

Leucine-rich repeat kinase 2 (LRRK2), a gene linked to autosomal-dominantly inherited and sporadic Parkinson’s disease (PD) as a risk factor, encodes a large and complex protein with a dual enzymatic activity. LRRK2 contains several domains involved in protein-protein interactions, however, the presence of both a kinase and GTPase domain points to intracellular signaling functions (Marín, 2006). While LRRK2 has been linked to several molecular pathways important for neuronal activity (Martin et al., 2014), the observation that its expression is high in microglia has attracted the attention of different groups to understand whether LRRK2 dysfunctions in these cells may impact neuronal activity as secondary event. In this regard, since 2012 numerous studies have demonstrated that LRRK2 controls microglia activation and plays important roles in these cells (Russo et al., 2014). Microglia are highly specialized macrophages responsible for mediating innate immune defense in the brain and scavenging debris or misfolded/aggregated proteins. They are considered main actors upon an inflammatory stimulus, and although a well-regulated inflammatory response is crucial for tissue repair and brain homeostasis, an excessive and prolonged neuroinflammation can lead to overproduction of toxic molecules, which results in deleterious cellular damage, as observed in different neurodegenerative diseases including PD (Tansey and Goldberg, 2010).
The role of LRRK2 on the protein kinase A (PKA)-nuclear factor kappa B (NFκB) pathway: Our group and others have shown that LRRK2 is a positive mediator of microglial inflammation triggered by lipopolysaccharide or, more importantly, by α-synuclein pre-formed fibrils (Russo et al., 2014, 2015). By dissecting the underlying molecular mechanism, we demonstrated that LRRK2 impacts PKA-NFκB pathway at level of NFκB p50 inhibitory signaling (Russo et al., 2015). NFκB transcription factor, classically composed by p65:p50 subunits as activator or p50:p50 homodimer as repressor, is one of the main regulator of pro-inflammatory mediators induction (Tak and Firestein, 2001). Specifically, we found that primary microglia with Lrrk2 genetic deletion and immortalized BV2 microglia with LRRK2 pharmacological inhibition exhibited increased PKA-mediated serine 337 phosphorylation and nuclear NFκB p50 subunit. In resting/unstimulated cells NFκB p50:p50 homodimers phosphorylated by PKA are constitutively imported into the nucleus, where they bind and repress NFκB target gene, whereas, the p50/p65 heterodimers are confined in the cytoplasm in an inactive form by forming a complex with IκB proteins (Guan et al., 2005). Then, upon stimulation, activated p50:p65 heterodimers translocate into the nucleus, compete and displace DNA-bound p50:p50 to initiate inflammatory-induced NFκB target genes transcription. Our results are supported also by Kim and colleagues, who showed that BV2 cells with LRRK2 knocked-down reported reduced NFκB transcription activity after lipopolysaccharide priming and increased p50:p50 homodimer bound to NFκB target site compared to wild-type (WT) cells (Kim et al., 2012), further indicating that LRRK2 controls NFκB inhibitory signaling. Overall, we hypothesized that the enhanced phosphorylation of NFκB p50 inhibitory subunit bound to DNA may hamper the binding of NFκB p65:p50 and the activation of gene transcription upon inflammatory priming, thus resulting in the reduction of inflammatory response in Lrrk2 knock-out or pharmacologically inhibited microglia cells. In agreement with these results, in our last and follow-up study (Russo et al., 2018) we found that brain lysates from LRRK2 G2019S KI mice, which mutation increases the kinase activity of the protein, exhibited reduced NFκB p50 S337 phosphorylation compared to WT mice. Moreover, of relevant interest, we demonstrated that primary microglia isolated from LRRK2 G2019S KI mice reported increased inflammatory response compared to WT microglia upon stimulation with α-synuclein pre-formed fibrils. Taken together, these observations indicate that LRRK2 kinase activity regulates microglial inflammation through control of PKA-NFκB p50 inhibitory signaling. Moreover, our findings suggest that LRRK2 G2019S, as well as all other pathological mutations that confer increased kinase activity, favors the transition of microglia toward a pro-inflammatory state, which, in turn, may result in an exacerbated neuroinflammation in LRRK2-related PD patients. Supporting this hypothesis, Dzamko et al., (2016) showed that LRRK2 G2019S carriers exhibit higher levels of peripheral NFκB-dependent inflammatory cytokines compared to control subjects (Dzamko et al., 2016).
Successively, in the attempt of gaining more insights into LRRK2-PKA axis, we used a combination of in vitro and ex vivo systems with hyperactive or inactive LRRK2 as well as different readouts of PKA activity to specifically evaluate the impact of LRRK2 on the PKA activation state. PKA enzyme, composed by a dimeric regulatory subunit (R) bound to two catalytic (C) subunits, is part of a multifunctional complex, which includes scaffold A-kinase anchoring proteins (AKAP) important in the compartmentalization of cAMP signaling, phosphatases and phosphodiesterases crucial for regulating the magnitude and the duration of PKA signaling. In this context, LRRK2 has been proposed as a neuronal AKAP, since it interacts with PKA RIIβ subunit and neurons with Lrrk2 knock-out reported RIIβ dispersed in the dendritic spines (Greggio et al., 2017). Of interest, by exploring LRRK2-dependent modulation of PKA activity, we found that LRRK2 G2019S interacts more with RIIβ compared to WT protein in HEK293T cells and brain lysates from LRRK2 G2019S KI mice exhibit increased phosphorylation of RIIβ compared to WT mice. Auto-phosphorylation of RII controls the interaction between RII dimer and C subunits and the binding of RII with AKAP, all steps important for the activation/inactivation state of PKA (Greggio et al., 2017). Whereas, loss of LRRK2 or its kinase inhibition results in a decrease of RIIβ phosphorylation and of LRRK2-RIIβ interaction. Taken together, these findings provide additional evidence that LRRK2 kinase activity regulates the on/off state of PKA and reveal LRRK2 G2019S as a brake and LRRK2 inhibition as an activator of PKA signaling.
Given the crucial role of phosphodiesterases in controlling the magnitude/duration of PKA signaling and the well-established link between PDE4 and the inflammatory responses in microglia, we then investigated whether LRRK2 activity affects cAMP levels. Interestingly, we found that microglia treated with LRRK2 kinase inhibitor display increased levels of cAMP compared to control cells, indicating that LRRK2 activity impacts cAMP degradation (Russo et al., 2018). In support of these results, we also observed that pharmacological inhibition of PDE4 activity affects PKA-mediated NFκB p50 phosphorylation and the combined treatment of PDE4 and LRRK2 inhibitors results in a similar increment of NFκB p50 phosphorylation compared to cells treated with PDE4 inhibitor alone, suggesting that LRRK2 kinase inhibition increases cAMP levels by inhibiting PDE4 activity. In this regard, PDE4B, the most PDE4 expressed in microglia cells, represents a promising modulator of microglia activation and neuroinflammation, and intriguingly, it has been proposed as a pharmacological target to reduce neuroinflammation (Pearse and Hughes, 2016). Evidence of therapeutic efficacy of PDE4 selective inhibitors has been demonstrated experimentally in a wide range of neurological diseases and injury models where inflammation plays an important role.
Conclusions: Overall, our findings, even if mainly performed in murine models, clearly indicate that LRRK2 kinase activity is a regulator of PKA-NFκB pathway and suggest PDE4 as a novel LRRK2 effector protein in microglia cells. Future studies directed at understanding LRRK2-dependent regulation of PDE4 will offer a more defined scenario of LRRK2 biology and pathobiology in these cells. Of relevance, our observations suggest that LRRK2 pathological mutations with increased kinase activity may stimulate PDE4-mediated cAMP degradation and consequently inhibit PKA-NFκB p50 pathway (Figure 1), thus promoting the transition of microglia toward an overactive state. Therefore, it is tempting to speculate that LRRK2 activity contributes to sustainment of neuroinflammation and that pharmacological inhibition treatments may be effective at attenuating chronic neuroinflammation and neurodegeneration in LRRK2-related PD patients.
Figure 1.
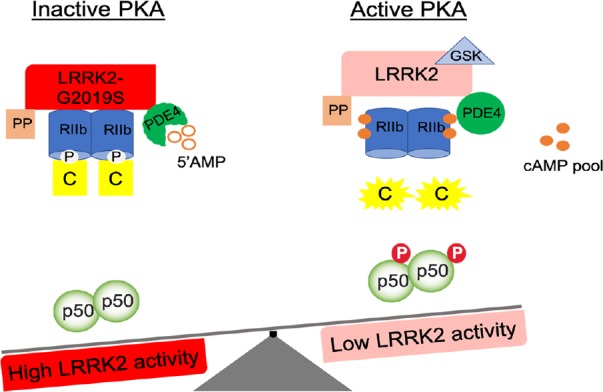
LRRK2 kinase activity regulates PKA-NFκB p50 inhibitory pathway.
Microglial cells with LRRK2-G2019S exhibit increased LRRK2-RIIβ interaction and RIIβ phosphorylation state and a reduction of PKA-mediated NFκB p50 phosphorylation level, indicative of an inhibition state of PKA. Whereas, microglia cells with LRRK2 pharmacological inhibition (GSK) exhibit reduced LRRK2-RIIβ interaction and RIIβ phosphorylation state, increased cAMP and PKA-mediated NFκB p50 phosphorylation levels, all indicative of an activation state of PKA. LRRK2: Leucine-rich repeat kinase; PKA: protein kinase A; NFκB: nuclear factor kappa B.
This work was supported by Michael J Fox Foundation, InCure (EU Joint Programme – Neurogenerative Disease Research, JPND), by the University of Padova (STARS Grants, LRRKing-Role of the Parkinson’s disease kinase LRRK2 in shaping neurites and synapses), by the Intramural Research program of the NIH and Umberto Veronesi post-doctoral fellowship year 2015 and 2017 Award (693 and 1395).
This work has been presented at Society for Neuroscience 2014 and LRRK2: Ten years along the road to therapeutic intervention 2016 as oral presentation and at 4th Venusberg Meeting on Neuroinflammation 2015 as poster.
Additional file: Open peer review report 1 (100KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Tetsuro Ishii, University of Tsukuba, Japan.
P-Reviewer: Ishii T; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Dzamko N, Rowe DB, Halliday GM. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord. 2016;31:889–897. doi: 10.1002/mds.26529. [DOI] [PubMed] [Google Scholar]
- 2.Greggio E, Bubacco L, Russo I. Cross-talk between LRRK2 and PKA: implication for Parkinson’s disease? Biochem Soc Trans. 2017;45:261–267. doi: 10.1042/BST20160396. [DOI] [PubMed] [Google Scholar]
- 3.Guan H, Hou S, Ricciardi RP. DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J Biol Chem. 2005;280:9957–9962. doi: 10.1074/jbc.M412180200. [DOI] [PubMed] [Google Scholar]
- 4.Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe EH. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One. 2012;7:e34693. doi: 10.1371/journal.pone.0034693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marín I. The Parkinson disease gene Lrrk2: evolutionary and structural insights. Mol Biol Evol. 2006;23:2423–2433. doi: 10.1093/molbev/msl114. [DOI] [PubMed] [Google Scholar]
- 6.Martin I, Kim JW, Dawson VL, Dawson TM. Lrrk2 pathobiology in Parkinson’s disease. J Neurochem. 2014;131:554–565. doi: 10.1111/jnc.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearse DD, Hughes ZA. PDE4B as a microglia target to reduce neuroinflammation. Glia. 2016;64:1698–1709. doi: 10.1002/glia.22986. [DOI] [PubMed] [Google Scholar]
- 8.Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, Greggio E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflammation. 2015;12:230. doi: 10.1186/s12974-015-0449-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo I, Bubacco L, Greggio E. Lrrk2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation. 2014;11:52. doi: 10.1186/1742-2094-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russo I, Di Benedetto G, Kaganovich A, Ding J, Mercatelli D, Morari M, Cookson MR, Bubacco L, Greggio E. Leucine-rich repeat kinase 2 controls protein kinase A activation state through phosphodiesterase 4. J Neuroinflammation. 2018;15:297. doi: 10.1186/s12974-018-1337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.