

Abstract
Natural Killer (NK) cells suppress tumor initiation and metastasis. Most carcinomas are heterogeneous mixtures of epithelial, mesenchymal and hybrid tumor cells, but the relationships of these phenotypes to NK susceptibility are understood incompletely. Grainyhead-like-2 (GRHL2) is a master programmer of the epithelial phenotype, that is obligatorily down-regulated during experimentally induced Epithelial-Mesenchymal Transition (EMT). Here, we utilize GRHL2 re-expression to discover unifying molecular mechanisms that link the epithelial phenotype with NK-sensitivity. GRHL2 enhanced the expression of ICAM-1, augmenting NK-target cell synaptogenesis and NK killing of target cells. The expression of multiple interferon response genes, including ICAM1, anti-correlated with EMT. We identified two novel GRHL2-interacting proteins, the histone methyltransferases KMT2C and KMT2D. Mesenchymal-epithelial transition, NK-sensitization and ICAM-1 expression were promoted by GRHL2-KMT2C/D interactions and by GRHL2 inhibition of p300, revealing novel and potentially targetable epigenetic mechanisms connecting the epithelial phenotype with target cell susceptibility to NK killing.
Keywords: Grainyhead-like-2, NK cells, ICAM-1, mesenchymal-to-epithelial transition, KMT2C, KMT2D, epigenetic, p300
Introduction
Adaptive and innate immune rejection of tumors involves a complex interplay between dynamically changing tumor cells and immune cells. Under microenvironmental stress, including that induced by the immune system itself, tumor cells can rapidly diversify their phenotypes so as to generate immuno-resistant variants, a phenomenon called immunoediting 20. Numerous mechanisms have been identified for tumor cell escape from T-cell mediated immunity, including down-regulation of MHCI or antigen processing components for antigen presentation, defects in IFN-γ signaling or long term immunosuppressive effects of IFN-γ, antigen loss, expression of immune checkpoint ligands, depletion of tryptophan or the expression of TGF-β 3, 13, 20, 30, 42, 51, 59, 74.
NK cells play crucial roles in the rejection of metastatic/circulating tumor cells 48, 56. NK cells can kill these directly, through multiple NK ligand-NK receptor interactions and the target cell adhesion molecule ICAM-1 (CD54) interaction with the NK cell integrin LFA-1 48, 54, 56. NK-target cell interaction and killing are promoted by the presence of antibodies against the target cell that bridge them with CD16 on the NK cell, an important contributor to tumor rejection by therapeutic antibodies 43, 86. Direct NK killing is also promoted by IFN-γ–mediated induction of target cell ICAM-1 expression and by type I interferons (from many cell types) and IL-15 (from dendritic cells), that aid in NK cell activation 14, 55, 58, 84.
NK cells also support T-cell mediated tumor rejection, via dendritic cell activation, enhancing T-cell based responses including checkpoint inhibitor therapy 4, 6, 73. Correspondingly, tumor incidence and progression are suppressed by NK cells, in proportion to both NK cell number and their cytotoxic competence 48, 54, 56. Tumor cells can, however, evade NK cell surveillance by down-regulating (or shedding) ligands for activating NK receptors (e.g., MICA, MICB, ULBP1-6,PVR), up-regulating inhibitory ligands (e.g., HLA-G, PD-L1, soluble NKG2D decoys), over-expressing IDO, resisting TNF cytotoxicity, down-regulating IFN I genes, up-regulating autophagy or through (poorly understood) NK cell exhaustion 1, 14, 40, 48, 54, 56, 58.
One common tumor cell phenotype accompanying tumor heterogeneity is the adoption, in a subpopulation of tumor cells, of a partial or complete epithelial-mesenchymal transition (EMT8). Reciprocally, EMT-driving transcription factors, in conjunction with the loss of checkpoint tumor suppressors, create “cellular pliancy” 65, permitting rapid diversification of phenotype, principally through epigenetic reprogramming. In the appropriate microenvironment, cells in this state may further transition to stemness 23, 80. Pioneering early studies in mouse models clearly showed that EMT provides a path to immunoediting and tumor escape and that both processes can be accelerated by cytokines 42, 70. Subsequent studies in mouse and cell culture models confirmed that EMT can promote tumor immune evasion 1, 18, 46, 75, 76. Accordingly, an EMT gene signature was identified in patients responding inefficiently to immune checkpoint inhibition 36. EMT phenotypes are diverse, however, which is reflected in the correspondingly diverse mechanisms by which epithelial vs. mesenchymal phenotypes regulate sensitivity to immune cells, confounding efforts to discover unifying principles (see Discussion).
In this study, we utilize a factor that uniformly programs the epithelial phenotype to discover underlying molecular mechanisms linking this phenotype with NK-sensitivity. The transcription factor Grainyhead-like-2 (GRHL2) is a master programmer of the epithelial phenotype in developmental, homeostatic and cancer-related contexts. Developmentally, GRHL2 is a pioneer transcription factor that pre-activates epithelial gene enhancers, promoting the embryonic stem-cell to epiblast transition 12, 37. Previously, we reported that GRHL2 suppresses EMT, in part, through several mechanisms 16, 17, 27, 63, including the (unique) inhibition of the histone acetyltransferase activity of the co-activator protein p300, the repression of ZEB1 expression and the inhibition of TGF-β signaling 16, 17, 27, 63. In addition to histone acetylation, histone methylation plays an important role in normal vs. tumor transcription programs 50. In particular, the histone methyltransferases KMT2C and KMT2D (MLL3 and MLL4) mono-methylate H3K4 at enhancers, marking them for activation; they have been characterized primarily as co-factors for nuclear receptors (e.g., androgen receptor, estrogen receptor) and the pioneer transcription factor FOXA139, 57, 64, 79. KMT2C and KMT2D are mutated frequently in human cancer21, 28, 62, 83, 87.
GRHL2 provides an unprecedented opportunity to discover mechanisms by which the enforcement of an epithelial phenotype affects tumor cell sensitivity to immune-mediated cytotoxicity. Herein, we report that GRHL2 sensitized cells to NK killing at the level of NK-target cell synaptogenesis, mediated by the up-regulation of ICAM1. GRHL2 protein interacted directly with KMT2C and KMT2D. GRHL2-KMT2C/D interactions and p300 inhibition were found to be important for MET, ICAM1 up-regulation and NK- sensitization effects, indicating that GRHL2 enforces an NK-sensitive epithelial gene expression program through functional interactions with epigenetic modifiers.
Results
GRHL2 increases sensitivity to NK killing
We generated and characterized mesenchymal cell lines that were reverted to an epithelial phenotype by retroviral expression of an epithelial master programming factor, Grainyhead-like-2 (GRHL2): HT1080 fibrosarcoma, Mesenchymal Subpopulation (MSP) cells (the CD44hlghCD24low EMT-like subpopulation of HMLE cells) and MCF10aneoT cells treated with TGF–β 16, 17, 22, 63. To address the effect of GRHL2-mediated Mesenchymal-Epithelial Transition (MET) upon NK killing, we utilized the immortalized human NK cell line, NK92-MI. This cell line (abbreviated NK92 here) is mutant for the Killer-Cell Immunoglobulin (KIR) receptor, thus obviating them to target cell MHCI expression; it also bears a transgene encoding autocrine IL-2. NK92/NK92-MI has been useful for diverse NK studies in vitro and in vivo 41.
We compared mesenchymal vs. epithelial cell lines within each isogenic pair for sensitivity to NK killing, by pre-loading the target cells with a fluorescent dye, 5-chloromethylfluorescein diacetate (CMFDA) and assaying for the loss of the fluorescent signal. For the three pairs of cell lines above, the GRHL2-expressing MET-like cells were more sensitive to NK killing than the parental mesenchymal cells (figure 1A). Similar results were obtained by assaying target cells that had been pre-labeled by lentiviral expression of nano-luciferase for the release of this enzyme into the culture supernatant (figure S1). Two additional experiments were performed to rule out a potential GRHL2 over-expression artifact (figure S2): a. the CD44hiCD24lowGRHL2low (i.e., MSP) subpopulation of HMLE cells were compared against the CD44lowCD24highGRHL2high subpopulation for NK sensitization; the latter was significantly more sensitive; b. MCF10aneoT cells with CRISPR/cas9-mediated deletion of both alleles of GRHL2 were refractory to NK killing at 12 h and 24 h; this was reversed by GRHL2.
Figure 1. GRHL2 expression sensitizes cell lines to NK killing.
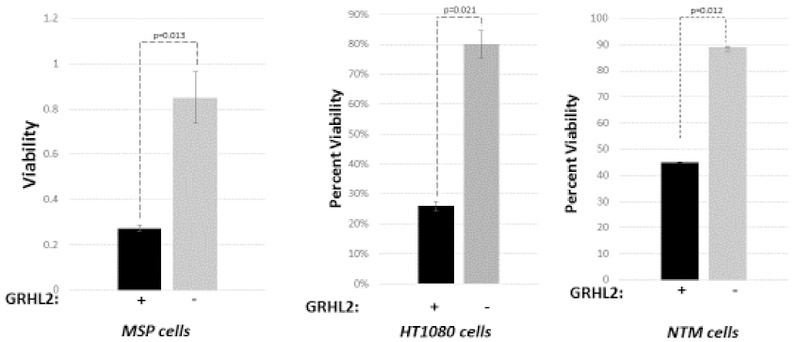

(A). GRHL2low mesenchymal cell lines become more susceptible to NK killing upon restored GRHL2 expression. Viability assays after incubation with or without NK92 cells were performed as in Materials and Methods, using three mesenchymal cell lines: Mesenchymal Sub-Population (MSP) cells derived by flow sorting of the parental HMLE mammary epithelial cell line 71, HT1080 fibrosarcoma cells and MCF10aneoT cells that were induced transiently with TGF-β (NTM) to undergo EMT. Characterization of MET in these cell lines by GRHL2 re-expression was published previously 16, 17, 22, 63. (B). MET induced by inducible mir200c re-expression (BT549 cells) or adenovirus E1a (HT1080 cells) sensitized cell lines to NK killing (nano-luciferase assays); MET in these cell lines was characterized previously 24, 67. Control cell lines had empty retroviral vector and were drug-selected.
To determine whether the NK sensitization effect was a general response to MET, two other MET inducers, adenovirus-5 E1a 24 and mir200c 34, 67 were assayed and found to enhance NK sensitivity as well (figure 1B). The results suggested that the reversion of mesenchymal tumor cell lines to an epithelial phenotype by three different genes sensitized the cells to NK killing, although it is not clear whether these three genes necessarily induced MET by entirely independent mechanisms (see Discussion).
NK cells kill target cells primarily through death ligand-death receptor interactions and through the perforin-mediated target cell internalization of NK-derived granzyme B 48. We tested the effect of GRHL2 on killing by each of these cytotoxins, and found that GRHL2 sensitized cells to both FASL and granzyme B. The death receptor pathway did not contribute significantly to NK killing in our system, however, because siRNA-mediated knockdown of FADD had a minimal effect, implicating granzyme B as the major cytotoxic mechanism in our cell lines (figure S3).
These results indicated that GRHL2 induced an epithelial gene program (reviewed in 27 that conferred target cell sensitivity to NK killing.
GRHL2 promotes ICAM1-mediated NK-target cell conjugation
The formation of an NK-target cell immune synapse (“conjugation”) is critical for optimal NK activation and target cell killing 47. Two-color fluorescent labeling/conjugation experiments indicated that NK92 cells conjugated with epithelial cell lines more efficiently than the corresponding mesenchymal cell lines, both in MCF10a- and HMLE-based contexts, the latter representing a cell line without GRHL2-over-expression (figure 2A).
Figure 2. GRHL2 expression enhances ICAM1-mediated NK-target cell conjugation.


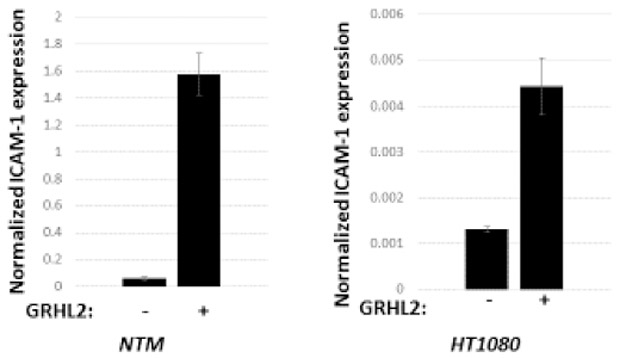

(A) NK conjugation is enhanced by GRHL2. Cell lines without significant GRHL2 expression (NTM or MSP) were compared with corresponding GRHL2-expressing cell lines (MSP+GRHL2 or HMLE) for conjugation with fluorescently labeled NK cells (see Materials and Methods for details). Average NK cells bound per target cell are shown graphically. (B) NK conjugation is ICAM1-dependent. Target cells indicated HT1080+GRHL2+nano-luc or HMLE+nano-luc) were pre-blocked with ICAM-1 antibody or control prior to NK killing assays based on nano-luciferase release. (C). GRHL2 induces basal ICAM-1 mRNA expression. Normalized results of qRT-PCR assays are shown.(NTM=MCF10aneoT/TGF-b-induced; normalized to CBX internal control; HT1080+/−GRHL2 cells, normalized to beta2-microglobulin internal control). (D) GRHL2 enhances IFN-γ induction of ICAM-1 mRNA (qRT-PCR data, left panels) and ICAM-1 protein (western blot data, right panels).
The target cell adhesion molecule ICAM-1 is important for conjugation of both NK and T-cells to their target cells 84. Conjugation and NK killing were suppressed by an ICAM-1 blocking antibody, confirming the critical role of ICAM-1 for immune synapse formation in our system (figure 2B). Interestingly, GRHL2 increased the basal expression of ICAM-1 mRNA (figure 2C). ICAM-1 expression is rapidly induced by IFN-γ derived from activated NK or T-cells, generating a feed-forward loop for immune rejection 84. GRHL2 promoted the IFN-γ induction of ICAM-1 protein and mRNA, suggesting a potential mechanism for enhanced conjugation and NK sensitivity (figure 2D). Two other genes involved in NK activation following synapse formation -- the NKG2D ligand, RAETL1/ULBP6 and the DNAM ligand CD155-- were also induced by GRHL2, suggesting that additional, ICAM-1 independent sensitization mechanisms might occur -- while other NK ligands for activating or inhibitory receptors were not affected significantly (figure S4 and Table S1). Collectively, these results indicated that the up-regulation of ICAM-1 expression and NK-target conjugation contributed significantly to the enhancement of NK sensitivity by GRHL2 expression.
GRHL2 protein regulates gene expression through p300 and by direct interactions with KMT2C and KMT2D
Previously, we reported that GRHL2 inhibits the histone acetyltransferase/co-activator protein p300, contributing to MET 63. To identify additional transcription factors that interact with GRHL2, a yeast-two hybrid assay was performed, using full-length GRHL2 as bait. The two hits of highest confidence were the related histone methyltransferases, KMT2C and KMT2D. These histone methyltransferases (formerly called MLL3 and MLL4, respectively) are enzymatic core components of the COMPASS complex that selectively mono-methylate histone-3 lysine-4 (H3K4) at enhancer elements, in conjunction with nuclear receptors or the pioneer factor FOXA1 39, 57, 64, 79. The sequences of the yeast two-hybrid “prey fragments” corresponding to KMT2C and KMT2D were homologous and occurred outside of known functional domains (figure 3A).
Figure 3. GRHL2 interacts functionally with KMT2C/D.




(A) Sequences of the interaction sites on KMT2C and KMT2D for GRHL2 protein interaction, deduced from minimal sequences of yeast two-hybrid clones. Domains on KMT2D are indicated. (B) Confirmation of GRHL2-KMT2C/D interactions, (upper panel): Co-transfection/co-immunoprecipitation/western blotting of GRHL2-S-tag protein precipitated from cell lysates with S-protein agarose and western blotted for FLAG-KMT2C or FLAG-KMT2D sequences corresponding to yeast-two hybrid clones, (lower panel): map of peptides corresponding to KMT2C and KMT2D proteins identified by RIME using GRHL2 antibody. (C) GRHL2 E32D33 are required for interaction with KMT2C/D. Full-length wild-type or E32D33->A32A33 mutant forms of GRHL2 were analyzed for interaction with KMT2C/D as in (B). (D). GRHL2-KMT2C/D interaction is important for activation of the Rab25 promoter. HT1080 cells were co-transfected with the indicated amounts of GRHL2-WT or GRHL2-ED→AA mutant (in pcDNA3.1) together with a Rab25 promoter-luciferase construct and assayed for luciferase activity (with normalization to beta-galactosidase expressed using co-transfected pTK-lacZ).
The interaction of GRHL2 protein with KMT2C/D proteins was confirmed initially by co-transfection of the yeast two hybrid prey fragments of KMT2C/D with full-length GRHL2 followed by co-immunoprecipitation/western blotting (figure 3B). Additionally, Rapid Immunoprecipitation Mass spectrometry of Endogenous proteins (RIME) using GRHL2 antibody identified both KMT2C and KMT2D among the top interactors, confirming that this interaction occurred on chromatin (table S2).
To test the functional importance of the GRHL2-KMT2C/D interactions for transcription, we mapped the domain on GRHL2 protein responsible for the interaction with KMT2C/D using interactions of recombinant proteins. This domain was within the N-terminal transactivation domain of GRHL2, and fine-mapping by mutagenesis of adjacent pairs of amino acids revealed that E32D33 were critical for the interaction (figure 3C). A GRHL2 mutant in which E32 and D33 were changed to A32A33 was then compared to wild-type GRHL2 for its ability to activate the Rab25 gene promoter -- a direct target for GRHL2 transactivation 5 -- in transient reporter assays. E32D33 and, by inference, GRHL2-KMT2C/D interactions, were critical for transactivation (figure 3D).
To test the function of the GRHL2-KMT2C/D interactions further, we expressed similar levels of wild-type GRHL2, GRHL2-ED→AA and GRHL2-Δ425-437 in parental cell lines of an EMT-like phenotype. Wild-type GRHL2 reverted mesenchymal cells (e.g., NTM) to an epithelial-like morphology, but expression of the non-KMT2C/D-interacting point mutant (ED→AA) or, as previously shown, the non-p300-inhibitory mutant, Δ425-437, produced more modest effects (figure 4A). Correspondingly, wild-type GRHL2 -- but neither of the GRHL2 mutants -- induced E-cadherin expression efficiently in two mesenchymal cell lines (MSP and NTM), suggesting that both mutants were at least partially defective for MET induction (figure 4A).
Figure 4. GRHL2 interaction with KMT2C/D and GRHL2 inhibition of p300 are important for MET.





(A) GRHL2-KMT2C/D interaction and GRHL2-p300 inhibition are important for endogenous E-cadherin induction (upper panel) and conversion to an epithelial morphology (lower panel). (B) GRHL2-KMT2C/D interaction and GRHL2-p300 inhibition are important for the induction of epithelial/GRHL2 target genes (qRT-PCR). (C) GRHL2-KMT2C/D interaction and GRHL2-p300 inhibition are important for the repression of mesenchymal (GRHL2-repressed) genes (qRT-PCR). (D) GRHL2 interaction with KMT2C/D is important for the recruitment of KMT2C to epithelial gene promoters. Cells (NTM) expressing GRHL2-WT, GRHL2-ED→AA or no GRHL2 were subjected to CHIP analysis using CHIP-validated KMT2C antibody 19. Control IgG values were essentially zero (not shown). (E) Double knockout of KMT2C (homozygous) and KMT2D (heterozygous) induces EMT. Cells (derived from MCF10a) of the indicated genotypes adopted the morphologies shown in the top panel. Epithelial and mesenchymal gene expression was assayed by qRT-PCR, with results shown in the lower left and lower right panels.
Quantitative RT-PCR analysis of the induction of selected epithelial marker/GRHL2-induced genes and repression of selected mesenchymal/GRHL2-repressed genes in NTM cells revealed that the non-KMT2C/D mutant of GRHL2 (ED→AA) indeed failed to induce MET (figure 4B, 4C).
These results suggested that GRHL2 may recruit KMT2C/D to target GRHL2-induced enhancers through direct interaction. To test this, we compared the effect of GRHL2-WT vs. GRHL2-ED→AA on the recruitment of KMT2C by CHIP, using a CHIP-validated KMT2C antibody 19. On Rab25, CDH1 (intron 2), CDH1 (distal), OVOL2 and TP63 regulatory elements (targeting GRHL2 binding sites extracted from published CHIP-seq), wild-type GRHL2 recruited KMT2C more efficiently than the non-KMT2C/D-interacting ED→AA mutant (figure 4D). These results indicated that GRHL2 recruited KMT2C/D to target promoters through direct GRHL2-KMT2C/D interaction, to induce the expression of epithelial genes.
Based on the importance of the GRHL2-KMT2C/D interaction for an epithelial phenotype, we tested the effect of CRISPR/cas9-mediated knockout of KMT2C, or both KMT2C and KMT2D, on epithelial vs. mesenchymal phenotypes in parental MCF10a cells. While the knockout of both KMT2C alleles had no effect on the morphology of MCF10a cells, the combined KMT2C−/−/KMT2D−/+ knockout converted the cells to an EMT-like morphology, accompanied by the loss of epithelial gene expression, including ICAM-1 (figure 4E) and, interestingly, GRHL2 itself, indicating the importance of KMT2C/D for the maintenance of an epithelial phenotype.
GRHL2 regulates NK sensitivity through KMT2C/D and p300.
GRHL2 regulates epithelial specific transcription uniquely, by inhibiting p300 acetyltransferase activity (contrasting with other factors, which utilize p300 acetyltransferase activity as a co-activator) and by recruiting KMT2C/D. Cell lines that expressed similar levels of wild-type vs. mutant GRHL2 proteins were assayed to address the roles of these epigenetic modifiers in NK-sensitization. The deletion of the p300-inhibitory sequence (amino acids 425-437), or the point mutation of the KMT2C/D-interacting sequence (E32D33→AA) attenuated the NK-sensitization effect of GRHL2 (figure 5A). The MCF10a cells with KMT2C−/−;KMT2D+/− genotype (described above) were significantly more resistant to NK killing than control KMT2C−/− cells (figure 5A), confirming the important role of KMT2 C/D in the epigenetic control of NK sensitivity. Correspondingly, wild-type GRHL2 induced ICAM-1 mRNA and protein expression more efficiently than did either GRHL2 mutant, under stimulated or IFN-γ-stimulated conditions (figure 5B). These results indicated that GRHL2 protein induced MET and sensitized (formerly) mesenchymal cells to NK killing through epigenetic regulation of gene expression. These effects involved the (biologically unique) inhibition of the histone methyltransferase co-activator p300 – presumably, an indirect effect via other factors -- and the interaction and promoter recruitment of the (nuclear receptor related) histone methyltransferases KMT2C and KMT2D.
Figure 5. GRHL2 interaction with KMT2C/D and GRHL2 inhibition of p300 are important for sensitization to NK killing.



(A) (left panel): NTM cells with wild-type or mutant forms of GRHL2 indicated were assayed for NK killing using the CMFDA labeling assay, (right panel): MCF10a cells with KMT2C knockout or KMT2C/D (heterozygous) knockout were assayed for NK killing by the CMFDA labeling assay. (B). GRHL2 interaction with KMT2C/D and GRHL2 inhibition of p300 are important for the expression of ICAM-1, both under basal and IFN-γ-induced conditions. (C) GRHL2 interaction with KMT2C/D is important for the recruitment of KMT2C to the ICAM-1 gene promoter, (left panel): screen shot of GRHL2, KMT2C binding sites at the 5’ end of the ICAM-1 gene, aligned with H3K27Ac and DNase hypersensitivity peaks. Data were published by 39 via UC Santa Cruz Genome Browser. (Right panel): Cells (NTM) expressing GRHL2-WT, GRHL2-ED→AA or no GRHL2 were subjected to CHIP analysis using CHIP-validated KMT2C antibody 19 and primers corresponding the indicated region of the ICAM-1 gene. Control IgG values were essentially zero (not shown).
Inspection of CHIP-seq data 39 (via University of California Santa Cruz Genome Browser) revealed overlapping binding sites for GRHL2 and KMT2C at the 3’ end of exon 1/5’ end of intron 1 boundary of the ICAM-1 gene, where peaks of H3K27Ac and DNase hypersensitivity indicated transcriptional activity (figure 5C). Comparison of KMT2C recruitment in cells that expressed no GRHL2, wild-type GRHL2 or GRHL2-ED→AA revealed that the GRHL2-KMT2C/D interaction was important for the recruitment of KMT2C to this binding site in the ICAM-1 gene (figure 5C), suggesting that the enhanced expression of ICAM-1 by GRHL2 is potentially direct, warranting further investigation.
IFN response gene expression anti-correlates with EMT
These data predicted that the tumor epithelial phenotype would correlate with the expression of ICAM1 and perhaps other IFN pathway genes. We focused on lung cancer, in light of the extensive immune infiltration and availability of expression data in this tumor type. To test this, we assigned EMT scores to lung adenocarcinoma tumor samples from The Cancer Genome Atlas (TCGA) using the validated pancancer 16 gene signature 32. These were compared with the expression of various IFN response genes shown in figure 6, after normalization for IFN-gamma transcript, to account for differences in tumor-immune cell infiltration. This normalization precluded the use of standard Pearson-correlation (R-value) calculations. Thus, we distributed both EMT scores and normalized gene expression values into quartiles (Q1=lowest, Q4=highest) and calculated the deviation from random distribution (0.5:0.5) upon comparison of the EMT Q1 quartile (i.e., essentially epithelial phenotype) vs. the low (Q1) or high (Q4) expression of the IFN response gene. The results (Fig. 6) indicated that the IFN response genes distributed significantly/nonrandomly with low EMT score. The data suggested that EMT suppresses, and the epithelial phenotype promotes the IFN signaling pathway, respectively.
Figure 6. IFN response gene expression anti-correlates with EMT.

Pattern of co-occurrence between high/low EMT scores in lung adenocarcinoma samples vs. high/low expression levels of the indicated IFN response genes. For high and low subsets of each EMT signature (top and bottom quartile, correspondingly), the fraction of the subset with high or low mRNA expression levels (after normalization for IFN-gamma transcript levels) was calculated for each gene of interest. The deviation from the 0.5:0.5 random distribution ratio is color-coded: Red=fraction higher than random; blue= fraction lower than randomly expected. The statistical significance (p-value) for each difference is indicated. Data for GRHL2 are shown as a positive control for low EMT score.
Discussion
GRHL2 protein, recently found to be a factor that can convert closed chromatin to open chromatin de novo – a “pioneer transcription factor” -- drives the embryonic stem cell to epiblast transition. As such, it is a quintessential licensing factor for epithelial gene expression 12, 37. GRHL2 has the properties of a factor responsible for establishing the “epithelial default phenotype” -- the generic epithelial cell phenotype occurring in the absence of differentiation-inductive factors 25. Consequently, GRHL2 expression suppresses and reverses the oncogenic EMT 16, 17, 27.
Several previous reports have shown that EMT can confer immune evasion with respect to T-cells or NK cells. Indeed, a mesenchymal gene expression profile associates with tumor resistance to checkpoint inhibitor therapy 59. However, EMT programs are diverse 11, 61. Correspondingly, diverse mechanisms of EMT-related immune evasion have been reported, including PD-L1 induction 13, autophagic degradation of granzyme B 82, failure of CDK1 to phosphorylate nuclear lamins for degradation 35, induction of indoleamine 2,3- dioxygenase (IDO) 66, induction of tryptophan 2,3-dioxygenase 33, defects in tyrosine phosphorylation at immune synapses 75, down-regulation of MHCI 18, defective antigen presentation machinery 49, antigen loss 42, thrombospondin up-regulation 46 and up-regulation of the matricellular protein SPARC 69.
We have taken advantage of GRHL2 as a core component of the epithelial phenotype to identify unifying molecular links between this phenotype and cellular susceptibility to direct killing by NK cells. In particular, we discovered previously that the functional inhibition of the co-activator p300 by GRHL2 plays an important role in the maintenance of an epithelial phenotype, including sensitivity to anoikis 63. Here, we have shown that this effect on p300 is additionally important for NK sensitivity. In this regard, GRHL2 has striking similarities to another p300-inhibitory protein, adenovirus E1a, which also reverses EMT and sensitizes cells to NK killing 24, 26, 52.
We have also shown here that KMT2C and KMT2D histone methyltransferases interact directly and functionally with GRHL2. While characterizing this interaction (originating from our yeast two-hybrid screen), GRHL2 was reported to frequently co-occupy promoters with FOXA1 and/or KMT2C, and FOXA1 was shown to interact directly with KMT2C, contributing to estrogen receptor function 39. This suggested a model in which FOXA1, acting as a pioneer transcription factor through the recruitment of KMT2C, promoted estrogen receptor function 39. Subsequent study demonstrated that GRHL2 is another pioneer transcription factor that also increases H3K4me1 marks at epithelial gene enhancers, landmarking them for induction in epiblasts 12, 37. Moreover, GRHL2 is a critical co-factor for androgen receptor function 57. When viewed collectively, these data suggest that GRHL2 is a pioneer transcription factor that establishes the “default” epithelial phenotype, permitting more specialized factors to induce tissue-specific epithelial genes downstream of the nuclear receptors and perhaps other factors. Under this model, our current data suggest that the GRHL2-KMT2C/D interaction and the GRHL2-mediated inhibition of p300 (the latter almost certainly occurring only on a subset of p300-dependent genes) contribute crucially to the establishment of an epithelial phenotype, which includes sensitization to NK killing. This makes sense, as NK cells are uniquely adapted to target epithelial cells that have undergone infection with viral pathogens that down-regulate MHCI 78. This connection between epithelial phenotype and NK sensitivity is further supported by our data demonstrating that two other MET inducing genes, mir200c, and adenovirus E1a, also augment NK sensitivity. It should be noted, however, that GRHL2 protein can engage an epithelial phenotype both directly and indirectly, by inducing mir200 family expression 15, 16. In addition, adenovirus E1a protein also inhibits p300, and p300 functions in a complex that contains a KMT2D-UTX-p300 heterotrimer85. Thus, the mir200c miRNA and E1a protein may function through pathways that intersect with GRHL2, which remains to be tested.
In this report, we have demonstrated that GRHL2 enhances NK-target cell interaction by up-regulating ICAM-1 expression. GRHL2, and multiple IFN response genes, including ICAM-1, anti-correlated with EMT score. In this connection, lung cancers frequently express a soluble ICAM-1 decoy receptor, due to a splice site mutation, which correlates with worse prognosis 45, 77, suggesting that classical ICAM-1 suppresses tumor progression. Combined with our current data, a pivotal role for ICAM-1 expression in NK sensitivity of epithelial cells is indicated. We have also observed that the extracellular matrix can affect NK conjugation and target cell killing, suggesting that extracellular matrix may modulate the availability of ICAM on tumor cells for NK cell interaction (manuscript in preparation), an additional factor to consider. Note also that T-cells and NK cells kill target cells by very similar mechanisms, possibly extending this concept to T-cell-mediated tumor rejection as well 7, 78.
EMT may protect tumor cells against NK killing by additional and/or complementary mechanisms, as noted above. One of these is increased autophagy 1. We observed increased autophagic flux in our mesenchymal cell lines compared with GRHL2-expressing epithelial cells, but, in contrast with previous reports using other cell types, there was no effect of beclin-1 or ATG5 knockdown on NK sensitivity (data not shown), suggesting context-dependent effects of autophagy. It has not escaped our notice that GRHL2 also sensitized our cell lines to granzyme B and FASL, which would presumably kill target cells independently of ICAM-1. In itself, this suggests that ICAM-1 either plays a previously unsuspected role in apoptosis, or that additional, intrinsic mechanisms protect EMT-derived tumor cells against apoptosis. In this connection, we did not observe significant regulation of genes encoding the Bcl-2 family (except Bok, which did not, however, prove functionally important), the caspase family, the caspase-inhibitor (IAP) family, or CDK1. In addition to ICAM-1 up-regulation, GRHL2 may overcome a super-imposed, intrinsic resistance mechanism that has yet to be identified.
Pharmacologic intervention to up-regulate GRHL2 and/or induce MET can be achieved by Axl inhibition in relevant tumor cells 2, or, in principle, by cAMP-elevating agents 60, potentially useful therapeutic strategies in these contexts. ICAM-1 is up-regulated by γ-irradiation or the COX-2 inhibitor celexicob, with accompanying sensitization of tumor cells to NK killing 38, 72, suggesting novel strategies. In addition, EZH2 inhibitors, which compensate for the lack of UTX/KMT2C/D recruitment to promoters by suppressing H3K27me3 marks, can restore nearly-normal gene expression profiles can be restored in KMT2C-mutant tumor cells 83. The connection between NK sensitivity and KMT2C/D reported here suggests that EZH2 inhibitors may prove useful for restoring immune rejection of tumors.
Materials and Methods
Cell lines
MCF10a-derived, HMLE-derived and HT1080-derived cell lines were obtained and cultured as described previously 16, 17, 22, 63. MCF10aneoT cells were induced to undergo EMT by treatment with 20ng/ml TGF-β for three days, and then outgrowth in the absence of TGF-β. Mesenchymal cell lines (MCF10aneoT+ TGF-β, abbreviated as NTM, the Mesenchymal Subpopulation derived from HMLE, abbreviated as MSP, HT1080 fibrosarcoma cells and BT549 triple negative breast cancer cells) were reverted to an epithelial phenotype by retroviral expression of GRHL2 in the vector pMXS-IRES-puro, as described previously 16, 17, 22, 63, or by expression of mir200c (see below). NK92-MI cells 41 (American Type Culture Collection) were cultured in Alpha Minimum Essential medium without ribonucleosides and deoxyribonucleosides but with 2 mM L-glutamine and 1.5 g/L sodium bicarbonate (Life Technologies) containing 0.2 mM myo-inositol, 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid, 10% horse serum, 10% fetal bovine serum and 5 ng/ml IL-2 (R and D Systems). BT549 cells (grown in RPMI-1640+5% fetal bovine serum+10 μg/ml insulin+penicillin-streptomycin-glutamine) with inducible mir200c expression were constructed by subcloning the mir200c sequence into pTRIPZ, packaging lentiviruses, infecting BT549 cells and selecting for puromycin-resistance 67. The CRISPR/cas9 lentiviral constructs for knocking out human GRHL2 were kindly provided by Dr. B. Hogan 31, which were packaged in 293T cells and infected into target cell lines, as we described previously 63. GRHL2 knockout (>95%) was verified by Western blotting total puromycin-resistant populations; subcloning of knockout cells proved unnecessary. The CRISPR/cas9 mediated knockouts of KMT2C and KMT2D were generated by subcloning guide sequences (For KMT2C: M3CrE3-F: caccgGCGATCTGTGTCTGAGGAAT M3CrE3-R: a a a c ATT CCTCAG AC ACAG ATCG Cc. For KMT2D: M4CrE3-F: caccgTCCCGCTGCCCGTGTAGACT M4CrE3-R: aaacAGTCTACACGGGCAGCGGGAc) into the BsmBI sites of pLenti-Crispr-v2 or pLKO5.sgRNA.EFS.tRFP (Addgene). Lentiviruses were packaged and infected into MCF10a (subclone MCF10aP) cells as described, followed by sequential selection for puromycin-resistance and flow-sorting for RFP. Indels that caused reading frame shifts were identified by generating PCR fragments from genomic DNA, which were subcloned into pTOPOII (Life Technologies) and sequenced. MCF7 and MCF7-EMT cell lines 44 were kindly provided by Dr. Bruce White (University of Connecticut). HT1080+E1a cells were described previously 24. To generate target cells expressing nano-luciferase, cell lines were infected with the Nano-luciferase expressing lentiviral construct pLenti6.2-Nanoluc-ccdb (Addgene) and selected for blasticidin resistance (10 μg/ml).
NK cytotoxicity assays
Assay 1:
Target cells were plated in 12-well format at 3.5×l04 cells per well in triplicate. At least one day later, target cells were stained with 1μM CMFDA for 25 min in serum free medium, and washed 3x with complete medium. NK92-MI cells were added to wells in the appropriate target cell medium at 3:1 effector to target ratio. At the time points indicated, wells were gently washed 3x with complete medium, and cells were lysed in 300uL of 3% SDS solution; 100uL of this sample was read in a black opaque 96-well plate (492 excitation/530 emission) in a BioTek Synergy H1 Hybrid Reader. Signal from (+)NK wells divided by (−) NK wells to calculate viability.
Assay 2:
This assay was developed and validated for NK killing previously 68. Nano-luc expressing target cells (see above) were plated at 3.5×l04 cells in triplicate in a 12-well plate. The following day, NK92-MI cells were added in target cell medium, and, at the indicated time points, 25 μL aliquots of the culture supernatant were assayed using the Nano-Luc Assay System (Promega). To determine percentage of cells killed by NKs, a non-NK-treated well of target cells was lysed in 500 μL of Cell culture lysis buffer (Promega) to measure total nano-luciferase activity.
FASL cytotoxicity was assayed by plating cells at 4.0×l04 per well in triplicate in 12-well plate. The next day, Super-FASL (Enzo) was added to 50 ng/mL. At the indicated timepoints, cells were washed 2x with 1X PBS and assayed for caspase activation using the Caspase-glo system (Promega).
To assay cells for granzyme B cytotoxicity, target cells plated at 4.0×l04 cells per well in 12-well plate. The following day, cells were washed in serum-free medium. Cells were permeabilized to allow granzyme B entry using streptolysin-O (Sigma) at concentrations that were pre-optimized to produce low spontaneous death but promote granzyme B killing (typically, about 100 ng/ml)9. Granzyme B, purified as described previously from YT cells 81, was added to 400 ng/mL and wells were monitored for cell death. At the indicated time points, 0.1 volumes of Presto Blue Viability Reagent (Life Technologies) was added to wells and incubated 20min at 37° C. Aliquots (150 μL) were assayed for fluorescence (560nm excitation/ 590nm emission) in opaque, black 96-well plates. Presto blue signal from wells treated with streptolysin-O alone were used to calculate viability.
Rab25 promoter assay
The Rab25 promoter (~1200 bp) was subcloned into the XhoI-HindIII site of pGL3 basic using a genomic DNA fragment PCR amplified with Rab25-F1-Xho: ttaattCTCGAGTGTCAAGGAAGGGCAGAAGT and Rab25-R1-Hind: ttaattAAGCTTAGAGGACGGAAGCTGAGAAC. The reporter construct (1.25 μg) was co-transfected with 1.25 μg of TK-lacZ and the indicated amounts of GRHL2/pcDNA3.1 or empty pcDNA3.1 into HT1080 cells using Lipofectamine 2000 at 3:1 ratio. Thirty-six hours later, cells were lysed in Cell culture lysis buffer and assayed for luciferase and beta-galactosidase activity, using Promega assay reagents.
NK Conjugation Assay
Target cells were plated at 3.5×l04 cells/well in 12 well plate. 24hrs later, target cells were stained with 1 μM CMPTX in Serum Free medium for 25min and NK92-MI cells were stained with 1 μM CMFDA in serum Free medium for 25 minutes. Cells were washed 3X, then stained NK92-MI cells were resuspended and added to stained target cells in 3:1 effector/target ratio in the presence of 50 μM ZVAD-FMK. At time points, cells were gently washed 3x with complete medium to remove unconjugated NK92-MI cells, then fixed in 2% paraformaldehyde for 10 min and washed 3xX. The plates were then imaged at 10x and 20x magnification using an epifluorescence microscope (Zeiss Axiovert). GFP and Texas Red Channels were used to detect CMFDA and CMPTX stain, respectively. Images were then quantified for NK/target ratios using Image J software from the WVU Microscope Core Imaging Facility.
Yeast two-hybrid screening.
The yeast two-hybrid screen was performed by Hybrigenics, S.A., Paris, France, using proprietary methods.
qRT-PCR
Duplicate or triplicate total RNAs were prepared from cells in 60mm dishes using the Qiagen RNeasy Plus Mini kit. RNA (2.5 μg) were converted to single strand cDNA using the Superscript III kit (Life Technologies). One microliter of (3:1 diluted) cDNA was assayed in 20 uL reactions using the PowerUp SYBR green master mix (Applied Biosystems) on an Applied Biosystems 7500 thermocycler. GAPDH was used as the internal control for normalization.
The following primer sets were used:
| gene | Primer F | Primer R |
|---|---|---|
| DCN | agaagctctcctacatccgc | ccaagtgaagctccctcaga |
| SRGN | attttcccaccttgacacca | ttttatggccatgggaatat |
| ZEB1 | catacacctactcaactac | tttcatcctgatttccatt |
| Col8A1 | catttaccgccgagctaacc | tgtctgcgggttgtagttct |
| Snail | gaaaggccttcaactgcaaa | tgacatctgagtgggtctgg |
| Twist | cgggagtccgcagtctta | gcttgagggtctgaatcttg |
| Rab25-2 | cctaaccaagcaccagacct | gctgttctgtctctgcttgg |
| CDH1-2 | tgaagaaggaggcggagaag | ctcagactagcagcttcgga |
| ESRP1-2 | tttgaatccacgagcactgc | taagtccatcctcggttgca |
| MMP2-2 | aggaggagaaggctgtgttc | ctccagttaaaggcggcatc |
| TP63-2 | atgcccagactcaatttagtga | ttctgcgcgtggtctgtgt |
| OVOL2 | tgcaacgtctgcaataaagc | acgtgcaggtacaggtcctc |
| EPCAM | tgcagggtctaaaagctggt | ccctatgcatctcacccatc |
| ITGB6 | tgcgaccatcagtgaagaag | gacaaccccgatgagaagaa |
| CLDN4 | ccggccttatggtgatagtg | gcggagtaaggcttgtctgt |
| IRF6 | gtgaatgccatcttcttccttct | cccaggccaaatctccttct |
| ICAM1-2 | atacaacagcatttggggcc | ccacttcccctctcatcagg |
| Wnt5a | cccgatttagcagtgtcagc | tacacagtgccagtctcagg |
| RAET1L | cttctgttcctgctgttcgg | cgggtgtgactgtcttgttg |
| S100A4 | gagcaacttggacagcaaca | tcatttcttcctgggctgct |
| CD112-2 | ccatgtatgtgtgagctgcc | tttctctgggctcatgggtt |
| CD155-2 | tggggtgaaaatgtctgtgc | ggtttactccagcctccact |
| GAPDH | tgcaccaccaactgcttagc | ggcatggactgtggtcatgag |
Protein interactions
To test GRHL2 for interactions with KMT2 proteins in transiently transfected 293T, GRHL2-STAP was generated in pcDNA3.1. GRHL2-myc-SBP was PCR amplified from the vector pCeMM-CTAP containing GRHL2, Ligated to a synthetic oligonucleotide containing the S-tag (S-tag-F: tcgac GGC GGC AAG GAG ACC GCC GCT GCC AAG TTC GAG AGA CAG CAC ATG GAC TCC GGC GGC TAG S-tag-R: CTAGCCGCCGGAGTCCATGTGCTGTCTCTCGAACTTGGCAGCGGCGGTCTCCTTGCCGCCg),re-PCR amplified as a BglII-BglII fragment and subcloned into the BamHI site of pcDNA3.1.
This was co-transfected with FLAG-tagged KMT2C or KMT2D sequences corresponding to the minimal yeast two hybrid prey clones, which were generated using the following PCR primers:
(for KMT2D): K2DDQ-C-F: tttaat GCGGCCGCaATGcagcattccctactgtctgc
K2DDQ-C-R: TTAATT AGATCT ctactccttctgctgtttccggaC
(for KMT2C): K2C-C-F: atatat GCGGCCGCa atg acggaagtactgtctccaaattct
K2C-C-R: tatata ggatcc ctatggaaattcacgcccagcttT and subcloned into the vector p3XFLAG-CMV10 (Sigma).
Plasmids were co-transfected into 293T cells, which were lysed and subjected to pulldown/western blotting with S-protein agarose (Novagen) and FLAG antibody as described previously 63.
For mapping of the KMT2C/D interaction site on GRHL2, a recombinant fragments of KMT2C was subcloned into the BamHI-SalI site of pET30c using these primers: KMT2C-pET-F: Aattaaggatccaaacggaagtactgtctccaaatt ; KMT2C-pET-R: tatatactcgagctatactatggaaattcacgcccagctt. This 6X histidine-S-tag fragment was purified from E. coli BL21 DE3 using nickel-agarose. GST-GRHL2 fragments were subcloned into pGEX-6P3 and purified as described. Once the N-terminal transactivation domain of GRHL2 (cloned and expressed as previously described), two-amino acid point mutations (to ala-ala) were produced as GST fusion proteins in the pGEX-6p3 vector by the use of synthetic oligonucleotides (Geneblocks, Integrated DNA Technologies) containing the minimal KMT2C/D binding site and BamHI-SalI ends (e.g., the wild-type sequence was ggatcccctccattcaatacccgaagagcctacaccagtgaggatgaagcctggaagtcatacttg gagaatcccctgacagcagccaccaaggccatgatgagcattaatggtgatgaggacagt Gctgctgccctcggcctgctctatgactactacaaggttcctcgagacaagaggctgctgtaggtcgac)
RIME
RIME was carried out essentially as described 53. 6 × 107 MCF7 cells were subjected to immunoprecipitation with 10 μg of GRHL2 antibody (Sigma HPA004820) or 10 μg of rabbit IgG control. Three biological replicates were obtained from sequential passages of cells. For each replicate, proteins that appeared in the IgG controls were removed. Subsequently, a final list of GRHL2-interacting proteins was compiled by identifying those found in all 3 replicates.
Antibodies
| Antibody | Application | Company | Catalogue #/Ref. |
|---|---|---|---|
| E-Cadherin | Western Blot | BD Bioscience | 610181 |
| WDR5 | Western Blot | Cell Signaling | D9E1I |
| GRHL2 | CHIP/Western Blot | Sigma | AMab91226 |
| KMT2C | CHIP | J. Wysocka lab (Stanford) | 19 |
| FLAG | Western Blot | Sigma | F3165-1MG |
| ICAM1 | Western Blot | Santa Cruz | Sc-8439 |
| AKT (pan) | Western Blot | Cell Signaling | C67E7 |
CHIP
CHIP assays were performed as previously described, with the primers listed below.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Rab25 | GCTGTCTCTGAAGGTCCTGT | AGAGGACGGAAGCTGAGAAC |
| CDH1 (5’) | CCCCTATCAGTTAGCACCGT | AGGTAGGGTAGAGGAGGCTC |
| CDH1 (intron 2) | GGTTCAAAAGATCCCCTGCG | GTCCCCTTTCCTAAGCCACA |
| CLDN4 | TGGATGGACGGGTTTAGAGG | AGGACTCAGAGGGGATCAGT |
| OVOL2 | TGCCTTAAATCGCGAGTGAG | CAGGTAGCGAGCTTGTTGAC |
| TP63 | GGGGTGGGTGGTAAGGTATT | TTGGAAAGAACACTGCCTGC |
| ICAM1 | ATAAAGGATCACGCGCCCC | AGCCCCTCCTTCCCATAAAC |
Bioinformatics and statistics
Retrieval and analysis of the expression levels and EMT scores using publicly available data for lung cancer were achieved as follows. Raw expression values (counts) and Z-scores were retrieved for LUAD TCGA (provisional, freeze as of June 20,2018) dataset using cBioPortal 29. EMT scores were calculated from the signature described in 32 and used without normalization. For normalization of IFN response genes in figure 6 to the IFN-γ transcript levels, raw expression data were used (only samples with a detectable (non-zero) counts). For all samples spanning the EMT continuum, the high and low subsets were defined as top and bottom quartiles of the scores for each EMT signature. For each gene of interest, the subsets of samples with high and low mRNA expression (normalized for IFN-γ transcript levels) were also defined as top and bottom quartiles of the corresponding expression range. Statistical significance for the co-occurrence between EMT subsets and expression subsets was calculated based on binominal distribution; P-values were adjusted for multiple comparisons using the Benjamini-Hochberg method.
Supplementary Material
Highlights.
- --Grainyhead-like-2 is a master programmer of the epithelial phenotype
- GRHL2 provides an unprecedented opportunity to discover mechanisms by which the enforcement of an epithelial phenotype affects tumor cell sensitivity to immune-mediated cytotoxicity
- GRHL2 sensitized cells to NK killing at the level of NK-target cell synaptogenesis
- --This effect was mediated by the up-regulation of ICAM1
- GRHL2 protein interacted directly with the histone methyltransferases, KMT2C and KMT2D
- GRHL2-KMT2C/D interactions and p300 inhibition were found to be important for MET, ICAM1 up-regulation and NK- sensitization effects
- ICAM1 anti-correlated with EMT score significantly in lung cancer
Acknowledgements
We wish to thank Erica Golemis for supporting the bioinformatics work, Ms. Brittney Rogers for technical assistance, Joanne Wysocka (Stanford) for the KMT2C antibody, Amanda Ammer for imaging assistance, Paolo Fagone for preparing the KMT2C and KMT2D recombinant protein fragments and Jason Carroll for supporting the RIME experiment. We appreciate the additional bioinformatics help from Dr. James Denvir. Core facilities were partiallly supported by the NIGMSU54GM104942, to WVU and MCRCC COBRE grant GM103488. Transition grant support (to S.M.F. and I.M.) was provided by the WVUHSC Office of Research and Graduate Education.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of potential conflicts of interest
There are no conflicts of interest to disclose.
References
- 1.Akalay I, Janji B, Hasmim M, Noman MZ, Andre F, De Cremoux P et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer research 2013; 73: 2418–2427. [DOI] [PubMed] [Google Scholar]
- 2.Antony J, Huang RY. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer research 2017; 77: 3725–3732. [DOI] [PubMed] [Google Scholar]
- 3.Aqbi HF, Wallace M, Sappal S, Payne KK, Manjili MH. IFN-gamma orchestrates tumor elimination, tumor dormancy, tumor escape, and progression. J Leukoc Biol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arina A, Murillo O, Dubrot J, Azpilikueta A, Alfaro C, Perez-Gracia JL et al. Cellular liaisons of natural killer lymphocytes in immunology and immunotherapy of cancer. Expert Opin Biol Ther 2007; 7: 599–615. [DOI] [PubMed] [Google Scholar]
- 5.Aue A, Hinze C, Walentin K, Ruffert J, Yurtdas Y, Werth M et al. A Grainyhead-Like 2/Ovo-Like 2 Pathway Regulates Renal Epithelial Barrier Function and Lumen Expansion. Journal of the American Society of Nephrology : JASN 2015; 26: 2704–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basingab FS, Ahmadi M, Morgan DJ. IFNgamma-Dependent Interactions between ICAM-1 and LFA-1 Counteract Prostaglandin E2-Mediated Inhibition of Antitumor CTL Responses. Cancer Immunol Res 2016. [DOI] [PubMed] [Google Scholar]
- 8.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer 2018; 18: 128–134. [DOI] [PubMed] [Google Scholar]
- 9.Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Molecular and cellular biology 1999; 19: 8604–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013; 19: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev 2016; 35: 645–654. [DOI] [PubMed] [Google Scholar]
- 12.Chen AF, Liu AJ, Krishnakumar R, Freimer JW, DeVeale B, Blelloch R. GRHL2-Dependent Enhancer Switching Maintains a Pluripotent Stem Cell Transcriptional Subnetwork after Exit from Naive Pluripotency. Cell Stem Cell 2018; 23: 226–238 e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014; 5: 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol 2014; 41: 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT, Yang Z et al. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Scientific reports 2016; 6: 19943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cieply B, Riley Pt, Pifer PM, Widmeyer J, Addison JB, Ivanov AV et al. Suppression of the Epithelial-Mesenchymal Transition by Grainyhead-like-2. Cancer research 2012; 72: 2440–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cieply B, Farris J, Denvir J, Ford HL, Frisch SM. Epithelial-Mesenchymal Transition and Tumor Suppression Are Controlled by a Reciprocal Feedback Loop between ZEB1 and Grainyhead-like-2. Cancer research 2013; 73: 6299–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer research 2017; 77: 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N et al. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Molecular cell 2017; 66: 568–576 e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6: 836–848. [DOI] [PubMed] [Google Scholar]
- 21.Fantini D, Glaser AP, Rimar KJ, Wang Y, Schipma M, Varghese N et al. A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene 2018; 37: 1911–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farris JC, Pifer PM, Zheng L, Gottlieb E, Denvir J, Frisch SM. Grainyhead-like 2 Reverses the Metabolic Changes Induced by the Oncogenic Epithelial-mesenchymal Transition: Effects on Anoikis. Mol Cancer Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francart ME, Lambert J, Vanwynsberghe AM, Thompson EW, Bourcy M, Polette M et al. Epithelial-mesenchymal plasticity and circulating tumor cells: Travel companions to metastases. Dev Dyn 2018; 247: 432–450. [DOI] [PubMed] [Google Scholar]
- 24.Frisch SM. E1a induces the expression of epithelial characteristics. The Journal of cell biology 1994; 127: 1085–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frisch SM. The epithelial cell default-phenotype hypothesis and its implications for cancer. Bioessays 1997; 19: 705–709. [DOI] [PubMed] [Google Scholar]
- 26.Frisch SM, Mymryk JS. Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol 2002; 3: 441–452. [DOI] [PubMed] [Google Scholar]
- 27.Frisch SM, Farris JC, Pifer PM. Roles of Grainyhead-like transcription factors in cancer. Oncogene 2017; 36: 6067–6073. [DOI] [PubMed] [Google Scholar]
- 28.Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene 2017; 627: 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016; 167: 397–404 e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X, Bali AS, Randell SH, Hogan BL. GRHL2 coordinates regeneration of a polarized mucociliary epithelium from basal stem cells. The Journal of cell biology 2015; 211: 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibbons DL, Creighton CJ. Pan-cancer survey of epithelial-mesenchymal transition markers across the Cancer Genome Atlas. Dev Dyn 2018; 247: 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene LI, Bruno TC, Christenson JL, D'Alessandro A, Culp-Hill R, Torkko K et al. A Role for Tryptophan-2,3-dioxygenase in CD8 T Cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol Cancer Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601. [DOI] [PubMed] [Google Scholar]
- 35.Hamilton DH, Huang B, Fernando RI, Tsang KY, Palena C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer research 2014; 74: 2510–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017; 168: 542. [DOI] [PubMed] [Google Scholar]
- 37.Jacobs J, Atkins M, Davie K, Imrichova H, Romanelli L, Christiaens V et al. The transcription factor Grainy head primes epithelial enhancers for spatiotemporal activation by displacing nucleosomes. Nat Genet 2018; 50: 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong JU, Uong TNT, Chung WK, Nam TK, Ahn SJ, Song JY et al. Effect of irradiation-induced intercellular adhesion molecule-1 expression on natural killer cell-mediated cytotoxicity toward human cancer cells. Cytotherapy 2018; 20: 715–727. [DOI] [PubMed] [Google Scholar]
- 39.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell reports 2016; 17: 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol 2018; 3. [DOI] [PubMed] [Google Scholar]
- 41.Klingemann H, Boissel L, Toneguzzo F. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. Front Immunol 2016; 7: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knutson KL, Lu H, Stone B, Reiman JM, Behrens MD, Prosperi CM et al. Immunoediting of cancers may lead to epithelial to mesenchymal transition. J Immunol 2006; 177: 1526–1533. [DOI] [PubMed] [Google Scholar]
- 43.Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest 2014; 124: 2668–2682. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Kondaveeti Y, Guttilla Reed IK, White BA. Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer letters 2015; 364: 44–58. [DOI] [PubMed] [Google Scholar]
- 45.Kotteas EA, Gkiozos I, Tsagkouli S, Bastas A, Ntanos I, Saif MW et al. Soluble ICAM-1 levels in small-cell lung cancer: prognostic value for survival and predictive significance for response during chemotherapy. Medical oncology 2013; 30: 662. [DOI] [PubMed] [Google Scholar]
- 46.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer cell 2009; 15: 195–206. [DOI] [PubMed] [Google Scholar]
- 47.Lagrue K, Carisey A, Oszmiana A, Kennedy PR, Williamson DJ, Cartwright A et al. The central role of the cytoskeleton in mechanisms and functions of the NK cell immune synapse. Immunol Rev 2013; 256: 203–221. [DOI] [PubMed] [Google Scholar]
- 48.Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer cell 2017; 32: 135–154. [DOI] [PubMed] [Google Scholar]
- 49.Maccalli C, Parmiani G, Ferrone S. Immunomodulating and Immunoresistance Properties of Cancer-Initiating Cells: Implications for the Clinical Success of Immunotherapy. Immunol Invest 2017; 46: 221–238. [DOI] [PubMed] [Google Scholar]
- 50.McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacology & therapeutics 2015; 150: 1–22. [DOI] [PubMed] [Google Scholar]
- 51.Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 2016; 165: 272–275. [DOI] [PubMed] [Google Scholar]
- 52.Miura TA, Cook JL, Potter TA, Ryan S, Routes JM. The interaction of adenovirus E1A with p300 family members modulates cellular gene expression to reduce tumorigenicity. J Cell Biochem 2007; 100: 929–940. [DOI] [PubMed] [Google Scholar]
- 53.Mohammed H, Taylor C, Brown GD, Papachristou EK, Carroll JS, D'Santos CS. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc 2016; 11: 316–326. [DOI] [PubMed] [Google Scholar]
- 54.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 2016; 16: 7–19. [DOI] [PubMed] [Google Scholar]
- 55.Muller L, Aigner P, Stoiber D. Type I Interferons and Natural Killer Cell Regulation in Cancer. Front Immunol 2017; 8: 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pahl J, Cerwenka A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2017; 222: 11–20. [DOI] [PubMed] [Google Scholar]
- 57.Paltoglou S, Das R, Townley SL, Hickey TE, Tarulli GA, Coutinho I et al. Novel Androgen Receptor Coregulator GRHL2 Exerts Both Oncogenic and Antimetastatic Functions in Prostate Cancer. Cancer research 2017; 77: 3417–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016; 16: 131–144. [DOI] [PubMed] [Google Scholar]
- 59.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M et al. Identification of essential genes for cancer immunotherapy. Nature 2017; 548: 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016; 351: aad3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pattabiraman DR, Weinberg RA. Targeting the Epithelial-to-Mesenchymal Transition: The Case for Differentiation-Based Therapy. Cold Spring Harb Symp Quant Biol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016; 7: 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pifer PM, Farris JC, Thomas AL, Stoilov P, Denvir J, Smith DM et al. Grainyhead-like 2 inhibits the coactivator p300, suppressing tubulogenesis and the epithelial-mesenchymal transition. Molecular biology of the cell 2016; 27: 2479–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016; 352: aad9780. [DOI] [PubMed] [Google Scholar]
- 65.Puisieux A, Pommier RM, Morel AP, Lavial F. Cellular Pliancy and the Multistep Process of Tumorigenesis. Cancer cell 2018; 33: 164–172. [DOI] [PubMed] [Google Scholar]
- 66.Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M et al. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer 2015; 112: 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rogers T, Christenson JL, Greene LI, O”Neill KI, Williams MM, Gordon MA et al. Reversal of Triple-negative breast cancer EMT by mir-200c decreases tryptophan catabolism and a program of immune-suppression. Mol Cancer Res 2018; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rossignol A, Bonnaudet V, Clemenceau B, Vie H, Bretaudeau L. A high-performance, non-radioactive potency assay for measuring cytotoxicity: A full substitute of the chromium-release assay targeting the regulatory-compliance objective. MAbs 2017; 9: 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sangaletti S, Tripodo C, Santangelo A, Castioni N, Portararo P, Gulino A et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell reports 2016; 17: 233–248. [DOI] [PubMed] [Google Scholar]
- 70.Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer research 2009; 69: 2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell States in the breast. Cell 2011; 145: 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schellhorn M, Haustein M, Frank M, Linnebacher M, Hinz B. Celecoxib increases lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Oncotarget 2015; 6: 39342–39356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S et al. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin Cancer Res 2017; 23: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018; 554: 538–543. [DOI] [PubMed] [Google Scholar]
- 75.Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB et al. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology 2017; 6: e1271858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery JP et al. New insights into the role of EMT in tumor immune escape. Mol Oncol 2017; 11: 824–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thanopoulou E, Kotzamanis G, Pateras IS, Ziras N, Papalambros A, Mariolis-Sapsakos T et al. The single nucleotide polymorphism g.1548A >G (K469E) of the ICAM-1 gene is associated with worse prognosis in non-small cell lung cancer. Tumour Biol 2012; 33: 1429–1436. [DOI] [PubMed] [Google Scholar]
- 78.Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 2009; 128: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017; 355: 1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Varga J, Greten FR. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat Cell Biol 2017; 19: 1133–1141. [DOI] [PubMed] [Google Scholar]
- 81.Veugelers K, Motyka B, Frantz C, Shostak I, Sawchuk T, Bleackley RC. The granzyme B-serglycin complex from cytotoxic granules requires dynamin for endocytosis. Blood 2004; 103: 3845–3853. [DOI] [PubMed] [Google Scholar]
- 82.Viry E, Baginska J, Berchem G, Noman MZ, Medves S, Chouaib S et al. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014; 10: 173–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med 2018; 24: 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. Natural killer cell-produced IFN-gamma and TNF-alpha induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol 2012; 91: 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang SP, Tang Z, Chen CW, Shimada M, Koche RP, Wang LH et al. A UTX-MLL4-p300 Transcriptional Regulatory Network Coordinately Shapes Active Enhancer Landscapes for Eliciting Transcription. Molecular cell 2017; 67: 308–321 e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol 2015; 6: 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yilmaz AS, Ozer HG, Gillespie JL, Allain DC, Bernhardt MN, Furlan KC et al. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer 2017; 123: 1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.