

Abstract
Systems Bioinformatics is a relatively new approach, which lies in the intersection of systems biology and classical bioinformatics. It focuses on integrating information across different levels using a bottom-up approach as in systems biology with a data-driven top-down approach as in bioinformatics. The advent of omics technologies has provided the stepping-stone for the emergence of Systems Bioinformatics. These technologies provide a spectrum of information ranging from genomics, transcriptomics and proteomics to epigenomics, pharmacogenomics, metagenomics and metabolomics. Systems Bioinformatics is the framework in which systems approaches are applied to such data, setting the level of resolution as well as the boundary of the system of interest and studying the emerging properties of the system as a whole rather than the sum of the properties derived from the system’s individual components. A key approach in Systems Bioinformatics is the construction of multiple networks representing each level of the omics spectrum and their integration in a layered network that exchanges information within and between layers. Here, we provide evidence on how Systems Bioinformatics enhances computational therapeutics and diagnostics, hence paving the way to precision medicine. The aim of this review is to familiarize the reader with the emerging field of Systems Bioinformatics and to provide a comprehensive overview of its current state-of-the-art methods and technologies. Moreover, we provide examples of success stories and case studies that utilize such methods and tools to significantly advance research in the fields of systems biology and systems medicine.
Keywords: Systems Bioinformatics, precision medicine, computational diagnostics, computational therapeutics, network analysis, drug repurposing
Introduction
Biological data, either as large-scale omics or as classical biodata, are the footprints of biological mechanisms. These mechanisms consist of numerous synergistic effects emerging from various systems of interwoven biomolecules, cells and tissues. Therefore, it is necessary to explore them with a systemic approach to reveal the behaviour of the system as a whole rather than as the sum of its parts.
Systems biology provides a holistic perspective on biological mechanisms via the integration of information and knowledge from multiple interdisciplinary fields (such as biology, chemistry, mathematics, computer science and physics). It aims to elucidate synergistic relationships between multiple factors in contrast to representing them as single entities and can lead to the generation of complex molecular networks of interactions modelled by computational or mathematical approaches. Systems biology harnesses its power from technological advances in the field of ‘omics’ and the advent of next-generation sequencing. These technologies provide a spectrum of information ranging from genomics, transcriptomics and proteomics to epigenomics, pharmacogenomics, metagenomics and metabolomics.
Bioinformatics and computational biology have made significant breakthroughs towards the analysis and interpretation of the data obtained from the above-mentioned omics technologies. The sheer size of data generated by these high-throughput methodologies, coupled with the need to analyse, integrate and concurrently interpret this avalanche of information in a systemic way, has paved the way to the upcoming field of Systems Bioinformatics.
Systems Bioinformatics is a relatively new approach, which lies in the intersection of systems biology and classical bioinformatics. It focuses on integrating information across different levels using a bottom-up approach—as adopted in systems biology—with a data-driven top-down approach used in bioinformatics. The bottom-up approach in systems biology typically brings together information from molecular cells and tissues in the framework of mathematical models to generate insights on the function and dynamic behaviour of cells, organs and organisms. The top-down approach uses bioinformatics methods to extract and analyse information from ‘omics’ data generated through high-throughput techniques.
Initially, informatics approaches to systems biology focused mainly on modelling and simulation. However, owing to the lack of sufficient experimental data, these methods fell short in building reliable models. Subsequently, the explosion of multilevel data generation brought a plethora of new methods, tools and solutions capable of studying systemic properties. The application of systemic approaches such as information theory, statistical inference, probabilistic models, graph theory and further network science approaches in the analysis of biological data paved the way to the creation of a distinct field, namely, Systems Bioinformatics.
Depending on the availability, the quality and the comprehensiveness of the data, Systems Bioinformatics’ methods contribute significant benefit in narrowing down the gap between genotype to phenotype as well as providing additional information regarding biomarker and drug discovery. These methods are applied to classical biological data, clinical/patient data and omics data as well. They are suitable for extracting precise and personalized results, thus, facilitating systems medicine (medicine that is in bidirectional interaction with computational multiscale analysis and modelling of disease-related mechanisms) and more specifically P4 Medicine (medicine that is personal, participatory, predictive and preventive) [1, 2] (as illustrated in Figure 1).
Figure 1.
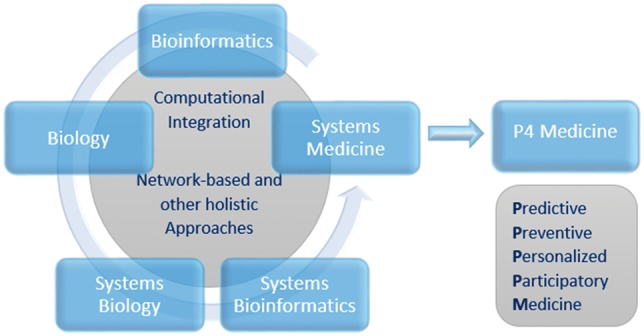
Systems Bioinformatics. A schematic representation of the emergence of Systems Bioinformatics as a distinct discipline among other interrelated and interdependent disciplines. The information provided by Bioinformatics, Biology and Systems Biology is integrated in the Systems Bioinformatics framework through computational integration and network-based and other holistic approaches to tackle challenges in Systems Medicine and in particular P4 Medicine.
The delivery of individually adapted medical care of high precision, based on multi-source patient information across various levels and in various scales, is the basic idea of modern medicine having various appellations depending on the emphasis given (e.g. translational/systems/P4/precision/personalized medicine). The omics spectrum offers the opportunity and the challenge for multiscale and multi-source analysis towards building a comprehensive profile of the Human System (Figure 2). The major challenges faced by Systems Bioinformatics towards this demanding form of medicine are: (i) the design and development of suitable bioinformatics pipelines to provide valid and sufficient biological information from the high-throughput molecular profiles of the patient, (ii) the development of robust information systems capable for data integration, information extraction and knowledge sharing, (iii) the construction of mathematical models to predict the evolution of a particular disease, its relation with the measured markers, its tolerance/resistance to various drug families and the existing risks to the patient. These challenges can be tackled with state-of-the-art computational methodologies and techniques, such as computational intelligence, machine learning, pattern recognition and data mining, modelling and simulation, network reconstruction and visualization, complex network analysis, deep learning, text mining/semantics and association analysis. Further to these, Systems Bioinformatics serves as the framework for the development of powerful computational methods and tools to create user-friendly platforms to visualize and analyse big and heterogeneous information in the form of a network.
Figure 2.

Network Integration. Multiscale and multisource data generated from the Human System can be represented in network form. These networks can be further analysed and, importantly, they can be integrated forming supernetworks and building a comprehensive profile of the Human System.
This review is structured in three main sections. In the first section on ‘Systems Bioinformatics’ we begin with an overview of the systems theory approach for complex biological problems. We then provide an in-depth summary of the network science approach in System Bioinformatics. We introduce certain basic network measures, which are used to analyse the components of a network, both locally and globally, and discuss the biological interpretation of such measures. We then describe in detail key biological network construction methods followed by a discussion on module-based approaches and network signatures. Finally, we describe network manipulation methods in the ‘Network controllability’ and ‘Network integration’ subsections. In the following subsection we provide a short summary regarding modelling and simulation approaches followed by a short discussion on the infrastructures and data management challenges in the field of Systems Bioinformatics. In the last two sections we provide an overview of methods and case studies with regards to the Systems Bioinformatics applications in biomarker and drug discovery.
Systems Bioinformatics
Systems approaches
Biological data have tremendously expanded both in size and complexity. Systems Bioinformatics focuses on the investigation of such vast and complex biological systems and their within interactions using a ‘holistic’ rather than a ‘reductionist’ approach, much like the systems biology field. A holistic approach to science and the analysis and description of a complex phenomenon emphasizes the whole and the interaction of its parts, whereas the reductionist approach focuses on the fundamental parts. In fact, the debate on reductionism versus holism has its roots in ancient years. According to reductionism proponents, the optimal method to understand any science is the decomposition in smaller components. Moreover, in its greedy form, reductionism may see the whole science as physics. Even in its layered-model form, reductionism considers human/health sciences as based on biology, biology based on chemistry and chemistry based on physics. On the other hand, a strong dissent has formulated a solid antireductionism trend. This trend has either epistemological or ontological origins, supporting that complete reductionism is technically impossible and that there are emergent laws that govern the system and cannot be derived from the laws governing the components of the system. Furthermore, it is supported that each system has a ‘buffering capacity’ where many micro-states correspond to fewer macro-states of the system, making reductionism to be considered as pointless after a certain decomposition level [3].
The reductionism’s approach in biology is epitomized by molecular biology, which in the past two decades has led to the generation of a plethora of omics data. These data provide information on the building blocks of the entire organism at different scales and for different types of cells, tissues and organs. Data on DNA fragments, genes, RNA fragments, peptides, proteins and metabolites measured in short time and space intervals provide a spatiotemporal distribution of these building blocks under various states of the organism. These interwoven building blocks control and are controlled by signals in a non-linear way. As a result, the understanding of the system requires something more than simply the bottom-up assembly of the system’ components.
Systems theory, which is a holistic approach, addresses the limitations from the reductionism’s point of view by considering the system as a whole, adopting a top-down approach [4]. Thus, it studies the emergent properties of the system such as homeostasis, adaptivity, tolerance, stability and modularity, through some basic overlying hierarchical principles such as entropy, positive and negative feedback control [4]. In the context of Systems approaches, the graph theory and the further science of networks have been successfully applied to the investigation of complex phenomena across a range of different scientific disciplines. The theoretical context of complex networks approach includes concepts that are derived from information theory, dynamical systems, statistical physics and topology approaches, as well as several mathematical methods suited for the analysis of the interaction of components in a complex network. In the following subsection we provide an in-depth overview of such network approaches and examples of their use in Systems Bioinformatics.
Networks
Biological network basics
Casting biological systems as networks and analysing their topology can be useful in understanding how such systems are organized. Graph theory provides a powerful mathematical framework for the understanding of the organization of such large and complex systems by considering them in the form of graphs [5, 6]. Graphs, also termed as networks, can be used to model the pairwise relations between objects. A network is a collection of nodes or vertices connected by edges, arcs or lines (as shown in Figure 3A). It may be undirected, meaning that there is no distinction between the two nodes associated with each edge, or its edges may be directed from one node to another (as shown in Figure 3B). In cell biology, nodes represent cellular components (e.g. proteins) and edges represent interactions or other relationships between these components [e.g. protein–protein interactions (PPIs)]. Basic network measures can be used to analyse the components of a network, both locally and globally, and facilitate the analysis and extraction of useful information from a biological network. The most elementary characteristic of a node is its degree, i.e. the number of edges connecting one node to its neighbours. The probability distribution of the degrees over the whole network is called degree distribution. In random networks, most nodes have a similar number of links and their degree distribution follows the Poisson distribution. In contrast, many real-world networks, including most biological networks, are scale free. This means that their degree distribution follows a power law, as most of the nodes have few links and only a few nodes are densely connected [7].
Figure 3.
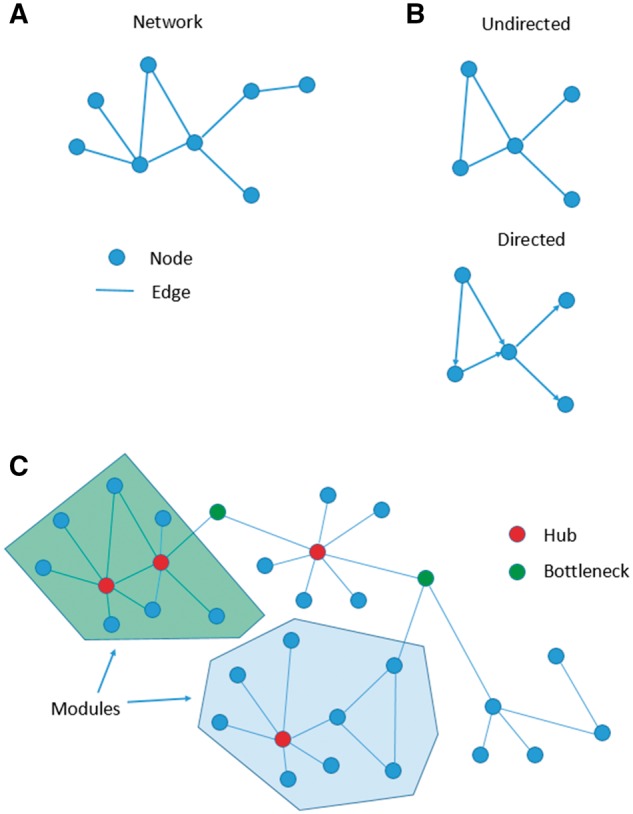
Network Basics. (A) The basic elements of a network are illustrated in this simple network where a circle indicates a node and a line indicates an edge. (B) Networks can be either undirected (upper panel) or directed (lower panel). (C) Hubs (red nodes – or dark grey nodes in Black & White printing) and bottlenecks (green nodes – or medium grey nodes in Black & White printing) are illustrated in this sample graph. Two example modules (green and blue areas – or shadowed areas in Black & White printing) are illustrated as subgroups of nodes and their respective edges.
Nodes with high degree, known as ‘hubs’ (illustrated in Figure 3C), can be key players in molecular mechanisms such as (i) a protein interacting with multiple other proteins, (ii) regulation of multiple genes by a key transcription factor or (iii) multipart regulation by other regulatory elements [i.e. microRNAs (miRNAs)]. All these cellular processes may be highly significant in determining the outcome or phenotype of a disease of interest. In human cells, hub genes have been found to indicate essential genes (i.e. critical for survival) rather than disease genes [8]. Another node feature is its betweenness, i.e. the extent to which a node participates in the shortest paths connecting other nodes. Nodes with high betweenness, known as ‘bottlenecks’, can be extremely influential in a network in the sense that they rest in critical junctions between hubs and can therefore represent bridges that allow groups of nodes to cross talk to each other (as illustrated in Figure 3C). Importantly, some of these bottleneck nodes represent key connections that if removed will result in the complete loss of connectivity between clusters of nodes, thus affecting greatly the overall topology and, as a result, the information propagation in the network. In molecular terms, an example of bottleneck nodes is that of proteins whose loss of function leads to deactivation of specific processes. In directed regulatory protein networks, betweenness was shown to be a good predictor of essentiality [9]. Various other network features can be calculated to provide insights into biological networks. Another measure is closeness (a measure of the average length of the shortest paths from one node to other nodes), which indicates important nodes that can communicate quickly with other nodes of the network. For example, in a protein signalling network closeness can be interpreted as the ‘probability’ of a protein to be functionally relevant for several other proteins. An example illustrating how network measures, such as network-efficiency and network-clustering, can be used as biomarkers is the recent study of Blain-Morales et al. [10] where a network was constructed using the alpha bandwidth (8–13 Hz) of the electroencephalogram recordings during anaesthesia in healthy humans. Global network efficiency quantifies the efficiency of information exchange across the whole network and is defined as the average inverse shortest path length over all pairs of nodes. The clustering-coefficient is a measure of the degree to which nodes in a network tend to cluster together (the global measure is calculated by averaging the local clustering-coefficients of all nodes). In Blain-Morales et al. [10] network efficiency was significantly decreased and network clustering-coefficient was significantly increased during anaesthesia-induced unconsciousness. These measures returned to baseline 3 h post-recovery, suggesting that they could be used as potential biomarkers for normal recovery brain networks post general anaesthesia induction.
Other network measures such as network size [11], density [11], PageRank versatility [12], path length [10] and modularity [10] can further be used to evaluate networks. For an extensive review of network measures, the reader is referred to [13]. Although topological properties from a graph-theory point of view do not always have a clear biological meaning, in many cases they can be good predictors of functional and disease modules (see [8] for further discussion).
Biological network construction methods
Biological networks can be split into two broad categories that best characterize their underlying nature: (i) evidence-based molecular networks that rely on experimental evidence for specific molecular interactions such as PPI networks, metabolic networks and regulatory networks (transcription factor—gene networks, non-coding RNA—gene networks) [14–18], (ii) statistically inferred networks, which are based on statistical inference that rely on interactions between components established by means of statistical analysis.
Evidence-based molecular networks : The information used to build networks of molecular interactions is obtained from small-, medium- or large-scale experimental data that are usually aggregated and available in online databases [19, 20]. A plethora of information can be derived from multiple resources including PPIs, gene regulatory relationships (including miRNAs) and metabolic pathways, using high-throughput (i.e. whole exome sequencing) and literature-curated data. In addition, valuable pre-compiled information can be derived from databases like Gene Ontology [21], REACTOME [22, 23] and literature-based annotations in Genome Recognition Analysis Internet Link [24].
The construction of biological interaction networks with the goal of uncovering causal relationships constitutes a major research topic in systems biology [25]. Many approaches have been developed to study the interactions among a large number of genes to highlight significant genes for each disease. Certain approaches utilize biological knowledge, to address many biological problems and find genes related to the disease of interest.
Construction of networks requires knowledge of PPIs, protein–DNA interactions (PDIs) and/or protein–metabolite interactions (PMIs). Such data can be obtained from open-access databases. For example, PPI data can be obtained from the Search Tool for Recurring Instances of Neighbouring Genes (STRING) [26], the Human Protein Reference Database (HPRD) [27], the Biomolecular Interaction Network Database (BIND) [28], the Molecular INTeraction database (MINT) [29] and the Biological General Repository for Interaction Datasets (BioGRID) [30]. For example, in a recent study, a network-based analysis of mass-spectrometry (MS)-based proteomics data of spinal nerves led to the identification of 19 biological processes to be involved in retrograde motoneurodegeneration and neuroprotection after axonal damage [31]. In this study, the authors used nine public PPI databases to obtain protein interaction data. Furthermore, PDI databases include the EdgeExpressDB (FANTOM4-EEDB) [32], the Transcriptional Regulatory Element Database [33], MSigDB [34], MultiNet [35] and the MetaCore [36]. The KEGG pathway database [37] can be used to obtain PMI data.
Nevertheless, as a large number of genes are not functionally characterized, these approaches are compromised owing to lack of available data [38]. Based on this limitation, many statistical network inference methods were developed to construct statistically inferred gene networks, based on omic data from high-throughput technologies, as they provide snapshots of the transcriptome under many tested experimental conditions [39].
Statistically inferred networks: A type of statistical inference network is the ‘co-expression network’, where genes are connected based on statistically significant correlated or anti-correlated (depending on the underlying question) expression profiles with respect to a disease of interest. Another type of statistically generated network is the ‘genetic network’ [40–42]. Sources like the BioGRID [43] database, allow researchers to investigate how the dysregulation of one gene affects the downstream response of another gene and, moreover, how this cascade of molecular functions influences specific disease phenotypes.
The basic idea behind the network inference methods is to search for sets of co-expressed genes. Depending on the metric that is used, these methods can be classified into three major categories [38]: (i) Mutual Information-based methods, (ii) Correlation-based methods and (iii) Tree-based methods.
Mutual information-based methods calculate the mutual information values of all pairs for a given gene expression profile, and if a pair’s corresponding value is larger than a given threshold then this pair of genes is considered as linked. The resulting network is constructed based on this threshold by including a weighted edge between two genes [44]. The weight can be calculated with several algorithms: ARACNE (Algorithm for the Reconstruction of Accurate Cellular Networks) [45], CLR (Context Likelihood or Relatedness Network) [46], MRNET (Maximum Relevance Minimum Redundancy) [47], MRNETB (Maximum Relevance Minimum Redundancy Backward) [47] and C3NET [48].
In the case of correlation-based methods, the different algorithms calculate the correlation or the partial correlation between pairs of genes. These methods are implemented through algorithms like GeneNet, a statistical learning algorithm which allows the assessment of Graphical Gaussian Models [49], and Weighted Correlation Network Analysis (WGCNA) [50], an algorithm which calculates correlations across each pair of genes. It computes an adjacency matrix using the Spearman correlation, Lasso—a shrinkage and selection method for linear regression [51]—and Adaptive Lasso—another version of Lasso modified to include penalty weights [52].
In the case of tree-based methods, algorithms use tree-based ensemble methods as feature selection techniques to solve a regression problem for each gene in the network. More specifically, the basic idea of tree-based methods in regression is to recursively divide the learning sample with binary tests based each on one input variable (the expression of one gene). These binary tests are optimized to minimize, in the largest amount possible, the variance of the output variable, namely, the expression of another gene from the remaining in the subsets of samples. Candidate divisions compare values from the input variable with a threshold, which is determined during the tree growing. Tree-based ensemble methods are more enhanced than single trees, as they estimate the average predictions of several trees. An example of a tree-based method is the GENIE3 algorithm [53], which emerged as the best performer in a significant network inference challenge [39].
In summary, statistical-inference methods are used to estimate the expression pattern relationships across all pairs of genes driving to co-expression network inference. However, correlation-based methods have a tendency to be algorithmically straightforward and computationally fast, but with the limitation that assume linear relationships among variables. In contrast, methods based on mutual information capture non-linear as well as linear interactions but they can be computationally expensive. From another point of view, tree-based methods are non-parametric and consequently, they do not need to make any assumption about the nature of the data. Tree-based methods can deal effectively with high-dimensionality data.
Module-based approaches and network signatures
Representing high-throughput data with networks often leads to complex and highly dense networks that cannot be easily interpreted by the human eye. To extract biologically meaningful information from these networks and establish links to disease, methods have been developed for scanning and parsing these networks. These methods allow for significant sub-networks to be highlighted in the sea of nodes and edges, often representing important ‘modules’ that are associated with a specific disease [8, 54, 55] (as illustrated in Figure 3C). This type of network ‘traversing’ can be performed between networks obtained from data from different phenotypes from the same disease (staging, subtyping) or from similar diseases (disease hierarchy). Through this way, common/shared modules can be identified by looking at network intersections. Alternatively, unique modules reflecting molecular signatures exclusive to specific conditions or phenotypes can be extracted. Depending on the integration mode, the identification of modules may lead to ‘active modules’ (by integrating molecular profiles and highlighting activity of nodes/interactions), ‘conserved modules’ (by comparing multiple species/states and concluding to conserved subnetworks), ‘differential modules’ (by comparing states and conditions and concluding to differentiated subnetworks) and ‘composite modules’ (by integrating multi-source information from complementary networks) [56].
Module identification can be performed using Systems Bioinformatics approaches and constitutes a powerful tool for delineating the systematic molecular basis of disease. Module identification thus abides by two assumptions: (i) modules that are specific to a disease of interest are expected to form dense clusters or hubs capable of detection by unsupervised network clustering algorithms, (ii) the functional relationship between the nodes residing within these clusters is expected to be similar with respect to underlying molecular mechanisms, biological processes and cellular/tissue localization [57].
Further functional significance of modules can be derived by using pathway enrichment analysis methods, either in the traditional sense (Fisher’s enrichment analysis) or by prioritizing/ranking pathways and genes based on network topological similarities to already validated disease network components. The latter case leads to another type of module functional annotation and assignment based on associations and connection to neighbouring nodes that are of known/validated functions. This approach has been used to elucidate functional and clinical significance of hazy areas of molecular networks for which associations between genes or proteins and specific disease phenotypes are not available in publicly available resources or through basic high-throughput analyses.
There are several different types of example cases that have used network methods to gain information on functional modules and network signatures. Novel information for genes, pathways and other molecular interactions involved in numerous disorders has been discovered using network module-based approaches. These include disorders such as type 2 diabetes mellitus [58–61], Alzheimer’s disease (AD) [62, 63], Parkinson’s disease [60, 64], cardiovascular diseases, asthma [61, 65–67] and a variety of tumours [68, 69]. When existing bioinformatics resources and databases fall short in shedding light on a specific disorder of interest, novel experiments have to be designed and conducted to successfully unravel the clinicopathological, genetic and molecular mechanisms underlying disease. Success stories include studies for spinocerebellar ataxia [70], Huntington’s disease [71] and schizophrenia [72–74].
For example, a recent study utilized large-scale expression data to extract/identify biologically significant modules from gene expression networks [75]. It has been hypothesized that disease tissue specificity is governed by the expression of a specific functional disease module (sub-network) in the tissue of disease manifestation [75]. The authors of this study adopted this hypothesis and used systems approaches to investigate the tissue-specific expression patterns of disease genes in the human interactome. They observed that genes expressed in a specific tissue shared a topological neighbourhood in the human interactome network, in contrast to genes expressed in different tissues. The authors further provided evidence that expression of all components of the tissue-specific disease module was necessary for determining the disease outcome. The construction of this tissue-specific disease network further allowed for predictions on novel disease–tissue relationships.
Network controllability
Network controllability is defined as the potential to steer a network from a given initial state to a final desired state within a finite time and with appropriate inputs/modifications. Such modifications are also known as ‘network attacks’. Another Systems Bioinformatics’ emerging research direction related to the dynamic properties of complex networks is the way in which these properties are spreading and/or reforming during network attacks. The attack process is usually based on specific mathematical models that decide which nodes-edges or even specific hubs to remove, to examine issues like controllability [76], error tolerance [77], attack vulnerability [77, 78], robustness [79, 80], topological characteristics [81] and control centrality [82]. Cascade attacks [83], degree and betweenness-based attacks and prominence-based attacks are commonly used types of attacks [84]. It has been shown that intentional attacks as well as random failures may easily affect (or even destroy) network functions such as connectivity and synchronization [84, 85], while a small range of node failures can affect the network controllability [86]. In the case of Systems Bioinformatics there are relatively limited, yet of great interest, studies that have used such an approach [87].
‘Driving nodes’ are highly important nodes in a network that governs its controllability. Control theory shows that to direct a complex network towards a desired state, there is a minimum number of driving nodes. Determining the minimum number of driving nodes can be demanding with respect to both computational resources and time, hence novel non-exhaustive algorithms for determining driving nodes are necessary. A recently published study [88] presents the actuation spectrum method that optimizes the trade-off between driving node prediction and time. The authors validate their methodology across numerous complex networks and show that a small number of driving nodes are sufficient to determine the state of a complex network. Another approach makes use of PPI networks and network controllability. By controlling the structure of human PPI networks, using the correct queues or inputs, it is possible to activate specific cellular processes that determine disease outcome (i.e. apoptosis). A recent study [89] utilized a PPI network of 6339 proteins and 34 813 interactions to perform classification of proteins with respect to their importance in the network. The authors quantified the effects of removing a specific protein from the network by calculating the number of remaining driving nodes. Results showed that the most important proteins according to this analysis were also the primary targets of disease-causing mutations, human viruses and drugs. This study showed that controllability of a network can provide crucial information for the shift between healthy and disease states, at the same time highlighting novel candidate drug targets.
Network integration
A key approach in Systems Bioinformatics is the construction of multiple networks representing each level of the omics spectrum and their integration in a layered network that exchanges information within and between layers (Figure 2). Different disease modules have been shown to act in synergy. Thus, to obtain a holistic picture of the complex mechanisms that underlie disease manifestation, it is necessary to construct networks of integrated disease modules. Such an integration can be achieved via various ways: (i) By investigating gene association it is possible to construct connections between different disease modules by looking at shared, common genes. This can reflect the genetic basis of diseases and provide associations between diseases of a common genetic background. (ii) Superimposing gene networks with gene expression data from RNA-Seq or microarray analysis can further enrich these networks of disease modules. (iii) A common genetic basis for disease modules can also be established by analysing genetic variants or polymorphisms (i.e. SNPs, indels). Networks of disease modules sharing genetic mutations can lead to important findings such as establishment of linkage and associations of variants as well as environmental factors with disease modules. (iv) Protein interaction network modules can also be merged using common PPIs between disease modules and moreover, overlaying this information with proteomics expression data can provide valuable insights into the proteome of diseases of interest. (v) Looking at common pathways between disease sub-networks can also provide valuable clues as to similarities and/or differences between diseases of interest. (vi) Metabolic pathways can also provide additional information towards the understanding of enzyme catalytic activity for different disease modules. Disorders that affect specific metabolic pathways (i.e. obesity) are more likely to share commonalties in the metabolic networks than diseases that share a genetic basis. (vii) Disease modules can also be linked using regulatory information such as shared miRNA regulators, thus highlighting important commonalities or differences between diseases.
Specific cellular components (or modules) associated with a disease are believed to share a topological neighbourhood within the human interactome [90]. In a recent study [90] the authors utilized novel mathematical conditions to map the topological relationships between diseases in the human interactome. They showed that diseases with common expression profiles, symptoms and comorbidity share overlapping modules in contrast to more phenotypically distinct diseases, which appear in distant topological neighbourhoods. These tools can provide valuable insights in predicting drug therapy for diseases with common phenotypes, even if they are genetically distinct.
Another recent study [91] adopted a novel, multiple-network-framework integration for epigenetic modules. This method utilized the Epigenetic Module based on Differential Networks (EMDN) algorithm, which simultaneously analyses DNA methylation and gene expression data [91]. Using The Cancer Genome Atlas (TCGA) breast cancer data, the authors reported that the EMDN algorithm could recognize positively and negatively correlated modules. These modules can serve as biomarkers to predict/diagnose breast cancer subtypes by using methylation profiles, where positively and negatively correlated modules are of equal importance in the classification of cancer subtypes. The authors of this study also showed that epigenetic modules also estimate the survival time of patients, and this factor is critical for cancer therapy.
Tools that analyse the structure and topology of these integrated networks are of extreme value and can provide insights into the synergistic role of multiple network components in diseases of interest. The methods for network analysis and integration we have discussed so far are mainly used to describe the topology of a biological network (or a set of networks). Although these methods capture the relationships between components, they fail to capture the dynamics, i.e. the time component is not modelled and, thus, simulations to obtain prediction of the evolution of the system cannot be performed. For example, static insights into the molecular basis of a disease do not provide a complete picture with regards to drug response without access to time-dependent data. Hence, the use of mathematical algorithms and computational tools for modelling the dynamics of these networks complements network analysis and is further detailed in the following section.
Systems modelling and simulation
To test the validity and predict the behaviour of complex biochemical systems, such as gene networks, it is often required to describe the effects of multiple, simultaneous and dynamic interactions within the components of the system that are too complex to interpret intuitively. Developing and simulating mathematical models is essential in investigating such complex biological systems. These complex systems can be further explored using mathematical models to describe the valid structure (i.e. the components of the system and their interactions based on experimental data) and identify the basic underlying principles of their function to predict behavioural responses to a certain perturbation [92].
Two types of models are commonly used to describe biological processes such as gene networks—‘quantitative’ and ‘logical’ [93, 94]. Quantitative models use differential equations to describe the non-linear dynamic interactions in a network, whereas logical models use the Boolean approach to describe dynamics in a qualitative way. Quantitative models provide precise information and can be directly compared with experiments including time-dependent data. However, they require sufficient knowledge of the mechanistic details and kinetic parameters and, thus, they are limited to applications to networks which are well characterized and are of small to moderate size. Logical models do not require such information and can be applied to large-scale networks with known structure, yet only provide limited information, as they cannot provide quantitative predictions and assist in choosing better alternative behaviours. In summary, each modelling approach has its advantages and disadvantages and recent work suggests that hybrid approaches might be optimal for challenges in systems biology (for a detailed discussion see [93]).
Mathematical models are indispensable in pharmacology and diagnostics. For example, spatio-temporal mathematical models of the blood coagulation network have been developed to aid drug development and diagnostics (as extensively reviewed in [95]). Another relevant application is the use of mathematical models of drug-targeted pathways (modelled with a set of differential equations based on the mass action law) to explore drug combinations [96]. Classical bioinformatics and systems biology can complement and strengthen each other in drug discovery and therapeutics where concrete predictions are required [92]. The value of combining high-throughput data with mathematical modelling is shown, for example, in devising personalized treatments in cancer (for extensive review see [97]). Integration of multi-omic data can be used as an additional constraint in constraint-based modelling in systems biology (to optimize parameter estimation and validation). For a recent survey summarizing constraint-based metabolomic modelling and multi-omic integration methods see [98].
Infrastructures and data management
It is important to highlight some of the modern computing trends that play an important role in driving research in Systems Bioinformatics and facilitate the transition to personalized medicine. The main limiting factor for research laboratories specializing in Systems Bioinformatics is computational power and resources. Significant investment is required to attain high-performance computer (HPC) servers or clusters, which have the capacity to store, manage and process the vast amount of data generated from high-throughput omics technologies. Often sheer maintenance of these machines can be a costly and a limiting factor that disallows the exploitation of the full potential of HPC. Cloud computing promises to solve major issues of system administration for these computer clusters by allowing for the exploitation of HPC, stored and managed in an expert environment, as virtual resources that are made available through the internet. Tool availability is also a major issue and having organized platforms with tools like CytoScape [99], GATK [100], BLAST [101], omics assemblers (i.e. IDBA-UD [102]) and programming languages and packages like R's Bioconductor library for expression data analysis [103], JAVA, Python, SQL and others is of major importance for scientists to facilitate dissemination of algorithms, data and results.
Systems bioinformatics applications
In this section we present the impact of Systems Bioinformatics on diagnostics and therapeutics by highlighting success stories and cases in a formatted manner: introductory text/data set collection/network construction/network analysis/findings and significance of research.
Systems bioinformatics applications in biomarker discovery
The use of networks in computational diagnostics via the detection of molecular biomarkers is one of the hallmarks of Systems Bioinformatics. Numerous recent state-of-the-art studies have made use of such networks to characterize cellular systems by simultaneously analysing thousands of genes, proteins, isoforms and complexes to address issues of computational diagnostics. Here, we highlight a few studies, showcasing the essence of networks’ contribution in precision diagnostics.
A recent study [62] used a machine learning approach, which integrates topological features from PPI networks, to identify candidate AD-associated genes.
Data set collection: Positive and negative data sets were collected from Entrez Gene database at the National Centre for Biotechnology Information (NCBI). The positive data set consisted of 458 genes known to be associated with AD. The negative data set consisted of the additional 55 947 Entrez genes, excluding the AD-associated genes.
Network construction: Human PPI data sets were extracted from a variety of sources including Online Predicted Human Interaction Database (OPID), STRING, MINT, BIND and InTAct databases.
Network analysis: By utilizing the PPI networks, the authors extracted topological features for the AD- and non-AD-associated genes. These features included nine topological properties of the PPI network for each gene, namely, the average shortest path length, betweenness centrality, closeness centrality, clustering coefficient, degree, eccentricity, neighbourhood connectivity, topological coefficient and radiality.
Findings and significance of research: The authors further combined sequence features and functional annotations features and concurrently performed feature selection using seven methods including gain-ratio-based attribute evaluation, oneR algorithm, chi-square-based selection, correlation-based selection, information gain-based attribute evaluation and relief-based selection. The most important features were fed into 11 machine learning algorithms to generate classifiers using the training data set capable of predicting AD- and non-AD-associated genes using the selected network, sequence and functional features. Methods included Naive Bayes (NB), NB Tree, Bayes Net, Decision table/NB hybrid classifier, Random Forest, J48, Functional Tree, Locally Weighted Learning (J48 + k-nearest neighbour), Logistic Regression and Support Vector Machine. Training of sophisticated machine learning classifiers using systemic properties can be a key feature in generating personalized medicine diagnostic approaches. The authors finally combined diagnostics with therapeutics by screening 45 known anti-Alzheimer drugs from DrugBank against novel predicted probable AD targets, obtained from their trained classifiers, using molecular docking. They further proposed a novel candidate untried drug, AL-108, with high affinity to potential therapeutic targets. Additional tools were also used to validate preliminary findings, including molecular dynamics simulations and MM/GBSA calculations on the docked complexes [62].
Another interesting study [104] used data from TCGA [105] to successfully construct a multidimensional subnetwork atlas for cancer prognosis. The authors addressed how multiple genetic and epigenetic factors (i.e. gene expression, copy number variation, miRNA expression and DNA methylation) affect molecular states of networks and patient survival.
Dat aset collection: The multidimensional cancer-associated data sets for 1027 patients for four cancer types were collected from TCGA Cancer Browser (https://genome-can cer.ucsc.edu/proj/site/hgHeatmap/). They contained clinical information, copy-number variation, promoter DNA methylation, mRNA-gene and miRNA expression data. They furthermore extracted PPI data from HPRD for network construction. To enrich these networks with additional miRNA-regulatory information the authors extracted miRNA and target gene information from two miRNA target databases [miRTarBase (Release 4.5) and TarBase v6], which provide experimentally validated miRNA–target interactions.
Network construction: PPI interaction network was constructed using data collected from HPRD.
Network analysis: The authors fitted a univariate Cox proportional hazards model between each molecular feature and patient survival time and thus scored each gene based on its significance to predict survival. Genes with a positive score were considered as survival-related genes. They next used this score (heat score) as the input into HotNet2, which uses a heat diffusion process and a statistical test-based algorithm to discover subnetwork signatures in the PPI network. Through this way subnetwork signatures of survival-related genes were determined both by the scores of their genes as well as gene topology in the PPI network.
Findings and significance of research: The authors then used Monte Carlo cross-validation and permutation testing procedure to assess predictive power of the subnetworks on patient overall survival. They used a Cox proportional hazards model with L1 penalized log partial likelihood (LASSO) for feature selection to train the models based on the molecular profile of individual subnetworks. Finally, the prognostic outcomes for the training set were used to determine the regression coefficients. These coefficients were then used in the testing model to predict outcomes for patients in the test set and calculate the concordance index (C-index). Results reveal novel PPI subnetworks with significant prognostic capabilities for a variety of cancer types. The authors further validated their subnetworks by performing prognostic impact evaluation, functional enrichment analysis, drug target annotation, tumour stratification and independent validation. They highlighted distinct pathways in the underlying subnetworks as potential new targets for therapeutic intervention for certain cancer types. This study integrated the protein interactome with cancer genomics data, thus allowing for a systemic analysis of the molecular mechanisms that underlie genesis of cancer and provides new directions in personalized cancer therapy [104].
Another recent study [106] adopted an approach that uses an enriched library of single-stranded oligodeoxynucleotides to profile complex biological samples. This method allows for the analysis of systemic native biomolecules. The authors defined their method as Adaptive Dynamic Artificial Poly-ligand Targeting and further utilized it as a diagnostic tool to profile plasma exosome of cancer patients. They achieved high classification accuracy in breast cancer patients by analysing the circulating exosomes in their blood [106].
The online database MelGene is yet another example of successful integration of Systems Bioinformatics approaches in current research for molecular diagnostics. This tool provides a comprehensive, regularly updated collection of data from genetic association studies in cutaneous melanoma, including random-effects meta-analysis results of all eligible polymorphisms [107]. The MelGene proposed network connections highlight potentially new loci in relation to melanoma risk.
Recent studies have shown that interpretation of proteomics data using network-based approaches can offer additional insights into the mechanistic and dynamics of protein assemblies, and hence into the molecular mechanisms underlying the system under study. Moreover, network-based approaches can be used to reconstruct a disease-perturbed cellular network model showing the interactions of identified differentially expressed proteins involved in selected cellular pathways related to the target pathophysiology. For example, Shirasaki et al. [108] have used affinity purification coupled to MS to investigate the proteome profile of Huntington’s disease. In particular, using a monoclonal antibody against huntingtin (Htt), they identified 747 proteins to be complexed with Htt. A systems-level view of Htt interactome was achieved by using WGCNA, which was used to construct weighted links between the Htt co-purifying proteins. Using topological overlap, the data were clustered into eight Htt-interactome modules that were related to distinct functional aspects such as brain region specificity, aging and protein aggregation modulation or Htt functions directly [108].
Moreover, several network-based approaches have been developed that can identify the cellular pathways which are altered under pathophysiological conditions, and can hence enrich biomarker discovery. For example, functional enrichment analysis of GO biological processes or KEGG pathways [37] of differentially expressed proteins can be performed using both free licence tools such as Database for Annotation, Visualization and Integrated Discovery (DAVID) [109], Protein ANalysis THrough Evolutionary Relationships (PANTHER) [110] and Gene Set Enrichment Analysis (GSEA) [34], as well as commercialized tools such as MetaCore [36] and Ingenuity Pathway Analysis. Furthermore, pathway topology approaches have been developed as alternative to enrichment analysis. For example, Signalling Pathway Impact Analysis [111] and Network Perturbation Amplitude [112] deliberate whether the proteins involved in functional modules defined by other databases interact with each other in cellular networks.
Various tools are currently available, which can aid the Systems Bioinformatics application in biomarker discovery. GWAB, a recent tool, makes use of systems approaches and computational methods to boost weak association signals for Genome Wide Association Studies (GWAS), a common problem when analysing this type of data. This tool works by incorporating publicly available data in the form of using GWAS summary statistics (p-values) for SNPs along with reference genes for a disease of interest. The authors demonstrated the feasibility of boosting GWAS disease associations using gene networks and further present a web server for GWAB, for the network-based boosting of human GWAS data [113]. Other tools like GeneMANIA [114] and PINTA [115] allow for gene prioritization and gene function prediction and can greatly aid in computational diagnostics.
Systems Bioinformatics applications in drug discovery
Systems Bioinformatics contributes in computational therapeutics by providing tools and algorithms for novel drug discovery. Research in this direction is often done in close collaboration with pharmaceutical companies. One of the main challenges faced by both the research community and the industry is the prediction of adverse drug effects, especially during the early stages of drug development. These types of predictions can lead to significant cost reductions by allowing for accurate drug assessment and discontinuation of development for drugs with severe adverse effects. The use of human genetic variation has been known to play an important role in drug response [116]; however, the effect of this factor alone is not sufficient to provide a complete perspective on the matter in hand. Systems pharmacology is a term that is widely used today in many high-calibre, recently published studies [117–119]. Systems pharmacology is a systems biology approach, which focuses on enhancing the understanding of drugs function in the human body at a systems’ level, described by several types of networks, rather than looking at the effect of single molecular components. It shifts away from traditional practice, which considers the effects of a drug with respect to its target protein and instead strives to address the effects of the drug by considering a network of drug–target interactions. Systems Bioinformatics is a precious field in the neighbourhood of systems pharmacology that provides important methods and tools for multi-source and multilevel integration of the omics spectrum with drug networks shedding light in the area of modern drug discovery.
A recent area of great interest where Systems Bioinformatics can be of substantial impact and value is the area of drug repurposing or repositioning [120]. This entails the use of Food and Drug Administration (FDA)-approved drugs to treat new diseases, which are different from the ones they were initially designed for. This allows for obvious shortcuts for pharmaceutical companies allowing them to by-pass the timely and costly process of FDA approval for novel drugs. Recent studies used gene expression data derived from microarrays or RNA-Seq data to obtain specific expression profiles for specific diseases of interest. By comparing these to collections of data sets from repositories such as CMap [121], Drugmap Central and more advanced versions like LINCS and the recent Drug Repurposing Hub [121, 122] allows for alternative drugs to be proposed for the treatment of diseases under investigation.
In a recent study [38], this approach was used to devise drug/target networks obtained from algorithms of mutual information and co-expression networks aiming to gain insights into the treatment of breast cancer subtypes.
Data collection: TCGA mRNA (microarray) gene expression data for Breast Invasive Carcinoma cases were obtained from Firehose (http://gdac.broadinstitute.org/). From a total of 587 samples (526 primary solid tumour samples and 61 primary solid normal samples—17.814 genes), the authors selected a subset of tumour data containing information regarding breast cancer staging, HER2, ER and PR status with their corresponding normal samples as well as breast cancer stages I, II, III and IV.
Network construction: The authors examined three major categories of statistical network inference methods: (i) mutual information-based methods, (ii) correlation-based methods and (iii) tree-based methods. They further utilized Biological information-based network methods and one ensemble scheme using all statistical network inference methods. They used the Cytoscape platform and more specifically the GeneMania plug-in to reconstruct the biological information-based gene network. This plug-in uses a large data set unifying functional networks comprising approximately 800 networks for six organisms including Homo sapiens. Using the H.sapiens network they constructed a sub-network for the top 1000 differentially expressed genes (DEGs) from the TCGA data set merging five Network types: Co-expression, Physical Interaction, Genetic interaction, Co-localization and Pathways.
Network analysis: The authors further performed gene re-ranking using the underlying networks. To investigate the influence of the reconstructed 17 gene networks (12 statistically and 5 biologically inferred) on gene prioritization, they applied a method that allows for a custom network selection combining the log fold change absolute values with the selected underlying network topology to re-rank the initial DEGs. The basic idea of the method is the reconciliation of the gene expression values taking into account the underlying gene network topological features such as degree and betweenness. The network patterns were further analysed to investigate their exclusive contribution with respect to breast cancer subtypes and stages. The authors then performed drug repurposing using the up- and down-regulated genes forming disease signatures by querying them in a well-established drug repurposing pipeline, namely, LINCS-L1000 (http://www.lincscloud.org/), an advanced version of CMap. In summary, the authors obtained 63 unique drugs for the breast cancer stages and 58 for the breast cancer subtypes. To further examine the resulting drugs, the authors constructed super networks by combining top drugs extracted from their analysis with the FDA-approved breast cancer drugs, connecting them with their target genes and superimposing these on the gene expression networks.
Findings and significance of research: The authors performed an analysis that concluded to eight network patterns, four for the stages (I, II, III and IV) and four for the subtypes (Triple Negative, Luminal A, Luminal B and HER2). These patterns were shown to highlight four exclusive stage-related pathways including phenylalanine metabolism for Stage II, peroxisome proliferator-activated signalling pathway and glycolysis and gluconeogenesis for Stage III and toll-like receptor signalling pathway for Stage IV. Finally, the authors performed drug repurposing to elucidate potential anti-breast-cancer properties for known drugs and they compared the molecular structure for their predicted re-purposed drugs against 25 FDA-approved drugs of clinical use. Two out of these 25 drugs (Gemcitabine and Palbociclib) were also found as repurposed drugs by the authors. In Stage I, two repurposed drugs, Clofarabine and Kinetin-riboside, were found to be structurally similar to Gemcitabine. Clofarabine seems to have potential efficacy in epigenetic therapy of solid tumours, especially at early stages of carcinogenesis.
Another recent line of work [64] performed network-based in silico drug efficacy screening by exploiting network-based approaches. The authors investigated the association between drug targets and diseases, presenting a drug–disease proximity measure [64].
Data collection: The authors used 1489 diseases defined by Medical Subject Headings (MeSH) compiled in a recent study [90]. For each disease, the disease–gene associations were collected from OMIM and GWAS catalogue.
For each disease, the authors extracted information on FDA-approved drugs from DrugBank and matched 79 of these diseases with at least one drug using tools like MEDI-HPS and Metab2Mesh resulting in 238 unique drugs and 384 targets. The authors took information published by [90] that contained experimentally documented human protein physical interactions from TRANSFAC, IntAct, MINT, BioGRID, HPRD, KEGG, BIGG, CORUM, PhosphoSitePlus and a large-scale signalling network.
Network construction: The human PPI network was compiled using information extracted from the databases described above, to generate an elaborate human interactome. The largest connected component of this interactome was consequently used in their analysis, consisting of 141 150 interactions between 13 329 proteins. Entrez Gene IDs were used to map disease-associated genes to the corresponding proteins in the interactome.
Network analysis: The proximity between a disease and a drug was evaluated using various distance measures that take into account the path lengths between drug targets and disease proteins. The authors focused on two types of network-based proximity relationships between drugs and disease proteins: (i) the most straightforward measure is the average shortest path length between all targets of a drug and the proteins involved in the same disease; (ii) the second proximity is the closest measure, representing the average shortest path length between the drug’s targets and the nearest disease protein.
Findings and significance of research: The authors validated their approach and optimized their proximity thresholds by assessing how well relative proximity discriminates 402 known drug–disease pairs from the 18 162 unknown drug–disease pairs by comparing the area under Receiver Operating Characteristic curve for different distance measures. Based on these results the authors showed that network proximity delineates therapeutic effects of a drug. This approach of utilizing network proximity in the interactome for drug targets and diseases, allowed for increased understanding in the therapeutic effect of drugs. They made use of cases from Parkinson’s disease and several inflammatory disorders to further substantiate findings. This approach can potentially have significant applications in drug discovery, drug repurposing and assessment of drug adverse effects.
Another study [123] led to the development of a current state-of-the-art tool that addresses computational therapeutics from a network perspective, the TCM-Mesh system. This tool allows for the high-throughput network pharmacology analysis for Traditional Chinese Medicine (TCM) [123].
Data set collection: TCM utilizes data curated from collections of 6235 herbs, 383 840 compounds, 14 298 genes, 6204 diseases, 144 723 gene–disease associations, 3 440 231 pairs of gene interactions, 163 221 side-effect records and 71 toxic records (data as of April 2017). The information for traditional Chinese herbs and traditional Chinese medicine preparation was extracted from TCM Database@Taiwan, TCMID, information of compounds and their targets; diseases and their related proteins were obtained from STITCH and OMIM, respectively; the protein interactions were obtained from STRING; the toxic and side-effect records of compounds were derived from TOXNET and SIDER.
Network construction: The authors used Cytoscape as well as a web-based software to facilitate visualization of a compound–gene–disease network construction between TCM and treated diseases.
Network analysis: The authors based their network analysis and scored their compounds using the combined score as defined and obtained from the STITCH database. This score represents the strength of the links between the compounds and their associated proteins.
Findings and significance of research: The authors used 1293 FDA-approved drugs, as well as compounds from a herbal material Panax ginseng and a patented drug Liuwei Dihuang Wan for evaluating their database. By comparison of different databases, as well as checking against literature, they demonstrated the completeness, effectiveness and accuracy of the TCM-Mesh database and further aided in increased understanding of the molecular mechanisms of TCM action.
Various tools are currently available, which can aid the Systems Bioinformatics application in drug discovery. For example, tools like Substructure-Drug-Target Network-Based Inference SDTNBI [124], C(2) Maps [125], Chem2Bio2RDF [126] and PROMISCUOUS [127] cumulatively provide integrated systems and pharmacology databases for chemoinformatics analysis, drug-target prediction, networks of disease–gene–drug connectivity relationships as well as drug repositioning analysis.
For a full list of tools and databases adopting or supporting Systems Bioinformatics methodologies, see Table 1. A more comprehensive list of related tools and databases going back to 2010 can be found in Supplementary Table S1.
Table 1.
Tools and databases for systems bioinformatics approaches in therapeutics, diagnostics, network visualization/analysis, integration and systems modelling
| Tool category/description | Publication year | Link | Reference |
|---|---|---|---|
| Network-based therapeutics | |||
| TCM-Mesh: The database and analytical system for network pharmacology analysis for TCM preparations | 2017 | http://mesh.tcm.microbioinformatics.org/ | [123] |
| SDTNBI: an integrated network and chemoinformatics tool for systematic prediction of drug–target interactions and drug repositioning | 2017 | The program is available on request | [124] |
| A protein network descriptor server and its use in studying protein, disease, metabolic and drug-targeted networks | 2016 | http://bidd2.nus.edu.sg/cgi-bin/profeat2016/main.cgi | [128] |
| systemsDock: a web server for network pharmacology-based prediction and analysis | 2016 | http://systemsdock.unit.oist.jp/iddp/home/index | [129] |
| BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology | 2016 | https://www.bindingdb.org/bind/index.jsp | [130] |
| NFFinder: an online bioinformatics tool for searching similar transcriptomics experiments in the context of drug repositioning | 2015 | http://nffinder.cnb.csic.es/ | [131] |
| NutriChem: a systems chemical biology resource to explore the medicinal value of plant-based foods | 2015 | http://sbb.hku.hk/services/NutriChem-2.0/FoodDisease.php | [132] |
| TIMMA-R: an R package for predicting synergistic multi-targeted drug combinations in cancer cell lines or patient-derived samples | 2015 | https://cran.r-project.org/web/packages/timma/ | [133] |
| Network-based diagnostics | |||
| GWAB: a web server for the network-based boosting of human genome-wide association data | 2017 | http://www.inetbio.org/gwab/ | [113] |
| Netter: re-ranking gene network inference predictions using structural network properties | 2016 | https://github.com/JRuyssinck/netter | [134] |
| MetaNetVar: Pipeline for applying network analysis tools for genomic variants analysis | 2016 | https://github.com/NCBI-Hackathons/Network_SNPs | [135] |
| GenomeRunner web server: regulatory similarity and differences define the functional impact of SNP sets | 2016 | http://www.integrativegenomics.org/ | [136] |
| NetDecoder: a network biology platform that decodes context-specific biological networks and gene activities | 2016 | http://netdecoder.hms.harvard.edu/ | [137] |
| MUFFINN: cancer gene discovery via network analysis of somatic mutation data | 2016 | http://www.inetbio.org/muffinn/ | [138] |
| HitWalker2: visual analytics for precision medicine and beyond | 2016 | https://github.com/biodev/HitWalker2 | [139] |
| NCG 5.0: updates of a manually curated repository of cancer genes and associated properties from cancer mutational screenings | 2016 | http://ncg.kcl.ac.uk/ | [140] |
| dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation | 2015 | http://140.138.144.145/∼dbSNO/index.php | [141] |
| Causal biological network database: a comprehensive platform of causal biological network models focused on the pulmonary and vascular systems | 2015 | http://causalbionet.com/ | [142] |
| Network reconstruction-visualization-analysis | |||
| MotifNet: a web-server for network motif analysis | 2017 | http://netbio.bgu.ac.il/motifnet/ | [143] |
| cMapper: gene-centric connectivity mapper for EBI-RDF platform | 2017 | http://cmapper.ewostech.net/ | [144] |
| BRANE Clust: Cluster-assisted gene regulatory network inference refinement | 2017 | http://www-syscom.univ-mlv.fr/∼pirayre/Codes-GRN-BRANE-clust.html | [145] |
| vcfr: a package to manipulate and visualize variant call format data in R | 2017 | https://cran.r-project.org/web/packages/vcfR/index.html | [146] |
| shinyheatmap: Ultra-fast low-memory heatmap web interface for big data genomics | 2017 | http://shinyheatmap.com/ | [147] |
| PROXiMATE: a database of mutant protein–protein complex thermodynamics and kinetics | 2017 | http://www.iitm.ac.in/bioinfo/PROXiMATE/ | [148] |
| Recon2Neo4j: applying graph database technologies for managing comprehensive genome-scale networks | 2017 | https://github.com/ibalaur/MetabolicFramework | [149] |
| RAIN: RNA–protein association and interaction networks | 2017 | http://rth.dk/resources/rain/ | [150] |
| Phenopolis: an open platform for harmonization and analysis of genetic and phenotypic data | 2017 | https://uclex.cs.ucl.ac.uk/ | [151] |
| Pheno4J: a gene to phenotype graph database | 2017 | https://github.com/phenopolis/pheno4j | [152] |
| SigMod: an exact and efficient method to identify a strongly interconnected disease-associated module in a gene network | 2017 | https://github.com/YuanlongLiu/SigMod | [153] |
| iRegNet3D: three-dimensional integrated regulatory network for the genomic analysis of coding and non-coding disease mutations | 2017 | http://iregnet3d.yulab.org/index/ | [154] |
| JDINAC: joint density-based non-parametric differential interaction network analysis and classification using high-dimensional sparse omics data | 2017 | https://github.com/jijiadong/JDINAC | [155] |
| SmartR: An open-source platform for interactive visual analytics for translational research data | 2017 | https://github.com/transmart/SmartR | [156] |
| D-Map: random walking on gene network inference maps towards differential avenue discovery | 2017 | http://bioserver-3.bioacademy.gr/Bioserver/DMap/index.php | [157] |
| TRaCE+: Ensemble inference of gene regulatory networks from transcriptional expression profiles of gene knock-out experiments | 2016 | http://www.cabsel.ethz.ch/tools/trace.html | [158] |
| The Network Library: a framework to rapidly integrate network biology resources | 2016 | https://github.com/gsummer | |
| Web-based network analysis and visualization using CellMaps | 2016 | http://cellmaps.babelomics.org/ | [159] |
| PathwAX: a web server for network crosstalk based pathway annotation | 2016 | http://pathwax.sbc.su.se/ | [160] |
| Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology | 2016 | http://brg.ai.sri.com/ptools/ | [161] |
| NAPS: Network analysis of protein structures | 2016 | http://bioinf.iiit.ac.in/NAPS/ | [162] |
| UbiNet: an online resource for exploring the functional associations and regulatory networks of protein ubiquitylation | 2016 | http://140.138.144.145/∼ubinet/index.php | [163] |
| MET network in PubMed: a text-mined network visualization and curation system | 2016 | http://btm.tmu.edu.tw/metastasisway | [164] |
| QuIN: a web server for querying and visualizing chromatin interaction networks | 2016 | https://quin.jax.org/ | [165] |
| NET-GE: a web server for NETwork-based human gene enrichment | 2016 | http://net-ge.biocomp.unibo.it/enrich | [166] |
| IIIDB: a database for isoform–isoform interactions and isoform network modules | 2015 | http://syslab.nchu.edu.tw/IIIDB/ | [167] |
| cyNeo4j: connecting Neo4j and Cytoscape | 2015 | http://apps.cytoscape.org/apps/cyneo4j | [168] |
| BRANE Cut: biologically related a priori network enhancement with graph cuts for gene regulatory network inference | 2015 | http://www-syscom.univ-mlv.fr/∼pirayre/Codes-GRN-BRANE-cut.html | [169] |
| NetExplore: a web server for modelling small network motifs | 2015 | http://line.bioinfolab.net/nex/NetExplore.htm | [170] |
| COXPRESdb in 2015: coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems | 2015 | http://coxpresdb.jp/ | [171] |
| NAIL: a software toolset for inferring, analysing and visualizing regulatory networks | 2015 | https://sourceforge.net/projects/nailsystemsbiology/ | [172] |
| LncReg: a reference resource for lncRNA-associated regulatory networks | 2015 | http://bioinformatics.ustc.edu.cn/lncreg/ | [173] |
| TeloPIN: a database of telomeric proteins interaction network in mammalian cells | 2015 | http://songyanglab.sysu.edu.cn/telopin/ | [174] |
| MIsoMine: a genome-scale high-resolution data portal of expression, function and networks at the splice isoform level in the mouse | 2015 | http://guanlab.ccmb.med.umich.edu/misomine/ | [175] |
| CerebralWeb: a Cytoscape.js plug-in to visualize networks stratified by subcellular localization | 2015 | http://www.innatedb.ca/CerebralWeb/ | [176] |
| Network-based integration | |||
| NaviCom: a web application to create interactive molecular network portraits using multilevel omics data | 2017 | https://navicom.curie.fr/bridge.php | [177] |
| KeyPathwayMinerWeb: online multi-omics network enrichment | 2016 | https://keypathwayminer.compbio.sdu.dk/keypathwayminer/ | [178] |
| Visual Omics Explorer (VOE): a cross-platform portal for interactive data visualization | 2016 | http://bcil.github.io/VOE/ | [179] |
| ModuleAlign: module-based global alignment of PPI networks | 2016 | http://ttic.uchicago.edu/∼hashemifar/ModuleAlign.html | [180] |
| Fuse: multiple network alignment via data fusion | 2016 | http://www0.cs.ucl.ac.uk/staff/natasa/FUSE/index.html | [181] |
| The SMAL web server: global multiple network alignment from pairwise alignments | 2016 | http://haddock6.sfsu.edu/smal/ | [182] |
| Mergeomics: a web server for identifying pathological pathways, networks and key regulators via multidimensional data integration | 2016 | http://mergeomics.research.idre.ucla.edu/ | [183] |
| MAGNA ++: Maximizing accuracy in global network alignment via both node and edge conservation | 2015 | http://www3.nd.edu/∼cone/MAGNA±+/ | [184] |
| ZoomOut: analysing multiple networks as single nodes | 2015 | http://bioserver-3.bioacademy.gr/Bioserver/ZoomOut/ | [185] |
| RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse | 2015 | http://www.regnetworkweb.org/ | [186] |
| Systems biology and modelling | |||
| FAIRDOMHub: a repository and collaboration environment for sharing systems biology research | 2017 | https://fair-dom.org/publication/fairdomhub-a-repository-and-collaboration-environment-for-sharing-systems-biology-research/ | [187] |
| The systems biology format converter | 2016 | https://www.ebi.ac.uk/biomodels/tools/converters/ | [188] |
| SBtab: a flexible table format for data exchange in systems biology | 2016 | https://www.sbtab.net/ | |
| PeTTSy: a computational tool for perturbation analysis of complex systems biology models | 2016 | http://www2.warwick.ac.uk/fac/sci/systemsbiology/research/software/ | [189] |
| AMIGO2: a toolbox for dynamic modelling, optimization and control in systems biology | 2016 | https://sites.google.com/site/amigo2toolbox/ | [190] |
| ComPPI: a cellular compartment-specific database for PPI network analysis | 2015 | http://comppi.linkgroup.hu/ | [191] |
| JSBML 1.0: providing a smorgasbord of options to encode systems biology models | 2015 | http://sbml.org/Software/JSBML | [192] |
| MpTheory Java library: a multi-platform Java library for systems biology based on the Metabolic P theory | 2015 | http://mptheory.scienze.univr.it/ | [193] |
| SYSBIONS: nested sampling for systems biology | 2015 | http://www.theosysbio.bio.ic.ac.uk/resources/sysbions/ | [194] |
| Dizzy-Beats: a Bayesian evidence analysis tool for systems biology | 2015 | https://sourceforge.net/p/bayesevidence/home/Home/ | [195] |
Discussion
The concept of utilizing networks to visualize the complex interaction of mechanisms implicated in disease has been around for several years. However, two important breakthroughs separate previous network-based approaches and are currently driving the state-of-the-art in Systems Bioinformatics: (i) construction of multiple networks representing each level of the omics spectrum and the integration of these in a layered network that exchanges information within and between layers [62, 67, 97] and (ii) the advent of novel techniques and methodologies for analysing and understanding these networks using mathematical algorithms and approaches derived from graph theory and information theory [6]. Using these methods for extracting biologically meaningful information from multiple levels of the omics spectrum can provide the integrated systemic knowledge for the development of a comprehensive Human System profile, which increases diagnostic accuracy and concurrently allows for novel therapeutic advances and assess response to therapy.
Nevertheless, the network-based approaches, either for evidence-based or for statistically inferred molecular networks, have a number of limitations. Specifically, networks based on experimental evidence are not complete as experiments are only a snapshot of the real biological world. Moreover, statistically inferred networks represent an undetermined computational problem because the number of the inferred relationships is much larger than the number of the independent measurements [196]. Owing to the lack of sufficient ground truth to validate the reconstructed molecular networks, special attention must be given when choosing benchmark data sets (e.g. existing curated databases with experimental information, medium-throughput experimental information and simulated data sets that mimic real data). To this extent, the DREAM (Dialogue on Reverse Engineering Assessments and Methods) initiative facilitates researchers from the Systems Bioinformatics field to assess the validity of the networks they are using and proceed with optimization and parameter-tuning regarding network reconstruction [197]. A subsequent limitation of the network-based approaches is the low overlap that the various network reconstruction approaches have, and the inadequacy in selecting the proper network each time. It is likely that computational approaches checking and exploiting complementarities and providing ensemble solutions of a network construction consensus will maximize the information content.
In our opinion, Systems Bioinformatics might currently appear rather aspirational, yet, considering its potential it is likely to have a major impact on medicine and pharmacology in the next decade. The field of medicine is expected to benefit from the invaluable knowledge attained from Systems Bioinformatics methodologies. The molecular basis of complex, polygenic diseases is highly heterogeneous and affected by multiple factors simultaneously. These include genetic predisposition, multipart molecular mechanisms and effects of the environment, diet, drug administration/response and numerous other factors. Although it might not be possible to replace the use of traditional approaches for therapeutics and diagnosis with computational methods, yet, it is likely that Systems Bioinformatics will provide revolutionary approaches and tools to clinicians in order to demystify the complex nature of these diseases. Computational diagnostics and therapeutics, enhanced by Systems Bioinformatics approaches, will not only aid clinicians in patient consultation and care but will also catalyse significant breakthroughs in prognostic measures, detection of disease at an early onset and overall disease prevention.
Supplementary Material
Anastasis Oulas is a Postdoctoral Research Fellow at the Bioinformatics ERA Chair and Bioinformatics Group of the Cyprus Institute of Neurology and Genetics.
George Minadakis is a Postdoctoral Research Fellow at the Bioinformatics ERA Chair and Bioinformatics Group of the Cyprus Institute of Neurology and Genetics.
Margarita Zachariou is a Postdoctoral Research Fellow at the Bioinformatics ERA Chair and Bioinformatics Group of the Cyprus Institute of Neurology and Genetics.
Kleitos Sokratous is a Postdoctoral Research Fellow at the Bioinformatics ERA Chair and Bioinformatics Group of the Cyprus Institute of Neurology and Genetics.
Marilena M. Bourdakou is a Visiting Scientist at the Bioinformatics ERA Chair and Bioinformatics Group of the Cyprus Institute of Neurology and Genetics.
George M. Spyrou holds the Bioinformatics ERA Chair and is the Head of the Bioinformatics Group at the Cyprus Institute of Neurology and Genetics.
Key Points
Systems Βioinformatics is an emerging field, which integrates information across different levels by combining the systems biology bottom-up approach with a data-driven top-down approach as in classical bioinformatics.
The advent of omics technologies has provided the stepping-stone for the emergence of Systems Bioinformatics as a holistic and systems approach in investigating complex biological systems.
The key approach in Systems Bioinformatics is the construction of multiple networks representing each level of the omics spectrum and their integration in a layered network that exchanges information within and between layers.
The network approach in Systems Bioinformatics comes with limitations including the lack of ground truth for the constructed biological network.
Systems Bioinformatics methods can enhance computational therapeutics and diagnostics hence, paving the way to precision medicine.
Funding
Anastasis Oulas, George Minadakis, Margarita Zachariou, Kleitos Sokratous and George M. Spyrou are funded by the European Commission Research Executive Agency Grant BIORISE (No. 669026), under the Spreading Excellence, Widening Participation, Science with and for Society Framework. This work was partly supported by H2020-WIDESPREAD-04-2017-Teaming Phase 1, Grant Agreement 763781, Integrated Precision Medicine Technologies.
References
- 1. Hood L, Friend SH.. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol 2011;8(3):184–7. 10.1038/nrclinonc.2010.227 [DOI] [PubMed] [Google Scholar]
- 2. Tian Q, Price ND, Hood L.. Systems cancer medicine: towards realization of predictive, preventive, personalized and participatory (P4) medicine. J Intern Med 2012;271(2):111–21. 10.1111/j.1365-2796.2011.02498.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatherer D. So what do we really mean when we say that systems biology is holistic? BMC Syst Biol 2010;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berlin R, Gruen R, Best J.. Systems medicine-complexity within, simplicity without. J Healthc Inform Res 2017;1(1):119–37. 10.1007/s41666-017-0002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emmert-Streib F, Dehmer M.. Networks for systems biology: conceptual connection of data and function. IET Syst Biol 2011;5(3):185–207. 10.1049/iet-syb.2010.0025 [DOI] [PubMed] [Google Scholar]
- 6. Najafi A, Bidkhori G, Bozorgmehr JH, et al. Genome scale modeling in systems biology: algorithms and resources. Curr Genomics 2014;15(2):130–59. 10.2174/1389202915666140319002221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barabasi AL, Albert R.. Emergence of scaling in random networks. Science 1999;286(5439):509–12. 10.1126/science.286.5439.509 [DOI] [PubMed] [Google Scholar]
- 8. Barabasi AL, Gulbahce N, Loscalzo J.. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12(1):56–68. 10.1038/nrg2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu H, Kim PM, Sprecher E, et al. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol 2007;3(4):e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blain-Moraes S, Tarnal V, Vanini G, et al. Network efficiency and posterior alpha patterns are markers of recovery from general anesthesia: a high-density electroencephalography study in healthy volunteers. Front Hum Neurosci 2017;11:328 10.3389/fnhum.2017.00328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farrar DC, Mian AZ, Budson AE, et al. Retained executive abilities in mild cognitive impairment are associated with increased white matter network connectivity. Eur Radiol 2017, doi: 10.1007/s00330-017-4951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao ZK, Dang WD, Li S, et al. PageRank versatility analysis of multilayer modality-based network for exploring the evolution of oil-water slug flow. Sci Rep 2017;7(1):5493 10.1038/s41598-017-05890-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barabasi AL, Oltvai ZN.. Network biology: understanding the cell's functional organization. Nat Rev Genet 2004;5(2):101–13. 10.1038/nrg1272 [DOI] [PubMed] [Google Scholar]
- 14. Rual JF, Venkatesan K, Hao T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005;437(7062):1173–8. 10.1038/nature04209 [DOI] [PubMed] [Google Scholar]
- 15. Lewis BP, Burge CB, Bartel DP.. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120(1):15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 16. Carninci P, Kasukawa T, Katayama S, et al. The transcriptional landscape of the mammalian genome. Science 2005;309(5740):1559–63. 10.1126/science.1112014 [DOI] [PubMed] [Google Scholar]
- 17. Jeong H, Tombor B, Albert R, et al. The large-scale organization of metabolic networks. Nature 2000;407(6804):651–4. 10.1038/35036627 [DOI] [PubMed] [Google Scholar]
- 18. Rolland T, Taşan M, Charloteaux B, et al. A proteome-scale map of the human interactome network. Cell 2014;159(5):1212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kapushesky M, Emam I, Holloway E, et al. Gene expression atlas at the European bioinformatics institute. Nucleic Acids Res 2010;38:D690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X, Yu X, Zack DJ, et al. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics 2008;9:271 10.1186/1471-2105-9-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harris MA, Clark J, Ireland A, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 2004;32:D258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Croft D, Mundo AF, Haw R, et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2014;42:D472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabregat A, Sidiropoulos K, Garapati P, et al. The Reactome pathway Knowledgebase. Nucleic Acids Res 2016;44(D1):D481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raychaudhuri S, Plenge R, Rossin E, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet 2009;5:e100534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Siegenthaler C, Gunawan R.. Assessment of network inference methods: how to cope with an underdetermined problem. PLoS One 2014;9(3):e90481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43(D1):D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peri S, Navarro JD, Kristiansen TZ, et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res 2004;32(90001):D497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bader GD, Betel D, Hogue CWV.. BIND: the biomolecular interaction network database. Nucleic Acids Res 2003;31(1):248–50. 10.1093/nar/gkg056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chatr-aryamontri A, Ceol A, Palazzi LM, et al. MINT: the molecular INTeraction database. Nucleic Acids Res 2007;35:D572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stark C, Breitkreutz B-J, Reguly T, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006;34(90001):D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casas C, Isus L, Herrando-Grabulosa M, et al. Network-based proteomic approaches reveal the neurodegenerative, neuroprotective and pain-related mechanisms involved after retrograde axonal damage. Sci Rep 2015;5:9185 10.1038/srep09185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Severin J, Waterhouse AM, Kawaji H, et al. FANTOM4 EdgeExpressDB: an integrated database of promoters, genes, microRNAs, expression dynamics and regulatory interactions. Genome Biol 2009;10(4):R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang C, Xuan Z, Zhao F, et al. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res 2007;35:D137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102(43):15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feist P, Hummon A.. Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int J Mol Sci 2015;16(2):3537 10.3390/ijms16023537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ekins S, Nikolsky Y, Bugrim A, et al. Pathway mapping tools for analysis of high content data In: Taylor DL, Haskins JR, Giuliano KA (eds). High Content Screening: A Powerful Approach to Systems Cell Biology and Drug Discovery. Totowa, NJ: Humana Press, 2006, 319–50. [Google Scholar]
- 37. Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012;40:D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bourdakou MM, Athanasiadis EI, Spyrou GM.. Discovering gene re-ranking efficiency and conserved gene-gene relationships derived from gene co-expression network analysis on breast cancer data. Sci Rep 2016;6(1):20518 10.1038/srep20518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marbach D, Costello JC, Kuffner R, et al. Wisdom of crowds for robust gene network inference. Nat Methods 2012;9(8):796–804. 10.1038/nmeth.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beltrao P, Cagney G, Krogan NJ.. Quantitative genetic interactions reveal biological modularity. Cell 2010;141(5):739–45. 10.1016/j.cell.2010.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boone C, Bussey H, Andrews BJ.. Exploring genetic interactions and networks with yeast. Nat Rev Genet 2007;8(6):437–49. 10.1038/nrg2085 [DOI] [PubMed] [Google Scholar]
- 42. Stuart JM, Segal E, Koller D, et al. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003;302(5643):249–55. 10.1126/science.1087447 [DOI] [PubMed] [Google Scholar]
- 43. Chatr-Aryamontri A, Oughtred R, Boucher L, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res 2017;45(D1):D369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kraskov A, Stogbauer H, Grassberger P.. Estimating mutual information. Phys Rev E Stat Nonlin Soft Matter Phys 2004;69(6 Pt 2):066138. [DOI] [PubMed] [Google Scholar]
- 45. Margolin AA, Nemenman I, Basso K, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 2006;7(Suppl 1):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faith JJ, Hayete B, Thaden JT, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol 2007;5(1):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meyer PE, Kontos K, Lafitte F, et al. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J Bioinform Syst Biol 2007;2007:79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Altay G, Emmert-Streib F.. Inferring the conservative causal core of gene regulatory networks. BMC Syst Biol 2010;4:132 10.1186/1752-0509-4-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Opgen-Rhein R, Strimmer K.. From correlation to causation networks: a simple approximate learning algorithm and its application to high-dimensional plant gene expression data. BMC Syst Biol 2007;1(1):37 10.1186/1752-0509-1-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Langfelder P, Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Friedman J, Hastie T, Tibshirani R.. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 2008;9(3):432–41. 10.1093/biostatistics/kxm045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kramer N, Schafer J, Boulesteix AL.. Regularized estimation of large-scale gene association networks using graphical Gaussian models. BMC Bioinformatics 2009;10:384 10.1186/1471-2105-10-384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huynh-Thu VA, Irrthum A, Wehenkel L, et al. Inferring regulatory networks from expression data using tree-based methods. PLoS One 2010;5(9):e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goh KI, Choi IG.. Exploring the human diseasome: the human disease network. Brief Funct Genomics 2012;11(6):533–42. 10.1093/bfgp/els032 [DOI] [PubMed] [Google Scholar]
- 55. Goh KI, Cusick ME, Valle D, et al. The human disease network. Proc Natl Acad Sci USA 2007;104(21):8685–90. 10.1073/pnas.0701361104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mitra K, Carvunis AR, Ramesh SK, et al. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet 2013;14(10):719–32. 10.1038/nrg3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Loscalzo J, Barabasi AL.. Systems biology and the future of medicine. Wiley Interdiscip Rev Syst Biol Med 2011;3(6):619–27. 10.1002/wsbm.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hart T, Dider S, Han W, et al. Toward repurposing metformin as a precision anti-cancer therapy using structural systems pharmacology. Sci Rep 2016;6(1):20441 10.1038/srep20441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim BY, Song KH, Lim CY, et al. Therapeutic properties of Scutellaria baicalensis in db/db mice evaluated using connectivity map and network pharmacology. Sci Rep 2017;7:41711 10.1038/srep41711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moran LB, Graeber MB.. Towards a pathway definition of Parkinson's disease: a complex disorder with links to cancer, diabetes and inflammation. Neurogenetics 2008;9(1):1–13. 10.1007/s10048-007-0116-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mukhopadhyay S, Saha R, Palanisamy A, et al. A systems biology pipeline identifies new immune and disease related molecular signatures and networks in human cells during microgravity exposure. Sci Rep 2016;6(1):25975 10.1038/srep25975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jamal S, Goyal S, Shanker A, et al. Integrating network, sequence and functional features using machine learning approaches towards identification of novel Alzheimer genes. BMC Genomics 2016;17(1):807 10.1186/s12864-016-3108-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ray M, Ruan J, Zhang W.. Variations in the transcriptome of Alzheimer's disease reveal molecular networks involved in cardiovascular diseases. Genome Biol 2008;9(10):R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guney E, Menche J, Vidal M, et al. Network-based in silico drug efficacy screening. Nat Commun 2016;7:10331 10.1038/ncomms10331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Breuer K, Foroushani AK, Laird MR, et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res 2013;41(D1):D1228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hwang S, Son SW, Kim SC, et al. A protein interaction network associated with asthma. J Theor Biol 2008;252(4):722–31. 10.1016/j.jtbi.2008.02.011 [DOI] [PubMed] [Google Scholar]
- 67. Menche J, Guney E, Sharma A, et al. Integrating personalized gene expression profiles into predictive disease-associated gene pools. NPJ Syst Biol Appl 2017;3(1):10 10.1038/s41540-017-0009-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cheng F, Hong H, Yang S, et al. Individualized network-based drug repositioning infrastructure for precision oncology in the panomics era. Brief Bioinform 2017;18:682–97. [DOI] [PubMed] [Google Scholar]
- 69. Lee E, Jung H, Radivojac P, et al. Analysis of AML genes in dysregulated molecular networks. BMC Bioinformatics 2009;10(Suppl 9):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lim J, Hao T, Shaw C, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 2006;125(4):801–14. 10.1016/j.cell.2006.03.032 [DOI] [PubMed] [Google Scholar]
- 71. Goehler H, Lalowski M, Stelzl U, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell 2004;15(6):853–65. 10.1016/j.molcel.2004.09.016 [DOI] [PubMed] [Google Scholar]
- 72. Camargo LM, Collura V, Rain JC, et al. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry 2007;12(1):74–86. 10.1038/sj.mp.4001880 [DOI] [PubMed] [Google Scholar]
- 73. Simoes SN, Martins DC Jr, Pereira CA, et al. NERI: network-medicine based integrative approach for disease gene prioritization by relative importance. BMC Bioinformatics 2015;16(Suppl 19):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang H, Guo W, Liu F, et al. Patients with first-episode, drug-naive schizophrenia and subjects at ultra-high risk of psychosis shared increased cerebellar-default mode network connectivity at rest. Sci Rep 2016;6(1):26124 10.1038/srep26124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kitsak M, Sharma A, Menche J, et al. Tissue specificity of human disease module. Sci Rep 2016;6:35241 10.1038/srep35241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang W-X, Ni X, Lai Y-C, et al. Optimizing controllability of complex networks by minimum structural perturbations. Phys Rev E 2012;85(2):026115 10.1103/PhysRevE.85.026115 [DOI] [PubMed] [Google Scholar]
- 77. Albert R, Jeong H, Barabasi AL.. Error and attack tolerance of complex networks. Nature 2000;406(6794):378–82. 10.1038/35019019 [DOI] [PubMed] [Google Scholar]
- 78. Lu ZM, Li XF.. Attack vulnerability of network controllability. PLoS One 2016;11(9):e0162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dong G, Gao J, Du R, et al. Robustness of network of networks under targeted attack. Phys Rev E Stat Nonlin Soft Matter Phys 2013;87(5):052804 10.1103/PhysRevE.87.052804 [DOI] [PubMed] [Google Scholar]
- 80. Samay N, Morrison S, Husseini A.. Network Science Communities. Cambridge: Cambridge University Press, 2014. [Google Scholar]
- 81. Pósfai M, Liu Y-Y, Slotine J-J, et al. Effect of correlations on network controllability. Sci Rep 2013;3(1):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu Y-Y, Slotine J-J, Barabási A-L.. Control centrality and hierarchical structure in complex networks. PLoS One 2012;7(9):e44459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Motter AE, Lai YC.. Cascade-based attacks on complex networks. Phys Rev E Stat Nonlin Soft Matter Phys 2002;66(6 Pt 2):065102. [DOI] [PubMed] [Google Scholar]
- 84. Holme P, Kim BJ, Yoon CN, et al. Attack vulnerability of complex networks. Phys Rev E 2002;65(5 Pt 2):056109. [DOI] [PubMed] [Google Scholar]
- 85. Wang XF, Chen G.. Synchronization in scale-free dynamical networks: robustness and fragility. IEEE Trans Circuits Syst I Fundam Theory Appl 2002;49:54–62. 10.1109/81.974874 [DOI] [Google Scholar]
- 86. Pu C-L, Pei W-J, Michaelson A.. Robustness analysis of network controllability. Physica A 2012;391(18):4420–5. 10.1016/j.physa.2012.04.019 [DOI] [Google Scholar]
- 87. Han J-DJ, Bertin N, Hao T, et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 2004;430:88–93. 10.1038/nature02555 [DOI] [PubMed] [Google Scholar]
- 88. Pequito S, Preciado VM, Barabasi AL, et al. Trade-offs between driving nodes and time-to-control in complex networks. Sci Rep 2017;7:39978 10.1038/srep39978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vinayagam A, Gibson TE, Lee HJ, et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc Natl Acad Sci USA 2016;113(18):4976–81. 10.1073/pnas.1603992113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Menche J, Sharma A, Kitsak M, et al. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015;347(6224):1257601 10.1126/science.1257601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ma X, Liu Z, Zhang Z, et al. Multiple network algorithm for epigenetic modules via the integration of genome-wide DNA methylation and gene expression data. BMC Bioinformatics 2017;18(1):72 10.1186/s12859-017-1490-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kitano H. Computational systems biology. Nature 2002;420(6912):206–10. 10.1038/nature01254 [DOI] [PubMed] [Google Scholar]
- 93. Le Novere N. Quantitative and logic modelling of molecular and gene networks. Nat Rev Genet 2015;16:146–58. 10.1038/nrg3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Samaga R, Klamt S.. Modeling approaches for qualitative and semi-quantitative analysis of cellular signaling networks. Cell Commun Signal 2013;11(1):43 10.1186/1478-811X-11-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shibeko AM, Panteleev MA.. Untangling the complexity of blood coagulation network: use of computational modelling in pharmacology and diagnostics. Brief Bioinform 2016;17(3):429–39. 10.1093/bib/bbv040 [DOI] [PubMed] [Google Scholar]
- 96. Huang L, Jiang Y, Chen Y.. Predicting drug combination index and simulating the network-regulation dynamics by mathematical modeling of drug-targeted EGFR-ERK signaling pathway. Sci Rep 2017;7:40752 10.1038/srep40752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ram PT, Mendelsohn J, Mills GB.. Bioinformatics and systems biology. Mol Oncol 2012;6(2):147–54. 10.1016/j.molonc.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Vijayakumar S, Conway M, Lio P, et al. Seeing the wood for the trees: a forest of methods for optimization and omic-network integration in metabolic modelling. Brief Bioinform 2017, doi: 10.1093/bib/bbx053. [DOI] [PubMed] [Google Scholar]
- 99. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13(11):2498–504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215(3):403–10. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 102. Peng Y, Leung HC, Yiu SM, et al. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012;28(11):1420–8. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- 103. Ihaka R, Gentleman R.. R: a language for data analysis and graphics. J Comput Graph Stat 1996;5(3):299. [Google Scholar]
- 104. Zhang F, Ren C, Lau KK, et al. A network medicine approach to build a comprehensive atlas for the prognosis of human cancer. Brief Bioinform 2016;17:1044–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Weinstein JN, Collisson EA, Mills GB, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013;45(10):1113–20. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Domenyuk V, Zhong Z, Stark A, et al. Plasma exosome profiling of cancer patients by a next generation systems biology approach. Sci Rep 2017;7:42741 10.1038/srep42741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Antonopoulou K, Stefanaki I, Lill CM, et al. Updated field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma: the MelGene database. J Invest Dermatol 2015;135(4):1074–9. 10.1038/jid.2014.491 [DOI] [PubMed] [Google Scholar]
- 108. Shirasaki DI, Greiner ER, Al-Ramahi I, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012;75(1):41–57. 10.1016/j.neuron.2012.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Huang DW, Sherman BT, Lempicki RA.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols 2008;4(1):44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 110. Mi H, Lazareva-Ulitsky B, Loo R, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 2005;33:D284–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Xu G, Paige JS, Jaffrey SR.. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotech 2010;28(8):868–73. 10.1038/nbt.1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kim W, Bennett Eric J, Huttlin Edward L, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011;44(2):325–40. 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shim JE, Bang C, Yang S, et al. GWAB: a web server for the network-based boosting of human genome-wide association data. Nucleic Acids Res 2017, doi: 10.1093/nar/gkx284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010;38(Suppl 2):W214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nitsch D, Tranchevent LC, Goncalves JP, et al. PINTA: a web server for network-based gene prioritization from expression data. Nucleic Acids Res 2011;39:W334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Madian AG, Wheeler HE, Jones RB, et al. Relating human genetic variation to variation in drug responses. Trends Genet 2012;28(10):487–95. 10.1016/j.tig.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yu W, Li Z, Long F, et al. A systems pharmacology approach to determine active compounds and action mechanisms of Xipayi KuiJie'an enema for treatment of ulcerative colitis. Sci Rep 2017;7(1):1189 10.1038/s41598-017-01335-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang J, Liu R, Liu B, et al. Systems Pharmacology-based strategy to screen new adjuvant for hepatitis B vaccine from Traditional Chinese Medicine Ophiocordyceps sinensis. Sci Rep 2017;7:44788 10.1038/srep44788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Irurzun-Arana I, Pastor JM, Troconiz IF, et al. Advanced Boolean modeling of biological networks applied to systems pharmacology. Bioinformatics 2017;33:1040–8. [DOI] [PubMed] [Google Scholar]
- 120. Lotfi Shahreza M, Ghadiri N, Mousavi SR, et al. A review of network-based approaches to drug repositioning. Brief Bioinform 2017, doi: 10.1093/bib/bbx017. [DOI] [PubMed] [Google Scholar]
- 121. Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313(5795):1929–35. 10.1126/science.1132939 [DOI] [PubMed] [Google Scholar]
- 122. Corsello SM, Bittker JA, Liu Z, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 2017;23(4):405–8. 10.1038/nm.4306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang RZ, Yu SJ, Bai H, et al. TCM-Mesh: the database and analytical system for network pharmacology analysis for TCM preparations. Sci Rep 2017;7(1):2821 10.1038/s41598-017-03039-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wu Z, Cheng F, Li J, et al. SDTNBI: an integrated network and chemoinformatics tool for systematic prediction of drug-target interactions and drug repositioning. Brief Bioinform 2017;18:333–47. [DOI] [PubMed] [Google Scholar]
- 125. Huang H, Wu X, Pandey R, et al. C(2)Maps: a network pharmacology database with comprehensive disease-gene-drug connectivity relationships. BMC Genomics 2012;13(Suppl 6):S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chen B, Dong X, Jiao D, et al. Chem2Bio2RDF: a semantic framework for linking and data mining chemogenomic and systems chemical biology data. BMC Bioinformatics 2010;11(1):255 10.1186/1471-2105-11-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. von Eichborn J, Murgueitio MS, Dunkel M, et al. PROMISCUOUS: a database for network-based drug-repositioning. Nucleic Acids Res 2011;39:D1060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhang P, Tao L, Zeng X, et al. A protein network descriptor server and its use in studying protein, disease, metabolic and drug targeted networks. Brief Bioinform 2016, doi: 10.1093/bib/bbw071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hsin KY, Matsuoka Y, Asai Y, et al. systemsDock: a web server for network pharmacology-based prediction and analysis. Nucleic Acids Res 2016;44(W1):W507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gilson MK, Liu T, Baitaluk M, et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res 2016;44(D1):D1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Setoain J, Franch M, Martinez M, et al. NFFinder: an online bioinformatics tool for searching similar transcriptomics experiments in the context of drug repositioning. Nucleic Acids Res 2015;43(W1):W193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Jensen K, Panagiotou G, Kouskoumvekaki I.. NutriChem: a systems chemical biology resource to explore the medicinal value of plant-based foods. Nucleic Acids Res 2015;43:D940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. He L, Wennerberg K, Aittokallio T, et al. TIMMA-R: an R package for predicting synergistic multi-targeted drug combinations in cancer cell lines or patient-derived samples. Bioinformatics 2015;31(11):1866–8. 10.1093/bioinformatics/btv067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ruyssinck J, Demeester P, Dhaene T, et al. Netter: re-ranking gene network inference predictions using structural network properties. BMC Bioinformatics 2016;17(1):76 10.1186/s12859-016-0913-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Moyer E, Hagenauer M, Lesko M, et al. MetaNetVar: pipeline for applying network analysis tools for genomic variants analysis. F1000Res 2016;5:674 10.12688/f1000research.8288.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dozmorov MG, Cara LR, Giles CB, et al. GenomeRunner web server: regulatory similarity and differences define the functional impact of SNP sets. Bioinformatics 2016;32(15):2256–63. 10.1093/bioinformatics/btw169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. da Rocha EL, Ung CY, McGehee CD, et al. NetDecoder: a network biology platform that decodes context-specific biological networks and gene activities. Nucleic Acids Res 2016;44:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cho A, Shim JE, Kim E, et al. MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biol 2016;17(1):129 10.1186/s13059-016-0989-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bottomly D, McWeeney SK, Wilmot B.. HitWalker2: visual analytics for precision medicine and beyond. Bioinformatics 2016;32(8):1253–5. 10.1093/bioinformatics/btv739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. An O, Dall'Olio GM, Mourikis TP, et al. NCG 5.0: updates of a manually curated repository of cancer genes and associated properties from cancer mutational screenings. Nucleic Acids Res 2016;44(D1):D992–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chen YJ, Lu CT, Su MG, et al. dbSNO 2.0: a resource for exploring structural environment, functional and disease association and regulatory network of protein S-nitrosylation. Nucleic Acids Res 2015;43:D503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Boue S, Talikka M, Westra JW, et al. Causal biological network database: a comprehensive platform of causal biological network models focused on the pulmonary and vascular systems. Database 2015;2015:bav030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Smoly IY, Lerman E, Ziv-Ukelson M, et al. MotifNet: a web-server for network motif analysis. Bioinformatics 2017;33:1907–9. 10.1093/bioinformatics/btx056 [DOI] [PubMed] [Google Scholar]
- 144. Shoaib M, Ansari AA, Ahn SM.. cMapper: gene-centric connectivity mapper for EBI-RDF platform. Bioinformatics 2017;33(2):266–71. 10.1093/bioinformatics/btw612 [DOI] [PubMed] [Google Scholar]
- 145. Pirayre A, Couprie C, Duval L, et al. BRANE clust: cluster-assisted gene regulatory network inference refinement. IEEE/ACM Trans Comput Biol Bioinform 2017, doi: 10.1109/TCBB.2017.2688355. [DOI] [PubMed] [Google Scholar]
- 146. Knaus BJ, Grunwald NJ.. vcfr: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour 2017;17(1):44–53. 10.1111/1755-0998.12549 [DOI] [PubMed] [Google Scholar]
- 147. Khomtchouk BB, Hennessy JR, Wahlestedt C.. Shinyheatmap: ultra fast low memory heatmap web interface for big data genomics. PLoS One 2017;12(5):e0176334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Jemimah S, Yugandhar K, Gromiha MM.. PROXiMATE: a database of mutant protein-protein complex thermodynamics and kinetics. Bioinformatics 2017;33:2787–8. 10.1093/bioinformatics/btx312 [DOI] [PubMed] [Google Scholar]
- 149. Balaur I, Mazein A, Saqi M, et al. Recon2Neo4j: applying graph database technologies for managing comprehensive genome-scale networks. Bioinformatics 2017;33:1096–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Junge A, Refsgaard JC, Garde C, et al. RAIN: RNA-protein association and interaction networks. Database 2017;2017:baw167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Pontikos N, Yu J, Moghul I, et al. Phenopolis: an open platform for harmonization and analysis of genetic and phenotypic data. Bioinformatics 2017;33:2421–3. [DOI] [PubMed] [Google Scholar]
- 152. Mughal S, Moghul I, Yu J, et al. Pheno4J: a gene to phenotype graph database. Bioinformatics 2017;33:3317–19. [DOI] [PubMed] [Google Scholar]
- 153. Liu Y, Brossard M, Roqueiro D, et al. SigMod: an exact and efficient method to identify a strongly interconnected disease-associated module in a gene network. Bioinformatics 2017;33:1536–44. [DOI] [PubMed] [Google Scholar]
- 154. Liang S, Tippens ND, Zhou Y, et al. iRegNet3D: three-dimensional integrated regulatory network for the genomic analysis of coding and non-coding disease mutations. Genome Biol 2017;18(1):10 10.1186/s13059-016-1138-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ji J, He D, Feng Y, et al. JDINAC: joint density-based non-parametric differential interaction network analysis and classification using high-dimensional sparse omics data. Bioinformatics 2017;33:3080–7. 10.1093/bioinformatics/btx360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Herzinger S, Gu W, Satagopam V, et al. SmartR: an open-source platform for interactive visual analytics for translational research data. Bioinformatics 2017;33;2229–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Athanasiadis E, Bourdakou M, Spyrou G.. D-Map: random walking on gene network inference maps towards differential avenue discovery. IEEE/ACM Trans Comput Biol Bioinform 2017;14(2):484–90. 10.1109/TCBB.2016.2535267 [DOI] [PubMed] [Google Scholar]
- 158. Ud-Dean SM, Heise S, Klamt S, et al. TRaCE+: ensemble inference of gene regulatory networks from transcriptional expression profiles of gene knock-out experiments. BMC Bioinformatics 2016;17(1):252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Summer G, Kelder T, Radonjic M, et al. The Network Library: a framework to rapidly integrate network biology resources. Bioinformatics 2016;32(17):i473–8. [DOI] [PubMed] [Google Scholar]
- 160. Ogris C, Helleday T, Sonnhammer EL.. PathwAX: a web server for network crosstalk based pathway annotation. Nucleic Acids Res 2016;44(W1):W105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Karp PD, Latendresse M, Paley SM, et al. Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology. Brief Bioinform 2016;17:877–90. 10.1093/bib/bbv079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Chakrabarty B, Parekh N.. NAPS: network analysis of protein structures. Nucleic Acids Res 2016;44(W1):W375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Nguyen VN, Huang KY, Weng JT, et al. UbiNet: an online resource for exploring the functional associations and regulatory networks of protein ubiquitylation. Database 2016;2016:baw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dai HJ, Su CH, Lai PT, et al. MET network in PubMed: a text-mined network visualization and curation system. Database 2016;2016:baw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Thibodeau A, Marquez EJ, Luo O, et al. QuIN: a web server for querying and visualizing chromatin interaction networks. PLoS Comput Biol 2016;12(6):e1004809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bovo S, Di Lena P, Martelli PL, et al. NET-GE: a web-server for NETwork-based human gene enrichment. Bioinformatics 2016;32(22):3489–91. [DOI] [PubMed] [Google Scholar]
- 167. Tseng YT, Li W, Chen CH, et al. IIIDB: a database for isoform-isoform interactions and isoform network modules. BMC Genomics 2015;16(Suppl 2):S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Summer G, Kelder T, Ono K, et al. cyNeo4j: connecting Neo4j and Cytoscape. Bioinformatics 2015;31(23):3868–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Pirayre A, Couprie C, Bidard F, et al. BRANE Cut: biologically-related a priori network enhancement with graph cuts for gene regulatory network inference. BMC Bioinformatics 2015;16(1):368 10.1186/s12859-015-0754-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Papatsenko D, Lemischka IR.. NetExplore: a web server for modeling small network motifs. Bioinformatics 2015;31(11):1860–2. 10.1093/bioinformatics/btv058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Okamura Y, Aoki Y, Obayashi T, et al. COXPRESdb in 2015: coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res 2015;43:D82–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hurley DG, Cursons J, Wang YK, et al. NAIL, a software toolset for inferring, analyzing and visualizing regulatory networks. Bioinformatics 2015;31(13):277–8. [DOI] [PubMed] [Google Scholar]
- 173. Zhou Z, Shen Y, Khan MR, et al. LncReg: a reference resource for lncRNA-associated regulatory networks. Database 2015;2015:bav083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Luo Z, Dai Z, Xie X, et al. TeloPIN: a database of telomeric proteins interaction network in mammalian cells. Database 2015;2015:bav018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Li HD, Omenn GS, Guan Y.. MIsoMine: a genome-scale high-resolution data portal of expression, function and networks at the splice isoform level in the mouse. Database 2015;2015:bav045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Frias S, Bryan K, Brinkman FS, et al. CerebralWeb: a Cytoscape.js plug-in to visualize networks stratified by subcellular localization. Database 2015;2015:bav041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Dorel M, Viara E, Barillot E, et al. NaviCom: a web application to create interactive molecular network portraits using multi-level omics data. Database 2017, doi: 10.1093/database/bax026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. List M, Alcaraz N, Dissing-Hansen M, et al. KeyPathwayMinerWeb: online multi-omics network enrichment. Nucleic Acids Res 2016;44(W1):W98–W104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kim B, Ali T, Hosmer S, et al. Visual Omics Explorer (VOE): a cross-platform portal for interactive data visualization. Bioinformatics 2016;32(13):2050–2. 10.1093/bioinformatics/btw119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hashemifar S, Ma J, Naveed H, et al. ModuleAlign: module-based global alignment of protein-protein interaction networks. Bioinformatics 2016;32(17):i658–64. [DOI] [PubMed] [Google Scholar]
- 181. Gligorijevic V, Malod-Dognin N, Przulj N.. Fuse: multiple network alignment via data fusion. Bioinformatics 2016;32(8):1195–203. 10.1093/bioinformatics/btv731 [DOI] [PubMed] [Google Scholar]
- 182. Dohrmann J, Singh R.. The SMAL web server: global multiple network alignment from pairwise alignments. Bioinformatics 2016;32(21):3330–2. 10.1093/bioinformatics/btw402 [DOI] [PubMed] [Google Scholar]
- 183. Arneson D, Bhattacharya A, Shu L, et al. Mergeomics: a web server for identifying pathological pathways, networks, and key regulators via multidimensional data integration. BMC Genomics 2016;17(1):722 10.1186/s12864-016-3057-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Vijayan V, Saraph V, Milenkovic T.. MAGNA ++: maximizing accuracy in global network alignment via both node and edge conservation. Bioinformatics 2015;31(14):2409–11. [DOI] [PubMed] [Google Scholar]
- 185. Athanasiadis EI, Bourdakou MM, Spyrou GM.. ZoomOut: analyzing multiple networks as single nodes. IEEE/ACM Trans Comput Biol Bioinform 2015;12(5):1213–6. 10.1109/TCBB.2015.2424411 [DOI] [PubMed] [Google Scholar]
- 186. Liu ZP, Wu C, Miao H, et al. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015;2015:bav095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Wolstencroft K, Krebs O, Snoep JL, et al. FAIRDOMHub: a repository and collaboration environment for sharing systems biology research. Nucleic Acids Res 2017;45(D1):D404–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Rodriguez N, Pettit JB, Dalle Pezze P, et al. The systems biology format converter. BMC Bioinformatics 2016;17:154 10.1186/s12859-016-1000-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Lubitz T, Hahn J, Bergmann FT, et al. SBtab: a flexible table format for data exchange in systems biology. Bioinformatics 2016;32(16):2559–61. 10.1093/bioinformatics/btw179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Balsa-Canto E, Henriques D, Gabor A, et al. AMIGO2, a toolbox for dynamic modeling, optimization and control in systems biology. Bioinformatics 2016;32(21):3357–9. 10.1093/bioinformatics/btw411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Veres DV, Gyurko DM, Thaler B, et al. ComPPI: a cellular compartment-specific database for protein-protein interaction network analysis. Nucleic Acids Res 2015;43:D485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Rodriguez N, Thomas A, Watanabe L, et al. JSBML 1.0: providing a smorgasbord of options to encode systems biology models. Bioinformatics 2015;31:3383–6. 10.1093/bioinformatics/btv341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Marchetti L, Manca V.. MpTheory Java library: a multi-platform Java library for systems biology based on the Metabolic P theory. Bioinformatics 2015;31(8):1328–30. 10.1093/bioinformatics/btu814 [DOI] [PubMed] [Google Scholar]
- 194. Johnson R, Kirk P, Stumpf MP.. SYSBIONS: nested sampling for systems biology. Bioinformatics 2015;31(4):604–5. 10.1093/bioinformatics/btu675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Aitken S, Kilpatrick AM, Akman OE.. Dizzy-Beats: a Bayesian evidence analysis tool for systems biology. Bioinformatics 2015;31(11):1863–5. 10.1093/bioinformatics/btv062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. De Smet R, Marchal K.. Advantages and limitations of current network inference methods. Nat Rev Microbiol 2010;8(10):717–29. [DOI] [PubMed] [Google Scholar]
- 197. Stolovitzky G, Monroe D, Califano A.. Dialogue on reverse-engineering assessment and methods: the DREAM of high-throughput pathway inference. Ann N Y Acad Sci 2007;1115(1):1–22. 10.1196/annals.1407.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.