

Abstract
Measurement of the electron transfer cascade (ETC) enzyme activities and their kinetic profiles is important in assessing mitochondrial function in the nervous system in health and disease, or following exposure to toxic agents. The optimization of enzymatic assays for brain tissues and neuronal cells are critical to the development of high-throughput assay formats. We describe a step-by-step protocol for reliable and reproducible assessment of the ETC enzyme kinetics (Complex-I through Complex-IV) for mitochondria from small quantities of tissue from different brain regions, such as the hippocampus, cerebellum, and frontal cortex, or from neuronal cells in culture. Methods for differential and density gradient centrifugation are detailed for cell body and synaptic mitochondria isolation from brain, as are those for measurement of ETC activities in micro-well plate or single-cuvette format using spectrophotometric methods. We have constructed easy-to follow assay layouts and useful tips to perform these assays under 3 hours.
Keywords: Brain subcellular fractionation, Primary neuronal cultures, Neuroblastoma cultures, Electron transport chain enzymes, mitochondria
INTRODUCTION
As summarized in the historical accounting by Ernster and Schatz (Ernster & Schatz, 1981), investigations into the role of mitochondria in cell growth, differentiation, and viability began in 1890 with the first description of mitochondria as subcellular structures in eukaryotic cells.
Mitochondria in eukaryotic cells vary in number and location according to the cell type. Human cells contain about 1,000–2,000 mitochondria per cell depending on the cellular demands for chemical energy in the form of ATP, cellular calcium (Ca2+) buffering capacity, cell signaling through the generation of reactive oxygen species (ROS), or import and degradation of damaged proteins. However, some of these processes, such as oxidative phosphorylation, if partially inhibited, would lead to an increase in ROS formation, cell protein damage, and possible cell death (Kohen & Nyska, 2002). It is well established that mitochondria are key to the phenomenon of programmed cell death or apoptosis occurring under conditions of cell stress, such as tissue ischemia and reperfusion (Ravagnan, Roumier, & Kroemer, 2002).
From the perspective of toxicological processes that might involve altered mitochondrial function and cell damage, especially in the nervous system, the example of Parkinson’s disease (PD) is the most prominent one. Models of this disease have been produced by inhibiting mitochondrial enzymes (Complex I) of oxidative phosphorylation (OXPHOS) by pesticides or other chemical agents (Nicklas, Vyas, & Heikkila, 1985; Sherer et al., 2007). Substantial evidence has also been accumulating pointing to the relationship between mitochondrial stress or malfunction and the incidence of Alzheimer’s disease (Swerdlow, 2012). Loss of activity or amyloid β (Aβ)-induced inhibition of enzymes involved in OXPHOS, in particular Complex I (NADH:ubiquinone oxidoreductase) and Complex IV (cytochrome oxidase), are characteristic of AD or AD cell models (Bobba et al., 2013).
Based on the observations pointing to the importance of mitochondria in brain toxicology and human disease, especially that OXPHOS enzymes are targets of toxic agents, it is important to describe techniques that would be useful in the study of the OXPHOS enzymes. Also, as it has been shown that mitochondria isolated from different brain regions or different compartments of neurons (synaptic vs. somatic mitochondria) may exhibit different susceptibilities to damaging agents (Wang et al., 2016), it is important to use techniques that allow for the study of both types of mitochondria.
BASIC PROTOCOL 1
Purification of somatic and synaptic mitochondria from rodent brain
There are differential characteristics in mitochondria derived from soma and the synaptic nerve endings (e.g. axonal and dendritic). Development of subcellular fractionation techniques with brain tissue has made it possible to obtain highly enriched preparations of synaptic terminals (Cotman & Matthews, 1971; Cotman & Taylor, 1972; De Robertis, 1967; Gurd, Jones, Mahler, & Moore, 1974) from which it is possible to isolate mitochondria of the synaptic region. During homogenization of brain tissue in an iso-osmotic medium, the axon terminals are sheared off from the axons. These nerve-ending particles (synaptosomes) reseal into microscopic structures containing the organelles present at nerve terminals and postsynaptic dendrites, including the mitochondria within these processes (Cotman & Taylor, 1972).
Materials
Brain tissue homogenization Buffer (see Reagents and Solutions)
Resuspension Buffer (see Reagents and Solutions)
Ficoll Density Gradient Solution (see Reagents and Solutions)
Mitochondrial Storage Buffer (see Reagents and Solutions)
Synaptosome Lysis Buffer (see Reagents and Solutions)
Sucrose Density Gradient Buffer for isolation of synaptic mitochondria (see Reagents and Solutions)
Equipment
Small animal guillotine with sharp blade
Sharp, straight edge surgical scissors
Small bone cutters
Curved forceps or tweezers with teeth
Curved forceps
Small sharp scissors
Potter-Elvehjem glass homogenizer tubes of various volumes (3 – 45 mL Thomas Scientific)
Teflon pestles (0.13 mm clearance)
Adjustable speed motor for brain homogenization—Eberbach homogenizer for Teflon-glass Potter-Elvehjem homogenization pestle and tube (Thomas Scientific).
Polycarbonate 50 mL tubes (8 or more) with screw-caps
A rotor that can accommodate tubes that hold 20 mL or larger volumes such as a JA-20 rotor, Beckman-Coulter
A refrigerated preparative centrifuge that can create a centrifugal force of 40 – 50,000 × g’s such as a Beckman-Coulter Avanti JXN-26 centrifuge.
Thin wall polyallomer 38.5 mL ultracentrifuge tubes (Beckman-Coulter).
An ultracentrifuge that can create 65,000 – 500,000 × g’s with a swinging bucket rotor for the
Ficoll density gradients such as Beckman-Coulter Optima XE ultracentrifuge with an SW28 rotor, or Thermo-Sorvall WX ultracentrifuge with the same rotor.
Isolation of somatic (free) mitochondria
-
1
Fully anesthetize a lab rodent and use a small animal guillotine (decapitator, Stoelting) to decapitate it.
Handle the animals according to the protocol approved by your institutional animal care and use committee (IACUC). The animals should be fully anesthetized by CO2 inhalation prior to initiation of the procedure. Use a small animal guillotine (decapitator, Stoelting) to decapitate it. Maintain the guillotine free of blood or hair and, periodically, have the blade sharpened professionally as recommended by the animal care unit of your institution.
-
2
Rinse the blood from the neck region with ice-cold homogenization buffer
-
3
Use straight edge scissors to cut under the scalp at the posterior end of the head and reflect the scalp in the direction of the eyes
-
4
Use the tips of the surgical bone cutters and cut the bone surrounding the point of insertion of the spinal cord to the brainstem—cut laterally moving towards the side of each ear lobe.
-
5
Open the skull to expose the dorsal surface of the brain using a forceps or tweezers with teeth by lifting the skull in the direction of the nose.
-
6
Scoop the brain out of the skull by using a curved clamp
-
7
Quickly weigh the brain and transfer it to a beaker containing homogenization buffer kept on ice. If there is too much blood that the buffer in the beaker turns to dark pink color, then remove the buffer and replace it with fresh buffer until the buffer color remains either colorless or very faint pink color.
-
8
Using a small sharp scissors mince each brain tissue in a small volume of the Homogenization Buffer in a beaker on ice.
-
9
Transfer the tissue to a cold glass Potter-Elvehjem homogenization tube with a Teflon pestle.
Homogenize each minced brain tissue in the Homogenization Buffer at a ratio of 10 mL Homogenization Buffer per gram of tissue weight. Use 8–10 strokes up and down with the pestle driven at 1,000–1,500 rpm. The sheering action leads to complete homogenization. Intermittently put the homogenization tube into ice to prevent heating of the vessel and denaturing proteins.
Brain tissue samples should always be kept at 0°C-to-4°C throughout the sub-fractionation procedure. Pre-cool the rotors, tubes, and centrifuges before use. Keeping the tissue cold and adding protease inhibitors prevents activation of proteases. It is advisable to include a phosphatase inhibitor cocktail in the preparation steps as well.
-
10
When homogenization is complete, measure, record the volume of the homogenate, and remove a small aliquot for protein determination later.
It is advisable at each step in the isolation of the subfractions to keep track of the total volume of each fraction and to remove a small aliquot of the sample for measurement of the protein concentration (mg/mL). By multiplying protein concentration times, the total volume of each fraction ([protein] mg/ml X Vol. (ml) ), you may monitor the protein recovery at each step of the purification procedure and assess the reproducibility of the isolation method.
-
11
Transfer the homogenates to pre-cooled centrifuge tubes and centrifuge at 1,000 × g for 5 min at 4°C in a preparative centrifuge (see equipment above) to remove cell nuclei and cell debris.
-
12
Collect the supernatant from each tube and transfer to new centrifuge tubes pre-cooled at 0°C-to-4° C.
-
13
Wash the pellet with about 2 mL of Homogenization Buffer and re-suspend the pellet with a pre-cooled pestle followed by vortexing
-
14
Add to the re-suspended pellet Homogenization Buffer at the rate of 5 mL per gram of original brain tissue
-
15
Centrifuge at 1,000 × g for 5 min at 4°C and combine the supernatants from the two centrifugations
-
16
Centrifuge the supernatants at 13,000 × g for 10 min at 4oC in a preparative centrifuge to obtain the pellet of crude mitochondrial fraction that contains free somatic mitochondria and synaptosomes (Discard the supernatant)
-
17
Add 2 mL of the Resuspension Buffer to each pellet, vortex and gently re-suspend the pellet by using a Pasteur pipette to triturate the pellet
Isolation of synaptosomes and synaptic mitochondria involves several steps in which subfractions are precipitated under high centrifugal force, and the pellets must be resuspended in fresh medium. The re-suspended fractions must be homogeneous, no chunks or particles. Gently dislodging the pellet from the centrifuge tube by adding a small amount of the medium, mildly vortexing the tube, transferring the sample to a small Teflon-glass homogenizer, and hand homogenizing the sample gently is an effective way of preparing each subfraction for the next step. Alternatively, you may use a Pasteur pipette and gently triturate the sample up and down until it becomes homogeneous.
-
18
Measure and record the volume and save a small aliquot for protein determination, then transfer the resuspension to graduated cylinders and add more Resuspension Buffer to bring the suspensions to a volume of 17.5 mL per brain preparation
-
19
Prepare the Ficoll gradients by adding 10 mL of the 14% Ficoll solution to the bottom of a polyallomer ultracentrifuge tube (38.5 mL capacity). Layer 10 mL of the 8% Ficoll solution on top of the 14% Ficoll. Layer 17.5 mL of the resuspended crude mitochondrial sample on top of the 8% Ficoll (see Fig. 1)
Ficoll rather than sucrose is used for preparing the discontinuous density gradient to isolate the synaptosomes because it preserves their structure (Cotman & Matthews, 1971).
Ficoll has a much higher molecular mass than sucrose, therefore, a much lower molar concentration of Ficoll is needed to achieve the same density as sucrose. An alternative procedure would be to use density gradients formed by Percoll (Dunkley, Jarvie, Heath, Kidd, & Rostas, 1986).
-
20
Add Resuspension Buffer as needed to balance the weight of each tube and to nearly fill the tubes for the centrifugation.
-
21
The samples are now ready to perform discontinuous Ficoll density gradient centrifugation in an ultracentrifuge (Thermo Sorvall WX ultracentrifuge, or Beckman-Coulter Optima XE with a swinging bucket rotor such as SW28).
-
22
Centrifuge at 63,000 × g for 50 min—the synaptosomes form a band at the interface of 8 and 14% Ficoll whereas the somatic (free) mitochondria from both neurons and glia are in the pellet (see Fig. 1).
-
23
Use a Pasteur pipette to aspirate the upper band and the liquid above it
-
24
Collect the synaptosome band by introducing the tip of a clean Pasteur pipette near the wall of the tube at the interface of the two Ficoll layers, and transfer the synaptosomes to a tube for a fixed angle rotor (e.g. Beckman-Coulter Type 70 Ti)
-
25
After removing all Ficoll solution from each tube, re-suspend the pellet of neuronal/glial somatic mitochondria in 0.25-to-0.5 mL of Mitochondrial Storage Buffer
-
26
Obtain a small sample (3x2013;10 μL) of the re-suspended mitochondria for protein determination to be performed later.
-
27
The mitochondrial suspension in Mitochondrial Storage Buffer may be snap frozen for later use—Dilute the suspension to a protein concentration of 2–10 mg/mL, divide into aliquots of 100x2013;250 x00B5;L, freeze rapidly in liquid nitrogen, and store at −80°C
Fig.1.
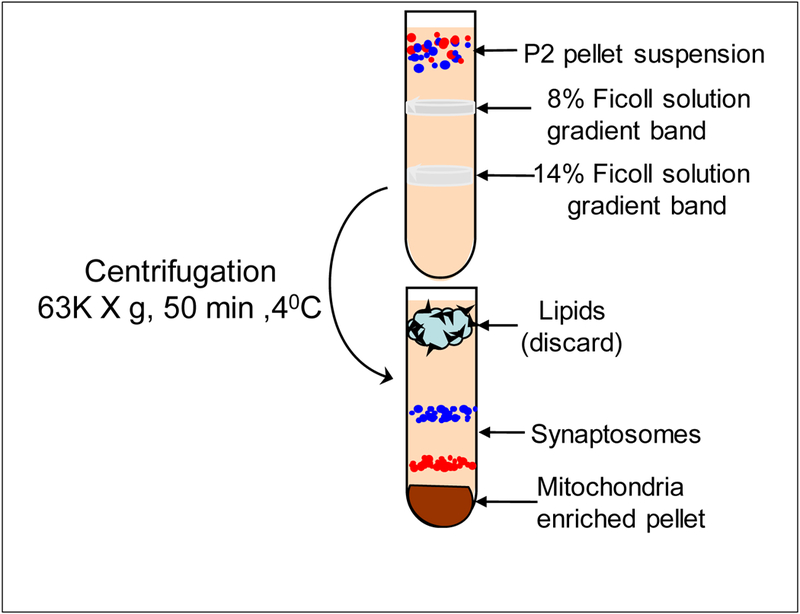
Isolation of mitochondria-enriched pellet by Ficoll density gradient centrifugation. First, add 8% Ficoll solution into centrifugation tube. Second, add 14% Ficoll solution. From bottom of the tube without mixing with 8% Ficoll gradient. Finally, add P2 pellet. Resuspension on the top of the centrifugation tube without mixing the gradient.
Isolation of synaptic mitochondria
-
28
Dilute the synaptosome fractions with 4 volumes of the Resuspension Buffer or enough volume to fill the tube to a level recommended by the manufacturer
-
29
Centrifuge at 145,000 x g for 17 min at 4°C (Beckman-Coulter Optima xE or Thermo Sorvall Wx ultracentrifuge) to remove the Ficoll and obtain the isolated synaptosomes as the pellet.
-
30
Discard the supernatant and retain the pellet. For the isolation of synaptic mitochondria, add approximately 0.5 mL of the Resuspension Buffer to the synaptosome pellet
-
31
Then add 5 volumes of the hypotonic Lysis Buffer to each aliquot and transfer to a centrifuge tube. Invert the tubes several times to mix well. Allow the tubes to sit on ice for 20 min with intermittent gentle vortexing to allow for complete lysis of the synaptosomes and release of synaptic mitochondria
-
32
Centrifuge the samples at 45,000 x g for 14 min at 4°C in a fixed angle rotor (e.g. Type 70 Ti). The pellet represents a mixture of synaptic membranes and synaptic mitochondria and this preparation may be used for enzyme assays of partially purified neuronal/synaptic mitochondria
-
33
To obtain more highly purified neuronal/synaptic mitochondria preparation, you need to perform sucrose density centrifugation to separate myelin and plasma membrane contaminants from the mitochondria— Start by discarding the supernatant and re-suspending the pellet in 13 mL of the 34% sucrose buffer.
-
34
Prepare a discontinuous sucrose density gradient by first introducing 12 mL of the 10% sucrose solution at the bottom of each polyallomer centrifuge tube (38.5 mL capacity) for a fixed angle rotor (e.g., Beckman-Coulter Type 70 Ti)
-
35
Using a long 18-gauge needle and syringe that reaches the bottom of the tube, slowly introduce 12 mL of the 28.5% sucrose solution to the bottom of the tube under the 10% sucrose to form a discontinuous density gradient of 10 and 28.5% sucrose
-
36
To complete formation of the sucrose density gradient, use a long needle to introduce the suspension of synaptic membranes and synaptic mitochondria in 34% sucrose to the bottom of the centrifuge tubes
-
37
Centrifuge the gradient of 10%, 28.5%, and 34% sucrose at 90,000 x g for 35 min in the Type 70 Ti rotor. In this flotation / sedimentation condition any myelin contaminants float to the interface between 10 and 28.5% sucrose, synaptic membranes float to the interface between 28.5 and 34% sucrose, and neuronal/synaptic mitochondria sediment in the 34% sucrose layer as a pellet at the bottom of the tube
-
38
Using an 18-gauge needle and a syringe, remove the 10% sucrose and the myelin band at the interface of 10 and 28.5% sucrose, then remove the band of synaptic plasma membranes at the interface of the 28.5% and 34% sucrose, then remove the remaining 34% sucrose solution. Add a small volume (0.25–0.50 mL) of Resuspension Buffer, re-suspend the purified synaptic mitochondrial pellet, and take a small aliquot (3–10 μL) for protein determination. If the synaptic mitochondrial subfraction is to be stored for later use, the pellet may be re- suspended in Mitochondrial Storage Buffer at a protein concentration of approximately 2–10 mg/mL, then divided into aliquots of 0.10 to 0.25 mL snap frozen in liquid nitrogen, and stored at −80°C.
BASIC PROTOCOL 2
Dissection of select brain regions
Normal aging, aging relevant neurodegenerative disease, and protein misfolding-relevant abnormalities are linked to the decreased capacity of ATP production. Differential mitochondrial ETC enzyme activities are expected to be different depending on the brain region. The procedures described in this section will serve as a guide for collection of tissue for preparation of mitochondria-enriched preparation.
Materials
Dissection pane
Surgical blades with handles or single-edged razors
A pair of large dissection scissors
A dissection microscope with a low-heat light source. Or, a large (4–5-inch diameter) 2,3 x powered magnifier with a stand.
Two pairs of small dissection scissors
Four or more small, thin metal spatulas
Two pairs of curved fine surgical forceps
One pair of large forceps
Animal sacrifice:
Follow the steps in BASIC PROTOCOL 1
Dissection Protocols for Particular Brain Regions Cerebellum
-
1
Place the brain with the ventral side facing the dissection pane surface, exposing the cerebellum (Fig.2A)
-
2
Make a transverse cut through the brain stem at the level of the colliculi [Fig.2A (1)]
-
3
Make a second transverse cut posterior to the cerebellum, through the underlying brain stem [Fig.2A (2)]
-
4
Place this block of tissue with one of the cut surfaces down, and cut the cerebellar peduncles with a razor blade and separate the cerebellum from the underlying brain stem [Fig.2B (1 and 2)]
Fig.2.
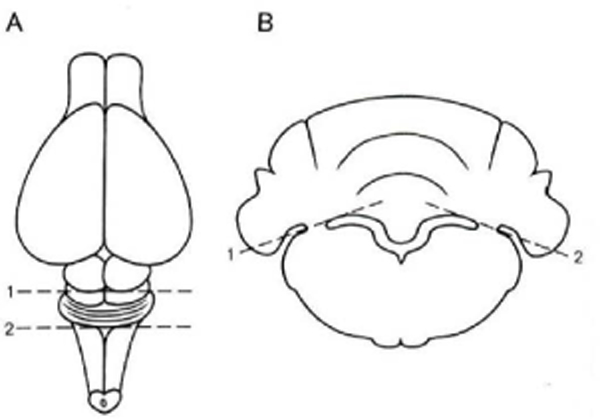
Dissection steps for slices of cerebellum. A: View of the dorsal surface of the rat brain. Positions of transverse cuts (1 and 2; dashed lines) to isolate cerebellum and pons are indicated. B: View of block of tissue from A. Positions of slanting cuts (1 and 2; dashed lines) to separate the cerebellum from underlying pons are indicated (Robertson, Zimmer, & Gahwiler, 1989).
Hippocampus
The following steps describe a 2–3 minutes-rapid dissection of a rodent hippocampus.
-
5
Remove the whole brain from the calvarium by opening the lateral sides of the skull with a pair of scissors as described elsewhere (Palkovits & Brownstein, 1983)
-
6
Make a lineal incision from posterotemporal pole of one hemisphere to the anterior pole of the frontal lobe at about 30 degree angle (Fig. 3A).
Fig.3.
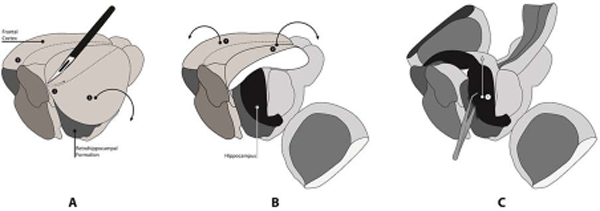
Schematic surgical procedure for the fast removal of hippocampus from rat brain. A: Two parasagittal incision at a 30° angle (1 and 2) . B: Separation of the central cortex area (1 and 2). C: Removal of two hippocampal formations (1). This illustration was adopted and professionally re-drawn (Navarro et al., 2008)
Adjust the incision depth to only the cortex and subjacent corpus callosum. Practice before harvesting the experimental tissues to get a sense of incision depth. Use either dissection microscope or magnifying glass for better view.
Prefrontal Cortex
-
7
After removal of the hippocampus, use the large forceps to fold back the cortex into the original position (Fig. 3A in hippocampus dissection).
-
8
Place the brain with the ventral side facing the dissection plate
-
9
Cut off the olfactory bulb with a sharp razor blade (Fig. 4A)
Fig.4.

Schematic representation of Prefrontal Cortex dissection. A: Removal of olfactory bulb. B: Dissection of prefrontal cortex. This section mainly contains motor cortex.
The anterior commissure is well visible at this point
Make the section as illustrated in Fig. 4B that contains mainly motor cortex
More detailed and colored photographs for the sectioning can be viewed in the literature (Chiu, Lau, Lau, So, & Chang, 2007; Spijker, 2011)
BASIC PROTOCOL 3
Mitochondria-enriched tissue Preparation from brain regions
Mitochondrial respiratory chain enzyme activities vary in different regions of the brain, depending on the energy demand of the regions. The more synaptic activities requires more energy, hence more mitochondria population (Leong, Lai, Lim, & Clark, 1984; Mukherjee et al., 2016). Due to the size of the regional tissue in rodent brain is limited; to isolate near-pure mitochondria population may not be sufficient to analyze enzymatic studies. Therefore, this protocol will explain how to prepare functional mitochondria-enriched preparations from three different regions of a mouse brain (i.e. frontal cortex, hippocampus, and cerebellum).
Materials
Glass-glass pestle Dounce homogenizer
Small table top centrifuge
Pipettes with assorted volumes
Conical bottom microfuge tubes (1mL, 0.5 mL)
Mitochondria storage buffer (MSB) (see Reagents and Solutions)
Protein assay kit (Pierce TM BCA assay kit; Thermo Scientific; Cat#23227)
UV/Vis Spectrophotometer
Cryobox
-
Homogenize each brain tissue harvested from selected regions (i.e. hippocampus, frontal cortex, and cerebellum) in a pre-chilled glass-glass pestle Dounce homogenizer (Fig. 1) (2 ml volume) with MSB in an ice-bucket. Homogenize the tissue by hand. 25–30 strokes would be sufficient. Do not allow froth during the homogenization.
Keep tissue: MPB ratio as ~1 g wet tissue: 9 ml MSB for a thorough homogenization.
-
Mitochondria-enriched sample is prepared by differential centrifugation method (Fig. 1). Centrifuge the homogenate in a small tabletop centrifuge for 10 min at 800 x g at 4°C. Save the supernatant.
Use a refrigerated tabletop centrifuge such as Eppendorf 54174R. Other option is to keep one tabletop refrigerator either in a cold-box or in a cold room)
Resuspend the pellet (P1) with 1–2 mL (depending on pellet size) of cold homogenization buffer and repeat the centrifugation described in Step-2. Discard the P1 pellet. Save the supernatant (S1).
Combine the supernatants in a conical bottom centrifugation tube.
-
Centrifuge the combined supernatants for 10 min at 11,000 x g at 4°C. Save the mitochondria- enriched pellet (P2) and the supernatant (S2).
Save a small aliquot (~ 25 μL) of S2 to determine the presence of Cytochrome c oxidase as a measure of the damage level of mitochondria during the homogenization process. This test will help to assess the fragility of mitochondria obtained from different regions of the brain.
Resuspend the mitochondria-enriched pellet in a small volume of mitochondria storage buffer (MSB) (~100–200 μL), measure the protein concentration by BCA protein assay, and readjust the protein concentration to 20 mg/mL if necessary. Divide the mitochondria-enriched homogenate into 20–25 μL aliquots, snap freeze in a bucket containing liquid nitrogen, and store the aliquots at −80°C.
Fig.5.
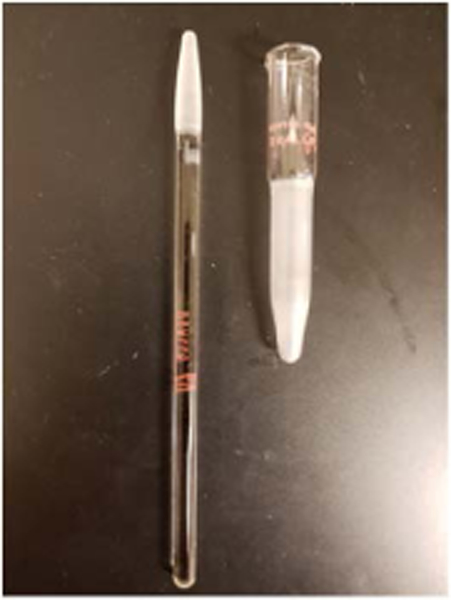
. Glass-Glass paste Dounce homogenizer with 2 ml volume. This homogenizer is suitable for mitochondria-enriched pellet (P2) homogenization without damaging the integrity of the mitochondria. A gentle hand-driven homogenization is recommended.
Fig.6.
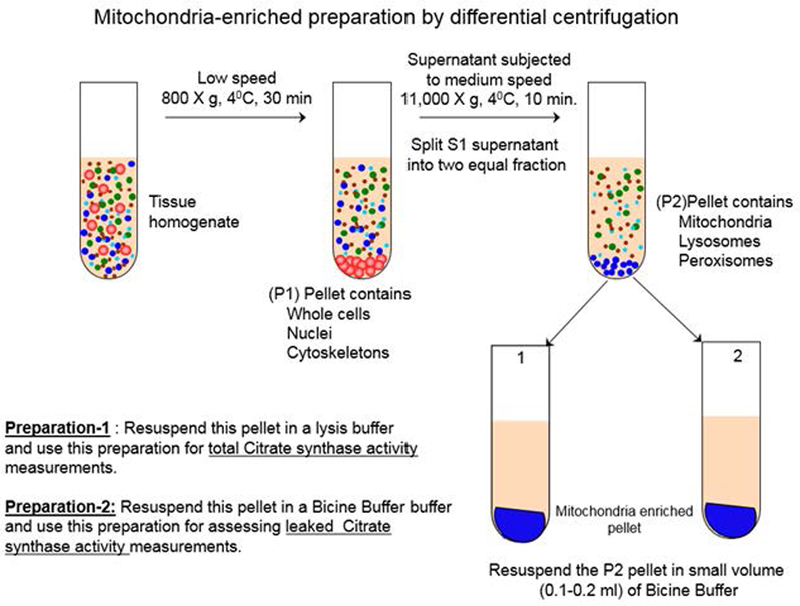
Schematic illustration of mitochondria-enriched sample preparation by differential centrifugation method.
It is advisable to store the mitochondria-enriched preparation as aliquots of high protein concentration in order to minimize protein degradation.
Use low retention 0.5 mL microfuge tubes that withstand cooling to −80°C for a long time. (VWR Cat# 20170–038; 0.5 ml conical bottom micro centrifuge tube). Label the tubes beforehand (preferably a day before) with a colored narrow-width tape. Use fine tip black color permanent marker. Store the aliquots in 10x10 or 9x10 cryobox and label the “front” with arrow if not already labeled by the manufacturer. Alternatively, Tubewriter™ 360 can be used for direct writing on the tube ( www.tubewriter.com).
SUPPORT PROTOCOL 1
Mitochondrial swelling assay
An increase in mitochondrial volume due to an influx of fluid; it occurs in hypotonic solutions due to osmotic pressure and in isotonic solutions as a result of altered permeability of the membranes of respiring mitochondria. Dilute suspensions of mitochondria are turbid, and absorb and scatter visible light. A direct estimation of mitochondrial size by light scattering indicates the physical intactness of mitochondria (Navarro & Boveris, 2004). Old or damaged mitochondria swell more than intact mitochondria; hence, less light scattering occurs. Therefore, less light absorption should be observed. Young, healthy, and intact mitochondria resist swelling. This is a simple and inexpensive method to get a sense of mitochondria quality during the isolation process. A typical mitochondrial light scattering pattern is shown in Fig. 1).
Fig.7.
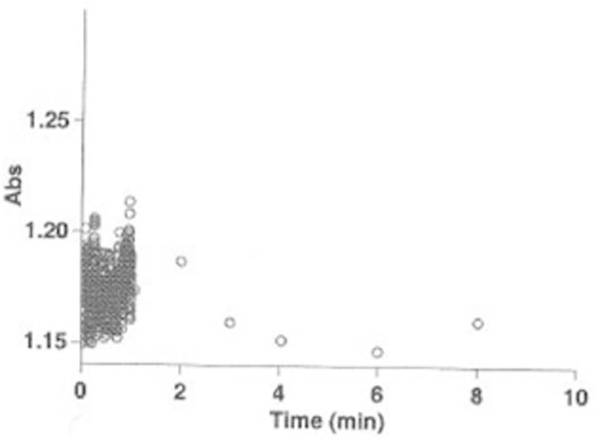
Mitochondria Swelling. Mitochondria scatter light in cells and tissue at visible wavelength (λ=540 nm). Simple optical transmission measurements are sensitive to changes in mitochondria intactness during the preparation. Majority of mitochondria during the preparation did not swell in this test as the absorbance did not dramatically change (ΔA540nm = 0.05).
Materials
Microfuge tube (1.5 mL)
Disposable cuvette (1 mL; BRAND disposable semi-micro plastic cuvette, VWR Cat# 759075D)
UV/Vis Spectrophotometer
Hypotonic buffer (HB) (see Reagents and Solutions)
Take an aliquot of mitochondria-enriched homogenate and adjust the protein concentration to 1 mg/mL in HB reagent.
Gently mix the suspension by inverting the tube 2x2013;3 times.
Place the suspension into a 1 ml disposable micro cuvette and place in the cuvette holder of the spectrophotometer
Set the wavelength of the spectrophotometer at 540 nm
Place a x201C;blankx201D; cuvette containing 1 ml HB reagent and “zero” the spectrophotometer.
Place the cuvette containing the mitochondria suspension in the spectrophotometer
Monitor and record the absorbance decrease for 10 min. Absorbance change within ΔA540nm =0.05 indicates the intactness of mitochondria for 10 min.
BASIC PROTOCOL 4
Assays of Mitochondrial Respiratory Enzyme Complexes
Complex I (NADH:ubiquinone oxidoreductase) activity measurement
Spectrophotometric assays of complex I can utilize either the oxidation of NADH or the reduction of ubiquinone analogs (Trounce, Kim, Jun, & Wallace, 1996). This assay utilizes decylubiquinone as the electron acceptor and NADH as the electron donor.
Materials
Sonicator with microtip (Branson)
96-well microtiter plate
Disposable cuvette (1 ml; BRAND disposable semi-micro plastic cuvette, VWR Cat# 759075D)
UV/Vis Spectrophotometer with thermostat
Microplate reader with thermostat
Mitochondria rupturing hypotonic solution (see Reagents and Solutions)
0.2 M KCN
5 mM NADH
1 mM Rotenone
1.6 mM Decylubiquinone
96-well microtiter plate assay format
Take out the sufficient frozen samples from −80°C and place them in an ice bath. Allow the samples gradually thaw.
Disrupt the mitochondria-enriched preparation by sonication with 6 pulses, power level 5 and 20% duty cycle, using a Branson sonicator with microtip (Trounce et al., 1996)
Follow the assay flow chart (Table 1) step-1 through 8 the reagent additions into each well. Discard the leftover samples.
Assay parameters:
Table 1:
Complex-I assay flow chart
| REAGENTS | BLANK | SAMPLE (−) Rotenone |
SAMPLE (+) Rotenone |
|---|---|---|---|
| (1) 50 mM KP Buffer | 241 μL | 241 μL – x μL | 238.5 μL – x μL |
| (2) Protein solution | - | X μL | X μL |
| (3) 0.2 M KCN | 2.5 μL | 2.5 μL | 2.5 μL |
| (4) 5 mM NADH | 2.5 μL | 2.5 μL | 2.5 μL |
| (5) 1 mM Rotenone | 2.5 μL | - | 2.5 μL |
| (6) Read the baseline A340nm for 2 minutes | |||
| (7) Start the reaction by adding 1.6 mM decylubiquinone | 1.5 μL | 1.5 μL | 1.5 μL |
| (8) Read the absorbance at A340nm for 2–3 minutes | |||
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Protein solution: 10 μg /well
Volume: 250 μL
Temperature: 30°C
Absorbance: 340 nm
The pathlength: (cm). To be determined (see SUPPORT PROTOCOL 3)
ε for Decylubiquinone: 6.2 (mM-1 cm-1)
The user should adjust the buffer volume based on the protein concentration of the mitochondria enriched preparation. It is advisable to use a concentrated mitochondria-enriched preparation that will provide 10 μg protein in 250 μL final reaction volume in a microplate-well.
Use ΔA340 for calculating specific Complex-I activity
(i.e., ΔA340 = A340 (−) Rotenone – A340 (+) Rotenone)
Decylubiquinone (an analog of ubiquinone) is sparingly soluble in aqueous buffers. For maximum solubility in aqueous buffers, dilute the ethanolic solution of decylubiquinone with the aqueous buffer of choice. Decylubiquinone has a solubility of approximately 0.3 mg/ml in a 1:3 solution of ethanol:PBS (pH 7.2) using this method. We do not recommend storing the aqueous solution for more than one day.
Do not use refrozen sonicated samples for measuring enzyme activity.
Refrain from isolating mitochondria from frozen tissues or cell pellets as such preparations often produce low complex-I activity (Zheng, Shoffner, Voljavec, & Wallace, 1990)
ALTERNATE PROTOCOL 1
Single Cuvette Complex I (NADH:ubiquinone oxidoreductase) activity measurement
[adapted from Spinazzi et al., (Spinazzi, Casarin, Pertegato, Salviati, & Angelini, 2012) with minor modifications].
This single cuvette assay format provides an alternative format for those experimentalists who either have fewer samples to analyze or do not have microplate reader in their laboratory.
Materials
Micro centrifuge tubes (1.5 ml)
Disposable cuvette (1 ml; BRAND disposable semi-micro plastic cuvette, VWR Cat# 759075D)
UV/Vis Spectrophotometer with thermostat
Microplate reader with thermostat
Parafilm
Mitochondria rupturing hypotonic solution (see Reagents and Solutions)
Potassium phosphate buffer (0.5 M, pH 7.5)
Fatty acid-free BSA (50 mg/ml)
10 mM KCN
10 mM NADH
1 mM Rotenone
10 mM Ubiquinone
Single cuvette assay format
Add 20 – 50 μg of mitochondria-enriched preparation or isolated mitochondria to 700 μL of hypotonic buffer (20 mM HEPES/KOH, pH 7.2) in a 1.5 ml micro centrifuge tube. Lyse mitochondria by three cycles of freeze-thawing in this buffer. Transfer the lysed mitochondrial fraction to a 1- ml cuvette.
To the lysed mitochondrial fraction in the 1-ml cuvette add 100 μL of potassium phosphate buffer (0.5 M, pH 7.5), 60 μL of fatty acid-free BSA (50 mg/mL 30 μL of KCN (10 mM) and 10 μL of NADH (10 mM).
Prepare, in parallel, a separate cuvette containing the same quantity of reagents and mitochondrial sample. To this, add 10 μL of 1 mM rotenone solution.
Cover both cuvettes with parafilm and mix by inverting. Read the baseline absorbance in a spectrophotometer at 340 nm for 2 min.
Start the reaction by adding 6 μL of ubiquinone (10 mM) and mix by inverting the cuvette (after covering the cuvette using parafilm) and follow the decrease in absorbance at 340 nm for 2 min.
Use ΔA340 nm for calculating the rotenone sensitive specific complex-I activity.
SUPPORT PROTOCOL 2: Addressing the turbidity problem in reaction mixture
Sometimes the poor solubility of Coenzyme-Q analogs (i.e., ubiquinone, decylubiquinol and others) and aqueous reaction mixture give rise to a turbidity caused by relatively high concentration of tissue extract, which interferes with the measurement of complex-I activity. This alternative method is based on measuring 2,6-dichloroindopehenol (DCIP) reduction by electrons accepted from decylubiquinol, following oxidation of NADH by complex-I. This approach was developed by Janssen et al., (Janssen et al., 2007) and successfully applied to measuring complex-I activities in mitochondrial fractions obtained from muscle and cultured fibroblasts.
Materials
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Dimethylsulfoxide (DMSO)
10 mM NADH
1mM Rotenone
Reaction mixture for complex-I (see Reagents and Solutions)
Add 2.5 – 10 μL mitochondria-enriched suspension from tissue or 20 μL mitochondria enriched fraction from cells into the cuvette containing the reaction mixture for complex-I
Gently mix the cuvette content by manually inverting few times
Incubate the cuvette content for 3 min in a 37°C temperature-regulated spectrophotometer
Add 20 μL of 10 mM NADH (final concentration of NADH would be 0.2 mM) into cuvette.
Measure the absorbance at 30 second intervals at A600nm for 4 min at 37°C
Add 1 μL rotenone from 1 mM stock solution prepared in DMSO
Measure the absorbance again at 3- second intervals at A600nm for 4 min.
Use the formula as in the basic protocol to calculate the amount of DCPIP. The molar absorptivity of DCPIP at 600 nm is ε =19.1 mM-1cm-1
Decylubiquinone, antimycinA, and rotenone are not water soluble and should be prepared in DMSO. This protocol can be performed in 96-well microtiter plate format as well.We used Cary/Varian 100 Bio UV-Vis spectrophotometer with thermostat
BASIC PROTOCOL 5
COMPLEx I-III (NADH-induced cytochrome-c oxidoreductase) activity measurement
In case of limited tissue or cell amounts, the mitochondrial respiration studies may not be possible. In this case, supercomplex activity measurements would be a useful approach.
Combined complex-I and -III assay can provide a useful information on altered inner mitochondrial membrane function and/or mitochondrial fragility (Trounce, Kim, Jun, & Wallace, 1996). This assay measures the absorbance increase at 550 nm caused by reduced cytochrome c (see Figure 1). Oxidized form of cytochrome c (Cyt c) functions as the electron acceptor and NADH as donor.
Fig.8.
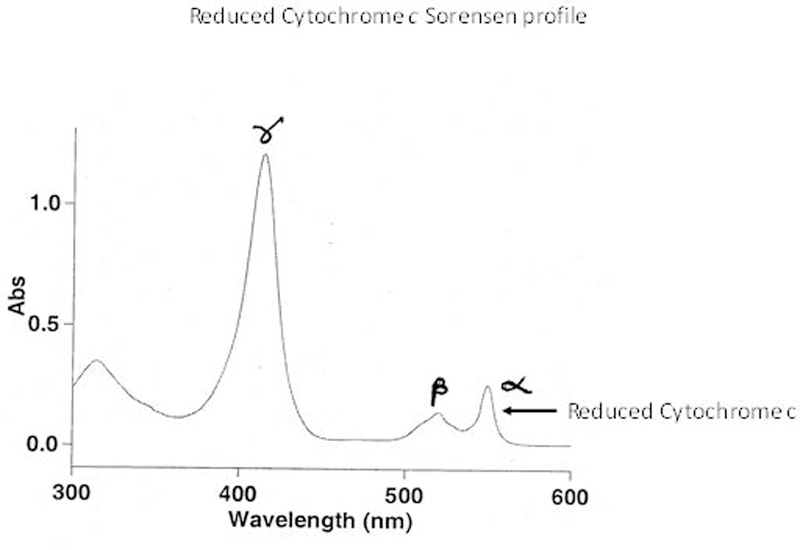
A representative scan for demonstrating the formation of reduced Cytochrome c
Materials
Sonicator with microtip (Branson)
96-well micro titer plate (clear bottom)
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Microplate reader with thermostat
50 mM Potassium phosphate buffer (KP) (see Reagents and Solutions)
0.2 mM KCN
4 mM Cyt c (oxidized)
5 mM NADH
1mM Rotenone
Reaction mixture for complex-I (see Reagents and Solutions)
96-well microtiter plate assay format:
Follow the BASIC PROTOCOL 4 step 1 through 2
Follow the assay flow chart (Table 2) step-1 through 7 for the reagent additions into each well. Discard the leftover samples.
-
Assay parameters:
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Volume: 250 μL
Protein solution: 10 g/well Temperature: 30°C
Pathlength: (cm) To be determined (See SUPPORT PROTOCOL- 4 / Step 7)
ε for Cyt-c (oxidized): 8.4 mM-1 cm-1Manufacturer provided data sheet should have the “ε” value. If not, contact their technical support. We used oxidized Cytochrome c from equine heart (Sigma-Aldrich; Cat# C2506)
First, add 235 μL KP Buffer in all wells labeled as “BLANK and SAMPLES (−) rotenone “and 232.5 μL in wells labeled as “SAMPLES (+) rotenone “. Remove the volume (μL) that will be replaced with protein samples. Small protein sample volumes will adhere to the base of the well. This way, the minute amount of protein solution will be mixed well in the larger volume of KP buffer.
Design the 96-well microtiter plate layout in a fashion that both (+) Rotenone and (−) Rotenone reaction mixtures can be assayed in parallel.
The rotenone sensitive activity due to complex-I generally varies between 30 and 80% of the total NADH-cytochrome-c reductase activity in mitochondria enriched preparations from different tissues or cell lines (Trounce et al., 1996)
12-channel multi pipette is recommended for simultaneously adding NADH to start the reaction
Always run the assay in triplicates
Crosscheck your micro plate light path value from the specification sheet or call the technical service department of the vendor.
The BioTek Synergy-HT plate reader has an auto sampler unit for addition of the reaction starter simultaneously; however, the reservoir of the auto sampler requires a large volume and it is not cost-effective if a few daily kinetic studies are performed. Instead, spot 5 μL of 1 mM NADH on the side wall of a well (see critical parameters in COMMENTARY section). Make sure that the program used for the plate reader includes “plate shaking before measurement”. Plate shaking allows the spotted NADH to mix rapidly with the reagents in the wells thus allowing for near “zero” time measurement to be obtained.
For the preparation of the NADH solution, weigh the required amount of β- NADH. First, add 100 μL 0.01 N NaOH to dissolve completely the β-NADH. Finally, adjust the final volume with distilled water. Prepare this solution fresh daily and discard any remaining solution upon completion of the assay.
Table 2:
Complex I-III assay flow-chart
| REAGENTS | BLANK | SAMPLE (+) Rotenone |
SAMPLE (−) Rotenone) |
|---|---|---|---|
| (1) 50 mM KP Buffer | 235 μL | 232.5 – x μL | 235 –x μL |
| (2)Protein solution | - | X μL | X μL |
| (3)0.2 mM KCN | 2.5 μL | 2.5 μL | 2.5 μL |
| (4)4 mM Cyt-c (oxidized) | 5 μL | 5 μL | 5 μL |
| (5)1.0 mM Rotenone | 2.5 μL | - | 2.5 μL |
| (6) START the reaction by adding 5 mM NADH | 5 μL | 5 μL | 5 μL |
| (7) Measure the absorbance at A550nm for 10 min | |||
ALTERNATIVE PROTOCOL 2
Single Cuvette COMPLEx I-III (NADH-induced cytochrome-c oxidoreductase) activity measurement
This single cuvette assay format provides an alternative format for those experimentalists who either have fewer samples to analyze or do not have microplate reader in their laboratory.
Materials
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Hypotonic buffer (see Reagents and Solutions)
KP Buffer (see Reagents and Solutions)
Fatty acid-free BSA
10 mM KCN
10 mM NADH
1mM Rotenone
1 mM Cyto c (oxidized)
Single cuvette assay format (adapted from Spinazzi et al., (Spinazzi et al., 2012) with minor modifications)
Incubate the mitochondria-enriched preparation ([protein] = 1–6 μg) for 2 min in in 700 μL of hypotonic buffer (20 mM HEPES/KOH, pH 7.2) in a 1-ml cuvette to induce an osmotic shock
Add 100 μL of potassium phosphate buffer (0.5 M, pH 7.5), 20 μL of fatty acid-free BSA (50 mg/ml), 30 μL of 10 mM KCN and 50 μL of 1.0 mM oxidized cytochrome c. Adjust the volume to 980 μL with distilled water.
Prepare, in parallel, a separate cuvette containing the same quantity of reagents and sample plus 10 μL of 1 mM rotenone solution.
Mix by inverting the cuvette and monitor the baseline absorbance at A550 nm for 2 min.
Start the reaction by adding 20 μL of 10 mM NADH, mix by inversion using parafilm, and then follow the increase of absorbance at A550 nm for 2 min.
Specific Complex I+III activity can be determined by ΔA550nm, which reflects the rotenone-sensitive activity.
BASIC PROTOCOL 6
COMPLEx-II (Succinate Dehydrogenase; Succinate-Ubiquinone oxidoreductase) activity measurement
Complex II consist of only four peptides and it is the simplest of all the complexes of the electron transfer chain. The nature of the association of complex II with the mitochondrial matrix membrane makes complex-II a useful mitochondrial marker enzyme (Scheffler, 2008a). The citrate synthase partially leaks out during the isolation of mitochondria from the frozen tissues (Janssen et al., 2007); therefore, citrate synthase may not be a useful mitochondrial marker enzyme. The following assay measures the reduction of DCPIP when coupled to complex-II catalyzed reduction of decylubiquinone (DBH2).
Materials
Sonicator with microtip (Branson)
Microplate reader with thermostat
96-well micro titer plate (clear bottom)
Reagent mixture-I and -II (see Reagents and Solutions)
1M Malonate
2.5 mM DBH2
96-well microtiter plate assay format
Follow the BASIC PROTOCOL 4 step 1 through 2
Follow the assay flow chart (Table 3) step 1 through 8 for the reagent additions into each well. Discard the leftover samples.
Assay parameters:
Table 3:
Complex II assay flow-chart
| REAGENTS | BLANK | SAMPLE (−) Malonate |
SAMPLE (+) Malonate |
|---|---|---|---|
| (1) Water | 85 μL | 95 – x μL | 85-x μL |
| (2) Protein solution | - | X μL | X μL |
| (3) Reagent Mixture-I | 100 μL | 100 μL | 100 μL |
| (4) Incubate the plate at 300C for 10 min with a gentle agitation | |||
| (5) Reagent Mixture-II with a brief mixing | 50 μL | 50 μL | 50 μL |
| (6) Add 1M Malonate | 10 μL | - | 10 μL |
| (7) Start reaction by adding 2.5 mM DB(8) Measure the absorbance at A600nm for 3–5 min | 5 μL | 5 μL | 5 μL |
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Assay volume: 250 μL / well
Protein solution: 10 μg /well
Temperature: 30°C
Light path length: , (cm) To be determined (See SUPPORT PROTOCOL-3 )
ε for DCPIP: 19.1 mM-1 cm-1
Use 12-channel multichannel pipette to minimize the pipetting intervals between rows.
DCPIP color should be blue and scanning graph should show maximal peak at 600 nm (Fig. 1). If the color changes to purple, properly discard DCPIP and prepare a fresh solution.
Fig.9.
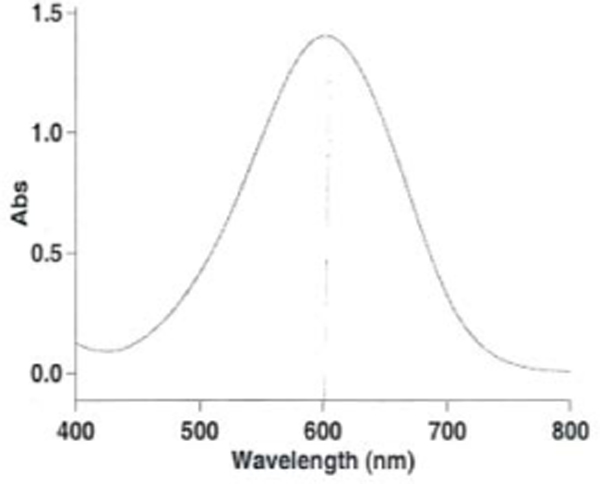
DCPIP scanning graph. The maximal peak at 600 nm indicates chemically stable DCPIP.
ALTERNATIVE PROTOCOL 3
Single Cuvette COMPLEx-II (Succinate Dehydrogenase; Succinate-Ubiquinone oxidoreductase) activity measurement
This single cuvette assay format provides an alternative format for those experimentalists who either have fewer samples to analyze or do not have microplate reader in their laboratory.
Materials
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Parafilm
Hypotonic buffer (se Reagents and Solutions)
KP Buffer (seeReagents and Solutions)
Fatty acid-free BSA
400 mM succinate
12.5 mM Decylubiquinone (DBH2)
50 mM 2-thenoyltrifluoroacetone (TTFA)
Single cuvette assay format (adapted from Spinazzi et al. (Spinazzi et al., 2012) with minor modifications)
Add 20 – 50 μg of mitochondria enriched protein sample to 700 μL of hypotonic buffer (20 mM HEPES/KOH, pH 7.2) in a 1.5 ml micro centrifuge tube. Lyse mitochondria by three cycles of freeze-thawing in this buffer. Transfer the lysed mitochondrial fraction to a 1-ml cuvette.
To the lysed mitochondria in the 1-ml cuvette, add 50 μL of potassium phosphate buffer (0.5 M, pH 7.5), 20 μL of fatty acid-free BSA (50 mg /ml), 30 μL of 10 mM KCN, 50 μL of 400 mM succinate and 145 μL of 0.015 % DCPIP (wt/vol). Adjust the volume to 996 μL with distilled water.
Cover the cuvette with parafilm and mix by inverting the cuvette and incubate inside the spectrophotometer at 37°C for 10 min.
Read the baseline activity at 600 nm for the last 2 min.
Start the reaction by adding 4 μL of 12.5 mM DBH2, mix by inversion and follow the decrease in absorbance at 600 nm for 3 min.
Check the specificity of complex II activity by running the assay after the addition of 10 μL of either 1 M malonate or 50 mM TTFA before starting the reaction. The degree of inhibition usually exceeds 85% with TTFA and 95% with malonate, indicating a high specificity of the reaction.
BASIC PROTOCOL 7
COMPLEx II-III (Succinate-Cytochrome-c Oxidoreductase) activity measurement
The succinate-induced cytochrome- c reduction by complex-III is the basis of this reaction. The assay involves oxidized cytochrome-c as the electron acceptor and succinate as donor. In this reaction setting, the fully activated complex-II is critical; therefore, the user must pre-incubate the mitochondria-enriched preparation with the succinate (Barrientos, Fontanesi, & Diaz, 2009; Spinazzi, Casarin, Pertegato, Salviati, & Angelini, 2012) as indicated below in the experimental layout.
96-well microtiter plate assay format
Materials
Sonicator with microtip (Branson)
Microplate reader with thermostat
96-well micro titer plate (clear bottom)
50 μM KCN
0.5 M Succinate
1 M Malonate
mM Cytochrome c (oxidized)
Follow the BASIC PROTOCOL 4 step-1 through 2
Follow the assay flow chart (Table 4) step 1 through 8 for the reagent additions into each well. Discard the leftover samples.
Assay parameters
Table 4:
Complex II-III assay flow-chart
| REAGENTS | BLANK | SAMPLE (−) Malonate |
SAMPLE (+) Malonate |
|---|---|---|---|
| (1) 0.1 M KP buffer, pH 7.4 | 217 μL | 227-x μL | 217-x μL |
| (2) Protein solution | - | X μL | X μL |
| (3) 50 μM KCN | 10 μL | 10 μL | 10 μL |
| (4) 0.5M Succinate | 10 μL | 10 μL | 10 μL |
| (5) Mix and pre-incubate the plate at 30°C for 20 min | |||
| (6) 1M Malonate | 10 μL | - | 10 μL |
| (7) Start reaction by adding 2.5 mM cytochrome-c (oxidized) | 3μL | 3μL | 3μL |
| (8) Measure the absorbance at A550nm for 3–5 minutes | |||
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Volume: 250 μL
Protein solution: 10 μg/well
Temperature: 30°C
Light pathlength: , (cm) To be determined (see SUPPORT PROTOCOL 3)
ε for Cyt c (oxidase): 8.4 mM-1 cm-1
It is a necessary step to preincubate the micro well content with succinate for 20 min at 37°C is to fully activate the Succinate –Cyt c oxidoreductase enzyme.
The volume of the protein solution range would be 1–3 μL that gives 10 μg protein/well.
It is difficult to subtract 1–3 μL volume from buffer solution to meet the final volume of 250 μL. The best way is to place the same volume of buffer in all wells; then remove the volume, which will replace with protein solution. (Let’s say you need to add 1.3 μL protein solution. Remove 1.3-μL buffer from the well, add 1.3 μL of protein solution, and wash your pipet tip 2–3 times with the buffer.)
User may also use 1 mg/mL AntimycinA instead of 1M Malonate
ALTERNATIVE PROTOCOL 4
Single cuvette assay format COMPLEx II-III (Succinate-Cytochrome-c Oxidoreductase) activity measurement
(adapted from Spinazzi et al. (Spinazzi et al., 2012) with minor modifications)
This single cuvette assay format provides an alternative format for those experimentalists who either have fewer samples to analyze or do not have microplate reader in their laboratory.
Materials
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Parafilm
Hypotonic buffer (see Reagents and Solutions)
10 mM KCN
400 mM succinate
KP Buffer (see Reagents and Solutions)
1 mM Cyto c (oxidized)
1 M Malonate
mg/ml AntimycinA
To a 1-ml cuvette, add 800 μL of distilled water, 40 μL of potassium phosphate buffer (0.5 M, pH 7.5), 30 μL of KCN (10mM), 25 μL of succinate (400 mM) and 1–2 μg of mitochondria-enriched suspension. Adjust the volume to 950 μL with distilled water
Mix by inverting the cuvette and incubate for 10 min inside the spectrophotometer at 37⁰C.
Start the reaction by adding 50 μL of oxidized cytochrome c (1 mM), mix by inverting the cuvette, and then follow the increase of absorbance at A550 nm for 3 min.
Check the specificity of this assay by adding 10 μL of 1 M malonate or 1 mg /ml AntimycinA in a separate cuvette prepared as described in steps above.
BASIC PROTOCOL 8
COMPLEx-III (Ubiquinol: Ferricytochrome-c Oxidoreductase) activity measurement
This assay measures the reduction of cytochrome-c (oxidized) catalyzed by complex-III. Complex-III is ubiquinone-cytochrome-c oxidoreductase and is often named the bc1 complex after the two cytochromes found within it; one also finds the simpler name cytochrome c reductase in the literature (Scheffler, 2008b). The assay involves oxidized cytochrome-c as the electron acceptor and decylubiquinone (DBH2) as donor.
96-well microtiter plate assay format
Materials
Sonicator with microtip (Branson)
96-well micro titer plate (clear bottom)
Microplate reader with thermostat
50 mM KCN
1.25 mM Cytochrome c (oxidized)
1 mM AntimycinA
5mM DBH2
Reaction Buffer for Complex-III (see Reagents and Solutions)
Follow the BASIC PROTOCOL 4 step 1 through 2
Follow the assay flow chart (Table 5) step 1 through 8 for the reagent additions into each well. Discard the leftover samples.
Assay Parameters
Table 5:
Complex III assay flow-chart
| REAGENTS | BLANK | SAMPLE (−) AntimycinA |
SAMPLE (+) AntimycinA |
|---|---|---|---|
| (1) Reaction Buffer for Complex-III | 225 μL | 227.5 – x μL | 225 – x μL |
| (2) 50 mM KCN | 10 μL | 10 μL | 10 μL |
| (3) 1.25 mM Cyt c (oxd.) | 10 μL | 10 μL | 10 μL |
| (4) 1 mM AntimycinA | 2.5 μL | - | 2.5 μL |
| (5) Protein solution | - | X μL | X μL |
| (6) Incubate the plate with a gentle mixing for 10 at 30°C | |||
| (7) Start the reaction by adding 5 mM DBH2 | 2.5 μL | 2.5 μL | 2.5 μL |
| (8) Measure the absorbance at A550nm for 2–3 minutes | |||
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Volume: 250 μL
Protein solution: 10 μg/well
Temperature: 30°C
Light pathlength: ,(cm) To be determined (See SUPPORT PROTOCOL-4 / Step 7)
ε for Cyt c (reduced): 29.5 mM-1 cm-1
BASIC PROTOCOL 9
COMPLEx-IV (Cytochrome-c oxidase) activity measurement
Complex IV or cytochrome-c oxidase activity is a simple assay and can be easily measured by following the oxidation of reduced cytochrome c at A500 nm. The specificity of complex IV activity is measured by the percent inhibition following addition of potassium cyanide (Spinazzi et al., 2011)
96-well micro titrate plate assay format
Materials
Sonicator with microtip (Branson)
96-well micro titer plate (clear bottom)
KP Buffer (see Reagents and Solutions)
50 mM KCN
mM Cyt c (reduced) (see Reagents and Solutions)
Follow the BASIC PROTOCOL 4 step-1 through 2
Follow the assay flow chart (Table 6) step 1 through 4 for the reagent additions into each well. Discard the leftover samples.
Assay Parameters:
Table-6:
Complex IV assay flow-chart
| REAGENTS | BLANK | SAMPLE (−) KCN |
SAMPLE (+) KCN |
|---|---|---|---|
| (1)O.1 M KP Buffer (pH 7.4) | 230 μL | 240 μL– | 230 μL –xμL |
| (2)Protein solution | - | X μL | X μL |
| (3)50 mM KCN | 10 μL | - | 10 μL |
| (3) Start the reaction by adding 0.5 mM Cyt-c (reduced) | 10 μL | 10 μL | 10 μL |
| (4) Measure the absorbance at A550nmfor 10 min | |||
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Volume: 250 μL / well
Protein solution: 10 μg/ well
Temperature: 30°C
Light pathlength: ,(cm) To be determined (See SUPPORT PROTOCOLE-4 / Step 7)
ε for Cyt-c (reduced) : 29.5 mM-1 cm-1
[Cyt c (reduced)] = 20 μM /well. The dispensing volumes are small and pipetting fractional volumes are not practical; therefore, it is a better practice to adjust the concentration rather than pipet volume to provide desired concentration per reaction well. In case of observing low enzyme activity, include 0.025% laurylmaltoside in reaction media. This detergent approximately doubles the specific activity of cytochrome-c oxidase (Birch-Machin & Turnbull, 2001). An excessive mechanical disruption during the sample preparation may inactivate the complex IV enzyme (Zheng, Shoffner, Voljavec, & Wallace, 1990).
ALTERNATIVE PROTOCOL 5
Single Cuvette COMPLEx-IV (Cytochrome-c oxidase) activity measurement
(adapted from Spinazzi et al., (Spinazzi et al., 2012) with minor modifications)
Materials
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Parafilm
10 mM KCN
KP Buffer (see Reagents and Solutions)
1 mM Cytochrome c (reduced) (see Reagents and Solutions)
To a 1-ml cuvette, add 400 μL of distilled water, 50 μL of potassium phosphate buffer (100 mM, pH 7), reduced Cytochrome c (1 mM) and read the baseline activity at 550 nm for the last 2 min. (see REAGENTS section how to prepare reduced Cytochrome c solution)
Adjust the volume to 995 μL with distilled water.
Start the reaction by adding 1–2 μg of mitochondria-enriched preparation (in a final volume of 5 μL) mix by inverting and monitor the decrease in absorbance at 550 nm. This takes approximately 3 min.
To check the specific activity of complex IV, add 30 μL of 10 mM of KCN in a separate reaction prepared as described above.
SUPPORT PROTOCOL 3
CITRATE SYNTHASE ACTIVITY MEASUREMENT AND PATHLENGTH DETERMINATION
Citrate synthase (CS), a Kreb’s cycle enzyme, is commonly used mitochondrial matrix marker enzyme (Barrientos et al., 2009; Trounce et al., 1996). CS activity is interpreted as a reference mitochondrial enzyme with relation to the total sample protein and to the activity of mitochondrial ETC enzymes.
The purpose of including CS activity measurement is to normalize other ETC enzyme activities. The CS activity measurement will also determine whether the sample aliquot may have deteriorated, resulting in nonspecific lowering of the activities of ETC enzymes (Kerr, Grahame, & Nakouzi, 2012). Normalization of ETC enzymes based on citrate synthase activity is best when fresh mitochondria preparation is used. The detection of citrate synthase activity in frozen tissue or cell pack preparation may not be useful due to leakage of citrate synthase through compromised mitochondria membrane.
Materials
Sonicator with microtip (Branson)
96-well micro titer plate (clear bottom)
Microplate reader with thermostat
10 mM oxaloacetic acid
Reaction mixture for CS (see Reagents and Solutions)
96-well micro titrate plate assay format
Follow the BASIC PROTOCOL 4 step 1 through 2
Follow the assay flow chart (Table 7) step 1 through 5 for the reagent additions into each well. Discard the leftover samples.
Assay Parameters:
Table 7.
Citrate synthase assay flow-chart
| REAGENTS | BLANK | SAMPLE |
|---|---|---|
| (1) Water | 197.5 μL | 197.5 μL – x μL |
| (2) Reaction mixture for citrate synthase reaction | 40 μL | 40 μL |
| (3) Protein solution Incubate the plate for 5 min at 30°C | - | X μL |
| (4) Start the reaction by adding 10 mM oxaloacetic acid | 12.5 μL | 12.5 μL |
| (5) Measure the absorbance at A412nm for 3–4 min. | ||
x (Volume (μL) of mitochondria suspension): Mitochondria-enriched preparation (indicated as SAMPLE in assay layout)
Volume: 250 μL
Protein solution: 10μg/well
Temperature: 30°C
Light pathlength: ,(cm) To be determined (See SUPPORT PROTOCOLE-4 / Step 7)
ε for DTNB: 13.6 mM-1cm-1
Freshly prepare this reaction mixture for each assay (1200 μL; good for 30 wells)
Normalize ETC enzyme activities by ETC enzyme activity rate / citrate synthase activity rate.
The ratios may be used to compare enzymatic activities in different samples that will offset the variation born from sample preparation and pipetting.
How to measure the Pathlength [l ,cm] of your sample in a 96-well microtiter plate?
When you use a 1 cm cuvette, the Pathlength is not affected by the volume of your sample as the measurement is from the side. In the case of a 96-well microtiter plate, the measurement is from the top and the pathlength changes with changes in the volume. The following protocol describes two ways to calculate the pathlength (Shah, 2014):
Mathematical way:
Pathlength = 4 * volume / 3.14 * d2 , where “d” is diameter of a single well
- Experimental way:
- Pathlength = (A977nm for water) – (A900nm for water) /0.18
- Add 250 μL of water to five wells
- Wait for 20 minutes at ambient temperature or gently shake for 2–3 minutes
- Read the absorbance at 977nm and 900 nm
- Average the readings and place them in the above formula
- Divide the result by 0.18
- The final number is the light path length () in centimeter
The value 0.18 corresponds to (A977nm –A900nm) of water in a 1-cm cuvette and is accepted as default value. If you use a BIO TEK microplate reader, BIO TEK/Synergy
HT/Gen5 2.0 software has pre-loaded information about micro-well plates from different brand names based on the buffer and kind of 96-well microtiter plate used in the assay.
ALTERNATIVE PROTOCOL 6
Single cuvette citrate synthase activity measurement
(adapted from Spinazzi et al., (Spinazzi et al., 2012) with minor modifications)
Materials
1-mL disposable cuvette (10-mm acryl-cuvettes from Sarstedt)
UV/Vis spectrophotometer with thermostat
Parafilm
200 mM Tris Buffer (see Reagents and Solutions)
Triton x-100
0.1 mM DTNB
10 mM Acetyl coenzyme A (acetyl-CoA)
M Oxaloacetic acid
To a 1-ml cuvette, add 300 μL of distilled water, 500 μL of Tris (200 mM, pH 8.0) with triton x-100 (0.2%[vol/vol]), 100 μL of 5,5’-dithiobis (2-nitrobenzoic acid) [DTNB], 30 μL of acetyl-CoA (10 mM) and the sample (1–2 μg of mitochondria); Adjust the volume to 950 μL with distilled water
Mix by inverting the cuvette and read the baseline activity at 412 nm for 2 min.
Start the reaction by adding 50 μL of 10 mM oxaloacetic acid, mix by inverting, and then monitor the increase in absorbance at 412 nm for 3 min.
Reagents and Solutions
Use 18 mohm (mΩ) megapure water for the reagents and solution preparation
4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) (0.1 M): Dissolve 1.18 g 4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride in 35 ml water and adjust the volume to 50 ml with water. Aliquot into small volumes and store at −20°C for 1–2 years.
AntimycinA (10 mg/ml): Dissolve the entire vial content (25.0 mg) in 2.5 ml of 95% ethanol, ether, DMSO, and chloroform. Store 4 months at 4°C, 6 months at −20°C, and longer (up to a year) at 80°C. Dilute the stock solution to 1 mg/ml by adding 10 μL of stock to 90 μL of ethanol on the day of assay.
-
Acetyl-CoA (50 mg/ml): Dissolve the entire vial content (5.0 mg) in 200 μL of water, (It is more practical to dissolve the bottle content). Aliquot in 10–20 μL and store at −80°C for several months.
Solubility: 100 mg/ml water. Add an additional DMSO in case of complete dissolving is not achieved
Benzamide (0.2M): Dissolve 0.24 g benzamide in 5 ml of 100% ethanol and adjust the volume to 10 ml with ethanol. Store at −20°C for several years.
Benzamidine.HCL (0.2 M): Dissolve 0.31 g benzamidine.HCl in 5 ml of 100% ethanol and adjust the final volume to 10 ml with ethanol. Store at −20°C for several years.
Brain Tissue Homogenization Buffer: 0.32 M sucrose, 0.5 mM MgSO4, 0.1 mM EGTA, 10 mM HEPES. Adjust pH to 7.4. Just before use, add benzamide, benzamidine hydrochloride (HCl), and 4-(2- aminoethyl)benzenesulfonyl fluoride-HCl (AEBSF) to the buffer to reach a final concentration of 1.1mM for each. Also, add a commercial protease inhibitor cocktail that contains pepstatin, bestatin, leupeptin, and aprotinin, as per the manufacturer’s instructions. Prepare fresh solution, keep at 4°C and discard the leftover.
BSA (fat-free) (50 mg/ml). Dissolve 0.25 g of defatted BSA in 5 ml of distilled water and store it in 1-ml aliquots at 4⁰C for up to 1 month or until signs of microbial contamination are seen.
-
Cytochrome-c (oxidized) (2 mM): Dissolve the entire vial (50mg) in 2.0 ml of water. Prepare 25–50 μL aliquots and store at −20oC for 6months-1 year
Cytochrome-c is sold in “oxidized” form and soluble in water (pH 7±1)
The reagent is light sensitive; therefore, use dark-color vial/tube [Argos/ Cat# T7100BK (1.5 ml); Cat# T7456–001(0.5 ml)].
-
DCPIP [2,6-Dichlorophenol indophenol-sodium salt hydrate] (10 mM): Dissolve 3.0 mg DCPIP.H2O in 1 ml of water. The color of this reagent must be blue. Store at 4oC for several weeks.
Solubility: 30 mg/ml water
If the color turns from blue to purple and aggregate forms due to storage, prepare a fresh solution.
-
DTNB [5,5’-Dithiobis(2-nitrobenzoic acid)](50 mM):Dissolve 4.0 mg DTNB in 1.0 ml of ethanol. Store at 4oC for 2 months
Solubility: 8 mg/ml ethanol
Alternatively, 5 mM DTNB can be made in about a 100 mM sodium or potassium phosphate buffer solution, pH 7.2 containing 0.10 mM EDTA. This solution is stable for more than two months if stored in the dark at 4°C up to 6 month.
Decylubiquinone (DBH2) stock solution (10mM). Add a few grains of potassium borohydride to 250 μL of 10 mM DBH2 in 100% ethanol. Add 5-μL aliquots of 0.1 M HCl until the solution becomes colorless. Briefly spin the solution at 10,000 x g for 1 min and transfer the solution into a new 500-μL tube, avoiding any potassium borohydride crystals. Adjust the pH of the solution between 2 and 3 with 5-μL aliquots of 1 M HCl and keep the solution on ice protected from light. Store at −20°C for 3 months
EDTA (0.5 M): Dissolve 18.6 g EDTA disodium salt dehydrate in about 75 ml of water. Adjust the pH to 7.5 with sodium hydroxide and adjust the final volume to 100 ml with water. Store at 4°C for at least 1 year.
EGTA (0.1 M): Dissolve 0.47 g EGTA in 10 ml of water. Aliquot into small volumes and store at −20°C for 1–2 years.
Ficoll Density Gradient Solution: Dissolve 14.0 g Ficoll in 100 mL of Resuspension Buffer overnight at room temperature. Then, add all protease inhibitors. Prepare sufficient volumes of an 8% w/v and a 14% w/v Ficoll solution to have 10 mL of each solution for each gradient centrifuge tube. Store at −20°C for several years.
Glutamate stock solution (0.2 M): Weigh 59 mg L-Glutamic acid. Add three drops of 5 N NaOH. Mix well. Adjust the volume to 2ml with 50 mM potassium phosphate buffer (pH 7.4). Aliquot to 25–50 μL and store at −20oC for 1 year.
HEPES (0.1 M): Dissolve 2.38 g HEPES in about 75 ml of water. Adjust pH 7.2 with 1M KOH. Adjust the final volume to 100 ml with water. Store at 4oC for 1 year.
17.Hypotonic Buffer (HB): 90 mM KCL + 20 mM MOPS-KOH, pH 7.2. Store at 4°C for 1–2 years.
KCl (1 M): Dissolve 7.46 g KCl in 100 ml water. Store at room temperature for indefinitely.
KCN stock solution (10 mM). Dissolve 6.50 mg of KCN in 10 ml of distilled water under a fume hood. Store at 4°C for 1–2 years.
Malate [DL-maleic acid di-sodium salt](0.2 M): Dissolve 0.36 g malate in 10 ml of nanopure water. Aliquot in small volumes and store at 4°C for 1 year.
Malonate (malonic acid) stock solution (1M): Dissolve 10.4 g malonic acid in 1N NaOH and adjust the final volume to 100 mL with distilled water. If a cloudy appearance observed during the volume adjustment, add little more 1N NaOH. The final reagent should look clear to slightly hazy. Store at −20°C for 1 year.
-
Mannitol (1 M): Dissolve 19.2 g Mannitol in 100 ml of water. Store at −20oC for 1–2 years
Solubility of Mannitol: 182 g/liter at room temperature.
MOPS/KOH Buffer (pH 7.2)(0.4 M) : Dissolve 4.19 g MOPS in approximately 40 ml of nanopure water. Adjust pH to 7.2 with 1 M potassium hydroxide. Adjust the final volume to 50 ml with water. Store at 4°C for 1 year. Check pH before use.
Mitochondria rupturing hypotonic solution: 25mM K2PO4, pH 7.2; 5 mM MgCl2 . Store at 4°C for 1 year. Visually check before use. If a turbidity observed, discard it and prepare fresh batch.
Mitochondrial Storage Buffer (MSB): Prepare a solution containing 230 mM Mannitol, 70 mM sucrose, 1 mM EGTA, 10 mM HEPES; pH 7.2, and add freshly, before use, a protease inhibitor cocktail following the instructions of the manufacturer of the cocktail. Store at 4°C for 6 months.
NADH stock solution (10mg/ml): Dissolve 10.0 mg NADH (anhydrous) in 1 ml of Na-bicarbonate buffer, pH 9.0. The molar based concentration is 14.0 mM Store at 2–8°C for 1 month.
-
NaOH (5 N): Dissolve 20.0 g sodium hydroxide in a 60–70 ml of nanopure water. Completely dissolve the pellets. Apply heat if necessary. Adjust the volume to 100 ml with water. Store at 4°C indefinitely.
Sodium hydroxide is a corrosive chemical. Wear your personal protective equipment (i.e., lab coat, gloves, and goggle).
-
Oligomycin (10mg/ml): Dissolve the entire vial (50 mg) in 0.5 ml of ethanol. Aliquot in 20 μL and store at −70oC.
Stability of oligomycin in different temperature settings: 2–3 weeks at 4oC. Several months at −20oC. Longer storage period at −70oC
Oligomycin can be dissolved in ethanol, ether, DMSO, or acetone. The solubility is 250 mg/mL
OAA (Oxaloacetic acid) stock solution (100mM): Weigh 6.60 mg OAA and dissolve in 500 μL of water. Keep on ice and prepare daily since oxaloacetic acid undergoes decarboxcylation in solution to form pyruvate and is therefore unstable.
Phenazine metasulfate [N-methylphenazonium methyl sulfate] (0.1 M): Dissolve 31 mg phenazine metasulfate in 1 ml of nanopure water at room temperature. Store at −20°C for 3–4 months protected from light.
Protease inhibitor cocktails : Protease Inhibitor Cocktail Set III; Calbiochem( Cat# 539134). Following initial thaw, aliquot and store at-20°C for 2–3 years.
Potassium phosphate Buffer (KP) (0.5 M, pH 7.5): Follow the preparation protocol in “Short Protocols in Molecular Biology”, Fourth Ed. Page A1–30. Store at 4°C for 1 year.
Reaction Buffer for complex-III: 0.25 M sucrose; 1 mM EDTA; 50 mM Tris-HCl, pH 7.4 in 10 ml. Store on ice until use. Prepare daily.
Reaction mixture for citrate synthase (CS) reaction: 1M Tris-HCl, pH 7.8 (750 μL); 6 mM Acetyl-coA (375 μL); 10 mM DTNB (75 μL). Store on ice. Prepare daily.
Reaction mixture for complex-I assay: Prepare a reaction mixture in a 1-mL spectrophotometer disposable cuvette with final concentrations indicated (2.5 mM KP, pH 7.4; 3.5 g/L BSA; 60 μM DCPIP(2,6-dichlorophenol indophenol); 70 μM decylubiquinone; 1 μM AntimycinA) in 960 μL. Store on ice. Prepare daily.
-
Reagent Mixture-I for complex-II assay
Combine the following reagents in a 15 ml conical bottom tube
-
■
0.3 M KCl; 25 mM Malate; 25 mM KCL; 7.5 mM KP Buffer (pH 7.4); 25 mM HEPES;
-
■
1.25 mM Phenazinemethosulfate; 2.5 μg/ml oligomycin; 0.5 mM ADP; 0.125 mM DCPIP
-
■
50 mM Succinate
-
■
Prepare Reagent Mixture-I fresh daily in each assay.
-
■
10 mL of the reagent mixture-I may be sufficient for one 96-well microtiter plate; however, the user may reduce the volume based on the experimental design.
Store on ice. Prepare daily.
-
■
-
Reagent Mixture-II for complex-II assay
Combine 5 μg/ml AntimycinA; 5 μg/ml Rotenone; 5 mM KCN; 125 μM DCPIP in 15 ml conical bottom tube. Store on ice. Prepare daily.
- Reduced Cyt c
- Place 200 μL of 2 mM Cyt c(oxidized) in a 1.5 mL dark color microfuge tube. Calculate the molarity based on the data provided by the vendor.
- Add few crystals of sodium borohydride (NaBH4) to reduce Cyt c(oxidized)
- Incubate the mixture at 51oC for 1 hour
- Add 4 drops of 0.1 N HCl to remove the excess NaBH4
- Add 4 drops of 0.1 N NaOH to neutralize the mixture; mix well
- Measure the final volume by an adjustable pipet and recalculate the final concentration
- Add 10 μL of the mixture in a 1 mL of quartz cuvette and scan for the absorbance between 350 nm – 600 nm wavelength
- A typical scan is shown in Figure 1
- Divide Cyt c (reduced) preparation into 10–20 μL aliquots into 0.5 mL dark color microfuge tubes and store aliquots at −20oC for 1 year.
Resuspension Buffer: 0.32 M sucrose, 10 μM EGTA. Adjust pH to 7.5 with 1M Tris base. Just before use, add benzamide, benzamidine-HCl, and AEBSF to the buffer to reach a final concentration of 10 μM. Also, add the protease inhibitor cocktail Set III per instructions of the manufacturer. Store on ice. Prepare daily.
-
Rotenone stock solution (20 mM): Dissolve 26.8 mg Rotenone in 3.4 mL of acetone. Protect from light and store at 4°C for 4–6 months.
This reagent is extremely toxic (utilized as an insecticide). Solubility: 20 mg/ml. Although it is soluble in alcohol, acetone, and chloroform, we have found that acetone is the preferred solvent. Solution becomes slightly hazy at room temperature.
- Reduced cytochrome c solution (Spinazzi et al., 2012)
-
■Dissolve 110 mg of ascorbic acid in 1 mL of 10 mM potassium phosphate buffer (pH 7.0), and then add a few grains of Tris powder to obtain a pH of 6.5–6.8.
-
■Dissolve 250 mg of cytochrome c in 1.2 mL of 10 mM potassium phosphate buffer (pH 7.0), and then add 0.3 mL of ascorbic acid solution
-
■Incubate with agitation for 1 h at 4⁰C. The reduction of cytochrome c will result in a color shift from brown to pink-orange.
-
■Purify from excessive ascorbic acid in solution using a PD10 disposable desalting column previously equilibrated with 50 ml 1o 10 mM potassium phosphate buffer (pH 7.0). Discard the first three drops, and then collect the eluted solution of reduced cytochrome c in several 1.5 mL micro tubes. Store the vials on ice.
-
■To check the efficiency of cytochrome c reduction and to assess the absence of residual ascorbic acid in the solution, read for 2 min the absorbance of a 50 μM cytochrome c solution in a 1-mL cuvette at 550 nm. The decrease in absorbance should not exceed 0.005 per min.
-
■Dissolve a few crystals of potassium ferricyanide in 10 ml of water under a chemical hood. Add to the cuvette 1 μL of a diluted potassium ferricyanide solution. This will result in cytochrome c oxidation. Measure the absorbance at 550 nm for 3 min.
-
■Add a few crystals of sodium dithionite and read the absorbance again. The initial absorbance values before the addition of dithionite should be about 95% of the values after dithionate addition.
-
■Discard any vials showing signs of auto reduction and pool together the selected vials.
-
■Aliquots of this solution can be stored under the liquid nitrogen for several years
-
■
-
37
Sodium phosphate (dibasic)(100 mM)/ EDTA (5mM)buffer (pH 7.5): Dissolve 1.42 g sodium phosphate (dibasic) in approximately 50 mL of nanopure water. Add 1ml of 0.5 M EDTA. Adjust pH to 7.5 with 1N NaOH. Adjust the final volume to 100 mL Store at 4°C for 1–2 years. Check pH before use.
-
38
Succinate (0.5 M): Dissolve 0.81 g succinic acid (disodium salt) in 5 mL of water. Adjust pH to 7.4 with 3 M KOH. Adjust the volume to 10 mL with water. Store in 0.25 mL aliquots at 4oC for 4–6 months.
User can adjust the stock volume based on needs. Solubility of Succinic acid: 100 mg/ dL water at room temperature.
-
39
Sucrose (1 M): Dissolve 34.2 g sucrose in 100 mL of water. Aliquot in small volumes and store at −20°C for 1 year.
-
40
Sucrose Density Gradient Buffer: Prepare buffer containing 120 mM KCl, 20 mM Tris/HCl, 5 mM dihydrogen potassium phosphate, 2 mM EGTA, pH 7.4, and add sucrose to prepare solutions of 10%, 28.5%, and 34% w/v to be used in the discontinuous sucrose density gradients. Store at room temperature and prepare daily.
-
41
Synaptosome Lysis Buffer: 3 mM Tris-HCl, pH 8.5. Add benzamide, benzamidine-HCl, AEBSF, and protease inhibitor cocktail at the same concentrations as in the Resuspension Buffer. Store on ice and prepare daily.
-
42
Tris-HCL Buffer, pH 8.0 (1M): Dissolve 121 g Tris base in 800 mL distilled water. Adjust pH 8.0 by adding 58.4 mL 0.1M HCL at 25°C. Adjust final volume to 1L with distilled water. Filter the solution through a 0.45-μm filter and store at 4°C up to 1 year. Check pH before use.
-
40.
TTFA [2-Thenoyltrifluoroacetone] (50 mM): Dissolve 11.1 mg 2-Thenoyltrifluoroacetone in 1 mL of 95% ethanol. Although storing at 4°C for several weeks is reported, daily preparation is recommended. Alternatively, TTFA can be dissolved in DMSO.
-
41.
Tween 20 solution (2.5% vol/vol). Dissolve 200 μL of Tween-20 in 7.8 mL of distilled water. Store at room temperature until use. Daily preparation is recommended.
-
42.
42.Ubiquinone solution (10 mM). Dissolve 2.0 mg of ubiquinone in 0.8 mL of absolute ethanol and store it in 100 μL aliquots at −20⁰C for 4–6 months.
COMMENTARY
Background Information
Many human diseases are closely relevant to mitochondrial dysfunction. Technological advancement, new assay platforms, and newly discovered features of mitochondria homeostasis have placed the “cell bioenergetics” in a pivotal stage. These observations were followed by ones that linked mitochondria to cell respiration, characterized their structure by electron microscopy, purified them using techniques of cell fractionation, characterized the bioenergetic processes associated with these organelles, identified intra-mitochondrial DNA, RNA, and protein synthesis, and linked mitochondrial abnormalities to disease states or toxicological events (Trushina & McMurray, 2007)
The mitochondrial medicine as a new emerging discipline still owes its existence to pioneer studies in the late 1940s and 1950s. In spite of significant progress and advances in bioenergetics, major issues remain mainly related with experimental design and user friendly, cost effective homemade reagent-based protocols.
In this protocol, we aimed (1) to provide simple, straightforward, reproducible mitochondria- enriched sample preparation from the rodent brain tissue for enzymatic assays and (2) to offer two modes of reaction platform (i.e., 96-well microtiter plate (high-throughput) and single cuvette assay. Depending on the preference and availability of the laboratory equipment, the users will have the options to design and perform their assays.
Critical Parameters
Protease inhibitor cocktails must be included in mitochondria-enriched sample preparation. Protein aliquots should be more concentrated so that minimum volume (1–3 μL) would provide required protein concentration per well or 1–2 μg per cuvette.
Avoid repeated freezing-thawing cycle. Aliquot the samples into small volume. Label the leftover sample if user wants to use for some other purposes. The kinetic assays are temperature sensitive. Make sure that microplate reader and spectrophotometer are temperature controlled. pH is also dependent on the temperature. Measure the pH of the reaction buffers at the same temperature of reaction (e.g. if the kinetic assay measurement uses 30°C, then buffer pH should be adjusted at 30°C when prepared). In 96-well micro titrate plate modality; the sample assay replication should be measured at a minimum in “triplicates (3x)”. In some cases, “quintuplicate” (5x) readings should be considered. Use the same brand-named 96-well microtiter plate in the kinetic assay measurements. That will minimize plate-to-plate variation. The reagent addition order in protocols is important. Spot the starter reagent on the side shown in Figure 10. Set your microplate reader’s internal shaker for 5 second at “medium level”. This shaking will allow reagent droplets to be mixed in all 96-wells and the reaction starts at the same time.
Fig.10.
Pipetting the reagent on the side of the micro titer well. The reaction starter reagent either stays on the wall or slightly mixes with the well content.
Troubleshooting
Use low-binding quality pipet tips since small protein sample pipetting is critical Wash the pipet tips 3–4 times in the buffer solution in the well to make sure that all protein solution is delivered. Always run spectrum analysis and physically observe the “peak” for Cytochrome c (reduced) (Fig. 1), DCPIP (Fig. 1). Never assume that your preparation yields the expected product.
If inhibitors of mitochondria do not work in your first experiment, take the following actions: (a) Check your oligomycinA expiration date. OligomycinA is stable in either 100% ethanol or DMSO for only 6 months when kept at −20°C (b) If the concentration suggested in these protocols did not work, then run concentration titration. Minimum five- concentration points (i.e. two upper level concentration, two- lower level concentration, and one suggested concentration in this protocol). Never use the leftover reagents from the previous experiment; therefore, we suggest small volume aliquots. Discarding the 96-well microtiter plate and single disposable cuvettes is critical. Dispose them in biohazardous bin as they contain toxic mitochondria inhibitors.
Statistical Analyses
Although we did not present any data obtained from these protocols, users should be mindful to monitor coefficient of variation (CV%) of their inter-run assays. Reproducibility is important and users should establish their CV%. The formula we used is shown here as
CV = Standard Deviation (SD) / Mean (Ⱦ) * 100
We recommend the users to operate CV= 10–20% in this kind of assays.
Anticipated Results
These protocols provide easy to follow steps with sensitive and reproducible measurement of catalytic activities of mitochondria electron transfer cascade (ETC) enzymes. The user should calculate the enzymatic activities for each mitochondrial ETC according the equation provided under the corresponding protocols.
In mitochondria swelling assay, ΔA540nm = 0.5 indicates that mitochondrial membrane is leaky therefore mitochondria swelling is completed.
Users should calculate the pathlength (, cm) based on the brand of 96-well micro titrate plate. (see SUPPORT PROTOCOL 3). About 10 μg mitochondria-enriched protein solution provides a good enzymatic kinetic profile in 96-well micro titrate plate format. In case of somatic and synaptic mitochondria-enriched preparation, 30–40 μg protein can provide measurable enzyme kinetics. Specific activity of each enzyme complex is expressed as μmol min-1 mg-1 protein. The specificity of Complex-I and Complex I-III activities can be calculated by enzyme complex activity [Total, (−) rotenone] – enzyme complex activity [(+) rotenone]. Specificity of these complexes is expected to be around 85%. Complex-II reaction mixture (Reaction mixture-II) includes set of inhibitors (rotenone, AntimycinA and KCN) that differentiate total Complex-II specific activity. To include 10 mM (final) malonate of 50 mM (final) 2-theonyltrifluoroacetone (TTFA) will inhibit Complex-II. The specific activity of Complex-II with and without malonate (or TTFA) will determine the specificity of Complex-II. Complex II-III specificity is determined by including 10uM (final) malonate and 1mg/ml (final) AntimycinA in the reaction in a separate set of well in the same assay setting. Complex-III specific activity is calculated by subtracting total activity [(−) AntimycinA] from AntimycinA resistant activity [ (+) AntimycinA]. Specificity of the Complex-III is expected at around 80%. Complex-IV specificity is determined by inclusion or exclusion of KCN.
Time Considerations
The purification of somatic and synaptic mitochondria and mitochondria-enriched protein suspension from different regions of rodent brain is a lengthy process (a day-long process). Using this procedure, you may process the brains obtained from 6 to 8 rodents (rats or mice) in a given day. Each preparation requires careful handling as the preparation protocol requires multiple steps. The number of animals used in preparation is also critical. We do recommend users to process maximum 4 rodent brains in a preparation. Enzyme kinetic assays requires relatively short period including pipetting. 2 hours per assay is a reasonable time to spend.
Significance Statement.
Mitochondrial bioenergetics are critical for normal central nervous system (CNS) function. Abnormal bioenergetics due to malfunctioning of mitochondrial enzymes of the electron transfer cascade (ETC) are associated with both CNS diseases and the effects of CNS toxic agents. The cellular complexity of the CNS and the structural complexity of the ETC enzymes present challenges for those wishing to assess functional changes in CNS mitochondria.
Protocols for the isolation of mitochondria from distinct brain regions and subcellular compartments of neurons, as well as for the conduct of ETC enzyme assays in multiplex format at microliter levels employing new analyzers, software for data analysis, and advanced chemical formulations of substrates, should be of assistance to investigators planning to study CNS mitochondrial function.
Acknowledgments
M. Michaelis and E. Michaelis acknowledge funding of their work by grants from the National Institute on Aging, AG 12993 and AG 035982.
Footnotes
Internet Resources
Statistics how to https://www.statisticshowto.datasciencecentral.com/
Cytochrome c spectral properties. https://www.sigmaaldrich.com/content/dam/sigma- aldrich/docs/Sigma/Datasheet/c2506dat.pdf
LITERATURE CITED
- Barrientos A, Fontanesi F, & Diaz F (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet, Chapter 19, Unit19 13. doi: 10.1002/0471142905.hg1903s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobba A, Amadoro G, Valenti D, Corsetti V, Lassandro R, & Atlante A (2013). Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion, 13(4), 298–311. doi: 10.1016/j.mito.2013.03.008 [DOI] [PubMed] [Google Scholar]
- Chiu K, Lau WM, Lau HT, So KF, & Chang RC (2007). Micro-dissection of rat brain for RNA or protein extraction from specific brain region. J Vis Exp(7), 269. doi:10.379½69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, & Matthews DA (1971). Synaptic plasma membranes from rat brain synaptosomes: isolation and partial characterization. Biochim Biophys Acta, 249(2), 380–394. [DOI] [PubMed] [Google Scholar]
- Cotman CW, & Taylor D (1972). Isolation and structural studies on synaptic complexes from rat brain. J Cell Biol, 55(3), 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis E (1967). Ultrastructure and cytochemistry of the synaptic region. The macromolecular components involved in nerve transmission are being studied. Science, 156(3777), 907–914. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, & Rostas JA (1986). A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res, 372(1), 115–129. [DOI] [PubMed] [Google Scholar]
- Ernster L, & Schatz G (1981). Mitochondria: a historical review. J Cell Biol, 91(3 Pt 2), 227s-255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurd JW, Jones LR, Mahler HR, & Moore WJ (1974). Isolation and partial characterization of rat brain synaptic plasma membranes. J Neurochem, 22(2), 281–290. [DOI] [PubMed] [Google Scholar]
- Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, . . .Rodenburg RJ (2007). Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem, 53(4), 729–734. doi: 10.1373/clinchem.2006.078873 [DOI] [PubMed] [Google Scholar]
- Kerr D, Grahame G, & Nakouzi G (2012). Assays of pyruvate dehydrogenase complex and pyruvate carboxylase activity In Wong L-JC (Ed.), Mitochondrial Disorders, Biochemical and molecular analysis (Vol. 837, pp. 93–119). New York, Dordrecht, Heidelberg, London: Springer. [DOI] [PubMed] [Google Scholar]
- Kohen R, & Nyska A (2002). Invited Review: Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicologic Pathology, 30(6), 620–650. doi: 10.1080/01926230290166724 [DOI] [PubMed] [Google Scholar]
- Leong SF, Lai JC, Lim L, & Clark JB (1984). The activities of some energy-metabolising enzymes in nonsynaptic (free) and synaptic mitochondria derived from selected brain regions. J Neurochem, 42(5), 1306–1312. [DOI] [PubMed] [Google Scholar]
- Navarro A, & Boveris A (2004). Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol, 287(5), R1244–1249. doi: 10.1152/ajpregu.00226.2004 [DOI] [PubMed] [Google Scholar]
- Navarro A, Lopez-Cepero JM, Bandez MJ, Sanchez-Pino MJ, Gomez C, Cadenas E, & Boveris A (2008). Hippocampal mitochondrial dysfunction in rat aging. Am J Physiol Regul Integr Comp Physiol, 294(2), R501–509. doi: 10.1152/ajpregu.00492.2007 [DOI] [PubMed] [Google Scholar]
- Nicklas W, Vyas I, & Heikkila RE (1985). Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6- tetrahydropyridine. Life Sci, 36(26), 2503–2508. doi: 10.1016/0024-3205(85)90146-8 [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Clark HR, Chavan V, Benson EK, Kidd GJ, & Srivastava S (2016). Analysis of Brain Mitochondria Using Serial Block-Face Scanning Electron Microscopy. J Vis Exp(113). doi: 10.3791/54214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palkovits M, & Brownstein MJ (1983). Microdissection of brain areas by the punch technique In Brain Microdissection Technique (pp. 1–36). New York: Wiley J. [Google Scholar]
- Ravagnan L, Roumier T, & Kroemer G (2002). Mitochondria, the killer organelles and their weapons. Journal of Cellular Physiology, 192(2), 131–137. doi:doi: 10.1002/jcp.10111 [DOI] [PubMed] [Google Scholar]
- Robertson RT, Zimmer J, & Gahwiler BH (1989). Dissection procedures for preparation ofslice culture In A dissection and tissue culture manual of the system (Shahar A et al. ,eds., pp.1–15). New York: Alan R. Liss, Inc [Google Scholar]
- Scheffler IE (2008a). Mitochondria (second ed.). New Jersey, USA: John Wiley & Sons, Inc. [Google Scholar]
- Scheffler IE (2008b). Mitochondria. In Mitochondria (pp. 2168–2297). New Jersey: John Wiley. [Google Scholar]
- Shah N (2014). How do you measure the pathlength of your sample in a 96-well microtiter plate? Retrieved from https://www.researchgate.net/post/How_do_you_measure_the_pathlength_of_your_sample_in_a_96-well_microtiter_plate
- Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, . . . Greenamyre JT (2007). Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. Journal of Neurochemistry, 100(6), 1469–1479. doi:doi: 10.1111/j.1471-4159.2006.04333.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spijker S (2011). Dissection of Rodent Brain Regions In Wan K (Ed.), Neuroproteomics (Vol. 57, pp. 13– 26): Springer. [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Ermani M, Salviati L, & Angelini C (2011). Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion, 11(6), 893–904. doi: 10.1016/j.mito.2011.07.006 [DOI] [PubMed] [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Salviati L, & Angelini C (2012). Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc, 7(6), 1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH (2012). Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal, 16(12), 1434–1455. doi: 10.1089/ars.2011.4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, & Wallace DC (1996). Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol, 264, 484–509. [DOI] [PubMed] [Google Scholar]
- Trushina E, & McMurray CT (2007). Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience, 145(4), 1233–1248. doi: 10.1016/j.neuroscience.2006.10.056 [DOI] [PubMed] [Google Scholar]
- Wang L, Guo L, Lu L, Sun H, Shao M, Beck SJ, Du H (2016). Synaptosomal Mitochondrial Dysfunction in 5xFAD Mouse Model of Alzheimer’s Disease. PLoS One, 11(3), e0150441. doi: 10.1371/journal.pone.0150441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng x. x., Shoffner JM, Voljavec AS, & Wallace DC (1990). Evaluation of procedures for assaying oxidative phosphorylation enzyme activities in mitochondrial myopathy muscle biopsies. Biochim Biophys Acta, 1019(1), 1–10. [DOI] [PubMed] [Google Scholar]
- Leong SF, Lai JC, Lim L, & Clark JB (1984). The activities of some energy-metabolising enzymes in nonsynaptic (free) and synaptic mitochondria derived from selected brain regions. J Neurochem, 42(5), 1306–1312. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Clark HR, Chavan V, Benson EK, Kidd GJ, & Srivastava S (2016). Analysis of Brain Mitochondria Using Serial Block-Face Scanning Electron Microscopy. J Vis Exp(113). doi: 10.3791/54214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Lopez-Cepero JM, Bandez MJ, Sanchez-Pino MJ, Gomez C, Cadenas E, & Boveris A (2008). Hippocampal mitochondrial dysfunction in rat aging. Am J Physiol Regul Integr Comp Physiol, 294(2), R501–509. doi: 10.1152/ajpregu.00492.2007 [DOI] [PubMed] [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Ermani M, Salviati L, & Angelini C (2011). Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion, 11(6), 893–904. doi: 10.1016/j.mito.2011.07.006 [DOI] [PubMed] [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Salviati L, & Angelini C (2012). Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc, 7(6), 1235–1246. doi: 10.1038/nprot.2012.058 [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, & Wallace DC (1996). Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol, 264, 484–509. [DOI] [PubMed] [Google Scholar]
- Trushina E, & McMurray CT (2007). Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience, 145(4), 1233–1248. doi: 10.1016/j.neuroscience.2006.10.056 [DOI] [PubMed] [Google Scholar]
- Zheng x. x., Shoffner JM, Voljavec AS, & Wallace DC (1990). Evaluation of procedures for assaying oxidative phosphorylation enzyme activities in mitochondrial myopathy muscle biopsies. Biochim Biophys Acta, 1019(1), 1–10. [DOI] [PubMed] [Google Scholar]
Key Reference
- Barrientos A, Fontanesi F, & Diaz F (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet, Chapter 19, Unit19 13. doi: 10.1002/0471142905.hg1903s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spijker S (2011). Dissection of Rodent Brain Regions In Wan K (Ed.), Neuroproteomics (Vol. 57, pp. 13– 26): Springer. [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Salviati L, & Angelini C (2012). Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc, 7(6), 1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, & Wallace DC (1996). Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol, 264, 484–509. [DOI] [PubMed] [Google Scholar]