

Abstract
The innate immune system is a broad collection of critical intra- and extracellular processes that limit the infectivity of diverse pathogens. The 2’−5’-oligoadenylate synthetase (OAS) family of enzymes are important sensors of cytosolic double-stranded RNA (dsRNA) that play a critical role in limiting viral infection by activating the latent ribonuclease L (RNase L) to halt viral replication and establish an antiviral state. Attesting to the importance of the OAS/RNase L pathway, diverse viruses have developed numerous distinct strategies to evade the effects of OAS activation. How OAS proteins are regulated by viral or cellular RNAs is not fully understood but several recent studies have provided important new insights into the molecular mechanisms of OAS activation by dsRNA. Other studies have revealed unanticipated features of RNA sequence and structure that strongly enhance activation of at least one OAS family member. While these discoveries represent important advances, they also underscore the fact that much remains to be learned about RNA-mediated regulation of the OAS/RNase L pathway. In particular, defining the full complement of RNA molecular signatures that activate OAS is essential to our understanding of how these proteins maximize their protective role against pathogens while still accurately discriminating host molecules to avoid inadvertent activation by cellular RNAs. A more complete knowledge of OAS regulation may also serve as a foundation for the development of novel antiviral therapeutic strategies and lead the way to a deeper understanding of currently unappreciated cellular functions of the OAS/RNase L pathway in the absence of infection.
Keywords: innate immunity, virus, translational regulation, oligoadenylate synthetase (OAS)/ribonuclease L (RNase L), double-stranded RNA (dsRNA), RNA structure
Graphical Abstract
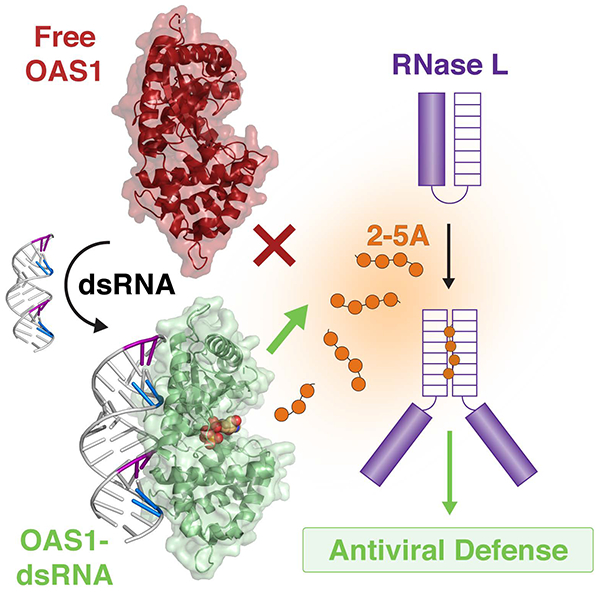
The 2’−5’-oligoadenylate synthetase/ribonuclease L (OAS/RNase L) pathway is a critical component of human innate immunity: double-stranded RNA (dsRNA) binding to OAS drives activation of 2’−5’-oligoadenylate (2–5A) synthesis to trigger the antiviral properties of RNase L.
INTRODUCTION
The human innate immune system comprises a large collection of cellular and molecular defenses that provide a critical front line of protection against microbial infection (see Sidebar: The innate immune system). Molecular innate immune sensors, such as the 2’−5’-oligoadenylate synthetase (OAS) family of enzymes, may be present at basal levels in some cells and their expression is strongly upregulated by interferon. As such, they are referred to as interferon-stimulated genes (ISGs) (Schneider, Chevillotte, & Rice, 2014). OAS enzymes are sensors of cytosolic double-stranded RNA (dsRNA), a potent indicator of viral infection (Sparrer & Gack, 2015; Takeuchi & Akira, 2010). dsRNA binding to OAS promotes synthesis of unique 2’−5’-linked oligoadenylate (2–5A) molecules that act as a secondary messenger to activate their only known downstream target: the latent ribonuclease L (RNase L) (Hovanessian & Justesen, 2007). RNase L is ubiquitously expressed and present in an inactive monomeric form in the cytoplasm; binding of 2–5A (≥3 nucleotides in length) drives RNase L dimerization and thus activation of its ribonuclease activity (Figure 1A) (Dong et al., 1994; Y. Han, Whitney, Donovan, & Korennykh, 2012; Huang et al., 2014). RNA degradation by RNase L establishes an antiviral state in several ways including direct cleavage of viral protein-encoding RNAs, degradation of cellular rRNA and tRNA which halts general translation while allowing expression of innate immune system proteins, and destabilization of the cellular transcriptome through cleavage of several specific mRNAs to suppress proliferation and adhesion (Banerjee et al., 2015; Chitrakar et al., 2019; Donovan, Rath, Kolet-Mandrikov, & Korennykh, 2017; Malathi, Dong, Gale, & Silverman, 2007; Rath et al., 2015; Siddiqui, Mukherjee, Manivannan, & Malathi, 2015). The OAS/RNase L pathway thus effectively halts viral replication and prevents viral spread to neighboring cells.
Sidebar: The innate immune system.
The cells in our body must identify foreign (non-self) molecules to detect and respond to infection. Beyond initial physical barriers, a front line of cellular and molecular defenses with broad specificity is provided by the innate immune system. This system directly protects against microbial replication, acts to warn neighboring cells, and primes the other (adaptive) arm of the immune system to respond to infection. A critical feature of innate immune proteins is that they must accurately identify molecular features of foreign molecules – referred to as pathogen-associated molecular patterns (PAMPs) – in order to rapidly respond to the threat of infection while avoiding unwanted self-activation (Schlee & Hartmann, 2016; Wu & Chen, 2014). The cellular proteins responsible for detecting PAMPs are known as pattern recognition receptors (PRRs). Double-stranded ribonucleic acid (dsRNA) is not normally present in uninfected cells but is present in some viral genomes or produced as a consequence of viral gene expression or replication. dsRNA is therefore an important and potent PAMP, detected by multiple cellular PRRs, and a strong signal for activation of the innate immune system (Hur, 2019). In addition to the OAS proteins, other RNA sensing PRRs limit infection by amplifying signaling cascades for interferon expression or by serving as direct effectors in their respective viral defense pathways (Hull & Bevilacqua, 2016; Loo & Gale, 2011; Samuel, 2011). Additionally, cellular processes are in place to avoid aberrant activation of PRRs by host RNAs (e.g. via RNA modification or editing). A recent study, for example, provided evidence that activation of the OAS/RNase L pathway by host dsRNA is controlled by the action of double-stranded RNA-specific adenosine deaminase 1 (ADAR1) (Li et al., 2017). As a result of these host defense systems, viruses have evolved diverse countermeasures to either mask their RNAs or to limit (or exploit) the effectiveness of specific PRRs and thus evade detection.
Figure 1. Overview of the OAS/RNase L pathway.

A. dsRNA binding to OAS (here shown as OAS1) promotes synthesis of 2’−5’-linked oligoadenylates (2–5A) which in turn induce dimerization and activation of the latent ribonuclease (RNase L). RNase L degrades viral and cellular RNA targets to promote antiviral responses. B. Summary of domain organization and activities of the four human OAS proteins: OAS1, OAS2, OAS3, and OASL. For the active OAS proteins (OAS1, OAS2, and OAS3), the domain responsible for 2–5A catalysis is shown in green; other OAS domains (grey) contribute to dsRNA binding. Splicing isoforms are indicated for OAS1 and OAS2 (OAS3 is expressed as a single isoform); these proteins differ in their C-terminus (green/white striped region). Human OASL contains a single non-catalytically active OAS domain appended with tandem ubiquitin-like domains (U1/U2; blue) and functions in antiviral innate immunity independent of RNase L.
The OAS proteins are members of an ancient class of template-independent RNA polymerases that also includes 3’-end specific polyadenosine polymerase (PAP) and tRNA CCA adding enzyme (CCA). OASs are thought to have diverged from these other RNA polymerases early in metazoan evolution (Torralba, Sojat, & Hartmann, 2008). All three RNA polymerases are also members of the wider nucleotidyl transferase superfamily which includes another innate immune sensor (of cytosolic DNA), cyclic GMP-AMP synthase (cGAS) (Hornung, Hartmann, Ablasser, & Hopfner, 2014; Sun, Wu, Du, Chen, & Chen, 2013). However, OAS is distinct from these structurally related proteins in several important ways. First, unlike PAP and CCA, OAS proteins do not require an RNA primer to initiate synthesis of polynucleotide chains from ATP. Second, OAS produces linear chains of unique 2’−5’-linked oligoadenylates, compared to the canonical 3’−5’ linkages synthesized by PAP and CCA, and the combination of 2’−5’ and 3’−5’ linkages that form the cyclic dinucleotide product of cGAS. Third, the OAS proteins are distinct from PAP and CCA in their absolute requirement for dsRNA as an allosteric activator of their activity. As described further below, dsRNA binding to OAS results in unique structural rearrangements necessary to form the enzyme active site and promote synthesis of 2–5A (Donovan, Dufner, & Korennykh, 2013). This requirement appears to be a relatively recent evolutionary adaptation driven specifically by OAS’s role as an innate immune sensor in higher organisms (Fish & Boissinot, 2016; Hu et al., 2018; Kjaer et al., 2009). In terms of its properties as an RNA-binding protein, OAS also appears quite distinct: it does not contain a canonical dsRNA-binding domain or other RNA recognition motif but instead recognizes dsRNA through a patch of positively charged amino acids located on the protein’s surface. Notably, this dsRNA binding surface is absent in OAS proteins in lower organisms that lack interferon signaling (Fish & Boissinot, 2016; Hu et al., 2018). This mode of dsRNA recognition by OAS is also distinct from other well-studied innate immune sensors of dsRNA, like retinoic acid-inducible gene I (RIG-I) or dsRNA-activated protein kinase (PKR) (Devarkar et al., 2016; Hornung et al., 2006; Manche, Green, Schmedt, & Mathews, 1992; Nallagatla, Toroney, & Bevilacqua, 2008, 2011), and suggests that OAS proteins might have only very weak discriminatory ability between different dsRNA sequences. This raises important questions about how OAS proteins specifically respond to viral RNAs and whether RNA sequence or structural motifs have any role to play in regulating OAS activity.
Recent evolutionary changes in OAS gene families add significant complexity in terms of the number of genes present, as well as their expression, cellular localization, and catalytic activity of their products, in a given organism. The human genome contains four distinct genes that encode a family of three catalytically active OAS enzymes (OAS1, OAS2, and OAS3) and one catalytically inactive member (OASL). All OAS proteins contain at least one copy of the globular OAS protein fold: OAS1, OAS2, and OAS3, contain one, two, and three OAS units, respectively, while OASL contains a single OAS unit embellished by two C-terminal copies of a ubiquitin-like domain (Figure 1B). Diversity of OAS expression is further increased by alternative splicing that results in at least ten different human protein isoforms (see Sidebar: Alternative splicing expands human OAS protein diversity). Comparisons between species adds yet further complexity. The murine genome, in particular, is notable for its extensive recent OAS gene duplication events (Kakuta, Shibata, & Iwakura, 2002). Further, while human OASL lacks enzymatic activity, mouse OASL2 and chicken OASL have retained their ability to produce 2–5A (Eskildsen, Justesen, Schierup, & Hartmann, 2003). Human OASL is nonetheless an important part of innate immunity and is thought to have evolved to exert its antiviral effect by binding RIG-I and enhancing its signaling (Ibsen et al., 2015; Zhu et al., 2014). Loss-of-function mutations in primate OAS1 have also been identified that substantially impair 2–5A synthesis or completely eliminate enzymatic activity altogether (Carey et al., 2019). Such species-specific differences in the OAS family presumably have their origins, at least in part, in the distinct viral challenges experienced by each species as well as the associated evolutionary “arms race” between host immune defense and viral countermeasure.
Sidebar: Alternative splicing expands human OAS protein diversity.
Alternative (or differential) splicing is a process that allows a single gene to code for multiple final protein products, greatly increasing the protein diversity encoded by the genome. During the process of mRNA transcription and maturation, specific exon(s) of a given gene can be included or excluded thereby increasing the pool of distinct mRNAs from a given gene to be directed for translation by the ribosome. All four human OAS genes are co-located in a single cluster on chromosome 12 and alternative splicing can generate a total of ten distinct mRNA products encoding OAS1 isoforms p42, p44, p46, p48 and p52, OAS2 isoforms p69 and p71, and OASL isoforms p39 and p59 (Hovnanian et al., 1998). OAS3 is expressed as a single isoform (p100) (Rebouillat, Hovnanian, David, Hovanessian, & Williams, 2000). For both OAS1 and OAS2, alternative splicing involves the final gene exon and thus changes the sequence present at the C-terminus of each protein (Bonnevie-Nielsen et al., 2005) (Figure 1B). While such changes in sequence have potential to alter protein function, a clear understanding of the impacts of alternative splicing of OAS genes are not currently available. OAS proteins are differentially induced by interferon and may localize to different subcellular fractions varying by cell type (Besse, Rebouillat, Marie, Puvion-Dutilleul, & Hovanessian, 1998; Chebath, Benech, Hovanessian, Galabru, & Revel, 1987; Hovanessian et al., 1987). Such observations suggest that regulation at the level of gene expression may have important consequences for OAS protein function both as a sensor of viral infection as well as in potential, currently unappreciated roles in the absence of infection.
Numerous studies of OAS proteins and the OAS/RNase L pathway over the last several decades have focused on their antiviral activity. Although the first X-ray crystal structure of an OAS protein was determined more than 15 years ago, relatively little was known regarding the molecular details of OAS regulation beyond the absolute requirement for a dsRNA activator and a rudimentary understanding of relative sensitivities of each OAS protein to dsRNA length, which increases from OAS1 (>17 base pairs (bp)) to OAS3 (>50 bp). However, as described in the sections that follow, recent structures of OAS1 and OAS3 domains bound to dsRNAs, combined with detailed biochemical analyses, have provided critical new molecular and functional insights into the mechanism of OAS regulation by dsRNA. Further, evidence has also begun to emerge that specific RNA sequences and structural motifs, which may be present in cellular or viral RNAs, have the potential to profoundly impact the extent of OAS activation. Defining the rules governing RNA regulation of OAS proteins represents a critical but largely unexplored frontier in our understanding of this important protein family and the full complement of strategies used by viruses to evade detection by the innate immune system.
MOLECULAR BASIS OF OAS 1–3 REGULATION BY dsRNA
OAS1 regulation by short dsRNAs
The first high-resolution structure of an OAS family member, that of porcine OAS1, was determined in 2003 and revealed the overall architecture of the OAS protein domain (Hartmann, Justesen, Sarkar, Sen, & Yee, 2003). OAS1 adopts a bilobed structure with the N-terminal (“N-lobe”) and C-terminal (“C-lobe”) regions forming the two major domains of the protein; these lobes are connected by a central linker and their interaction is further stabilized by an N-terminal extension that wraps around the C-lobe (Figure 2A). Despite the absence of either dsRNA activator or ATP substrate, structural comparisons and mutagenesis studies confirmed the expected similarity of OAS to other members of the nucleotidyl transferase superfamily as well as potentially conserved features of the catalytic activities of these enzymes. Further, though OAS1 was found to lack a previously characterized RNA binding domain or motif, residues critical for dsRNA binding were identified across an extended, positively charged surface between the N- and C-lobes on the opposite side of the protein from the ATP-binding cleft and catalytic center (Figure 2B) (Hartmann et al., 2003). The authors further speculated that the role of dsRNA binding might be to widen the catalytic center to facilitate ATP substrate binding for 2–5A synthesis. However, the molecular mechanism underpinning the absolute requirement for dsRNA activation of 2–5A synthesis by OAS1 still remained unclear.
Figure 2. Conformational changes induced by dsRNA binding promote synthesis of 2–5A by OAS1.

A. Cartoon of the OAS1 structure in its dsRNA-bound state (PDB code 4IG8). ATP binding sites and other structural features described in the main text are indicated. B. Electrostatic surface potential representation of OAS1 showing the positive (blue) charge of the dsRNA binding surface. C. Superposition of free porcine OAS1 (PDB code 1PX5; blue) and dsRNA-bound human OAS1 (green) with a zoomed view showing a secondary structure change induced by dsRNA binding. D. Reorganization of key residues upon dsRNA binding: a functionally critical positional switch in which K66 replaces R195 in an electrostatic interaction with E233 (right), helps promote reorganization of the OAS1 catalytic triad (D75, D77 and D148; dashed box, left).
An initial answer to this question came a decade later with the crystal structure of human OAS1 bound to a short (18 bp) dsRNA duplex in the presence of the substrate analog, dATP (Donovan et al., 2013). This first structure of an OAS1-dsRNA complex beautifully revealed the molecular basis for dsRNA recognition and the fundamental mechanism of OAS1 activation by dsRNA: dsRNA binding drives an essential structural reorganization within OAS1 that narrows, rather than widens, the ATP-binding cleft and repositions the catalytic residues to complete its active site. Subsequent structures of porcine OAS1-dsRNA complexes in the absence and presence of one or two molecules of AMPCPP (an ATP analog with a carbon atom linking the α and β phosphate groups) later revealed further important insights into the key drivers of dsRNA-induced conformational changes, the role of Mg2+ ions bound in the catalytic center, and the molecular basis for the specificity of 2’−5’-linkage formation by OAS1 (Lohofener et al., 2015).
As noted above, OAS1 lacks a canonical RNA-binding motif or domain and instead dsRNA interacts with a relatively flat surface of the protein on the opposite face from its catalytic site as previously predicted (Figure 2B). The OAS1-dsRNA complex structure revealed that this surface of OAS1 is distinct from that used by the structurally related but constitutively active CCA adding enzyme for tRNA binding (Tomita et al., 2004; Xiong & Steitz, 2004). The 18 bp dsRNA used in the co-crystal structure spans the full length of its OAS1 binding surface and is recognized in two consecutive minor grooves, separated by approximately 30 Å, by two clusters of predominantly positively charged residues (one each in of the two OAS1 lobes). This mode of recognition defines the minimum length of dsRNA needed to bind and activate OAS1 at ~17 bp. The majority of interactions are made to phosphate groups and to the ribose 2’-OH in the minor groove, in common with many other dsRNA binding proteins (Chang & Ramos, 2005; Masliah, Barraud, & Allain, 2013). The importance of these interactions is supported by prior mutagenesis studies showing loss of activity upon substitution of these OAS1 residues (Hartmann et al., 2003; Sarkar et al., 2005; Torralba et al., 2008), and the more general observations that dsRNA 2’-O-methylation significantly reduces OAS/RNase L pathway and recombinant OAS1 activation by dsRNA in cells and in vitro, respectively (Minks et al., 1980; Sarkar, Bandyopadhyay, Ghosh, & Sen, 1999). Further, the finding that dsRNA binding is predominantly driven by non-sequence specific interactions to phosphate and ribose groups is also consistent with the ability of OAS1 to bind and become activated by a diverse range of RNA sequences. However, the OAS1-dsRNA complex structure also provides some mechanistic explanation for the emerging idea that some specific RNA sequences are capable of more potently activating OAS1 (discussed further below). In contrast, the largely sequence non-specific nature of OAS1-dsRNA interaction less readily explains the finding that RNA modification via replacement of uridine with pseudouridine significantly reduces OAS1 activation by a given dsRNA (Anderson et al., 2011). Rather, the origin for this phenomenon might reside in the rigidifying effect of this base modification on the dsRNA helical structure: in the co-crystal structure, the dsRNA is significantly distorted from ideal A-form helical geometry which would be less readily accomplished with a pseudouridine-containing dsRNA.
Crystal structures of OAS1-dsRNA complexes also revealed how binding of dsRNA drives functionally critical conformational changes in OAS1 necessary for activation of 2–5A synthesis. When bound by dsRNA, large conformational changes occur in the N-lobe, including large movements (>10 Å) of some secondary structure elements, while maintaining the two OAS1 lobes in their same relative positions as in the free protein. Overall, these changes in the N-lobe narrow the ATP binding cleft on the opposite side of the protein with rearrangements in OAS1 propagating from the dsRNA binding surface through the protein core to its catalytic center. Most critically, two basic residues (K66 and R195) exchange their positions to maintain an electrostatic interaction with the functionally critical E233; this switch directs R195 to the dsRNA binding surface, drives formation of a new α-helical structure and organizes the OAS1 catalytic triad (D75, D77, and D148) and ATP donor and acceptor sites (Figure 2C,D). Thus, dsRNA binding allosterically induces the formation of the catalytically competent OAS1 active site with correctly positioned catalytic triad, bound Mg2+ ions, and ATP substrates (Donovan et al., 2013; Lohofener et al., 2015). It is noteworthy that the other structurally related nucleotidyl transferase enzymes PAP and CCA lack the potential for this charge switch and structural reorganization, and are locked in an “on” state, consistent with their constitutive (but primer-dependent) activity. Thus, dsRNA activation of OAS1’s polymerase activity is a unique adaptation necessitated by its role as a cytosolic dsRNA sensor within the innate immune system.
Basis of long dsRNA sensing by OAS3
OAS3 is the largest OAS variant and is composed of three OAS domains (DI, DII, and DIII; Figure 1B). Initial sequence-based analyses suggested DIII is the sole catalytically active domain while DI and DII are both “pseudoenzymes” that have lost the ability to synthesize 2–5A due to mutations of their catalytic aspartic acid residues (Rebouillat et al., 2000). The first functional studies on OAS3 suggested that it preferentially synthesizes 2–5A dimers, a species that would be unable to activate RNase L, leading to speculation that OAS3 could have roles independent of RNase L activation (Marie, Blanco, Rebouillat, & Hovanessian, 1997). However, more recent analyses have shown that OAS3 is activated at a lower concentration of dsRNA, compared to OAS1, and is in fact capable of synthesizing 2–5A long enough to activate RNase L (Ibsen et al., 2014). The same study used mutational analysis to confirm the exclusive role of the OAS3 DIII domain in catalysis, as predicted given the lack of a complete catalytic center in the two other domains (DI and DII). However, the functionally critical changes in DI and DII apparently extend beyond the catalytic center as restoration of altered catalytic residues by mutagenesis failed to restore enzymatic activity (Donovan, Whitney, Rath, & Korennykh, 2015).
These findings raise an important question: in the absence of catalytic activity, what functional role, if any, do DI and DII play? To address this question, Donovan et al. purified each OAS3 domain separately and tested their individual abilities to interact with dsRNA and produce 2–5A as compared to OAS1 (Donovan et al., 2015). While all three isolated domains retained their ability to bind dsRNA, none could synthesize 2–5A under previously determined conditions for OAS activity. Interestingly, however, isolated DIII was found to have some activity in the presence of Mn2+ as the divalent cation but the basis and potential relevance of this phenomenon is unclear. OAS3 DI was found to bind dsRNA ~14-fold more tightly than OAS1 which, together with the crystal structure determined of this domain bound to a 19 bp dsRNA, suggests that DI plays an essential role in RNA recognition (Donovan et al., 2015). The higher dsRNA affinity of OAS3 DI can likely be explained by the fact that while OAS1 possesses the enzyme’s catalytic activity and must therefore retain the ability to bind and release dsRNA for multiple rounds of catalysis, OAS3 DI is a pseudoenzyme that serves exclusively as a dsRNA binding platform for presentation of the activator to the catalytic DIII domain. A second study used small angle X-ray scattering (SAXS) to show that the individual domains of OAS3 form a linear arrangement which would allow them to work in concert to specifically sense long RNAs (Ibsen et al., 2014). Consistent with this idea, OAS3 DI and DIII appear to be most critical for RNA binding and catalysis, while DII binds dsRNA more weakly and may act as a spacer allowing OAS3 to expand the RNA length requirement beyond that of OAS1 and OAS2 (Donovan et al., 2015). This domain organization allows OAS3 to bind the largest dsRNA molecules (minimum RNA length requirement of ~50 bp), while short dsRNAs cannot simultaneously bind to each of the domains and thus induce the conformational changes in the catalytic DIII required for 2–5A production.
OAS2 is activated by dsRNA of intermediate length
Studies of OAS2 activity and structure have significantly lagged behind those of OAS1 and OAS3. In vitro studies in particular, have been hindered by the need to express OAS2 using insect (Sarkar et al., 1999) or mammalian (Koul et al., 2019; Marie, Rebouillat, & Hovanessian, 1999) expression systems in order to obtain catalytically active recombinant protein. The lack of activity in bacterially expressed OAS2 has been attributed to a requirement for post-translational modifications that are apparently unique to OAS2 among the active OAS proteins. OAS2 has four potential sites of glycosylation and a role for one or more of these was strongly suggested by the finding that inhibiting glycosylation in insect cells also leads to the production of inactive protein (Sarkar et al., 1999).
OAS2 functions as a homodimer and each protomer is comprised of two OAS domains: a catalytically inactive N-terminal domain (DI) and a catalytically active C-terminal domain (DII) (Figure 1B). Like OAS3, the individually expressed and purified OAS2 domains cannot produce 2–5A in isolation or when reconstituted (Koul et al., 2019; Marie et al., 1999). Isolated OAS2 DI has reduced affinity for dsRNA while DII can bind dsRNA with similar affinity to the full-length protein (Marie et al., 1999). These findings reveal that both OAS domains are required for OAS2 activity and suggest that the domains must work cooperatively, possibly to form OAS2 dimers or to induce the structural rearrangements necessary in DII for catalysis. Therefore, OAS2 gains a level of specificity as it can only be activated by RNAs long enough to bind and span both domains, in a similar structural arrangement as observed for OAS3.
Early studies suggested that OAS2 is activated by dsRNAs of an intermediate size between those of OAS1 and OAS3, with a minimum length requirement of 25 bp (Sarkar et al., 1999). More recently, OAS2 activation was assessed in vitro using a series of dsRNAs ranging from 19–123 bp in length and found to increase consistently with increasing dsRNA length above a slightly longer minimum of 35–40 bp (Koul et al., 2019). Consistent with these analyses, the authors also found that a range of structured viral RNAs lacking helical regions >35 bp all failed to activate OAS2. If OAS2 requires a minor groove interaction site for each lobe (like the interactions made by OAS1 and DI of OAS3) within each of its two OAS domains, then OAS2 would require four consecutive minor grooves for interaction, consistent with the experimentally determined 35–40 bp minimum dsRNA length requirement. Several fundamental questions still remain regarding OAS2 regulation by dsRNA. For example, since OAS2 appears to be a homodimer, is dimerization necessary and, if so, must dsRNA bind to both OAS2 proteins within the dimer for activation or do the two active domains function independently of each other? Defining the OAS2 dimer and domain organization is also a critical next step that will address whether each OAS2 protomer can bind a separate dsRNA molecule or if a single dsRNA binds simultaneously to both. With tools in place to produce active OAS2 in sufficient quantities (Koul et al., 2019) for detailed biochemical and biophysical studies, we can anticipate answers to such questions arriving in the near future.
ROLE OF RNA SEQUENCE AND STRUCTURE IN OAS ACTIVATION
The recent structural and functional studies discussed so far have provided a new rational basis for the observed sensitivities of each OAS protein to dsRNA of different lengths and their minimum requirements for activation. Despite these important advances, the new structures offer only limited clues on the role of RNA sequence specificity or how OAS proteins are potentially influenced by structured motifs within larger RNAs containing double-stranded regions, as might be found in viral or cellular RNAs. The following sections describe other recent advances centered on RNA sequence and structure determinants of OAS activation. Identifying and defining RNA molecular signatures for OAS activation (or inhibition) will be an essential step towards a complete understanding of OAS/RNase L pathway regulation by the host cell and in antiviral activity.
RNA sequence specificity in OAS activation
OAS1 “consensus” sequences
One emerging area has been to explore dsRNA features other than length, such as specific sequence, that potentially dictate the extent of OAS activation. One study tested a panel of short (19 bp) dsRNAs with distinct sequences to probe for differences in their ability to activate OAS1 in vitro and the OAS/RNase L pathway in human A549 cells (Kodym, Kodym, & Story, 2009). Sequence alignment of the dsRNAs that most strongly promoted OAS1 activity thus identified a consensus activation sequence WWN9WG (where W is A or U, and N is any nucleotide). Analysis of a larger panel of over 100 dsRNA sequences was then used to confirm that this consensus sequence strongly correlated with the ability of a given short dsRNA to activate OAS1. When mapped onto a dsRNA helix, the minor grooves of the WW and WG consensus dinucleotide sequences are spaced by the nine intervening base pairs and presented on the same face of the A-form helix for recognition by OAS1, as observed in the OAS1-dsRNA crystal structure (Donovan et al., 2013), which used an RNA duplex containing two copies of this sequence (Figure 3A). Further, the G nucleotide of the WWN9WG sequence presented to OAS1 is the only base specifically recognized via a hydrogen bond from OAS1 residue Ser56, revealing at least part of the basis for the specificity of this consensus sequence. To date, the effect of this consensus sequence has only been tested for OAS1; whether this or other specific sequence(s) can similarly enhance OAS2 or OAS3 activity remains to be determined.
Figure 3. Double-stranded and structured RNA activators of OAS.

Select RNAs discussed in the main text: A. 18 bp dsRNA used in the OAS1-dsRNA crystal structure and identification of the 3’-ssPy motif, B. Adenovirus (Ad2) VA RNAI with sites of Dicer cleavage indicated (black scissors), C. cellular nc886 RNA with location of OAS1 activating structural motif indicated (red shading), D. Example ss-dsRNA (43 bp) with sites of RNase cleavage to generate a minimal 3’-ssPy motif indicated (orange scissors), E. WNV 5’-terminal region, and F. WNV 3’-terminal region with OAS1 activating stem-loop highlighted (red shading). For all RNAs 3’-ssPy motifs are indicated by the orange text and the OAS1 consensus activation sequence WWN9WG (shown above the 18 bp dsRNA in panel A) by white text on blue background. The purple background for the 18 bp dsRNA top strand denotes the consensus sequence directly contacted by OAS1 in the OAS1-dsRNA crystal structure (inset in Panel A).
A second, bipartite OAS1 activating sequence was identified in a separate study that used systematic evolution of ligands by exponential enrichment (SELEX) to identify RNA aptamers able to bind OAS1 (Hartmann et al., 1998). The RNA aptamers were 103 nucleotides in length and contained 34 random internal nucleotides for the SELEX process. Single-strand RNA aptamers and, in some cases corresponding dsRNA created by annealing of a complementary RNA strand, were tested for their ability to activate OAS1 in vitro. Sequence alignment was then used to reveal commonalities among the most potent activators. Good OAS1 activators were found to have an overall high cytosine and low guanine content and contained the sequences APyAPy(N)nCC and UU(N)nACCC (where Py is C or U) in different parts of the RNA molecule (Hartmann et al., 1998). These findings further support the idea that nucleotide content (especially cytosine) and specific RNA sequences can play an important role in defining the extent of OAS1 activation. Most studies on the innate immune response use the synthetic compound poly(I:C) as a positive control for PRR activation, but how poly(I:C) activates OAS1 is poorly understood (Minks et al., 1980). The relative importance of cytosine in dsRNA identified in this study could be one explanation of why poly(I:C) is such a potent activator of OAS1.
Sequence and Structural Motifs: 3’-ssPy Motif and nc886 RNA
The non-coding adenoviral “virus associated” RNA I (VA RNAI; Figure 3B) possesses several pro-viral activities but is most widely recognized for its ability to inhibit PKR, relieving a cellular blockade on initiation of protein synthesis by this kinase (Vachon & Conn, 2016). Curiously, however, despite these multiple inhibitory actions against host antiviral and other cellular processes, VA RNAI was also reported to promote 2–5A synthesis by OAS1 (Desai et al., 1995). With our lab’s long-standing interests in VA RNAI structure and activity, we set out to further understand the molecular basis for this apparently paradoxical activity; remarkably, however, our in vitro transcribed VA RNAI failed to activate OAS1 to the extent previously reported (Vachon, Calderon, & Conn, 2015). The basis of this difference was ultimately traced to the absence of a 3’-end CUUU sequence in our in vitro construct, which would normally be present in VA RNAI as a result of RNA polymerase III termination (Nielsen, Yuzenkova, & Zenkin, 2013). This observation thus led to the serendipitous discovery of a novel motif for OAS1 activation, the 3’-end single-stranded pyrimidine (3’-ssPy) motif.
Studies using VA RNAI variants with altered 5’- and 3’-end sequences revealed that optimal OAS1 activation required a 3’-end pyrimidine-rich (C or U) single-stranded sequence (Vachon et al., 2015). While 3’-end single-stranded purine-rich (G or A) sequences promoted an intermediate level of activation, extension of the VA RNAI terminal stem with an equivalent length of base pairs failed to enhance OAS1 activation. Deletion of the equivalent 3’-end sequences from two other RNA polymerase III non-coding transcripts, Epstein-Barr virus EBER-1 RNA and human cellular nc886 RNA (Figure 3C), also reduced OAS1 activation by these RNAs, albeit to a significantly lesser extent in the latter case. To facilitate modeling of the effect of the 3’-ssPy motif, the 18 bp dsRNA duplex used in the OAS1-dsRNA crystal structure (Figure 3A) was also examined: addition of one 3’-end single-stranded U residue was sufficient to significantly enhance OAS1 activation by this model dsRNA. While the mechanism of 3’-ssPy motif action remains to be determined in detail, modeling using the OAS1-dsRNA structure was used to direct mutagenesis of surrounding conserved OAS1 residues. These studies identified one amino acid, G157, whose substitution with a bulkier residue did not impact overall OAS1 activity but fully eliminated sensitivity to the 3’-ssPy motif. G157 resides in a loop adjacent to the dsRNA binding surface suggesting that allosteric regulation of OAS1 by RNA may be more complex than currently appreciated (Figure 4). The implications for OAS1 antiviral activity, and in other potential roles in the uninfected cell, warrant further investigation. For example, given RNase L’s preferential cleavage at single-stranded UA or UU sequences, production of dsRNAs appended with these sequences from larger structured RNAs with single-stranded regions could produce additional OAS1-activating dsRNAs with 3’-ssPy motifs, thus potentiating signaling via the OAS/RNase L pathway (Malathi et al., 2010; Vachon et al., 2015; Wreschner, McCauley, Skehel, & Kerr, 1981). Additionally, OAS1 could play a role in sensing of aberrant 3’-end processing of cellular RNAs.
Figure 4. Model for enhancement of OAS1 activation by RNA sequence and structural motifs.

OAS1 requires at least ~17 bp dsRNA for binding, shown here on the left for the model 18 bp dsRNA duplex with OAS1 consensus activation sequence (purple) and right the nc886 central stem sequence. Once positioned by the OAS1 binding site, RNA signatures such as the 3’-ssPy motif (orange) or the tertiary structure of nc886 (red) can dramatically increase OAS1 activation. The mechanism of activation by these RNA features is currently unknown but modeling and mutagenesis analyses suggest the OAS1 loop containing G157 may be important (see main text for details).
A more recent study by McKenna and colleagues examined the impact of single-stranded 3’-end sequences on OAS2 activation (Koul et al., 2019). A series of RNA in vitro transcription constructs was created to produce dsRNAs of various lengths (19–123 bp), with both 3’-ends in each dsRNA appended with a 17-nucleotide single-stranded sequence (ss-dsRNA; Figure 3D). This 3’-end sequence was also truncated to a single U residue by RNase treatment to generate a minimal 3’-ssPy motif. While the 3’-ssPy motif had no effect on OAS2 activation, dsRNAs with 17 nucleotide 3’-overhangs (“ss-dsRNAs”) were consistently found to more strongly activate OAS2 once the helical region was >35 bp in length. This finding suggests OAS2 may have some selectivity for dsRNAs appended with longer single-stranded sequences. However, an additional element of complexity was revealed by parallel studies of OAS1 with the same dsRNA constructs: here, OAS1 was found to be insensitive to both the longer single-stranded sequence and the single 3’-end U residue (i.e. 3’-ssPy motif). This observation suggests that the context in which the 3’-ssPy is presented to OAS1 may be critical for the ability of this RNA signature to enhance 2–5A synthesis. Specifically, while our earlier studies used an 18 bp dsRNA containing an OAS1 activation consensus sequence (with the 3’-ssPy motif appended to the strand immediately following the G residue of the WWN9WG consensus), the dsRNAs used by Koul et al. lacked this sequence in a corresponding position (Figure 3D). These findings suggest that RNA signatures like the 3’-ssPy motif do not directly influence OAS1 (or possibly, OAS2) binding to dsRNAs but, when positioned optimally by other features of the adjacent dsRNA binding site, can serve to strongly enhance 2–5A synthesis.
Further support for this idea also comes from recent studies of the human cellular nc886 RNA. nc886 is a ubiquitously expressed cytosolic non-coding RNA that was first identified during studies of PKR’s proliferative role in some cancers (Kunkeaw et al., 2013; Lee et al., 2011) and has since been shown to directly bind and inhibit the kinase (Calderon & Conn, 2017; Jeon et al., 2012). nc886 adopts two distinct structures which differ in their migration on native polyacrylamide gels and their ability to inhibit PKR: the slower migrating form (“conformer 1”) is a potent PKR inhibitor while the faster migrating form (“conformer 2”) is a weak PKR activator that acts as a pseudoinhibitor at higher concentrations in the presence of another PKR-activator such as poly(I:C) RNA. As noted earlier, we also showed that nc886 can activate OAS1 in vitro as a mixture of conformers (Vachon et al., 2015). A more recent detailed study of OAS1 activation by nc886 in vitro and in A549 cells revealed that most activity is conferred by a unique tertiary structure motif present only in the apical stem-loop of nc886 conformer 1 (Calderon & Conn, 2018). Interestingly, this same conformer 1 structural feature is critical for PKR inhibition by nc886, suggesting the potential for cellular RNA-mediated communication between these two dsRNA-sensing elements of the innate immune system. Analysis of nc886 with truncated terminal and central stem structures showed that, although formation of the OAS1-activating structure requires only the apical stem-loop RNA sequence, this domain is not alone sufficient to activate OAS1. Rather, a predominantly base-paired region of the central stem, approximately 18 bp in length, is also required for OAS1 activation (Calderon & Conn, 2018). Our interpretation of these observations is that OAS1 binding to nc886 is dictated by the dsRNA region of the central stem, which optimally positions the apical stem-loop motif to potentiate OAS1 activation.
Collectively, these studies suggest a potential common theme for OAS1 activation by sequence and structural motifs appended to preferred OAS1 binding sites (Figure 4). The range of dsRNA sequences that preferentially bind OAS1, motifs that promote OAS1 activation once positioned optimally, and whether OAS2 or OAS3 are subject to similar regulation require further investigation.
Coding and non-coding viral RNAs containing dsRNA regions
Viruses may present dsRNA in the host cell through their genomes, during viral gene expression or replication, or through the expression of abundant non-coding RNA species like adenovirus VA RNAI. However, many important details about how the sequences and/or structures of these viral RNAs contribute to OAS regulation and host-virus interaction have remained elusive. One early analysis of OAS/RNase L pathway activation by a viral RNA was performed with Hepatitis C virus (HCV) genomic RNA (J. Q. Han & Barton, 2002). Specifically, the HCV RNA genome was found to activate OAS, leading to its cleavage by RNase L and thus limiting viral infection. In addition to this direct action of the OAS/RNase L pathway, a product of HCV RNA cleavage by RNase L can also act as a PAMP that activates the RIG-I pathway, propagating immune signaling and upregulation of interferon stimulated genes to prevent HCV replication (Malathi et al., 2010). Further supporting this idea, some HCV genotypes are deficient in the single-stranded UA and UU-containing sequences preferred by RNase L (J. Q. Han & Barton, 2002) suggesting that HCV has developed a novel viral OAS/RNase L pathway evasion strategy wherein sequences preferentially cleaved by RNase L are selected against. While OAS can still bind HCV RNA and produce 2–5A, the viral genome suffers minimal damage due to downstream RNase L activity thus allowing viral replication to persist. Differences observed in efficacy of interferon therapy for HCV infection may also be directly related to the sensitivity of the viral RNA to RNase L cleavage (J. Q. Han & Barton, 2002).
The 5’- and 3’-end regions of the West Nile virus (WNV) RNA genome (Figure 3E,F) have also been identified as potential activators of OAS1 (Deo et al., 2015; Deo et al., 2014). The isolated 5’-terminal region and mixture of the two terminal region RNAs activated OAS1 most robustly in vitro while the isolated 3’-terminal region and a single stem-loop structure from this region activated OAS1 to a lesser extent. These findings reveal both the potential for activation of the OAS/RNase L pathway by multiple regions of the WNV genome and that genome cyclization via interaction of the 5’- and 3’-terminal regions, a required step for viral replication, may not be sufficient for the virus to evade this detection (Deo et al., 2015). However, the full impact of OAS activation by WNV genomic RNA, its relevance to detection of infection, and potential for viral evasion all require further study. Both WNV RNA regions contain multiple WWN9WG consensus activation sequences though their specific contributions to OAS1 activation were not directly tested and most do not fall in dsRNA regions. However, strong OAS1 activation by the isolated WNV RNA regions clearly shows that bulges, non-canonical base pairs, and mismatches within the ~18 bp regions are readily tolerated and that OAS1 can be activated by a diverse group of structured RNAs.
As noted earlier, a number of abundantly expressed non-coding RNAs can activate OAS1 but not OAS2 (or presumably OAS3 given its even greater dsRNA length requirement). These RNAs include adenovirus VA RNAI, Epstein-Barr virus EBER-1 RNA, and human immunodeficiency virus TAR RNA (Desai et al., 1995; Koul et al., 2019; Maitra et al., 1994; Sharp et al., 1999). Our analysis revealed that the 3’-end sequence (3’-ssPy motif) is critical in the context of the wild-type full-length VA RNAI (Vachon et al., 2015), but this cannot be the complete picture of OAS1 regulation by this RNA transcript. First, in the original study that identified OAS1 activation by VA RNAI, increasing the Watson-Crick base pairing in both the terminal and apical stem was found to increase OAS1 activation, with the greatest effect observed for the latter region, which is most distant from the 3’-ssPy motif (Desai et al., 1995). Second, a shorter VA RNAI fragment, roughly corresponding to the product of VA RNAI cleavage by Dicer (Andersson et al., 2005; Wahid, Coventry, & Conn, 2008) (Figure 3B), binds OAS1 with higher affinity, but is a very poor activator (pseudoinhibitor) of OAS1 (Meng et al., 2012). OAS1 activation by EBER-1 and TAR RNAs have not been studied in detail but it is likely that the activity of these structured non-coding RNAs is similarly complex. All three non-coding RNAs contain more than one copy of the WWN9WG consensus activation sequence but, as for the WNV terminal region RNAs, their contribution to OAS1 activation has not been determined. Regardless of these uncertainties in detail, it is intriguing and worthy of further investigation that three viral non-coding RNAs, which possess other well-established antiviral activities, are capable of activating a second parallel host antiviral pathway. This could be a tolerated consequence or an activity that is exploited for pro-viral purposes at late stages of infection when these non-coding RNAs are highly abundant.
CONCLUSIONS
Viruses depend on the host cell translational machinery for protein synthesis and thus their replication. To accomplish this, viruses must expose their proteins and nucleic acids to the cellular milieu but avoid detection by innate immune antiviral factors capable of restricting viral replication. The OAS family of enzymes comprise a group of cytosolic dsRNA innate immune sensors whose importance in this process is underscored by the numerous ways diverse viruses have developed to evade detection. These include directly inhibiting OAS, sequestering dsRNA produced as a result of viral infection, producing 2–5A analogs, degrading 2–5A via viral-encoded phosphodiesterases, expression of protein or RNA inhibitors of 2–5A interaction with RNase L or its ribonuclease activity, or through selective pressure to remove RNase L target sites within viral RNAs (Cayley, Davies, McCullagh, & Kerr, 1984; Drappier et al., 2018; J. Q. Han & Barton, 2002; Keel, Jha, & Kieft, 2012; Silverman, 2007; Silverman & Weiss, 2014; Taguchi et al., 2004; Thornbrough et al., 2016; Townsend, Jha, Han, et al., 2008; Townsend, Jha, Silverman, & Barton, 2008; Zhang et al., 2013; Zhao et al., 2012). While there is strong evidence for critical roles for each OAS family member in at least some context, defining the protective effects of each family member is an active area of investigation. For example, a recent study indicated OAS3 may be the main effector of the OAS/RNase L pathway in response to a range of viruses including WNV and influenza A virus (IAV) (Li et al., 2016). In this study, knockout of OAS3, but not OAS1 or OAS2, prevented 2–5A synthesis and rRNA degradation resulting in significantly higher viral titers. However, deletion of OAS1 or OAS2 had no detectable effects on 2–5A or rRNA and exhibited only minor effects on viral titers suggesting OAS1 and OAS2 may exert their antiviral functions through alternative pathways. Further, another study in which each of the ten human OAS proteins was expressed in A549 cells revealed that only OAS1 (p42 and p46 isoforms) and OAS3 exhibited measurable antiviral effects against Dengue virus, leading to the conclusion that OAS gene family members contribute differently to antagonize this virus (Lin et al., 2009). Yet, other evidence from analysis of OAS single nucleotide polymorphisms (SNPs) also appears to complicate these conclusions: SNPs in both OAS1 and OAS3 are known to increase susceptibility to WNV infection (Bigham et al., 2011; Lim et al., 2009; Yakub et al., 2005), while two SNPs in OAS2 have been suggested to increase the susceptibility to Dengue virus (Alagarasu et al., 2013; Thamizhmani & Vijayachari, 2014). Delineating the specific, potentially overlapping, roles of each OAS protein family member is likely to continue to be an active area of investigation along with studies to define the nature of the viral RNAs detected by these proteins.
As discussed in this review, recent structural studies have revealed the molecular details of OAS allosteric regulation by dsRNA. Together with complementary biochemical and biophysical studies, this work has also provided a clear rationale for the observed dsRNA length requirements and the contribution of each OAS domain in dsRNA binding or catalysis (summarized in Figure 5). The OAS protein family’s ability to discriminate dsRNAs based on length is likely an essential adaptation that allows detection and response to a wider range of virus-derived dsRNA during infection. However, despite these important advances, we still understand relatively little about how distinct features of the dsRNA contribute to the extent of OAS activation. Studies identifying specific sequences and single-stranded or structured motifs that enhance OAS1 activation (Figure 5), as well as identification of viral and cellular non-coding or coding RNA regions that strongly activate OAS1, suggest that much remains to be learned. The case for larger RNAs with complex structures is likely further complicated by the potential for multiple OAS interaction sites (especially for the single domain OAS1). Even the “simple” 18 bp dsRNA used for structural studies (Donovan et al., 2013) contained two overlapping antiparallel copies of the WWN9WG consensus sequences, and yet the dsRNA bound in a single orientation and only one consensus was apparently sensed by OAS1. This issue is further complicated by studies of OAS1 activation by the same consensus sequence and 3’-ssPy motif which suggest that the context in which these features are presented is also a critical determinant of their ability to promote OAS activation. Thus, it may not be simply the presence or absence of an RNA feature, but also their placement within the RNA that determines how much or little each contributes to OAS activation. A final consideration is whether such molecular signatures act in concert or competition to affect the level of OAS1 activity. Our current studies suggest that there are preferred OAS binding sites with weak ability to activate OAS1 and that these can diminish activation by other sites within the same dsRNA. Collectively, these considerations likely have important implications for how OAS proteins are able to selectively sense viral RNAs and it will be important to tease apart the details of how individual and combinations of RNA features control OAS activation. Equally, selection within viral RNAs for OAS binding sites that mask or compete with activating features could potentially be a mechanism to allow viral replication to persist by acting as a “sponge” for OAS activity.
Figure 5. Summary of OAS protein activation by RNA.

Current understanding of OAS protein regulation by dsRNA, as discussed in the main text, is summarized in the three vertical panels for OAS1 (purple), OAS2 (blue), and OAS3 (grey). Top, minimum dsRNA lengths required to activate each OAS protein. Center, OAS activation by dsRNA with dsRNA induced organization of the catalytic center (green domain) denoted by the yellow star. OAS1 binds short dsRNAs and can also be strongly activated by longer dsRNAs with multiple binding sites. OAS2 functions as a dimer of currently unknown organization (two potential dimer interactions are shown; also possible are via DII-DII only and DI/DI-DII/DII head-head). Also unknown is whether both OAS2 proteins must bind to dsRNA and whether they function independently or cooperatively. OAS3 is activated only by longer dsRNAs due to the requirement to span three OAS domains; shorter RNAs are unable to induce the conformational change in DIII (inset). Bottom, other RNA sequences and motifs identified to enhance 2–5A synthesis.
While OAS’s major function in the antiviral OAS/RNase L pathway is well-established, differential OAS expression and induction by interferon in different cell types (Hovanessian & Justesen, 2007), as well as OAS conservation in organisms without interferon, collectively imply these proteins play important cellular roles outside of innate immunity. Additionally, SNPs associated with the human OAS1 gene have also been connected to diabetes (Field et al., 2005), multiple sclerosis (Fedetz et al., 2006; O’Brien et al., 2010), and prostate cancer (Mandal, Abebe, & Chaudhary, 2011) suggesting some role in processes that impact these human diseases. A complete understanding of how RNA controls OAS activity is critical to gain a deeper understanding of multiple facets of innate immunity, host-virus interactions, and fundamental mechanisms of cellular translational control.
Acknowledgments
We thank Drs. Christine M. Dunham, Daniel Reines, and Anice C. Lowen for discussions and comments on the manuscript. We are also grateful to Dr. Sean McKenna for sharing details of his group’s recent work on OAS2 ahead of publication to allow its inclusion in this review.
The authors declare there are no conflicts of interest.
Funding Information
This work was supported by National Institutes of Health grants R01-AI144067 (to GLC.), F31-AI133950 (to SLS) and T32-GM008367, the Emory University Research Council (to G.L.C.), and Emory University School of Medicine Bridge Funding Program (to G.L.C.). S.L.S. is also grateful for support from the Achievement Rewards for College Scientists (ARCS) Foundation, Atlanta Chapter.
Contributor Information
Samantha L. Schwartz, Department of Biochemistry, Emory University School of Medicine and Graduate Program in Biochemistry, Cell and Developmental Biology (BCDB)
Graeme L. Conn, Department of Biochemistry, Emory University School of Medicine and Graduate Program in Biochemistry, Cell and Developmental Biology (BCDB).
References
- Alagarasu K, Honap T, Damle IM, Mulay AP, Shah PS, & Cecilia D (2013). Polymorphisms in the oligoadenylate synthetase gene cluster and its association with clinical outcomes of dengue virus infection. Infect Genet Evol, 14, 390–395. doi: 10.1016/j.meegid.2012.12.021 [DOI] [PubMed] [Google Scholar]
- Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, & Kariko K (2011). Nucleoside modifications in RNA limit activation of 2’−5’-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res, 39(21), 9329–9338. doi: 10.1093/nar/gkr586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, & Akusjarvi G (2005). Suppression of RNA interference by adenovirus virus-associated RNA. J Virol, 79(15), 9556–9565. doi: 10.1128/jvi.79.15.9556-9565.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Li G, Li Y, Gaughan C, Baskar D, Parker Y, … Silverman RH (2015). RNase L is a negative regulator of cell migration. Oncotarget, 6(42), 44360–44372. doi: 10.18632/oncotarget.6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse S, Rebouillat D, Marie I, Puvion-Dutilleul F, & Hovanessian AG (1998). Ultrastructural localization of interferon-inducible double-stranded RNA-activated enzymes in human cells. Exp Cell Res, 239(2), 379–392. [DOI] [PubMed] [Google Scholar]
- Bigham AW, Buckingham KJ, Husain S, Emond MJ, Bofferding KM, Gildersleeve H, … Bamshad M (2011). Host genetic risk factors for West Nile virus infection and disease progression. PLoS One, 6(9), e24745. doi: 10.1371/journal.pone.0024745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnevie-Nielsen V, Field LL, Lu S, Zheng DJ, Li M, Martensen PM, … Pociot F (2005). Variation in antiviral 2’,5’-oligoadenylate synthetase (2’5’AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am J Hum Genet, 76(4), 623–633. doi: 10.1086/429391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon BM, & Conn GL (2017). Human noncoding RNA 886 (nc886) adopts two structurally distinct conformers that are functionally opposing regulators of PKR. RNA, 23(4), 557–566. doi: 10.1261/rna.060269.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon BM, & Conn GL (2018). A human cellular noncoding RNA activates the antiviral protein 2’−5’-oligoadenylate synthetase 1. J Biol Chem, 293(41), 16115–16124. doi: 10.1074/jbc.RA118.004747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey CM, Govande AA, Cooper JM, Hartley MK, Kranzusch PJ, & Elde NC (2019). Recurrent Loss-of-Function Mutations Reveal Costs to OAS1 Antiviral Activity in Primates. Cell Host Microbe, 25(2), 336–343.e334. doi: 10.1016/j.chom.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayley PJ, Davies JA, McCullagh KG, & Kerr IM (1984). Activation of the ppp(A2’p)nA system in interferon-treated, herpes simplex virus-infected cells and evidence for novel inhibitors of the ppp(A2’p)nA-dependent RNase. Eur J Biochem, 143(1), 165–174. [DOI] [PubMed] [Google Scholar]
- Chang KY, & Ramos A (2005). The double-stranded RNA-binding motif, a versatile macromolecular docking platform. Febs j, 272(9), 2109–2117. doi: 10.1111/j.1742-4658.2005.04652.x [DOI] [PubMed] [Google Scholar]
- Chebath J, Benech P, Hovanessian A, Galabru J, & Revel M (1987). Four different forms of interferon-induced 2’,5’-oligo(A) synthetase identified by immunoblotting in human cells. J Biol Chem, 262(8), 3852–3857. [PubMed] [Google Scholar]
- Chitrakar A, Rath S, Donovan J, Demarest K, Li Y, Sridhar RR, … Korennykh A (2019). Real-time 2–5A kinetics suggest that interferons beta and lambda evade global arrest of translation by RNase L. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1818363116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo S, Patel TR, Chojnowski G, Koul A, Dzananovic E, McEleney K, … McKenna SA (2015). Characterization of the termini of the West Nile virus genome and their interactions with the small isoform of the 2’ 5’-oligoadenylate synthetase family. J Struct Biol, 190(2), 236–249. doi: 10.1016/j.jsb.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Deo S, Patel TR, Dzananovic E, Booy EP, Zeid K, McEleney K, … McKenna SA (2014). Activation of 2’ 5’-oligoadenylate synthetase by stem loops at the 5’-end of the West Nile virus genome. PLoS One, 9(3), e92545. doi: 10.1371/journal.pone.0092545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SY, Patel RC, Sen GC, Malhotra P, Ghadge GD, & Thimmapaya B (1995). Activation of interferon-inducible 2’−5’ oligoadenylate synthetase by adenoviral VAI RNA. J Biol Chem, 270(7), 3454–3461. [DOI] [PubMed] [Google Scholar]
- Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, … Marcotrigiano J (2016). Structural basis for m7G recognition and 2’-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci U S A, 113(3), 596–601. doi: 10.1073/pnas.1515152113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Xu L, Zhou A, Hassel BA, Lee X, Torrence PF, & Silverman RH (1994). Intrinsic molecular activities of the interferon-induced 2–5A-dependent RNase. J Biol Chem, 269(19), 14153–14158. [PubMed] [Google Scholar]
- Donovan J, Dufner M, & Korennykh A (2013). Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1. Proc Natl Acad Sci U S A, 110(5), 1652–1657. doi: 10.1073/pnas.1218528110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J, Rath S, Kolet-Mandrikov D, & Korennykh A (2017). Rapid RNase L-driven arrest of protein synthesis in the dsRNA response without degradation of translation machinery. RNA, 23(11), 1660–1671. doi: 10.1261/rna.062000.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J, Whitney G, Rath S, & Korennykh A (2015). Structural mechanism of sensing long dsRNA via a noncatalytic domain in human oligoadenylate synthetase 3. Proc Natl Acad Sci U S A, 112(13), 3949–3954. doi: 10.1073/pnas.1419409112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drappier M, Jha BK, Stone S, Elliott R, Zhang R, Vertommen D, … Michiels T (2018). A novel mechanism of RNase L inhibition: Theiler’s virus L* protein prevents 2–5A from binding to RNase L. PLoS Pathog, 14(4), e1006989. doi: 10.1371/journal.ppat.1006989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskildsen S, Justesen J, Schierup MH, & Hartmann R (2003). Characterization of the 2’−5’-oligoadenylate synthetase ubiquitin-like family. Nucleic Acids Res, 31(12), 3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedetz M, Matesanz F, Caro-Maldonado A, Fernandez O, Tamayo JA, Guerrero M, … Alcina A (2006). OAS1 gene haplotype confers susceptibility to multiple sclerosis. Tissue Antigens, 68(5), 446–449. doi: 10.1111/j.1399-0039.2006.00694.x [DOI] [PubMed] [Google Scholar]
- Field LL, Bonnevie-Nielsen V, Pociot F, Lu S, Nielsen TB, & Beck-Nielsen H (2005). OAS1 splice site polymorphism controlling antiviral enzyme activity influences susceptibility to type 1 diabetes. Diabetes, 54(5), 1588–1591. [DOI] [PubMed] [Google Scholar]
- Fish I, & Boissinot S (2016). Functional evolution of the OAS1 viral sensor: Insights from old world primates. Infect Genet Evol, 44, 341–350. doi: 10.1016/j.meegid.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JQ, & Barton DJ (2002). Activation and evasion of the antiviral 2’−5’ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA, 8(4), 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Whitney G, Donovan J, & Korennykh A (2012). Innate immune messenger 2–5A tethers human RNase L into active high-order complexes. Cell Rep, 2(4), 902–913. doi: 10.1016/j.celrep.2012.09.004 [DOI] [PubMed] [Google Scholar]
- Hartmann R, Justesen J, Sarkar SN, Sen GC, & Yee VC (2003). Crystal structure of the 2’-specific and double-stranded RNA-activated interferon-induced antiviral protein 2’-5’-oligoadenylate synthetase. Mol Cell, 12(5), 1173–1185. doi:S1097276503004337 [pii] [DOI] [PubMed] [Google Scholar]
- Hartmann R, Norby PL, Martensen PM, Jorgensen P, James MC, Jacobsen C, … Justesen J (1998). Activation of 2’−5’ oligoadenylate synthetase by single-stranded and double-stranded RNA aptamers. J Biol Chem, 273(6), 3236–3246. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, … Hartmann G (2006). 5’-Triphosphate RNA is the ligand for RIG-I. Science, 314(5801), 994–997. doi: 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- Hornung V, Hartmann R, Ablasser A, & Hopfner KP (2014). OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol, 14(8), 521–528. doi: 10.1038/nri3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanessian AG, & Justesen J (2007). The human 2 ‘−5’-oligoadenylate synthetase family: Unique interferon-inducible enzymes catalyzing 2 ‘−5 ‘ instead of 3 ‘−5 ‘ phosphodiester bond formation. Biochimie, 89(6–7), 779–788. [DOI] [PubMed] [Google Scholar]
- Hovanessian AG, Laurent AG, Chebath J, Galabru J, Robert N, & Svab J (1987). Identification of 69-kd and 100-kd forms of 2–5A synthetase in interferon-treated human cells by specific monoclonal antibodies. EMBO J, 6(5), 1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovnanian A, Rebouillat D, Mattei MG, Levy ER, Marie I, Monaco AP, & Hovanessian AG (1998). The human 2’,5’-oligoadenylate synthetase locus is composed of three distinct genes clustered on chromosome 12q24.2 encoding the 100-, 69-, and 40-kDa forms. Genomics, 52(3), 267–277. doi: 10.1006/geno.1998.5443 [DOI] [PubMed] [Google Scholar]
- Hu J, Wang X, Xing Y, Rong E, Ning M, Smith J, & Huang Y (2018). Origin and development of oligoadenylate synthetase immune system. BMC Evol Biol, 18(1), 201. doi: 10.1186/s12862-018-1315-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, … Sicheri F (2014). Dimeric structure of pseudokinase RNase L bound to 2–5A reveals a basis for interferon-induced antiviral activity. Mol Cell, 53(2), 221–234. doi: 10.1016/j.molcel.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull CM, & Bevilacqua PC (2016). Discriminating Self and Non-Self by RNA: Roles for RNA Structure, Misfolding, and Modification in Regulating the Innate Immune Sensor PKR. Acc Chem Res, 49(6), 1242–1249. doi: 10.1021/acs.accounts.6b00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S (2019). Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu Rev Immunol. doi: 10.1146/annurev-immunol-042718-041356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen MS, Gad HH, Andersen LL, Hornung V, Julkunen I, Sarkar SN, & Hartmann R (2015). Structural and functional analysis reveals that human OASL binds dsRNA to enhance RIG-I signaling. Nucleic Acids Res, 43(10), 5236–5248. doi: 10.1093/nar/gkv389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen MS, Gad HH, Thavachelvam K, Boesen T, Despres P, & Hartmann R (2014). The 2’−5’-oligoadenylate synthetase 3 enzyme potently synthesizes the 2’−5’-oligoadenylates required for RNase L activation. J Virol, 88(24), 14222–14231. doi: 10.1128/JVI.01763-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SH, Lee K, Lee KS, Kunkeaw N, Johnson BH, Holthauzen LM, … Lee YS (2012). Characterization of the direct physical interaction of nc886, a cellular non-coding RNA, and PKR. FEBS Lett, 586(19), 3477–3484. doi: 10.1016/j.febslet.2012.07.076 [DOI] [PubMed] [Google Scholar]
- Kakuta S, Shibata S, & Iwakura Y (2002). Genomic structure of the mouse 2’,5’-oligoadenylate synthetase gene family. J Interferon Cytokine Res, 22(9), 981–993. doi: 10.1089/10799900260286696 [DOI] [PubMed] [Google Scholar]
- Keel AY, Jha BK, & Kieft JS (2012). Structural architecture of an RNA that competitively inhibits RNase L. RNA, 18(1), 88–99. doi: 10.1261/rna.030007.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaer KH, Poulsen JB, Reintamm T, Saby E, Martensen PM, Kelve M, & Justesen J (2009). Evolution of the 2’−5’-oligoadenylate synthetase family in eukaryotes and bacteria. J Mol Evol, 69(6), 612–624. doi: 10.1007/s00239-009-9299-1 [DOI] [PubMed] [Google Scholar]
- Kodym R, Kodym E, & Story MD (2009). 2’−5’-Oligoadenylate synthetase is activated by a specific RNA sequence motif. Biochem Biophys Res Commun, 388(2), 317–322. doi: 10.1016/j.bbrc.2009.07.167 [DOI] [PubMed] [Google Scholar]
- Koul A, Deo S, Booy EP, Orriss GL, Genung M, & McKenna SA (2019). Impact of double-stranded RNA characteristics on the activation of human 2’−5’-oligoadenylate synthetase 2 (OAS2). Scientific Reports (In press). [DOI] [PubMed] [Google Scholar]
- Kunkeaw N, Jeon SH, Lee K, Johnson BH, Tanasanvimon S, Javle M, … Lee YS (2013). Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene, 32(32), 3722–3731. doi: 10.1038/onc.2012.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Kunkeaw N, Jeon SH, Lee I, Johnson BH, Kang GY, … Lee YS (2011). Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA, 17(6), 1076–1089. doi: 10.1261/rna.2701111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, … Weiss SR (2017). Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife, 6. doi: 10.7554/eLife.25687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, … Weiss SR (2016). Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A, 113(8), 2241–2246. doi: 10.1073/pnas.1519657113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Lisco A, McDermott DH, Huynh L, Ward JM, Johnson B, … Murphy PM (2009). Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog, 5(2), e1000321. doi: 10.1371/journal.ppat.1000321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Yu HP, Chang BL, Tang WC, Liao CL, & Lin YL (2009). Distinct antiviral roles for human 2’,5’-oligoadenylate synthetase family members against dengue virus infection. J Immunol, 183(12), 8035–8043. doi: 10.4049/jimmunol.0902728 [DOI] [PubMed] [Google Scholar]
- Lohofener J, Steinke N, Kay-Fedorov P, Baruch P, Nikulin A, Tishchenko S, … Fedorov R (2015). The Activation Mechanism of 2’−5’-Oligoadenylate Synthetase Gives New Insights Into OAS/cGAS Triggers of Innate Immunity. Structure, 23(5), 851–862. doi: 10.1016/j.str.2015.03.012 [DOI] [PubMed] [Google Scholar]
- Loo YM, & Gale M Jr. (2011). Immune signaling by RIG-I-like receptors. Immunity, 34(5), 680–692. doi: 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra RK, McMillan NA, Desai S, McSwiggen J, Hovanessian AG, Sen G, … Silverman RH (1994). HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology, 204(2), 823–827. doi: 10.1006/viro.1994.1601 [DOI] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M Jr., & Silverman RH (2007). Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature, 448(7155), 816–819. doi: 10.1038/nature06042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Saito T, Crochet N, Barton DJ, Gale M Jr., & Silverman RH (2010). RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA, 16(11), 2108–2119. doi: 10.1261/rna.2244210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manche L, Green SR, Schmedt C, & Mathews MB (1992). Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol, 12(11), 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Abebe F, & Chaudhary J (2011). 2’−5’ oligoadenylate synthetase 1 polymorphism is associated with prostate cancer. Cancer, 117(24), 5509–5518. doi: 10.1002/cncr.26219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie I, Blanco J, Rebouillat D, & Hovanessian AG (1997). 69-kDa and 100-kDa isoforms of interferon-induced (2’−5’)oligoadenylate synthetase exhibit differential catalytic parameters. Eur J Biochem, 248(2), 558–566. [DOI] [PubMed] [Google Scholar]
- Marie I, Rebouillat D, & Hovanessian AG (1999). The expression of both domains of the 69/71 kDa 2’,5’ oligoadenylate synthetase generates a catalytically active enzyme and mediates an anti-viral response. Eur J Biochem, 262(1), 155–165. [DOI] [PubMed] [Google Scholar]
- Masliah G, Barraud P, & Allain FH (2013). RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci, 70(11), 1875–1895. doi: 10.1007/s00018-012-1119-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Deo S, Xiong S, Dzananovic E, Donald LJ, van Dijk CW, & McKenna SA (2012). Regulation of the interferon-inducible 2’−5’-oligoadenylate synthetases by adenovirus VA(I) RNA. Journal of Molecular Biology, 422(5), 635–649. doi: 10.1016/j.jmb.2012.06.017 [DOI] [PubMed] [Google Scholar]
- Minks MA, West DK, Benvin S, Greene JJ, Ts’o PO, & Baglioni C (1980). Activation of 2’,5’-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells by 2’-O-methylated poly (inosinic acid). poly(cytidylic acid), Correlations with interferon-inducing activity. J Biol Chem, 255(13), 6403–6407. [PubMed] [Google Scholar]
- Nallagatla SR, Toroney R, & Bevilacqua PC (2008). A brilliant disguise for self RNA: 5’- end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biol, 5(3), 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallagatla SR, Toroney R, & Bevilacqua PC (2011). Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr Opin Struct Biol, 21(1), 119–127. doi: 10.1016/j.sbi.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Yuzenkova Y, & Zenkin N (2013). Mechanism of eukaryotic RNA polymerase III transcription termination. Science, 340(6140), 1577–1580. doi: 10.1126/science.1237934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien M, Lonergan R, Costelloe L, O’Rourke K, Fletcher JM, Kinsella K, … Tubridy N (2010). OAS1: a multiple sclerosis susceptibility gene that influences disease severity. Neurology, 75(5), 411–418. doi: 10.1212/WNL.0b013e3181ebdd2b [DOI] [PubMed] [Google Scholar]
- Rath S, Donovan J, Whitney G, Chitrakar A, Wang W, & Korennykh A (2015). Human RNase L tunes gene expression by selectively destabilizing the microRNA-regulated transcriptome. Proc Natl Acad Sci U S A, 112(52), 15916–15921. doi: 10.1073/pnas.1513034112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouillat D, Hovnanian A, David G, Hovanessian AG, & Williams BR (2000). Characterization of the gene encoding the 100-kDa form of human 2’,5’ oligoadenylate synthetase. Genomics, 70(2), 232–240. doi: 10.1006/geno.2000.6382 [DOI] [PubMed] [Google Scholar]
- Samuel CE (2011). Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology, 411(2), 180–193. doi: 10.1016/j.virol.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Bandyopadhyay S, Ghosh A, & Sen GC (1999). Enzymatic characteristics of recombinant medium isozyme of 2’−5’ oligoadenylate synthetase. J Biol Chem, 274(3), 1848–1855. [DOI] [PubMed] [Google Scholar]
- Sarkar SN, Kessler SP, Rowe TM, Pandey M, Ghosh A, Elco CP, … Sen GC (2005). Natural mutations in a 2’−5’ oligoadenylate synthetase transgene revealed residues essential for enzyme activity. Biochemistry, 44(18), 6837–6843. doi: 10.1021/bi0502893 [DOI] [PubMed] [Google Scholar]
- Schlee M, & Hartmann G (2016). Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol, 16(9), 566–580. doi: 10.1038/nri.2016.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, & Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol, 32, 513–545. doi: 10.1146/annurev-immunol-032713-120231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Raine DA, Gewert DR, Joshi B, Jagus R, & Clemens MJ (1999). Activation of the interferon-inducible (2’−5’) oligoadenylate synthetase by the Epstein-Barr virus RNA, EBER-1. Virology, 257(2), 303–313. doi: 10.1006/viro.1999.9689 [DOI] [PubMed] [Google Scholar]
- Siddiqui MA, Mukherjee S, Manivannan P, & Malathi K (2015). RNase L Cleavage Products Promote Switch from Autophagy to Apoptosis by Caspase-Mediated Cleavage of Beclin-1. Int J Mol Sci, 16(8), 17611–17636. doi: 10.3390/ijms160817611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman RH (2007). Viral encounters with 2’,5’-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol, 81(23), 12720–12729. doi: 10.1128/JVI.01471-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman RH, & Weiss SR (2014). Viral Phosphodiesterases That Antagonize Double-Stranded RNA Signaling to RNase L by Degrading 2–5A. J Interferon Cytokine Res, 34(6), 455–463. doi: 10.1089/jir.2014.0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrer KM, & Gack MU (2015). Intracellular detection of viral nucleic acids. Curr Opin Microbiol, 26, 1–9. doi: 10.1016/j.mib.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, & Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science, 339(6121), 786–791. doi: 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi T, Nagano-Fujii M, Akutsu M, Kadoya H, Ohgimoto S, Ishido S, & Hotta H (2004). Hepatitis C virus NS5A protein interacts with 2’,5’-oligoadenylate synthetase and inhibits antiviral activity of IFN in an IFN sensitivity-determining region-independent manner. J Gen Virol, 85(Pt 4), 959–969. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, & Akira S (2010). Pattern recognition receptors and inflammation. Cell, 140(6), 805–820. doi: 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- Thamizhmani R, & Vijayachari P (2014). Association of dengue virus infection susceptibility with polymorphisms of 2’−5’-oligoadenylate synthetase genes: a case-control study. Braz J Infect Dis, 18(5), 548–550. doi: 10.1016/j.bjid.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornbrough JM, Jha BK, Yount B, Goldstein SA, Li Y, Elliott R, … Weiss SR (2016). Middle East Respiratory Syndrome Coronavirus NS4b Protein Inhibits Host RNase L Activation. MBio, 7(2), e00258. doi: 10.1128/mBio.00258-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Fukai S, Ishitani R, Ueda T, Takeuchi N, Vassylyev DG, & Nureki O (2004). Structural basis for template-independent RNA polymerization. Nature, 430(7000), 700–704. doi: 10.1038/nature02712 [DOI] [PubMed] [Google Scholar]
- Torralba S, Sojat J, & Hartmann R (2008). 2’−5’ oligoadenylate synthetase shares active site architecture with the archaeal CCA-adding enzyme. Cell Mol Life Sci, 65(16), 2613–2620. doi: 10.1007/s00018-008-8164-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend HL, Jha BK, Han JQ, Maluf NK, Silverman RH, & Barton DJ (2008). A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA, 14(6), 1026–1036. doi: 10.1261/rna.958908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend HL, Jha BK, Silverman RH, & Barton DJ (2008). A putative loop E motif and an H-H kissing loop interaction are conserved and functional features in a group C enterovirus RNA that inhibits ribonuclease L. RNA Biol, 5(4), 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon VK, Calderon BM, & Conn GL (2015). A novel RNA molecular signature for activation of 2’−5’ oligoadenylate synthetase-1. Nucleic Acids Res, 43(1), 544–552. doi: 10.1093/nar/gku1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon VK, & Conn GL (2016). Adenovirus VA RNA: An essential pro-viral non-coding RNA. Virus Research, 212, 39–52. doi: 10.1016/j.virusres.2015.06.018 [DOI] [PubMed] [Google Scholar]
- Wahid AM, Coventry VK, & Conn GL (2008). Systematic deletion of the adenovirus-associated RNAI terminal stem reveals a surprisingly active RNA inhibitor of double-stranded RNA-activated protein kinase. J Biol Chem, 283(25), 17485–17493. doi: 10.1074/jbc.M802300200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreschner DH, McCauley JW, Skehel JJ, & Kerr IM (1981). Interferon action--sequence specificity of the ppp(A2’p)nA-dependent ribonuclease. Nature, 289(5796), 414–417. [DOI] [PubMed] [Google Scholar]
- Wu J, & Chen ZJ (2014). Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol, 32, 461–488. doi: 10.1146/annurev-immunol-032713-120156 [DOI] [PubMed] [Google Scholar]
- Xiong Y, & Steitz TA (2004). Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature, 430(7000), 640–645. doi: 10.1038/nature02711 [DOI] [PubMed] [Google Scholar]
- Yakub I, Lillibridge KM, Moran A, Gonzalez OY, Belmont J, Gibbs RA, & Tweardy DJ (2005). Single nucleotide polymorphisms in genes for 2’−5’-oligoadenylate synthetase and RNase L inpatients hospitalized with West Nile virus infection. J Infect Dis, 192(10), 1741–1748. doi: 10.1086/497340 [DOI] [PubMed] [Google Scholar]
- Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, … Weiss SR (2013). Homologous 2’,5’-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci U S A, 110(32), 13114–13119. doi: 10.1073/pnas.1306917110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, … Weiss SR (2012). Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe, 11(6), 607–616. doi: 10.1016/j.chom.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, Dhar J, … Sarkar SN (2014). Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity, 40(6), 936–948. doi: 10.1016/j.immuni.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]