoy S, Haralick RM.. Feature normalization and likelihood-based similarity measures for image retrieval. Pattern Recog Lett 2001;22(5):563–82. 10.1016/S0167-8655(00)00112-4 [DOI] [Google Scholar]
- 51. Guyon I, Elisseeff A.. An introduction to variable and feature selection. J Mach Learn Res 2002;3:1157–82. [Google Scholar]
- 52. Shannon CE. A mathematical theory of communication: the bell system technical journal. Bell Syst Tech J 1948;27(3):3–55. [Google Scholar]
- 53. Peng H, Long F, Ding C.. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell 2005;27:1226–38. 10.1109/TPAMI.2005.159 [DOI] [PubMed] [Google Scholar]
- 54. Yi Z, Ding C, Tao L.. Gene selection algorithm by combining reliefF and mRMR. BMC Genomics 2008;9(Suppl 2):453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li BQ, Hu LL, Niu S, et al. Predict and analyze S-nitrosylation modification sites with the mRMR and IFS approaches. J Proteomics 2012;75(5):1654–65. 10.1016/j.jprot.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 56. Jing W, Zhang D, Jing L.. PREAL: prediction of allergenic protein by maximum Relevance Minimum Redundancy (mRMR) feature selection. BMC Syst Biol 2013;7(Suppl 5):5028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li Y, Wang M, Wang H, et al. Accurate in silico identification of species-specific acetylation sites by integrating protein sequence-derived and functional features. Sci Rep 2015;4(1):5765–5. 10.1038/srep05765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang H, Wang M, Tan H, et al. PredPPCrys: accurate prediction of sequence cloning, protein production, purification and crystallization propensity from protein sequences using multi-step heterogeneous feature fusion and selection. PLoS One 2014;9(8):e105902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang M, Zhao XM, Tan H, et al. Cascleave 2.0, a new approach for predicting caspase and granzyme cleavage targets. Bioinformatics 2014;30:71–80. 10.1093/bioinformatics/btt603 [DOI] [PubMed] [Google Scholar]
- 60. Friedman N, Dan G, Goldszmidt M.. Bayesian network classifiers. Mach Learn 1997;29(2/3):131–63. [Google Scholar]
- 61. Liang L, Djuric N, Guo Y, et al. MS- k NN: protein function prediction by integrating multiple data sources. BMC Bioinformatics 2013;14(Suppl 3):61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chou KC, Shen HB.. Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-Nearest Neighbor classifiers. J Proteome Res 2006;5(8):1888–97. 10.1021/pr060167c [DOI] [PubMed] [Google Scholar]
- 63. Shen H, Chou KC.. Using optimized evidence-theoretic K-nearest neighbor classifier and pseudo-amino acid composition to predict membrane protein types. Biochem Biophys Res Commun 2005;334(1):288–92. 10.1016/j.bbrc.2005.06.087 [DOI] [PubMed] [Google Scholar]
- 64. Kim S-J, Koh K, Lustig M, et al. An interior-point method for large-scale l 1-regularized logistic regression. IEEE J Sel Topics Sign Proces 2007;1(4):1519–55. [Google Scholar]
- 65. Zardo P, Collie A.. Predicting research use in a public health policy environment: results of a logistic regression analysis. Implement Sci 2014;9(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Breiman L. Random forest. Mach Learn 2001;45(1):5–32. 10.1023/A:1010933404324 [DOI] [Google Scholar]
- 67. Liaw A, Wiener M.. Classification and regression by randomforest. R News 2001;23. [Google Scholar]
- 68. Fern N-DM, Cernadas E, et al. Do we need hundreds of classifiers to solve real world classification problems? J Mach Learn Res 2014;15:3133–81. [Google Scholar]
- 69. Meyer D, Dimitriadou E, Hornik K, et al. Misc Functions of the Department of Statistics (e1071), TU Wien. R Package version 1.6-1, 2009.
- 70. Tu JV. Advantages and disadvantages of using artificial neural networks versus logistic regression for predicting medical outcomes. J Clin Epidemiol 1996;49(11):1225–31. 10.1016/S0895-4356(96)00002-9 [DOI] [PubMed] [Google Scholar]
- 71. Bergmeir C, Benitez JM.. Neural networks in R using the stuttgart neural network simulator: RSNNS. J Stat Softw 2012;46(7):1–26.22837731 [Google Scholar]
- 72. Petron E. Stuttgart neural network simulator: exploring connectionism and machine learning with SNNS. Linux J 1999;1999. [Google Scholar]
- 73. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 1975;405(2):442–51. 10.1016/0005-2795(75)90109-9 [DOI] [PubMed] [Google Scholar]
- 74. O'Shea JP, Church GM, Schwartz D, et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods 2013;10:1211–12. [DOI] [PubMed] [Google Scholar]
- 75. Jeong KC, Sutherland MC, Vogel JP.. Novel export control of a Legionella Dot/Icm substrate is mediated by dual, independent signal sequences. Mol Microbiol 2015;96(1):175–88. 10.1111/mmi.12928 [DOI] [PubMed] [Google Scholar]
- 76. Shah AD, Bartlett JW, Carpenter J, et al. Comparison of random forest and parametric imputation models for imputing missing data using MICE: a CALIBER study. Am J Epidemiol 2014;179:179–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Saeys Y, Inza I, Larrañaga P.. A review of feature selection techniques in bioinformatics. Bioinformatics 2007;23(19):2507–17. [DOI] [PubMed] [Google Scholar]
- 78. Forgy EW. Cluster analysis of multivariate data: efficiency versus interpretability of classifications. Biometrics 1965;21:768–9. [Google Scholar]
- 79. Maaten L, Hinton G.. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 80. Li W, Cowley A, Uludag M, et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 2015;43(W1):W580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Letunic I, Bork P.. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2016;44(W1):W242–5. 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schroeder GN, Petty NK, Mousnier A, et al. Legionella pneumophila strain 130b possesses a unique combination of type IV secretion systems and novel Dot/Icm secretion system effector proteins. J Bacteriol 2010;192(22):6001–16. 10.1128/JB.00778-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Darby A, Lertpiriyapong K, Sarkar U, et al. Cytotoxic and pathogenic properties of Klebsiella oxytoca isolated from laboratory animals. PLoS One 2014;9(7):e100542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fodah RA, Scott JB, Tam HH, et al. Correlation of Klebsiella pneumoniae comparative genetic analyses with virulence profiles in a murine respiratory disease model. PLoS One 2014;9(9):e107394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Luo Z-Q, Isberg RR.. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci USA 2004;101(3):841–6. 10.1073/pnas.0304916101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zusman T, Degtyar E, Segal G.. Identification of a hypervariable region containing new Legionella pneumophila Icm/Dot translocated substrates by using the conserved icmQ regulatory signature. Infect Immun 2008;76(10):4581–91. 10.1128/IAI.00337-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bardill JP, Miller JL, Vogel JP.. IcmS‐dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol Microbiol 2005;56(1):90–103. [DOI] [PubMed] [Google Scholar]
- 88. Juhas M, Crook DW, Hood DW.. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell Microbiol 2008;10(12):2377–86. 10.1111/j.1462-5822.2008.01187.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Burstein D, Amaro F, Zusman T, et al. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat Genet 2016;48(2):167–75. 10.1038/ng.3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Carver T, Thomson N, Bleasby A, et al. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 2009;25(1):119–20. 10.1093/bioinformatics/btn578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19(9):1639–45. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.