

Antibiotic resistance genes (ARGs) are frequently detected with high abundance in manure-applied soils. Anaerobic digestion is one of widely used processes for animal waste treatment. Thus, it is critical to understand the potential of anaerobic digestion to attenuate ARGs. Although some previous studies recommended thermophilic digestion for ARG removal, they did not get sufficient evidence to support this view. The antibiotics applied to animals are mostly excreted through feces and urine because of incomplete metabolism. It is indispensable to know whether residual antibiotics in manure will hinder ARG attenuation in anaerobic digesters. The significance of our research is in comprehensively understanding the evolution and mechanism of ARGs in anaerobic digestion of swine manure affected by temperature and residual antibiotics, which will allow the development of an ARG elimination strategy before their release into the environment.
KEYWORDS: anaerobic digestion, antibiotic resistance genes, high-throughput quantitative PCR, swine manure
ABSTRACT
This study employed high-throughput quantitative PCR and 16S rRNA sequencing to evaluate the effect of temperature and residual antibiotics on the dynamics of antibiotic resistance genes (ARGs) and microbial communities during anaerobic digestion of swine manure. The abundances of total ARGs and 16S rRNA genes significantly decreased in all of four treatments (25°C, 37°C, and 37°C with 50 mg of wet weight antibiotics of body weight, and 55°C). The abundances of most ARG types were significantly correlated with those of the 16S rRNA gene and transposase gene (P < 0.01). However, the abundances of total ARGs at 55°C were much higher than those of other treatments. Meanwhile, the microbial communities at 55°C, where the Streptococcus pathogen remained at a relatively high abundance and cellulose degraders and hydrogen producers, such as Ethanoligenens and Coprococcus bacteria, increased, were markedly different from those of other treatments. Redundancy analysis indicates that temperature, pH, and the genus Streptococcus had the highest explanation for ARG variation among experimental factors, chemical properties, and representative genera, respectively. Network analysis further showed that the genus Streptococcus contributed greatly to the higher ARG abundance at 55°C. The moderate antibiotic residue only caused a slight and transitory inhibition for microbially diverse populations and promotion for ARG abundance, probably due to the degradation of antibiotics and microbial adaptability. Our results clarify the cooperativity of gene transfer-related items on ARG variation and intensively prove that higher temperature cannot always achieve better ARG removal in anaerobic digestion unless pathogens and gene transfer elements are more efficiently inhibited.
IMPORTANCE Antibiotic resistance genes (ARGs) are frequently detected with high abundance in manure-applied soils. Anaerobic digestion is one of widely used processes for animal waste treatment. Thus, it is critical to understand the potential of anaerobic digestion to attenuate ARGs. Although some previous studies recommended thermophilic digestion for ARG removal, they did not get sufficient evidence to support this view. The antibiotics applied to animals are mostly excreted through feces and urine because of incomplete metabolism. It is indispensable to know whether residual antibiotics in manure will hinder ARG attenuation in anaerobic digesters. The significance of our research is in comprehensively understanding the evolution and mechanism of ARGs in anaerobic digestion of swine manure affected by temperature and residual antibiotics, which will allow the development of an ARG elimination strategy before their release into the environment.
INTRODUCTION
Since antibiotics are commonly used in the livestock industry in developing countries, animal waste has become an important reservoir of antibiotic resistance genes (ARGs). ARGs receive more public concern because they can transfer among different hosts by horizontal gene transfer (HGT) and pose a risk to human health (1). Evidence suggests that the long-term application of manure and manure-derived fertilizer can increase antibiotic resistance in agricultural soils by gene transfer from intestinal bacteria to indigenous counterparts (1, 2). These findings highlight the need to critically evaluate the potential of manure treatment processes to attenuate ARGs and limit their spread to the environment.
Prior to land application, biotechnologies such as aerobic composting and anaerobic digestion have been broadly employed to treat animal waste. Of them, anaerobic digestion not only degrades organic matter and removes pathogens effectively (3, 4) but also produces renewable energy economically (5, 6). Recent studies have begun to investigate the fate of ARGs during anaerobic digestion of manure (7–9). In anaerobic digestion, temperature is a critical variable which can affect the composition and function of the microbial community (10). The effect of temperature in anaerobic digestion on ARG variation has been conducted mostly for wastewater sludge (11–13) and rarely for livestock manure (7). Although some previous studies recommended thermophilic digestion for ARG attenuation, thermophilic digestion did not always get better ARG removal than mesophilic condition. A recent solid-state anaerobic digestion experiment found 23.7% higher ARGs in the thermophilic cattle manure digestion product than those in the mesophilic product (14). The reason for different ARG subtypes responding differently to digestion temperature remains unclear.
Most of the applied antibiotics are excreted through feces and urine because of incomplete metabolism and have been detected up to more than 100 milligrams per kilogram of manure (15). Previous investigations have found significant correlations between the concentration of antibiotics and ARGs in manure and compost samples (1, 16). Tylosin-amended soils have showed higher abundances of ARGs than soils treated with manure only (17). However, whether residual antibiotics in manure could act as a selective pressure for ARG proliferation in anaerobic digesters requires further investigation.
Sorensen et al. (18) suggested that the horizontal gene transfer between different bacterial hosts via mobile gene elements and vertical gene transfer due to the reproduction of bacterial hosts are two main processes of ARG proliferation. For horizontal gene transfer, integrons associated with transposons and plasmids carry gene cassettes coding for antibiotic resistance and are involved in spreading resistance (19). For vertical gene transfer, bacterial communities strongly affected the dynamics of ARGs during composting (20, 21) and anaerobic digestion (7). Bacterial abundance, usually presented by 16S rRNA gene copy, also contributes to the abundance of ARGs. It is generally accepted that although thermophilic condition decreases the diversity of bacterial communities in anaerobic digestion, it can enhance the degradation of organic matter and promote the growth of some microbial taxa, including, for example, those involved in biogas production (5, 22). Therefore, to interpret ARG variation during anaerobic digestion, the dynamics of horizontal gene transfer and vertical gene transfer-related items should be fully examined.
So far, conventional quantitative PCR is most frequently used for ARG quantification. However, this technology is time-consuming and costly to quantify many ARGs. That is why previous studies usually investigated a few ARGs. Although metagenomic analysis can cover all the ARGs that have been deposited in the Comprehensive Antibiotic Research Database (CARD), the costs of large-scale metagenomics are still prohibitive (13). Recently, high-throughput quantitative PCR (HT-qPCR) targeting almost all major classes of ARGs and mobile genetic elements (MGEs) have been gradually introduced to quantify ARGs in different environmental samples, including manure, compost, and soil (1, 20).
In this study, HT-qPCR was employed to characterize the dynamics of the ARG profile in anaerobic digestion of swine manure affected by different temperatures and moderate antibiotic residues (50 mg/kg). Meanwhile, the shift of microbial communities was comprehensively explored using Illumina sequencing of 16S rRNA gene. The contribution of experimental conditions, chemical properties, and microbial taxa to the variation of ARGs and the cooccurrence patterns between ARGs and microbial community were analyzed thereafter.
RESULTS
Abundance of 16S rRNA genes and ARGs.
Anaerobic digestion significantly decreased 16S rRNA gene abundance for all the treatments (P < 0.05) (Fig. 1A, right y axis). The 16S rRNA abundance in T55 was, however, significantly higher than those in other treatments on day 14 and significantly higher than that in T37A on day 30 (P < 0.05) (where T and A indicate the incubation temperature and antibiotic spiking, respectively). Antibiotic spiking did not significantly affect the abundance of 16S rRNA genes.
FIG 1.

Abundance of ARGs, MGEs, and 16S rRNA genes detected in manure slurry on days 0, 14, and 30. (Top) Left y axis indicates the relative copy numbers of MGEs and ARGs normalized to bacterial cell; right y axis indicates 16S rRNA gene abundance. (Bottom) The absolute copy numbers of MGEs and ARGs. In sample name, the letters “D,” “T,” and “A” indicate day, temperature, and antibiotics, respectively. The lowercase letters were used to indicate the differences among 16S rRNA data revealed by one-way analysis of variance (ANOVA) after multiple comparison.
The normalized ARG abundance also decreased during anaerobic digestion, in which the aminoglycoside, tetracycline, and macrolide-lincosamide-streptogramin B (MLSB) resistance genes were three dominant types (Fig. 1A). It is unanticipated that the normalized ARG abundance in T55 was also higher than those in other treatments on day 30. The normalized ARG abundance in T37A was higher than that in T37 on day 14, but they were comparable on day 30.
Calculated from ARG relative abundance multiplying with 16S rRNA abundance (equation 5), the ARG abundance per milliliter slurry significantly decreased in all treatments and were much higher in T55 than those in other treatments on days 14 and 30 (Fig. 1B). Besides abundance, the number of ARGs that can be detected in T55 was also higher than that of other treatments on day 30 (see Fig. S1 in the supplemental material).
Consistent with the trend of total ARG abundance, most ARG types decreased their abundances on day 14 and more seriously on day 30, except for vancomycin which was detected with low abundance (see Fig. S2 in the supplemental material). In addition, the sulfonamide, florfenicol-chloramphenicol-amphenicol-fluoroquinolone-quinolone (FCA), multidrug, MLSB, tetracycline, aminoglycoside, and other resistance genes had the lowest removal efficiencies in T55 compared with other treatments on days 14 and 30. It is noteworthy that among MGEs, the transposase genes decreased their abundances as most ARG types, whereas the integrase genes (intI) increased and were more abundant in T55 than in other treatments.
Characterization of microbial community.
A total of 7,853,250 high-quality sequences were obtained from 63 samples, with sequences per sample ranging from 43,511 to 212,575. At the 3% dissimilarity level, the observed species ranged from 1,337 to 2,109 per sample with all the reads normalized to 43,460. The alpha diversity indices of microbial communities in T55 decreased on day 3 and rebounded thereafter; a similar phenomenon but a weaker inhibition was observed for T37A (see Fig. S3 in the supplemental material). The overall shift pattern of microbial communities in all the treatments was driven by the sampling day; however, at the later stage, the microbial communities in T55 differed tremendously from those in other treatments (see Fig. S4 in the supplemental material).
There were 25 bacterial phyla and 3 archaeal phyla detected during anaerobic digestion, and Firmicutes was the most dominant phylum, with the detection frequencies ranging from 77.54% to 89.98% (see Fig. S5 in the supplemental material). No significant difference was observed for the same microbial phylum among the four treatments. However, the dominant genera in Firmicutes in T55 showed some differences compared with other treatments (Fig. 2). Ethanoligenens bacteria gradually increased in relative abundance and became the fourth dominant genus in T55. Coprococcus and Tepidimicrobium bacteria, two less abundant genera in manure, also were enriched in anaerobic digestion at 55°C. Streptococcus bacteria, the most dominant genus in manure, decreased the relative abundance from 35.42% to less than 3.5% in T25, T37, and T37A. Although this genus also decreased in T55, its relative abundance remained 22.12% in the end. Correspondingly, three genera (Clostridium, SMB53, and an unknown) belonging to family Clostridiaceae in T55 did not increase as much as those in other treatments.
FIG 2.

Average percentages of dominant genera belonging to phylum Firmicutes in manure slurry during anaerobic digestion. Genera with frequencies higher than 3% in at least one treatment are shown. In sample name, the letters “D,” “T,” and “A” indicate day, temperature, and antibiotics, respectively.
Figure S6 in the supplemental material shows the potential pathogenic bacteria detected in this study based on the information provided online (http://www.mgc.ac.cn/cgi-bin/VFs/jsif/main.cgi). In addition to the above-mentioned genera Streptococcus and Clostridium, Corynebacterium and Escherichia were the other two genera, with a frequency of >1% in the original slurry. Both of them decreased in all the treatments during anaerobic digestion.
Factors contributing to variation of ARGs.
Spearman correlation indicated that the abundances of most ARG types were significantly correlated with those of 16S rRNA genes and transposase genes, except that the abundance of vancomycin-resistant genes was significantly correlated with that of integrase gene (P < 0.01) (Table 1).
TABLE 1.
Spearman correlation between the abundances of ARG types and those of the 16S rRNA gene, integrase gene, and transposase gene
| ARG type | Spearman correlation of gene: |
||
|---|---|---|---|
| 16S rRNA | intI | Tn | |
| Aminoglycoside | 0.918a | 0.353 | 0.737a |
| Beta-lactam | 0.489a | 0.216 | 0.813a |
| FCA | 0.918a | 0.303 | 0.755a |
| MLSB | 0.885a | 0.305 | 0.684a |
| Multidrug | 0.914a | 0.318 | 0.714a |
| Other | 0.916a | 0.254 | 0.632a |
| Sulfonamide | 0.880a | 0.313 | 0.568a |
| Tetracycline | 0.903a | 0.339 | 0.714a |
| Vancomycin | 0.331 | 0.691a | 0.172 |
| Total | 0.926a | 0.33 | 0.745a |
Correlation is significant at the 0.01 level (2 tailed).
Table S1 in the supplemental material shows the detected chemical properties of samples in different treatments on days 0, 14, and 30. The pH values were reduced slightly in T25 and more deeply in T37 and T37A but kept relatively constant in T55. NO3− concentrations were reduced by more than 50%, whereas NH4+ concentrations almost doubled in all treatments. Similar trends were observed for total suspended solids (TSS) and volatile suspended solids (VSS), and the VSS/TSS ranged from 0.70 to 0.77. In T37A, the concentrations of oxytetracycline (OTC), sulfamethazine (SM2), and ciprofloxacin (CIP) decreased during anaerobic digestion, with their removal efficiencies up to 69.0%, 65.0%, and 37.0%, respectively (see Fig. S7 in the supplemental material). The water-soluble portion of SM2 was much higher than those of OTC and CIP.
Redundancy analysis (RDA) indicates that three experimental factors (temperature, antibiotics, and sampling day), five chemical properties (pH, NH4+, NO3−, TSS, and VSS), and four representative genera (Streptococcus, Clostridiaceae SMB53, Clostridiaceae unknown, and Ethanoligenens) showed significant contributions to the variation of ARGs (P < 0.01) (Table 2). Temperature, pH, and Streptococcus had the highest explanation among experimental factors, chemical properties, and representative genera, respectively.
TABLE 2.
The explanation and P value of experimental design, slurry chemical properties, and representative genera that showed significant correlations with the relative abundance of ARGs evaluated by redundancy analysis
| Name | Explanation (%) | Pseudo-F statistic | P value |
|---|---|---|---|
| Expt design | |||
| Temperature | 15.5 | 4 | 0.002 |
| Antibiotics | 11.8 | 2.9 | 0.003 |
| Sampling day | 14 | 3.6 | 0.002 |
| Chemical properties | |||
| pH | 13.3 | 3.4 | 0.002 |
| NH4+ | 10.5 | 2.6 | 0.006 |
| NO3− | 9.4 | 2.3 | 0.014 |
| TSS | 10.7 | 2.6 | 0.006 |
| VSS | 10.4 | 2.6 | 0.008 |
| Representative genera | |||
| Streptococcus | 18.8 | 5.1 | 0.001 |
| Clostridiaceae SMB53 | 16.6 | 4.4 | 0.002 |
| Clostridiaceae unknown | 14.9 | 3.9 | 0.001 |
| Ethanoligenens | 12.1 | 3 | 0.006 |
Cooccurrence of ARGs and microbial communities.
A mantel test showed that there was a significant correlation between ARG profiles and microbial community (at the operational taxonomic unit [OTU] level) based on Bray-Curtis distance (r = 0.38, P < 0.001). A Procrustes test visually displayed the relationship between ARG relative abundance and 16S rRNA gene OTUs of each sample and presented a goodness-of-fit test (sum of squares M2 = 0.5533, r = 0.6683, P < 0.0001) on the basis of Bray-Curtis dissimilarity metrics and 9,999 permutations (Fig. 3). For both ARGs and microbial communities, two samples in T55, namely, the original sample (D0) and the sample in T25 on day 14, clustered closely to each other, while other samples clustered more closely.
FIG 3.
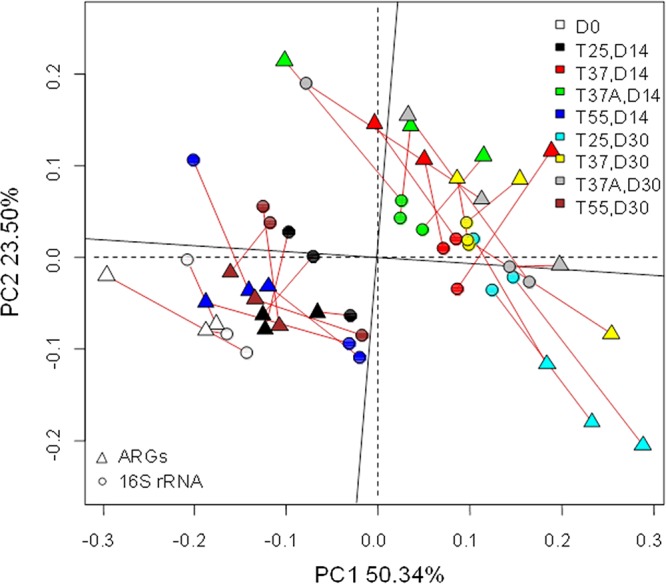
The significant correlation between ARG relative abundance and 16S rRNA gene OTUs revealed by Procrustes test on the basis of Bray-Curtis dissimilarity metrics and 9,999 permutations (sum of squares, M2 = 0.5533, r = 0.6683, P < 0.0001).
Network analysis was employed to find out the potential genera responsible for different relative abundances of ARGs among the four treatments. The microbial genera with a detection frequency of >0.01% in at least one treatment and the ARGs with a minimum cycle threshold (CT) value of ≤21 and higher relative abundance in T37, T37A, and T55 than in T25 (see Table S2 in the supplemental material) were selected for Spearman correlation analysis. A network consisting of 64 genera (source) and 35 ARGs (target) was thereafter constructed based on positive and significant correlations (P < 0.01) (Fig. 4). Five subtypes of MLSB resistance genes, 2 subtypes of tetracycline resistance genes, and 1 subtype of aminoglycoside and sulfonamide resistance genes had more than 20 sources and formed the center of the network. Meanwhile, 17 genera had over 10 target ARGs (see Table S3 in the supplemental material). Interestingly, the ARGs these genera strongly correlated with (ρ > 0.8) were no other than those in the center of the network and focused on MLSB and tetracycline resistance genes. Among these genera, Streptococcus had a maximum detection frequency up to 40.42%, with 16 significantly correlated target ARGs (P < 0.01) and 7 strongly correlated target ARGs (ρ > 0.8, P < 0.01). More importantly, Streptococcus bacteria were much more abundant in T55 than other treatments on day 30 (Fig. 2). Therefore, it is very likely that Streptococcus bacteria made a great contribution to higher ARG abundance in T55.
FIG 4.

Network analysis showing the significant correlation between 64 microbial genera (source) and 35 ARGs (target) (P < 0.01). The genera with frequency of >0.01% in at least one treatment, and the ARGs with minimum CT value of ≤21 and higher relative abundance in T55 and T37 than in T25 (Table S3) were involved in network analysis. The size of nodes is proportioned to the number of connections (the degree). The ARGs with connections more than 20 are labeled with primer name. The genera with more than 10 connections are labeled with code number and characterized in Table S4.
DISCUSSION
As a rapid and sensitive method, HT-qPCR covers a number of ARGs conferring resistance to almost all major classes of antibiotics to provide comprehensive profiles of ARGs. In the present study, HT-qPCR was introduced to evaluate the impact of temperature and residual antibiotics on the dynamics of ARGs during anaerobic digestion of swine manure. Consistent with those observed in previous studies, most ARG types gradually decreased their abundances in four different treatments.
However, thermophilic digestion did not achieve better removal of either relative or absolute abundance of total ARGs (Fig. 1), which is different from some previous investigations using conventional qPCR (7, 11). Three horizontal and vertical transfer-related items, including microbial abundance and composition and mobile genetic elements, were deeply discussed to explain this phenomenon.
First, the relatively high 16S rRNA gene copies in T55 indicated that there was higher bacterial abundance and activity in thermophilic digestion of swine manure (Fig. 1A). Microbial abundance, indicated by 16S rRNA gene copy number, is a direct contributor to the abundance of ARGs according to equation 5. That is why the abundances of most ARG types showed significant correlations with those of the 16S rRNA gene (Table 1). It should be mentioned that high temperature sometimes can promote microbial biomass in an anaerobic digestion system. A recent study also found higher 16S rRNA gene abundance in thermophilic digestion of dairy manure than in mesophilic and moderate conditions (7). Moset et al. (5) suggested that thermophilic anaerobic digestion of cattle manure showed higher pH, organic matter degradation (especially fiber), and methane yield than mesophilic conditions. This study used fresh swine manure as the sole substrate. Although the methanogen (mainly included in Euryarchaeota) was less abundant in all the treatments probably because of the high ammonia concentration (23), some genera, for example Ethanoligenens and Coprococcus which were involved in cellulose degradation or hydrogen production (24), increased their abundances during thermophilic digestion (Fig. 2).
Second, although decreased during anaerobic digestion in all the treatments, the total abundance of pathogenic bacteria was much higher in T55 (28.76%) than other treatments (≤10.54%) at the end, coincident with the trend of ARG variation (Fig. S6). The relative abundance of ARGs, which reflects the ratio of antibiotic resistance bacteria relative to total bacteria, is another direct contributor to the abundance of ARGs according to equation 5. Pathogenic bacteria are the potential hosts of ARGs. Hence, the detection frequency of pathogens is an important indicator for the relative abundance of ARGs. Tian et al. (25) indicated that the better reduction of ARG abundance by thermophilic digestion might be partially associated with the decrease of 18 possible hosts after the temperature increase. Among these pathogens, Streptococcus was one of the most dominant genera in swine gut microbiota (26) and these bacteria were highly abundant in our original slurry (Fig. 2). Streptococcus bacteria decreased because class Bacilli are obligate or facultative aerobes, whereas another pathogen, Clostridium spp., increased since class Clostridia are anaerobic (27). However, the decrease of Streptococcus spp. and the increase of Clostridium spp. at 55°C were not as much as those in other treatments. Whether some species of Streptococcus are tolerant to or like thermophilic conditions has not yet been documented. The network analysis further suggested that Streptococcus spp. might be responsible for the higher ARG abundance in T55. The abundance of ARGs may remain constant or even increases after anaerobic digestion if the total abundance of potential hosts does not decrease. A metagenomic survey found that although the abundance of various ARG types shifted from influent to effluent sludge, there was no measurable change for the abundance of total ARGs in either thermophilic or mesophilic treatment (13).
Third, different from the trend of the transposase genes, the abundance of integrase genes increased during anaerobic digestion and much more in T55 than in other treatments (Fig. S2). The horizontal transfer of ARGs among bacteria would enlarge the range of their hosts and further increase their relative abundances. Horizontal gene transfer (HGT) is usually mediated by mobile genetic elements, such as plasmids, integrons, and transposons. The abundances of integrase genes and transposase genes are, therefore, considered important indicators of HGT frequency (1, 28, 29). Zhu et al. (1) investigated diverse and abundant ARGs in manure, compost, and compost-applied soil and found a strong correlation between transposase genes and ARGs. Similarly, a positive correlation (P < 0.001) between the total abundance of transposase genes and ARGs was observed in the soil samples from dairy farms (29). In the current study, the abundance of transposase genes decreased during anaerobic digestion (Fig. S2) and also showed significant correlations with those of most ARG types (P < 0.01), indicating the important role of transposons on the variation of ARGs. Right now, the variation trend of class 1 integrase gene (intI1) in anaerobic digestion has not reached a consensus (11, 12, 30), as it is attributed to different substrates, inoculums, and operating conditions. In this study, the greater abundance of integrase genes at 55°C is partially responsible for the higher relative abundance of ARGs.
For the impact of residual antibiotics, the spiking of antibiotics (50 mg/kg in manure) in our study caused a slight and transitory inhibition for microbially diverse populations on day 3 (Fig. S3), relatively high abundance of total ARGs on day 14 (Fig. 1), and relatively low log removal of transposase gene on day 14 (Fig. S2). However, the abundance of the 16S rRNA gene (Fig. 1A) and the total detection frequency of the pathogen (Fig. S6) were comparable between T37 and T37A. Miller et al. (31) found that sulfamethoxazole addition (50 mg/liter) did not significantly change the abundance of tetracycline (tetO and tetW) and sulfonamide (sulI and sulII) ARGs during thermophilic digestion of sludge. More recently, Wang et al. (9) indicated that chlortetracycline (spiked at 500 mg/kg dry weight [DW]) alone or coupled with copper (spiked at 5,000 mg/kg DW) affected the evolution of the microbial community and hindered the removal of some tetracyclines and copper resistance genes during mesophilic digestion of swine manure. There might be a kind of dose-dependent response of ARGs to antibiotics. In this study, the residual antibiotics did not exert a longer influence on the shift of the microbial community and ARGs (including corresponding tetracycline, sulfonamide, and FCA resistance genes), probably because of the degradation of antibiotics and the adaptability of microorganisms to environmental pressures. Similarly, Selvam et al. (32) found that the addition of chlortetracycline, sulfadiazine, and ciprofloxacin (100, 20, and 20 mg/kg, respectively) led to transient perturbations of the bacterial community which recovered at the late stage of composting.
By exploring the correlation between microbial phyla and some specific ARGs, previous studies suggest that the microbial community is a key factor driving the change of ARGs in anaerobic digestion (7, 9). In our study, redundancy analysis revealed that some dominant genera in Firmicutes showed significant correlations with the variation of the antibiotic resistome (Table 2). A Mantel test and Procrustes test at the OTU level further evidenced the important role of the microbial community on the evolution of the antibiotic resistome (Fig. 3). Interestingly, the correlations between microbial community and antibiotic resistome were also observed in other processes like aerobic composting and land application of sludge, in which the explanation of microbial phyla was sometimes higher than that of MGEs (20, 33). All the horizontal and vertical transfer-related items discussed above are closely related to the succession of microbial communities and shaping the development of ARGs jointly.
In summary, anaerobic digestion is an effective process to reduce ARGs from swine manure. However, better ARG removal is not determined by thermophilic condition unless pathogens and gene transfer elements are more efficiently inhibited. Besides, the selective pressure of moderate antibiotic residues on ARG promotion is transitory and insignificant in anaerobic digestion. Anaerobic digestion is a complicated process which can be regulated for different purposes, such as producing acid, hydrogen, and methane. The substrates and operation conditions for anaerobic digestion are omnifarious. Further studies using high-throughput methods to reveal the evolution of ARGs under different setups are warranted.
MATERIALS AND METHODS
Anaerobic digestion setup.
Swine manure was taken from a farm in a suburb of Xiamen, China. Twelve 1-liter wide-month bottles with a sealed cover were used as the vessel for anaerobic digestion. Three hundred grams of fresh manure was added to each bottle. Of them, the manure in three bottles was spiked with oxytetracycline (OTC), sulfamethazine (SM2), and ciprofloxacin (CIP) (50 mg/kg wet weight for each antibiotic). These bottles were then added with 100 ml of inoculum and 200 ml of distilled water successively, gassed with N2 for 25 minutes, and caped. The inoculum was obtained by preincubating the swine manure with a sludge from wastewater treatment plant (WWTP) and distilled water (1:1:1) anaerobically at 37°C. Finally, the bottles containing antibiotics were incubated at 37°C; other bottles (three replicates per group) were incubated at 25°C, 37°C, and 55°C, respectively. Therefore, four treatments were set up, including T25, T37, T37A, and T55, where T and A mean the incubation temperature and antibiotic spiking, respectively (see Fig. S8 in the supplemental material). The digestion took 30 days. All digesters were gently mixed by hand and loosened to vent the gas every 2 days in 2 weeks and every 5 days thereafter. Manure slurries were separately sampled on days 0, 3, 7, 14, 21, and 30 and stored at −20°C before analysis. On day 0, only the samples in the antibiotic-spiked treatment were collected.
Physicochemical parameters.
Manure slurry pH was determined using a combination electrode and pH meter (Ohaus, USA). Total suspended solid (TSS) and volatile suspended solid (VSS) in slurry were obtained by desiccation at 105°C and calcination at 600°C. NH4+ and NO3− were extracted with 2 M KCl at a ratio of 1:10 (v:v) and determined by ion chromatography (ICS-3000; Dionex, Sunnyvale, CA, USA). The extraction and determination of antibiotics are described in the supplementary data (Text 1).
DNA extraction.
Two- to 3-ml aliquots of slurry subsamples were prepared to extract genomic DNA using a FastDNA SPIN kit for feces (MP Biomedicals, USA) following the manufacturer’s instructions. The DNA quality and concentration were determined by gel electrophoresis and Quant-iT PicoGreen double-stranded DNA (dsDNA) assay kit (Invitrogen, USA). The extracted DNA was stored at −20°C before further gene manipulation.
16S rRNA gene amplification, sequencing, and data analysis.
The 16S rRNA V4 region was selected as the taxonomy-specific fragment and amplified using primers 515F and 806R (34) with a unique six-base barcode for each sample. Three-fold 50-µl reactions were conducted using DreamTaq green PCR master mix (Thermo Scientific, USA) and pooled. Concentrations of PCR products were quantified using a Quant-iT PicoGreen dsDNA assay kit (Invitrogen, USA) and combined with equal amounts. The mixture was then purified using an Axygen PCR product purification kit and sequenced on an Illumina MiSeq platform at Beijing Genomics Institute (BGI, China).
High-quality sequences were obtained by trimming off barcodes and primers and filtering out low-quality and chimeric sequences using barcoded Illumina paired-end sequencing (BIPES) pipeline. The Quantitative Insights Into Microbial Ecology (QIIME; v1.9.0) toolkit was further employed to pick out operational taxonomic units (OTUs) at the 97% sequence similarity, assign taxonomies, make an OTU table, and calculate alpha diversity indices (35, 36).
High-throughput quantitative PCR.
The abundance of ARGs in samples on days 0, 14, and 30 were evaluated by high-throughput quantitative PCR (HT-qPCR) in a SmartChip real-time PCR (Warfergen, Inc., USA), as described previously with a slight modification (20). A total of 296 primer sets were used in this study (see Table S4 in the supplemental material). According to the antibiotics they confer resistance to, ARGs are classified into nine types, comprising genes with resistance to (i) vancomycin, (ii) tetracyclines, (iii) sulfonamides, (iv) macrolide-lincosamide-streptogramin B (MLSB), (v) florfenicol-chloramphenicol-amphenicol-fluoroquinolone-quinolone (FCA), (vi) beta-lactams, (vii) aminoglycosides, (viii) multiple drugs (multidrug), and (ix) others. In addition, 16S rRNA genes and MGEs, including 8 transposase genes and 2 integrase genes of class 1 were also detected. All the reactions were performed in triplicate. For each primer set, to confirm the amplification, three conditions needed to be met simultaneously, namely, (1) amplification efficiency ranging from 1.8 to 2.2, (2) CT values of samples higher than that of nontemplate control, and (3) more than two positive replicates. Based on the ΔΔCT method (37), the following equations were used to calculate their relative abundance normalized to the 16S rRNA gene (equation 3), the relative abundance per cell (equation 4), the absolute abundance (equation 5), and the fold change (FC) compared with the reference sample (equation 6).
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
Where CT is the threshold cycle, ARG is one of the 295 antibiotic resistance gene assays, 16S is the 16S rRNA gene assay, “target” is the experimental sample, “Ref” is the reference sample, 4.1 is the average 16S rRNA gene copy number per bacterium currently estimated based on rRNA Operon Copy Number Database (rrnDB version 4.3.3), 16Sa is the absolute abundance of 16S rRNA genes quantified by a QuantStudio 6 flex real-time PCR system (Applied Biosystems, USA). In calculation of the ΔCT, if there is no amplification or the CT is higher than 31, the detection limit CT (31) was used (38, 39). A gene was considered statistically enriched or attenuated if the range created by standard deviations of the mean fold-change was entirely >1 or <1, respectively (1).
Statistical analysis.
Averages, standard deviations, and fold-change values were calculated with Microsoft Excel 2007. Principal-component analysis (PCA) and redundancy analysis (RDA) were performed using Canoco 5.0 to decipher the dissimilarity of ARGs and the contributions of experimental setups, chemical properties, and biological factors. Correlation analysis between ARGs and bacterial communities (Procrustes test, Mantel test, and Spearman correlation coefficient [ρ] and P value) and the heatmap were conducted using R version 3.2.0 (40) with vegan (41) and the pheatmap package (42). The correlation coefficient (ρ) and false-discovery rate FDR method adjusted P value were filtered and then translated into an association network using Gephi 0.9.1 (43). Bivariate correlation and other significance tests were performed using SPSS v16.0. OriginPro 9.0 was used to generate other plots.
Data availability.
All sequences have been submitted to the National Center for Biotechnology Information Sequence Read Archive under accession no. PRJNA399621.
Supplementary Material
ACKNOWLEDGMENTS
We sincerely acknowledge the financial support from the National Natural Science Foundation of China (41877058, 21477122, and 51678553), the Science and Technology Planning Project of Fujian Province (2019T3102 and 2016T3006) and Xiamen City, China (3502Z20162005), the Knowledge Innovation Program of the Chinese Academy of Sciences (IUEQN201504), and the Key Research and Development Project of Zhejiang Province, China (2015C03009).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02878-18.
REFERENCES
- 1.Zhu YG, Johnson TA, Su JQ, Qiao M, Guo GX, Stedtfeld RD, Hashsham SA, Tiedje JM. 2013. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc Natl Acad Sci U S A 110:3435–3440. doi: 10.1073/pnas.1222743110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heuer H, Schmitt H, Smalla K. 2011. Antibiotic resistance gene spread due to manure application on agricultural fields. Curr Opin Microbiol 14:236–243. doi: 10.1016/j.mib.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Beneragama N, Iwasaki M, Lateef SA, Yamashiro T, Ihara I, Umetsu K. 2013. The survival of multidrug-resistant bacteria in thermophilic and mesophilic anaerobic co-digestion of dairy manure and waste milk. Anim Sci J 84:426–433. doi: 10.1111/asj.12017. [DOI] [PubMed] [Google Scholar]
- 4.Dennehy C, Lawlor PG, Gardiner GE, Jiang Y, Cormican P, McCabe MS, Zhan X. 2017. Process stability and microbial community composition in pig manure and food waste anaerobic co-digesters operated at low HRTs. Front Environ Sci Eng 11:4. doi: 10.1007/s11783-017-0923-9. [DOI] [Google Scholar]
- 5.Moset V, Poulsen M, Wahid R, Hojberg O, Moller HB. 2015. Mesophilic versus thermophilic anaerobic digestion of cattle manure: methane productivity and microbial ecology. Microb Biotechnol 8:787–800. doi: 10.1111/1751-7915.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dennehy C, Lawlor PG, Jiang Y, Gardiner GE, Xie S, Nghiem LD, Zhan X. 2017. Greenhouse gas emissions from different pig manure management techniques: a critical analysis. Front Environ Sci Eng 11:11. doi: 10.1007/s11783-017-0942-6. [DOI] [Google Scholar]
- 7.Sun W, Qian X, Gu J, Wang XJ, Duan ML. 2016. Mechanism and effect of temperature on variations in antibiotic resistance genes during anaerobic digestion of dairy manure. Sci Rep 6:30237. doi: 10.1038/srep30237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song W, Wang X, Gu J, Zhang S, Yin Y, Li Y, Qian X, Sun W. 2017. Effects of different swine manure to wheat straw ratios on antibiotic resistance genes and the microbial community structure during anaerobic digestion. Bioresour Technol 231:1–8. doi: 10.1016/j.biortech.2017.01.054. [DOI] [PubMed] [Google Scholar]
- 9.Wang R, Chen MX, Feng F, Zhang JY, Sui QW, Tong J, Wei YS, Wei DB. 2017. Effects of chlortetracycline and copper on tetracyclines and copper resistance genes and microbial community during swine manure anaerobic digestion. Bioresour Technol 238:57–69. doi: 10.1016/j.biortech.2017.03.134. [DOI] [PubMed] [Google Scholar]
- 10.Lin Q, De Vrieze J, Li JB, Li XZ. 2016. Temperature affects microbial abundance, activity and interactions in anaerobic digestion. Bioresour Technol 209:228–236. doi: 10.1016/j.biortech.2016.02.132. [DOI] [PubMed] [Google Scholar]
- 11.Diehl DL, LaPara TM. 2010. Effect of temperature on the fate of genes encoding tetracycline resistance and the integrase of class 1 integrons within anaerobic and aerobic digesters treating municipal wastewater solids. Environ Sci Technol 44:9128–9133. doi: 10.1021/es102765a. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y, Wilson CA, Novak JT, Riffat R, Aynur S, Murthy S, Pruden A. 2011. Effect of various sludge digestion conditions on sulfonamide, macrolide, and tetracycline resistance genes and class I integrons. Environ Sci Technol 45:7855–7861. doi: 10.1021/es200827t. [DOI] [PubMed] [Google Scholar]
- 13.Zhang T, Yang Y, Pruden A. 2015. Effect of temperature on removal of antibiotic resistance genes by anaerobic digestion of activated sludge revealed by metagenomic approach. Appl Microbiol Biotechnol 99:7771–7779. doi: 10.1007/s00253-015-6688-9. [DOI] [PubMed] [Google Scholar]
- 14.Sun W, Gu J, Wang X, Qian X, Peng H. 2019. Solid-state anaerobic digestion facilitates the removal of antibiotic resistance genes and mobile genetic elements from cattle manure. Bioresour Technol 274:287–295. doi: 10.1016/j.biortech.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 15.Joy SR, Bartelt-Hunt SL, Snow DD, Gilley JE, Woodbury BL, Parker DB, Marx DB, Li X. 2013. Fate and transport of antimicrobials and antimicrobial resistance genes in soil and runoff following land application of swine manure slurry. Environ Sci Technol 47:12081–12088. doi: 10.1021/es4026358. [DOI] [PubMed] [Google Scholar]
- 16.Xie WY, Yang XP, Li Q, Wu LH, Shen QR, Zhao FJ. 2016. Changes in antibiotic concentrations and antibiotic resistome during commercial composting of animal manures. Environ Pollut 219:182–190. doi: 10.1016/j.envpol.2016.10.044. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YJ, Hu HW, Gou M, Wang JT, Chen D, He JZ. 2017. Temporal succession of soil antibiotic resistance genes following application of swine, cattle and poultry manures spiked with or without antibiotics. Environ Pollut 231:1621–1632. doi: 10.1016/j.envpol.2017.09.074. [DOI] [PubMed] [Google Scholar]
- 18.Sorensen SJ, Bailey M, Hansen LH, Kroer N, Wuertz S. 2005. Studying plasmid horizontal transfer in situ: a critical review. Nat Rev Microbiol 3:700–710. doi: 10.1038/nrmicro1232. [DOI] [PubMed] [Google Scholar]
- 19.Partridge SR, Tsafnat G, Coiera E, Iredell JR. 2009. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol Rev 33:757–784. doi: 10.1111/j.1574-6976.2009.00175.x. [DOI] [PubMed] [Google Scholar]
- 20.Su JQ, Wei B, Ou-Yang WY, Huang FY, Zhao Y, Xu HJ, Zhu YG. 2015. Antibiotic resistome and its association with bacterial communities during sewage sludge composting. Environ Sci Technol 49:7356–7363. doi: 10.1021/acs.est.5b01012. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Sangwan N, Li HY, Su JQ, Oyang WY, Zhang ZJ, Gilbert JA, Zhu YG, Ping F, Zhang HL. 2017. The antibiotic resistome of swine manure is significantly altered by association with the Musca domestica larvae gut microbiome. ISME J 11:100–111. doi: 10.1038/ismej.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghasimi DSM, Tao Y, de Kreuk M, Abbas B, Zandvoort MH, van Lier JB. 2015. Digester performance and microbial community changes in thermophilic and mesophilic sequencing batch reactors fed with the fine sieved fraction of municipal sewage. Water Res 87:483–493. doi: 10.1016/j.watres.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 23.Zhou S, Nikolausz M, Zhang JN, Riya SH, Terada A, Hosomi M. 2016. Variation of the microbial community in thermophilic anaerobic digestion of pig manure mixed with different ratios of rice straw. J Biosci Bioeng 122:334–340. doi: 10.1016/j.jbiosc.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 24.Jia YY, Wilkins D, Lu HY, Cai MW, Lee PKH. 2015. Long-term enrichment on cellulose or xylan causes functional and taxonomic convergence of microbial communities from anaerobic digesters. Appl Environ Microbiol 82:1519–1529. doi: 10.1128/AEM.03360-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian Z, Zhang Y, Yu B, Yang M. 2016. Changes of resistome, mobilome and potential hosts of antibiotic resistance genes during the transformation of anaerobic digestion from mesophilic to thermophilic. Water Res 98:261–269. doi: 10.1016/j.watres.2016.04.031. [DOI] [PubMed] [Google Scholar]
- 26.Kim HB, Borewicz K, White BA, Singer RS, Sreevatsan S, Tu ZJ, Isaacson RE. 2012. Microbial shifts in the swine distal gut in response to the treatment with antimicrobial growth promoter, tylosin. Proc Natl Acad Sci U S A 109:15485–15490. doi: 10.1073/pnas.1205147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang JJ, Chou CH, Ho CY, Chen WE, Lay JJ, Huang CC. 2008. Syntrophic co-culture of aerobic Bacillus and anaerobic Clostridium for bio-fuels and bio-hydrogen production. Int J Hydrogen Energy 33:5137–5146. doi: 10.1016/j.ijhydene.2008.05.021. [DOI] [Google Scholar]
- 28.Johnson TA, Stedtfeld RD, Wang Q, Cole JR, Hashsham SA, Looft T, Zhu YG, Tiedje JM. 2016. Clusters of antibiotic resistance genes enriched together stay together in swine agriculture. mBio 7:e02214-15. doi: 10.1128/mBio.02214-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou B, Wang C, Zhao Q, Wang Y, Huo M, Wang J, Wang S. 2016. Prevalence and dissemination of antibiotic resistance genes and coselection of heavy metals in Chinese dairy farms. J Hazard Mater 320:10–17. doi: 10.1016/j.jhazmat.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Sui QW, Zhang JY, Chen MX, Tong J, Wang R, Wei YS. 2016. Distribution of antibiotic resistance genes (ARGs) in anaerobic digestion and land application of swine wastewater. Environ Pollut 213:751–759. doi: 10.1016/j.envpol.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 31.Miller JH, Novak JT, Knocke WR, Young K, Hong Y, Vikesland PJ, Hull MS, Pruden A. 2013. Effect of silver nanoparticles and antibiotics on antibiotic resistance genes in anaerobic digestion. Water Environ Res 85:411–421. doi: 10.2175/106143012X13373575831394. [DOI] [PubMed] [Google Scholar]
- 32.Selvam A, Xu D, Zhao Z, Wong JW. 2012. Fate of tetracycline, sulfonamide and fluoroquinolone resistance genes and the changes in bacterial diversity during composting of swine manure. Bioresour Technol 126:383–390. doi: 10.1016/j.biortech.2012.03.045. [DOI] [PubMed] [Google Scholar]
- 33.Chen Q, An X, Li H, Su J, Ma Y, Zhu YG. 2016. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ Int 92-93:1–10. doi: 10.1016/j.envint.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 34.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X, Zheng J, Liu C, Liu L, Liu Y, Fan H, Zhang T. 2017. Performance and bacterial community dynamics of vertical flow constructed wetlands during the treatment of antibiotics-enriched swine wastewater. Chem Eng J 316:727–735. doi: 10.1016/j.cej.2017.02.029. [DOI] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Ouyang WY, Huang FY, Zhao Y, Li H, Su JQ. 2015. Increased levels of antibiotic resistance in urban stream of Jiulongjiang River, China. Appl Microbiol Biotechnol 99:5697–5707. doi: 10.1007/s00253-015-6416-5. [DOI] [PubMed] [Google Scholar]
- 39.Wang FH, Qiao M, Su JQ, Chen Z, Zhou X, Zhu YG. 2014. High throughput profiling of antibiotic resistance genes in urban park soils with reclaimed water irrigation. Environ Sci Technol 48:9079–9085. doi: 10.1021/es502615e. [DOI] [PubMed] [Google Scholar]
- 40.R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 41.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Steven MHH, Wagner H. 2015. vegan: community ecology package. R package version 2.3-0 http://CRAN.R-project.org/package=vegan.
- 42.Kolde R. 2015. pheatmap: pretty heatmaps. R package version 1.0.2 http://CRAN.Rproject.org/package=pheatmap.
- 43.Bastian M, Heymann S, Jacomy M. 2009. Gephi: an open source software for exploring and manipulating networks. Proc Third Int ICWSM Conf 2:361–362. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequences have been submitted to the National Center for Biotechnology Information Sequence Read Archive under accession no. PRJNA399621.