

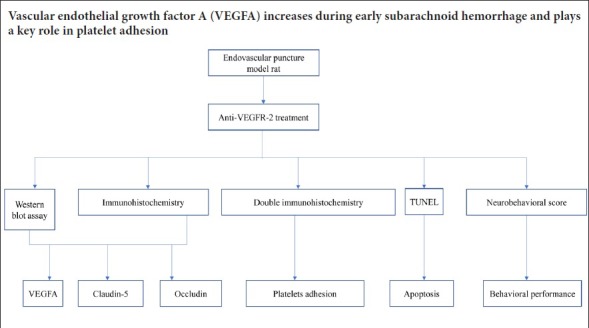
Keywords: nerve regeneration, vascular endothelial growth factor A, vascular endothelial growth factor receptor 2, subarachnoid hemorrhage, brain injuries, platelet adhesion, collagen, blood-brain barrier, neural regeneration
Abstract
The role of vascular endothelial growth factor A in platelet adhesion in cerebral microvessels in the early stage of subarachnoid hemorrhage remains unclear. In this study, the endovascular puncture method was used to produce a rat model of subarachnoid hemorrhage. Then, 30 minutes later, vascular endothelial growth factor A antagonist anti-vascular endothelial growth factor receptor 2 antibody, 10 μg, was injected into the right ventricle. Immunohistochemistry and western blot assay were used to assess expression of vascular endothelial growth factor A, occludin and claudin-5. Immunohistochemical double labeling was conducted to examine co-expression of GP Ia-II integrin and type IV collagen. TUNEL was used to detect apoptosis in the hippocampus. Neurological score was used to assess behavioral performance. After subarachnoid hemorrhage, the expression of vascular endothelial growth factor A increased in the hippocampus, while occludin and claudin-5 expression levels decreased. Co-expression of GP Ia-II integrin and type IV collagen and the number of apoptotic cells increased, whereas behavioral performance was markedly impaired. After treatment with anti-vascular endothelial growth factor receptor 2 antibody, occludin and claudin-5 expression recovered, while co-expression of GP Ia-II integrin and type IV collagen and the number of apoptotic cells decreased. Furthermore, behavioral performance improved notably. Our findings suggest that increased vascular endothelial growth factor A levels promote platelet adhesion and contribute to early brain injury after subarachnoid hemorrhage. This study was approved by the Biomedical Ethics Committee, Medical College of Xi’an Jiaotong University, China in December 2015.
Chinese Library Classification No. R456; R363; R741
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a severe cerebrovascular disease. Approximately half of SAH patients die in the acute phase, and most survivors suffer from delayed cerebral ischemia and neurological deficits, which dramatically reduce their quality of life (van Gijn et al., 2007). Numerous studies show that early brain injury, a group of pathophysiological processes that occur during the first 72 hours after SAH, contributes to high mortality and neurological deficits (Macdonald et al., 2011; Sehba et al., 2012; Suzuki et al., 2018). Over the past decade, the mechanisms of early brain injury have been studied extensively, revealing a complex process that includes cortical spreading depression, oxidative stress, inflammation, disruption of the blood-brain barrier and microthrombosis (Bederson et al., 1995; Piepgras et al., 1995; Britz et al., 2007; Busija et al., 2008; Kozniewska and Romaniuk, 2008; Yuan et al., 2010; Chrissobolis et al., 2011; Friedrich et al., 2012; Pisapia et al., 2012; Sabri et al., 2012; Sehba et al., 2012; Yuen et al., 2014; Li et al., 2015).
A large number of studies have focused on microthrombosis formation, a critical event in SAH (Sehba et al., 2005; Friedrich et al., 2012; Sabri et al., 2012; Naraoka et al., 2014). In a mouse SAH model, microthrombus formation was directly observed after pial arteriole constriction. The more severe the arterial contraction, the more extensive the thrombosis (Friedrich et al., 2012). Activation of the coagulation cascade, including increased platelet activating factor, Von Willebrand factor and tissue factor, is observed in patients after SAH, which may be an early predictor of the occurrence of delayed cerebral ischemia (Hirashima et al., 1997; Frijns et al., 2006; Vergouwen et al., 2008).
Our previous study showed that a rapid increase in vasopressin leads to enhanced platelet aggregation in SAH rats (Liu et al., 2016c). Inhibiting the interaction between vasopressin and V1a receptor could thus reduce microthrombosis and apoptosis, and thereby improve neurological function. However, how platelet adhesion (the primary step in microthrombosis) occurs and its role in early brain injury are poorly understood. Under physiological conditions, endothelial cells with their tight junctions play an important role in anti-thrombosis. In an SAH model, an impairment in endothelial function in the basilar artery leads to platelet adhesion and aggregation (Ohkuma et al., 1993). Therefore, an understanding of the pathocytological changes in endothelial cells after SAH is the key to unraveling the primary step in microthrombosis.
Vascular endothelial growth factor (VEGF), especially VEGFA, is known for its angiogenic activity and has been well studied in embryology and oncology (Ferrara et al., 2003). VEGF induces embryonic angiogenesis through its receptor (VEGFR), which is mainly located on endothelial cells (Ferrara et al., 2003). Using animal models of SAH, Kusaka et al. (2004) and Liu et al. (2016b) found that VEGF and VEGFR levels rose during early brain injury, and were related to an increase in blood-brain barrier permeability. Anti-VEGF treatment reduced the neurobehavioral impairments and brain edema.
We hypothesized that increased VEGFA might promote endothelial thrombotic damage at the early phase of SAH via interaction with VEGFR-2. In this study, we investigated if VEGFA is involved in platelet adhesion and whether anti-VEGFR-2 therapy is effective against early brain injury after SAH.
Materials and Methods
Animals
Healthy adult male Sprague-Dawley rats weighing 250–300 g were purchased from the Experimental Animal Center of Xi’an Jiaotong University, China (production license number: SCXK [Shaan] 2012-003). The rats were housed at constant temperature (20–25°C) and humidity (50–60%) under a 12-hour light/dark cycle and allowed free access to food and water. The rats were acclimated for 1 week before experiments.
This study strictly complied with the Recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH Publications No. 80-23, revised 1996). All protocols were approved by the Biomedical Ethics Committee, Medical College of Xi’an Jiaotong University, China in December 2015.
Rats were randomly divided into the following groups: sham (n = 20), SAH (n = 20), SAH + vehicle (n = 20), and SAH + antagonist (anti-VEGFR-2 antibody; n = 20).
Production of the SAH model
Endovascular puncture protocol was used to produce the SAH model (Bederson et al., 1995). The rats were anesthetized with an intraperitoneal injection of ketamine-xylazine (80 mg/kg) and positioned on the operating table. A sharpened 3.0 prolene suture was introduced into the right external carotid artery through the internal carotid artery. The suture was inserted into the intracranial internal carotid artery. When resistance was felt, the suture was pushed 3 mm further to penetrate the internal carotid artery near the bifurcation with the middle cerebral artery. Endovascular occlusion time was approximately 30 seconds. In the sham group, a similar procedure was carried out, except that puncturing of the bifurcation was not performed. During the operation, body temperature was kept at 37 ± 0.5°C.
Subarachnoid blood clots were assessed to reduce the impact of differences in SAH severity (Sugawara et al., 2008). Blood clots on a total of six segments of the basilar artery and the circle of Willis were graded from 0 to 3 (Sugawara et al., 2008). Grade 0 indicates no subarachnoid blood. Grade 1 indicates minimal subarachnoid blood. Grade 2 indicates moderate blood clot. Grade 3 indicates that blood clots covered all arteries within the segment. The score for each rat was the sum of the grades in the six segments. Severity of SAH was based on the score as follows: 0–7: mild SAH; 8–12: moderate SAH; and 13–18: severe SAH (Sugawara et al., 2008). Only rats with moderate and severe SAH were included in this study.
Drug administration
Anti-VEGFR-2 antibody was administered via an intracerebroventricular injection. Briefly, rats were positioned on a stereotaxic frame (Kent Scientific Co., Torrington, CT, USA). Anti-VEGFR-2 antibody (10 μg; 1 mg in 1 mL phosphate-buffered saline (PBS); murine; Abcam, Cambridge, UK) was injected into the right lateral ventricle with a microinjector (Gaoge, Shanghai, China) through a hole drilled in the skull (4.0 mm below the skull, 1.5 mm lateral and 0.8 mm posterior to the bregma). The injection speed was 1 μL/min. The injection was performed 30 minutes after SAH. PBS (0.01 M) was used as vehicle. The dose of anti-VEGFR-2 antibody and administration protocol were partially based on previous studies (Krum et al., 2008; Liu et al., 2016b). Normal mouse immunoglobulin G1 (IgG1, 10 μg; Abcam, Cambridge, UK) was injected as an isotype control to exclude the possibility of a nonspecific response to intracerebroventricular protein injection. After injection, the microinjector was removed and the incision was quickly sutured.
Brain tissue preparation
All rats were sacrificed 24 hours after SAH. After saline perfusion, the brains were removed immediately. For TUNEL and immunostaining, the brains were fixed in 4% paraformaldehyde overnight at 25°C. For western blot assay, the brains were frozen immediately and stored at −80°C for use. The brains were fixed in paraformaldehyde, dehydrated through a graded ethanol solution series, permeabilized with xylene, and embedded in paraffin. Then, 5–10-μm transverse serial sections were cut on a slicer. For every rat, five sections each (bregma −4.16 mm, including the hippocampus) were randomly chosen for TUNEL and immunostaining.
Immunohistochemistry
VEGFA, occludin and claudin-5 expression was assessed by immunohistochemistry. Sections were heated in an oven for approximately 20 minutes, deparaffinized with xylene, and rehydrated in an ethanol series. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 minutes. The sections were washed with PBS for 15 minutes, immersed in 10 mM citrate buffer (pH 6.0), heated at 95°C for 30 minutes in a microwave oven, cooled at 25°C for approximately 20 minutes, and then washed in PBS. Sections were subsequently blocked in 5% goat serum in PBS for 1 hour at 25°C, and then incubated with primary antibody (rabbit anti-VEGFA polyclonal antibody, 1:200, Beijing Biosynthesis; rabbit anti-occludin polyclonal antibody, 1:200, Invitrogen, Carlsbad, CA, USA; mouse anti-claudin-5 polyclonal antibody, 1:500, Invitrogen) overnight at 4°C. The sections were then washed for 15 minutes in PBS, incubated with biotinylated goat anti-rabbit IgG (Beijing Biosynthesis) for 60 minutes at room temperature, and washed for 15 minutes in PBS. Sections were incubated with horseradish peroxidase-labeled streptavidin (Beijing Biosynthesis) for 30 minutes at room temperature, and then with diaminobenzidine (Beijing Biosynthesis). Hematoxylin was used as a counterstain. The intensity of the signal was calculated using ImageJ software (Bethesda, MD, USA). The intensity of the light source and aperture on the microscope (Olympus, Tokyo, Japan) were kept constant. The images were transformed into grayscale images, and after thresholding, the percentage of the total stained area was calculated for VEGFA, occludin and claudin-5 in the hippocampus.
Western blot assay
The frozen brain tissues were solubilized in RIPA buffer on ice using a tissue homogenizer. Sodium dodecyl sulfate-polyacrylamide gel (10% or 12%) electrophoresis was used to separate the protein samples. The proteins were electrotransferred onto polyvinylidene fluoride membranes, and then blocked for 2 hours in a solution of nonfat milk and Tris-buffered saline-Tween 20. The membranes were then incubated with primary antibody against VEGFA (1:1000, rabbit; Abcam), occludin (1:1000, rabbit; Invitrogen), claudin-5 (1:1000, mouse; Invitrogen) or beta-actin (1:1000; Beijing Biosynthesis) overnight at 4°C. The membranes were washed with Tris-buffered saline-Tween 20 for 30 minutes, and then incubated with horseradish peroxidase-conjugated secondary antibody (1:10,000, mouse or rabbit; Jackson ImmunoResearch, West Grove, PA, USA) at room temperature for 1 hour. A chemiluminescence detection kit (Heliosense Biotechnologies Inc., Xiamen, China) and X-ray film (Heliosense Biotechnologies Inc.) were used to detect target protein signals. The protein signals were analyzed with Image Lab ver. 5.2 (Bio-Rad, Hercules, CA, USA).
Double immunolabeling
Polymer-horseradish peroxidase and alkaline phosphatase Kit (Beijing ZSGB-bio) was used to detect GP Ia-II and collagen IV, to assess platelet adhesion to collagen. Sections were dewaxed and rehydrated as described above. Alkaline phosphatase and endogenous peroxidase activities were blocked consecutively with 3% acetic acid and 3% hydrogen peroxide for 10 minutes, followed by a 15-minute wash in PBS. Sections were incubated with 0.2% trypsin for 5 minutes at room temperature, immersed in 1 mM Tris/EDTA buffer (pH 9.0), heated at 95°C for 30 minutes in a microwave oven, cooled at room temperature for 20 minutes, and then washed in PBS. Non-specific protein binding was blocked with 5% goat serum in PBS for 1 hour at room temperature. Sections were then incubated with primary antibodies (mouse anti-rat GP Ia-II integrin monoclonal antibody, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA; rabbit anti-rat collagen IV monoclonal antibody, 1:250, Abcam) overnight at 4°C, and washed in PBS for 15 minutes. Sections were then incubated with secondary antibodies (rabbit horseradish peroxidase polymer, Beijing ZSGB-bio; mouse alkaline phosphatase polymer, Beijing ZSGB-bio) for 60 minutes at room temperature, and washed in PBS for 15 minutes. Afterwards, the sections were incubated with diaminobenzidine and GBI-Permanent Red, and hematoxylin was used as a counterstain. The area fraction obtained with ImageJ was used to assess the interaction between collagen IV (brown) and platelets (red).
TUNEL assay
Apoptosis was detected using a TUNEL kit (Promega, Fitchburg, WI, USA). Briefly, sections were deparaffinized and rehydrated, rinsed in 0.1 M PBS for 10 minutes, and then reacted with proteinase K (20 μg/mL) for 10 minutes at 25°C. After washing in PBS, the sections were incubated with equilibration buffer (200 mM potassium cacodylate, 25 mM Tris-HCl, 0.2 mM DTT, 0.25 mg/mL bovine serum albumin, 2.5 mM cobalt chloride) for 10 minutes at room temperature. The specimens were then incubated in labeling reaction mix (terminal deoxynucleotidyl transferase and deoxynucleotides) for 1 hour at room temperature in the dark. The sections were then incubated with 2× saline citrate (300 mM NaCl, 30 mM sodium citrate) for 10 minutes at 25°C, washed in PBS, and reacted with 4′,6-diamidino-2-phenylindole for 15 minutes at 25°C in the dark. Fluorescence microscopy (Olympus) was used to acquire images. The positive control was produced by adding DNaseI for 5 minutes at 25°C. PBS instead of terminal deoxynucleotidyl transferase was used for the negative control.
Neurological scores
Neurological deficits were evaluated as described previously (Sugawara et al., 2008) 24 hours after operation. Spontaneous activity, movements of all limbs and movements of forelimbs were each scored from 0–3, while climbing the wall of the wire cage, reaction to touch on both sides of the trunk and response to vibrissae touch ranged from 1–3. The six parameters were summed, and the final score ranged from 3–18.
Statistical analysis
Statistical analyses were performed using SPSS 18.0 (SPSS Inc., Chicago, IL, USA). All data followed a normal distribution and were expressed as the mean ± SD. One-way analysis of variance was used for comparisons among multiple groups. Least significant difference test was used to compare two groups. P < 0.05 was considered statistically significant.
Results
SAH-induced death and brain tissue changes
A total of 114 rats were included in this study. The mortality was 0% in the sham group, and 22.3% in the SAH groups. Brains were photographed immediately after removal, and 13.8% of the brains with mild SAH were excluded from the study. The excluded rats were supplemented for each group. Large blood clots were observed on the brains of the SAH groups (Figure 1). There was no subarachnoid hemorrhage or blood clot in the sham group. Scores for the SAH brains ranged from 8 to 18, indicating moderate or severe SAH (Figure 1).
Figure 1.
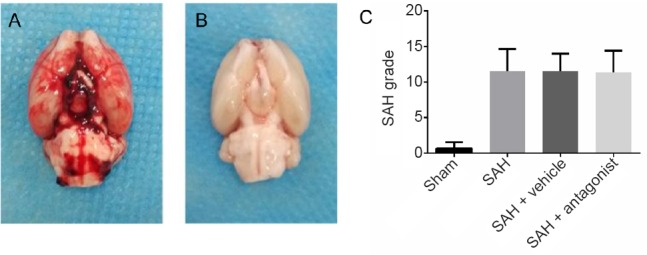
Brain gross observation and SAH grade in rats.
(A, B) The rat brains were harvested 24 hours after operation. The SAH brain was covered with blood clots (A), while the sham brain was not (B). (C) The grades of SAH ranged from 8 to 18, indicating moderate to severe SAH. Data are expressed as the mean ± SD (one-way analysis of variance followed by the least significant difference test). SAH: Subarachnoid hemorrhage.
Increased expression of VEGFA after SAH
Western blot assay and immunohistochemistry were used to assess the expression of VEGFA post-SAH in the hippocampus. Both western blot assay and immunohistochemistry indicated that compared with the sham group, VEGFA was significantly increased at 24 hours after SAH (P < 0.01; Figure 2).
Figure 2.
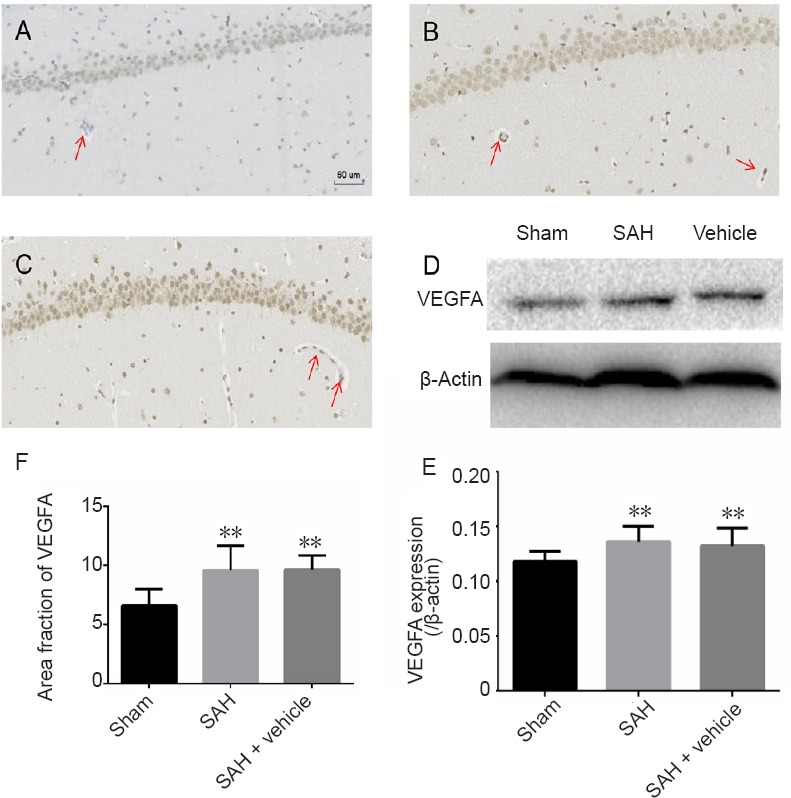
Immunohistochemistry and western blot assay for VEGFA in the hippocampus after SAH.
(A–C) VEGFA immunoreactivity in the hippocampus of the sham (A), SAH (B) and SAH + vehicle (C) groups. VEGFA-positive cells are stained brown (red arrows), and the area fraction was calculated in three regions of interest in each section, including the CA1–3 and dentate gyrus, using three coronal sections from each rat. (D) Western blot assay results for VEGFA expression. VEGFA is approximately 34 kDa and β-actin is approximately 42 kDa. (E) Protein expression levels of VEGFA are expressed as a ratio to β-actin levels for normalization. (F) Quantitation of VEGFA immunoreactivity. Western blot assay was performed in triplicate for each brain. Data are expressed as the mean ± SD (n = 5; one-way analysis of variance followed by the least significant difference test). **P < 0.01, vs. sham group. VEGFA: Vascular endothelial growth factor A; SAH: subarachnoid hemorrhage.
VEGFA antagonist increases the expression of occludin and claudin-5 in the hippocampus after SAH
Western blot assay and immunohistochemistry were used to assess the expression of occludin and claudin-5 after SAH in the hippocampus (Figures 3 and 4). Results for occludin and claudin-5 were similar. In sections from the sham group, microvessels were stained brown. The SAH and SAH + vehicle groups did not show visible coloration. In enlarged images, some of the microvessels appeared discontinuous. In rats administered anti-VEGFR-2 antibody, the endothelial connections were tight and expression of occludin and claudin-5 was increased. There was a significant difference between the sham and SAH groups (P < 0.01). The area fraction in the SAH + antagonist group was significantly increased compared with the SAH + vehicle group (P < 0.01). The western blot assay results were consistent with these findings. Compared with the sham group, expression of both occludin and claudin-5 was significantly reduced in the SAH and SAH + vehicle groups (P < 0.01). In comparison, in rats given anti-VEGFR-2 antibody treatment, the expression of occludin (P < 0.05) and claudin-5 (P < 0.01) was significantly increased.
Figure 3.
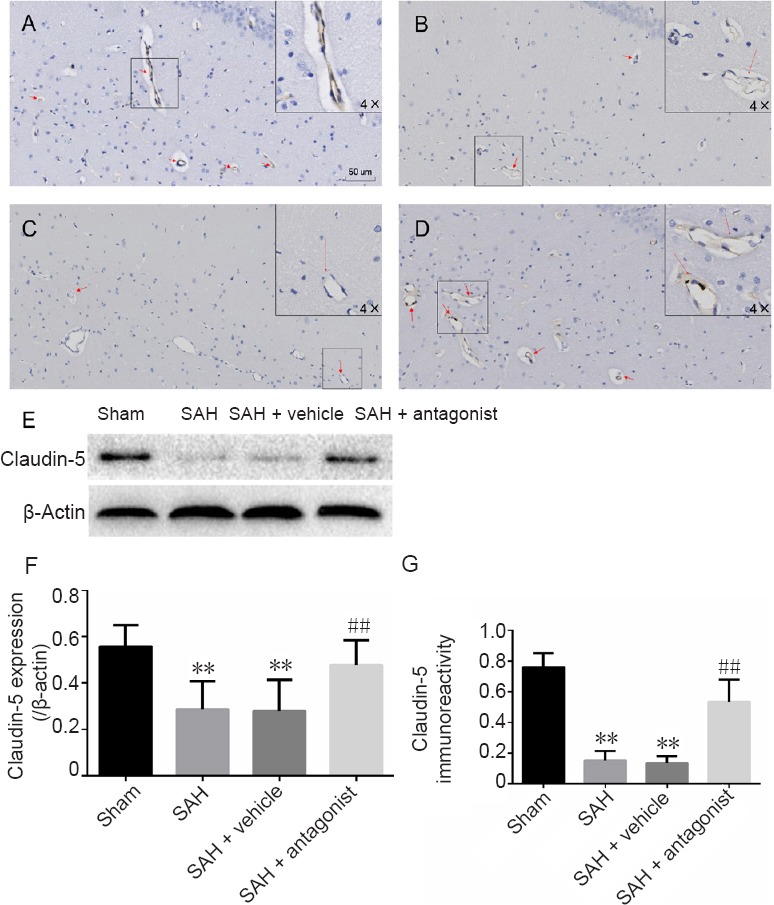
Effect of VEGFA on the immunoreactivity and expression of claudin-5 in the hippocampus after SAH.
(A–D) Claudin-5-positive cells in the hippocampus in the sham (A), SAH (B), SAH + vehicle (C) and SAH + antagonist (D) groups. Positive endothelium was stained brown (red arrow). Endothelial damage is shown in the enlarged image (4× greater magnification, B and C). Area fraction was calculated in three regions of interest in each section, including CA1–3 and the dentate gyrus, using three coronal sections from each rat. (E) Western blot assay for claudin-5 expression. Claudin-5 is approximately 23 kDa and β-actin is approximately 42 kDa. (F) Protein expression levels of claudin-5 are expressed as a ratio of claudin-5 to β-actin levels for normalization. (G) Quantitation of claudin-5 immunoreactivity. Data are expressed as the mean ± SD (n = 5; one-way analysis of variance followed by the least significant difference test). **P < 0.01, vs. sham group; ##P < 0.01, vs. SAH + vehicle group. VEGFA: Vascular endothelial growth factor A; SAH: subarachnoid hemorrhage.
Figure 4.

Effect of VEGFA antagonist on the immunoreactivity and expression of occludin in the hippocampus after SAH.
(A–D) Occludin immunoreacitivity in the hippocampus in the sham (A), SAH (B), SAH + vehicle (C) and SAH + antagonist (D) groups. Positive endothelium is stained brown (red arrow). Endothelial damage is shown in the enlarged image (4× higher magnification, B and C). Area fraction was calculated in three regions of interest in each section, including the CA1–3 and the dentate gyrus, with three coronal sections from each rat. (E) Western blot assay results for occludin expression. Occludin is approximately 65 kDa and β-actin is approximately 42 kDa. (F) Protein expression levels of occludin are expressed as a ratio of occludin to β-actin levels for normalization. (G) Quantitation of occludin immunoreactivity. Data are expressed as the mean ± SD (n = 5; one-way analysis of variance followed by the least significant difference test). **P < 0.01, vs. sham group; #P < 0.05, ##P < 0.01, vs. SAH + vehicle group. VEGFA: Vascular endothelial growth factor A; SAH: subarachnoid hemorrhage.
VEGFA antagonist decreases platelet adhesion in the hippocampus after SAH
Double immunolabeling was performed for GP Ia-II and collagen IV in the hippocampus to assess platelet adhesion to collagen (Figure 5). There were few or no platelets and microthrombi in microvessels in the sham group. However, numerous GP Ia-II structures were observed directly adhering to collagen IV in microvessels with or without incomplete endothelium in the SAH and SAH + vehicle groups (P < 0.01). The number of these structures was significantly reduced in the SAH + antagonist group (P < 0.01).
Figure 5.
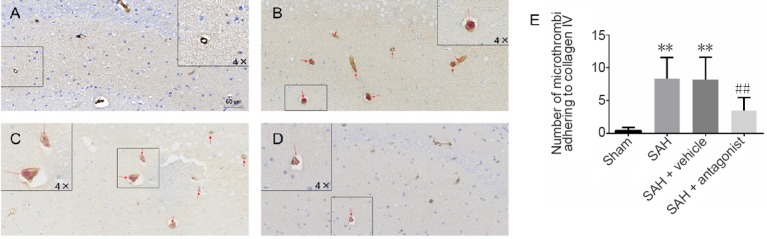
Effect of VEGFA antagonist on the colocalization of GP Ia-II integrin and collagen IV in the hippocampus after SAH.
(A–D) Double immunolabeling for GP Ia-II integrin and collagen IV in the sham (A), SAH (B), SAH + vehicle (C) and SAH + antagonist (D) groups. GP Ia-II is stained red, while collagen IV is stained brown. (E) The number of microthrombi adhering to collagen IV (red arrow) in the CA1–3 and dentate gyrus. Data are expressed as the mean ± SD (n = 5; one-way analysis of variance followed by the least significant difference test). **P < 0.01, vs. sham group; ##P < 0.01, vs. SAH + vehicle group. VEGFA: Vascular endothelial growth factor A; SAH: subarachnoid hemorrhage.
VEGFA antagonist decreases apoptosis in early brain injury after SAH
Early brain injury after SAH was assessed in the hippocampus (Figure 6). TUNEL assay showed that compared with the sham group, there was a significant increase in TUNEL-positive cells in the SAH and SAH + vehicle groups (P < 0.01). The number of these cells was significantly reduced in the SAH + antagonist group compared with the SAH + vehicle group (P < 0.01).
Figure 6.
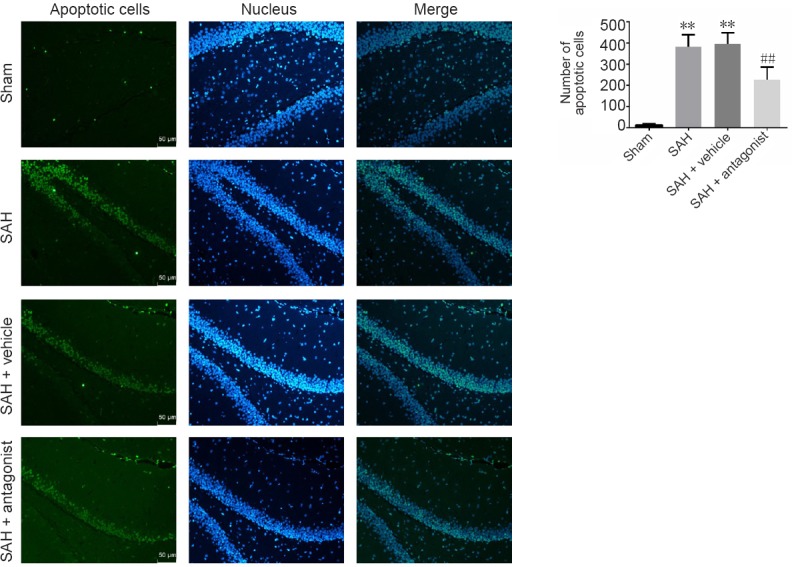
Effect of VEGFA antagonist on apoptotic cells in the rat hippocampus after SAH.
Nuclei are stained blue with 4′,6-diamidino-2-phenylindole, and TUNEL-positive nuclei are stained green. In merged images, cyan nuclei represent apoptotic cells. The number of apoptotic cells was calculated in three regions of interest (original magnification, 200×), including the CA1–3 and dentate gyrus, for three coronal sections from each brain. VEGFA: Vascular endothelial growth factor A; SAH: subarachnoid hemorrhage. Data are expressed as the mean ± SD (n = 5; one-way analysis of variance followed by the least significant difference test). **P < 0.01, vs. sham group; ##P < 0.01, vs. SAH + vehicle group.
VEGFA antagonist improves behavioral performance in rats with early brain injury after SAH
Neurological scoring was used to assess the neuroprotective effect of VEGFR-2 antagonist. Rats in the sham group showed no obvious abnormalities in behavior, while rats in the SAH groups exhibited varying degrees of neurological impairment. The scores in the SAH and SAH + vehicle groups were significantly reduced compared with the sham group (P < 0.01). In contrast, the score in the SAH + antagonist group was significantly improved compared with the SAH + vehicle group (P < 0.05; Figure 7).
Figure 7.
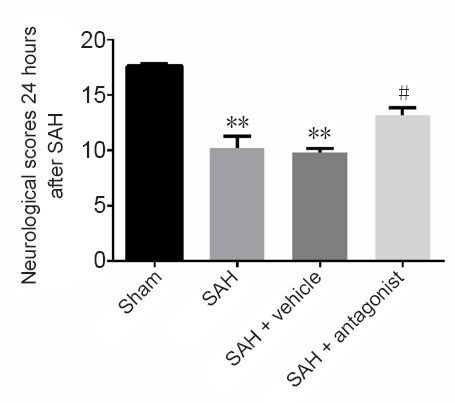
Effect of VEGFA antagonist on neurological scores 24 hours after SAH.
The data are presented as the mean ± SD (n = 5; one-way analysis of variance followed by the least significant difference test). **P < 0.01, vs. sham group; #P < 0.05, vs. SAH + vehicle group. VEGFA: Vascular endothelial growth factor A; SAH: subarachnoid hemorrhage.
Discussion
Early brain injury comprises many pathophysiological processes, which contribute to delayed ischemic neurological deficits post SAH. Microthrombosis plays an important role in early brain injury. In this study, we investigated an early step in this process, platelet adhesion to collagen IV in microvessels, in a rat endovascular perforation model of SAH. We found that VEGFA may contribute to endothelial tight junction damage and increase this adhesion during early brain injury. Anti-VEGFR-2 treatment reduced the adhesion and apoptosis, and improved neurological scores.
VEGFA expression increased in the rat hippocampus post SAH, in keeping with a previous study (Liu et al., 2016b). VEGFA is a key factor involved in revascularization, endothelial cell migration and proliferation. For these processes, connections between endothelial cells must first be weakened. Hence, up-regulated VEGFA might weaken vascular endothelial tight junctions via VEGFR-2 located on endothelial cells. Our results support this concept as anti-VEGFR-2 treatment inhibited the reduction in expression of occludin and claudin-5 following SAH. Moreover, some microvessels appeared disrupted following SAH, indicating that increased VEGFA reduces the integrity of microvessels by weakening tight junctions. Indeed, a number of previous studies have shown that the blood–brain barrier, including microvessels, pericytes, extracellular collagen matrix and astrocyte projections, are disrupted post SAH (Park et al., 2001, 2014; Badaut et al., 2002; Lo et al., 2003; Kusaka et al., 2004; Ostrowski et al., 2005; Frijns et al., 2006; Naval et al., 2006; Vergouwen et al., 2008; Lee et al., 2009; Guo et al., 2010; Wang et al., 2012; Fagiani and Christofori, 2013; Wong et al., 2013; Li et al., 2015; Liu et al., 2016a). Li et al. (2015) found that ZO-1 and occludin were markedly down-regulated in a Sprague-Dawley rat model of SAH. Liu et al. (2016a) found that increased VEGF expression in the cerebral cortex induced blood-brain barrier disruption, aggravated brain edema and impaired neural function via VEGFR-2. However, the role of platelet adhesion in microvascular disruption remained largely unknown. The endothelial tight junctions prevent platelets from adhering to the extracellular collagen, helping to maintain the hemostatic/thrombotic balance. VEGFA impairs microvascular integrity and disrupts the connections between endothelial cells during the acute phase of SAH, perturbing this balance. Extracellular collagen IV is then directly exposed to blood components, including platelets, resulting in adhesion.
GP Ia-II is a glycoprotein located on platelets and is responsible for adhesion between platelets and collagen (Hynes, 1992). Here, we observed platelet adhesion to collagen IV outside microvessels in the rat hippocampus. The number of microthrombi was markedly reduced in rats with SAH given anti-VEGFR-2 treatment. This indicates that the adhesion between platelets and collagen IV was antagonized. Simultaneously, the tight junction components occludin and claudin-5 were substantially increased in these animals. These findings indicate that the antagonist maintained microvessel integrity and reduced platelet adhesion.
Our TUNEL data revealed fewer apoptotic cells in the hippocampus in the SAH + antagonist group compared with the SAH group. Apoptosis is one of the major causes of neurological impairment in SAH (Sehba et al., 2012). Anti-VEGFR-2 treatment alleviated injury and increased neurological scores. These results suggest that VEGFA may contribute to early brain injury by promoting apoptosis and microthrombosis.
There are some limitations to this study. First, the endovascular puncture model was chosen to study early brain injury (Sehba et al., 2012). However, real-time detection of intracranial pressure was not included in the protocol. To avoid variability caused by differences in SAH intensity, we assessed the SAH grade, and only rats with moderate or severe SAH were included in the analyses. Second, some reports (Friedrich et al., 2011, 2013) found that collagen IV is reduced in SAH rats, which may hinder platelet adhesion. The reduction might be caused by inflammation and the release of collagenases by neutrophils (Friedrich et al., 2013). Obviously, microthrombosis is not the only causal factor in early brain injury. VEGFA-induced microvascular damage may precede inflammation. Further studies are needed to clarify the mechanisms of early brain injury in SAH.
Footnotes
Conflicts of interest: All of the authors claim that there are no conflicts of interest.
Financial support: This study was financially supported by the National Natural Science Foundation of China, No. 81471179 (to JNS). The funding body played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, or in the decision to submit the paper for publication.
Institutional review board statement: This study was approved by the Biomedical Ethics Committee, Medical College of Xi’an Jiaotong University, China in December 2015.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was financially supported by the National Natural Science Foundation of China, No. 81471179 (to JNS).
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Patel B, Raye W, Qiu Y, Song LP; T-Editor: Jia Y
References
- 1.Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26:1086–1091. doi: 10.1161/01.str.26.6.1086. discussion 1091-1092. [DOI] [PubMed] [Google Scholar]
- 3.Britz GW, Meno JR, Park IS, Abel TJ, Chowdhary A, Nguyen TS, Winn HR, Ngai AC. Time-dependent alterations in functional and pharmacological arteriolar reactivity after subarachnoid hemorrhage. Stroke. 2007;38:1329–1335. doi: 10.1161/01.STR.0000259853.43084.03. [DOI] [PubMed] [Google Scholar]
- 4.Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K. Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol. 2008;86:379–395. doi: 10.1016/j.pneurobio.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci (Landmark Ed) 2011;16:1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 6.Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. 2013;328:18–26. doi: 10.1016/j.canlet.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 7.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 8.Friedrich B, Muller F, Feiler S, Scholler K, Plesnila N. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: an in-vivo microscopy study. J Cereb Blood Flow Metab. 2012;32:447–455. doi: 10.1038/jcbfm.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedrich V, Bederson JB, Sehba FA. Gender influences the initial impact of subarachnoid hemorrhage: an experimental investigation. PLoS One. 2013;8:e80101. doi: 10.1371/journal.pone.0080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedrich V, Flores R, Muller A, Bi W, Peerschke EI, Sehba FA. Reduction of neutrophil activity decreases early microvascular injury after subarachnoid haemorrhage. J Neuroinflammation. 2011;8:103. doi: 10.1186/1742-2094-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frijns CJ, Kasius KM, Algra A, Fijnheer R, Rinkel GJ. Endothelial cell activation markers and delayed cerebral ischaemia in patients with subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2006;77:863–867. doi: 10.1136/jnnp.2005.081539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Z, Sun X, He Z, Jiang Y, Zhang X, Zhang JH. Matrix metalloproteinase-9 potentiates early brain injury after subarachnoid hemorrhage. Neurol Res. 2010;32:715–720. doi: 10.1179/016164109X12478302362491. [DOI] [PubMed] [Google Scholar]
- 13.Hirashima Y, Nakamura S, Suzuki M, Kurimoto M, Endo S, Ogawa A, Takaku A. Cerebrospinal fluid tissue factor and thrombin-antithrombin III complex as indicators of tissue injury after subarachnoid hemorrhage. Stroke. 1997;28:1666–1670. doi: 10.1161/01.str.28.9.1666. [DOI] [PubMed] [Google Scholar]
- 14.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 15.Kozniewska E, Romaniuk K. Vasopressin in vascular regulation and water homeostasis in the brain. J Physiol Pharmacol 59 Suppl. 2008;8:109–116. [PubMed] [Google Scholar]
- 16.Krum JM, Mani N, Rosenstein JM. Roles of the endogenous VEGF receptors flt-1 and flk-1 in astroglial and vascular remodeling after brain injury. Exp Neurol. 2008;212:108–117. doi: 10.1016/j.expneurol.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- 18.Lee JY, He Y, Sagher O, Keep R, Hua Y, Xi G. Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res. 2009;1287:126–135. doi: 10.1016/j.brainres.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Liang G, Ma T, Li J, Wang P, Liu L, Yu B, Liu Y, Xue Y. Blood-brain barrier permeability change and regulation mechanism after subarachnoid hemorrhage. Metab Brain Dis. 2015;30:597–603. doi: 10.1007/s11011-014-9609-1. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Fujimoto M, Kawakita F, Ichikawa N, Suzuki H. Vascular endothelial growth factor in brain edema formation after subarachnoid hemorrhage. Acta Neurochir Suppl. 2016a;121:173–177. doi: 10.1007/978-3-319-18497-5_31. [DOI] [PubMed] [Google Scholar]
- 21.Liu L, Fujimoto M, Kawakita F, Nakano F, Imanaka-Yoshida K, Yoshida T, Suzuki H. Anti-vascular endothelial growth factor treatment suppresses early brain injury after subarachnoid hemorrhage in mice. Mol Neurobiol. 2016b;53:4529–4538. doi: 10.1007/s12035-015-9386-9. [DOI] [PubMed] [Google Scholar]
- 22.Liu ZW, Gu H, Zhang BF, Zhao YH, Zhao JJ, Zhao YL, Ma XD, Song JN. Rapidly increased vasopressin promotes acute platelet aggregation and early brain injury after experimental subarachnoid hemorrhage in a rat model. Brain Res. 2016c;1639:108–119. doi: 10.1016/j.brainres.2016.02.038. [DOI] [PubMed] [Google Scholar]
- 23.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 24.Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Marr A, Roux S, Kassell N. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2) Lancet Neurol. 2011;10:618–625. doi: 10.1016/S1474-4422(11)70108-9. [DOI] [PubMed] [Google Scholar]
- 25.Naraoka M, Matsuda N, Shimamura N, Asano K, Ohkuma H. The role of arterioles and the microcirculation in the development of vasospasm after aneurysmal SAH. Biomed Res Int. 2014;2014:253746–253746. doi: 10.1155/2014/253746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naval NS, Stevens RD, Mirski MA, Bhardwaj A. Controversies in the management of aneurysmal subarachnoid hemorrhage. Crit Care Med. 2006;34:511–524. doi: 10.1097/01.ccm.0000198331.45998.85. [DOI] [PubMed] [Google Scholar]
- 27.Ohkuma H, Ogane K, Fujita S, Manabe H, Suzuki S. Impairment of anti-platelet-aggregating activity of endothelial cells after experimental subarachnoid hemorrhage. Stroke. 1993;24:1541–1546. doi: 10.1161/01.str.24.10.1541. [DOI] [PubMed] [Google Scholar]
- 28.Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- 29.Park KW, Metais C, Dai HB, Comunale ME, Sellke FW. Microvascular endothelial dysfunction and its mechanism in a rat model of subarachnoid hemorrhage. Anesth Analg. 2001;92:990–996. doi: 10.1097/00000539-200104000-00035. [DOI] [PubMed] [Google Scholar]
- 30.Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 31.Piepgras A, Thomé C, Schmiedek P. Characterization of an anterior circulation rat subarachnoid hemorrhage model. Stroke. 1995;26:2347–2352. doi: 10.1161/01.str.26.12.2347. [DOI] [PubMed] [Google Scholar]
- 32.Pisapia JM, Xu X, Kelly J, Yeung J, Carrion G, Tong H, Meghan S, El-Falaky OM, Grady MS, Smith DH, Zaitsev S, Muzykantov VR, Stiefel MF, Stein SC. Microthrombosis after experimental subarachnoid hemorrhage: time course and effect of red blood cell-bound thrombin-activated pro-urokinase and clazosentan. Exp Neurol. 2012;233:357–363. doi: 10.1016/j.expneurol.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabri M, Ai J, Lakovic K, D’Abbondanza J, Ilodigwe D, Macdonald RL. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience. 2012;224:26–37. doi: 10.1016/j.neuroscience.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Sehba FA, Mostafa G, Friedrich V, Jr, Bederson JB. Acute microvascular platelet aggregation after subarachnoid hemorrhage. J Neurosurg. 2005;102:1094–1100. doi: 10.3171/jns.2005.102.6.1094. [DOI] [PubMed] [Google Scholar]
- 35.Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97:14–37. doi: 10.1016/j.pneurobio.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki H, Nishikawa H, Kawakita F. Matricellular proteins as possible biomarkers for early brain injury after aneurysmal subarachnoid hemorrhage. Neural Regen Res. 2018;13:1175–1178. doi: 10.4103/1673-5374.235022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet. 2007;369:306–318. doi: 10.1016/S0140-6736(07)60153-6. [DOI] [PubMed] [Google Scholar]
- 39.Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1761–1770. doi: 10.1038/jcbfm.2008.74. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Meng CJ, Shen XM, Shu Z, Ma C, Zhu GQ, Liu HX, He WC, Sun XB, Huo L, Zhang J, Chen G. Potential contribution of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 to blood-brain barrier disruption and brain edema after experimental subarachnoid hemorrhage. J Mol Neurosci. 2012;48:273–280. doi: 10.1007/s12031-012-9769-6. [DOI] [PubMed] [Google Scholar]
- 41.Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC. The blood-brain barrier: an engineering perspective. Front Neuroeng. 2013;6:7. doi: 10.3389/fneng.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan ZH, Zhu JY, Huang WD, Jiang JK, Lu YQ, Xu M, Su W, Jiang TY. Early change of plasma and cerebrospinal fluid arginine vasopressin in traumatic subarachnoid hemorrhage. Chin J Traumatol. 2010;13:42–45. [PubMed] [Google Scholar]
- 43.Yuen N, Lam TI, Wallace BK, Klug NR, Anderson SE, O’Donnell ME. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: evidence for involvement of ERK1/2 MAP kinase. Am J Physiol Cell Physiol. 2014;306:C931–942. doi: 10.1152/ajpcell.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]