

Abstract
Model‐informed drug development (MIDD) was central to the development of the oral proteasome inhibitor ixazomib, facilitating internal decisions (switch from body surface area (BSA)‐based to fixed dosing, inclusive phase III trials, portfolio prioritization of ixazomib‐based combinations, phase III dose for maintenance treatment), regulatory review (model‐informed QT analysis, benefit–risk of 4 mg dose), and product labeling (absolute bioavailability and intrinsic/extrinsic factors). This review discusses the impact of MIDD in enabling patient‐centric therapeutic optimization during the development of ixazomib.
BACKGROUND
Ixazomib is the first and only approved orally administered proteasome inhibitor. In the United States (US) and European Union (EU), ixazomib is indicated as part of an all‐oral triplet regimen, in combination with lenalidomide and dexamethasone (Rd), for the treatment of patients with multiple myeloma (MM) who have received at least one prior therapy.1, 2 Ixazomib is also approved in multiple additional countries worldwide. These approvals were based on the randomized, double‐blind, phase III TOURMALINE‐MM1 study of ixazomib‐Rd vs. placebo‐Rd in patients with relapsed/refractory MM (RRMM) after 1–3 prior lines of therapy.3 In TOURMALINE‐MM1, 722 patients were randomized to receive ixazomib 4 mg or matching placebo on days 1, 8, and 15, plus lenalidomide 25 mg on days 1–21 and dexamethasone 40 mg on days 1, 8, 15, and 22, every 28 days until disease progression or unacceptable toxicity.3 Randomization was stratified by prior lines of therapy (1 vs. 2 or 3), International Staging System disease stage (I or II vs. III), and prior proteasome inhibitor exposure (yes vs. no). The results showed significantly improved progression‐free survival (PFS, primary endpoint; median 20.6 vs. 14.7 months, hazard ratio (HR) 0.74, P = 0.012) and higher response rates with ixazomib‐Rd compared to placebo‐Rd.3 Furthermore, ixazomib‐Rd was associated with limited additional toxicity compared to placebo‐Rd, with no adverse impact observed on patient‐reported quality of life.3
Ixazomib was approved by the US Food and Drug Administration (FDA) only 6 years after the first MM patients were treated in phase I clinical trials,4, 5, 6 with this time period encompassing comprehensive clinical pharmacology characterization of this novel agent. Importantly, unlike most small molecule anticancer drugs that are initially approved without complete information to guide use across clinical contexts (e.g., organ impairment, drug–drug interactions (DDI)),7 the prescribing information for ixazomib contained complete clinical pharmacology characterization with no postapproval requirements noted by the FDA. One of the enablers of the speed of development of ixazomib and the completeness of the initial prescribing information from a clinical pharmacology perspective was the prospective and strategic integration of extensive pharmacologic evaluation, including MIDD8 at all phases of the program, with a focus on patient‐centric therapeutic optimization across contexts of clinical use. MIDD is being increasingly recognized as a vital component of modern drug development, with specific opportunities identified in anticancer drug development to maximize benefit–risk across patient populations, probability of success, and overall drug development efficiency.9 Quantitative and systems pharmacology concepts and tools are becoming increasingly popular in the pharmaceutical industry for streamlining the development process and avoiding unnecessary costs,10 with numerous modeling techniques being employed to contribute to the overall benefit–risk analysis for a drug and in regulatory decision‐making.11, 12, 13, 14, 15
Several questions faced by the ixazomib development team are illustrative of common themes in oncology drug development that can benefit from model‐informed knowledge management. MIDD was used across the development continuum for ixazomib to optimize benefit–risk, inform drug development decisions, and facilitate regulatory review (Figure 1). The key questions by phase of development, associated MIDD approach, value gained from a model‐informed approach, and the level of impact assessed using the European Federation of Pharmaceutical Industries and Associations Model Informed Drug Discovery and Development (MID3) category are summarized in Table 1 . Pharmacokinetic (PK) and clinical safety and efficacy data from multiple studies, including the phase III TOURMALINE‐MM1 study,3 contributed to MIDD analyses, which incorporated a variety of modeling techniques. For example, population PK analyses were performed to quantitatively characterize the patient‐specific sources of variability in ixazomib systemic exposure,16, 17 a physiologically based PK (PBPK) model was developed to bridge clinical understanding of DDI risk and forecast risk for unstudied interactions,18 and the concentration–QTc relationship of ixazomib was assessed to quantitatively assess the risk for QT prolongation in lieu of a dedicated QT study in cancer patients.19 Further, exposure–response (ER) relationships were examined for efficacy and safety endpoints of clinical interest in order to quantitatively characterize the benefit–risk profile in support of the recommended posology,20, 21 and a model‐based meta‐analysis (MBMA) framework was developed to predict PFS from overall response rate (ORR) in RRMM and serve as a decision‐making tool in clinical development.22 This combination of tools resulted in the comprehensive characterization of the clinical pharmacology of ixazomib, and herein we review the wide‐ranging value of these analyses and the benefits accrued from using an MIDD approach during the development of ixazomib.
Figure 1.
MIDD across the development continuum for ixazomib.
Table 1.
Questions addressed via MIDD approaches during the development of ixazomib
| Development stage | Question | MIDD approach | Value | EFPIA impact category |
|---|---|---|---|---|
| Phase I | Is BSA‐based dosing necessary? | Population PK analysis of emerging phase I data | Switch from BSA‐based to fixed dosing, simplifying posology/manufacture and reducing risk of dosing errors | Medium |
| Can patients with mild or moderate renal impairment be enrolled in pivotal trials? | Population PK analysis of emerging phase I data |
Design of inclusive phase III trial without exclusion of MM patients with mild or moderate renal impairment Reduced study design for renal impairment study |
Medium | |
| What is the optimal dosing approach to maximize benefit–risk in the setting of maintenance treatment of MM? | Exposure–response analysis of emerging phase I safety and efficacy | Titration dosing regimen selected for phase III maintenance trial with a safety/tolerability profile to maximize adherence and decrease risk of poor compliance | Medium | |
| Phase II | Which ixazomib‐based combinations should be selected for pivotal development? | MBMA to predict PFS from ORR in RRMM | Enabled portfolio prioritization of ixazomib‐based combinations for lifecycle management | High |
| Does ixazomib prolong the QT interval? | Concentration–QTc analysis of phase I data | Concluded lack of effect on QTc, eliminating need for dedicated QT study and informing labeling | High | |
| Phase III |
How can the lack of effect of CYP3A inhibitors be reconciled with clinically meaningful effect of a strong CYP3A inducer? What is the absolute bioavailability of ixazomib? |
PBPK modeling and simulation Integrated population PK analysis of IV (phase I) and oral (phases I–III) data |
Provided quantitative mechanism‐based reconciliation of observed DDI outcomes facilitating regulatory review Absolute bioavailability estimated without need for dedicated study to meet specific regulatory requirement (TGA for approval in Australia), inform PBPK modeling, and contribute to labeling |
Low Medium |
| Does the 4 mg dose in combination with lenalidomide‐dexamethasone offer optimal benefit–risk for patients with RRMM? | Exposure–response analyses of safety and efficacy in phase III TOURMALINE‐MM1 study | Approval of 4 mg weekly ixazomib in combination with lenalidomide‐dexamethasone as an optimal dose for patients with RRMM in global regulatory review, also supporting proposed dose‐reduction guidelines for treatment‐emergent toxicities | Medium | |
| How can the benefit–risk profile be enhanced for the Japanese population? | Population PK and exposure–response analyses of safety and efficacy in phase III TOURMALINE‐MM1 study | Identified modestly higher systemic exposures in Japanese patients that impacted dose intensity of lenalidomide, counteracting the positive effects of higher ixazomib dose/exposure, thereby supporting dose‐reduction guidelines in a Japan phase II bridging study (NCT02917941) to maximize benefit–risk profile | Medium |
BSA, body surface area; CYP3A, cytochrome P450 3A; DDI, drug–drug interaction; EFPIA, European Federation of Pharmaceutical Industries and Associations; IV, intravenous; MBMA, model‐based meta‐analysis; MIDD, model‐informed drug development; MM, multiple myeloma; ORR, overall response rate; PBPK, physiologically based pharmacokinetic; PFS, progression‐free survival; PK, pharmacokinetic; RRMM, relapsed/refractory multiple myeloma; TGA, Therapeutic Goods Administration.
POPULATION PHARMACOKINETIC ANALYSIS AT THE END OF PHASE I TO SUPPORT THE SWITCH FROM BSA‐BASED TO FIXED DOSING
Ixazomib was initially investigated in early phase I studies in RRMM using BSA‐based dosing,4, 5 as commonly employed with anticancer drugs23 and as used for the other two proteasome inhibitors that are approved for the treatment of MM, bortezomib and carfilzomib.24, 25 However, reflecting suggestions that fixed dosing may be just as appropriate in controlling interpatient PK variability of anticancer drugs,23, 26 and with the aim of developing a simpler and more convenient dosing regimen for ixazomib, a population PK analysis was conducted using emerging PK data from patients enrolled across four phase I studies. This analysis aimed to evaluate the feasibility of switching from BSA‐based to fixed dosing for clinical development and to determine the influence of baseline patient characteristics on ixazomib PK.17
The analyses demonstrated that BSA (1.4–2.6 m2) had no significant effect on ixazomib clearance (Figure 2 a), indicating that total systemic exposures of ixazomib (area under the concentration–time curve; AUC) following fixed dosing should be independent of the individual patient's BSA; simulated AUC values after a fixed 4 mg oral dose using the final population PK model confirmed this. The model also estimated no impact of age, race, or mild/moderate renal impairment on ixazomib clearance or AUC, based on the available data.17
Figure 2.
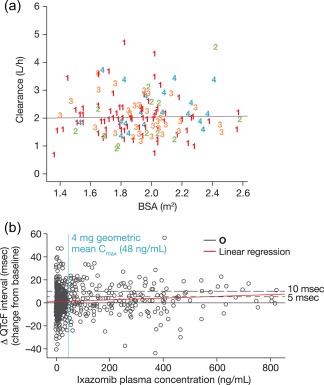
(a) Relationship between BSA and ixazomib clearance (numbers represent individual patients enrolled across four different phase I studies). (b) Relationship between ixazomib plasma concentrations and mean change from baseline in QTcF.
As a consequence of these findings, clinical development switched posology from BSA‐based to fixed dosing, simplifying capsule strength manufacture and dosing across clinical studies. Furthermore, the results of these analyses indicating the lack of an effect of mild or moderate renal impairment on ixazomib clearance supported the design of a more inclusive phase III trial in RRMM (patients with creatinine clearance of ≥30 mL/min were eligible for the TOURMALINE‐MM1 study).3 Of note, the model‐informed approach to characterizing the effects of mild or moderate renal impairment and mild hepatic impairment on ixazomib PK contributed to the overall efficiency in the registration process and simplified the conduct of organ impairment studies, eliminating the need for full designs and enabling appropriately reduced study designs.27, 28
This example supports a general framework for appropriate consideration of body size/weight‐related factors to inform dosing decisions in early clinical development of anticancer agents. While BSA‐based dosing has been historically the precedent in this therapeutic area, examples such as ixazomib indicate clearly that the impact of interpatient variability in body size/weight in an adult population of cancer patients is small relative to other known and unknown sources of variability in drug clearance. Especially for oral agents, for which variability in absorption and first‐pass metabolism can be substantial,29 the scientific rationale for individualizing doses based on patient‐specific body size/weight is weak. Accordingly, reflecting upon this example and viewed against the backdrop of other examples30, 31, 32 and expert opinion in this area,23, 26, 33, 34 it is recommended that first‐in‐human studies of anticancer agents be initiated with fixed dosing. A population PK analysis at the end of phase I should then be conducted to confirm the lack of meaningful effects of body size/weight on clearance in order to confirm the appropriate posology for phase II/III trials. We recommend this approach instead of the path taken in the case of ixazomib because the available prior studies suggest that the cases in which body size/weight has a clinically meaningful effect on anticancer drug clearance are expected to be limited. The more likely scenario is that even if body size/weight is identified as a statistically significant contributor to overall PK variability, the magnitude of the effect is likely not of clinical relevance to warrant body size‐based dosing. Not applying a scientifically guided approach to this important question and empirically taking forward body size‐based dosing in pivotal trials can lead to deleterious consequences (i.e., patients with higher BSA getting overdosed and smaller patients getting subtherapeutic exposures). This can adversely impact the benefit–risk profile in the population, as has been noted for the protein synthesis inhibitor omacetaxine,35 which is indicated for the treatment of adult patients with chronic‐ or accelerated‐phase chronic myeloid leukemia with resistance and/or intolerance to two or more tyrosine kinase inhibitors.36 While omacetaxine is approved for use at a BSA‐based dose, it is possible that results from a postmarketing commitment study could suggest that fixed dosing may be a viable approach.
CONCENTRATION–QTc ANALYSIS IN LIEU OF A DEDICATED QTc STUDY
Assessment of the risk of QT prolongation is a regulatory requirement for all new drugs, with a thorough QT (TQT) or dedicated QT study generally recommended.37 This assessment was of particular relevance for ixazomib, given the prior reports of cardiac toxicity in patients with MM with proteasome inhibitor therapy,38, 39, 40 and the inclusion of cardiac toxicities within the “Warnings and Precautions” section of the US prescribing information for other proteasome inhibitors.24, 25 However, in the context of the challenges of conducting TQT studies with some anticancer drugs and the increasing use and acceptability of concentration–QT analyses as an alternative approach of assessing QT prolongation risk,7, 41, 42, 43, 44 an integrated analysis of QT effects of ixazomib from nonclinical and early‐phase clinical data was conducted in lieu of a dedicated QT study.19
A linear mixed‐effects model that accounted for the day and time of ECG data acquisition45 was used to analyze the relationship between ixazomib plasma concentration and QTcF (QTc corrected for heart rate using Fridericia's method). The findings from this analysis showed that ixazomib did not prolong the QTc interval at clinically relevant exposures (Figure 2 b). At the 4 mg approved clinical dose, the mean change from baseline in QTcF was estimated to be 0.07 msec (90% confidence interval (CI): –0.22, 0.36) from the model‐based analysis. Furthermore, other findings showed that the upper limit of the 90% CI for QTcF changes from baseline was well below the regulatory threshold37 of 5 msec even at ixazomib plasma concentrations four times the mean for the 4 mg dose.19 These findings were included in the US prescribing information, which states: “NINLARO did not prolong the QTc interval at clinically relevant exposures based on pharmacokinetic–pharmacodynamic analysis of data from 245 patients.”1 The results of this concentration–QTc analysis were accepted in lieu of a dedicated QT or TQT study with ixazomib, with no postmarketing requirements or commitments issued by the FDA, and this analysis was also reviewed favorably by regulatory agencies worldwide, including the European Medicines Agency, SwissMedic, the Pharmaceutical and Medical Devices Agency in Japan, Health Canada, and the Therapeutic Goods Administration in Australia.
Viewed from a broader perspective, the example of ixazomib taken together with other similar examples7 and recommendations in the most recent (June 2017) update to the FDA ICH E14 guidance46 provide a strong rationale for prospective strategic integration of PK time‐matched ECGs into oncology phase I dose‐escalation studies. Collection of these data over a range of doses and associated exposures in patients across the escalation and expansion phases of these studies should enable model‐informed estimation of the effects on QTc, to underwrite proarrhythmic risk assessment, contextualized by in vitro and preclinical in vivo safety pharmacology data in a totality‐of‐evidence mindset, eliminating the need for expending valuable patient resources in redundant clinical investigation in a dedicated QTc study.
ROLE OF POPULATION PHARMACOKINETIC ANALYSIS IN GLOBAL REGULATORY REVIEW AND PRESCRIBING INFORMATION
The early population PK analysis was updated using data from 755 patients enrolled across 10 clinical studies,16 including the phase III TOURMALINE‐MM1 study.3 No significant covariates were identified on ixazomib clearance. Consequently, it was determined that no ixazomib dose adjustment is required for BSA, sex, age, race, mild/moderate renal impairment (creatinine clearance ≥30 mL/min), or mild hepatic impairment (Figure 3 a). These findings are reflected in the US prescribing information for ixazomib and EU Summary of Product Characteristics.1, 2 Labeling for use in special populations was informed by the totality of evidence based on population PK analysis and the reduced‐design renal/hepatic impairment studies, viewed in relation to dose/exposure–safety understanding of ixazomib. Importantly, the use of PK data from studies of ixazomib as a single agent and in combination with Rd enabled this aspect to be tested in the model. It was shown that Rd has no impact on the PK of ixazomib, a valuable finding in the context of ixazomib‐Rd being the approved regimen and numerous ongoing studies in MM of this and related regimens. Finally, the model‐based estimate of absolute bioavailability of 58% of oral ixazomib capsules from this joint population model of IV and oral dose PK data helped meet specific regulatory requirements for this PK parameter,47 informed subsequent PBPK model development (as reviewed in the next section), and contributed to the absorption, distribution, metabolism, and excretion characterization of ixazomib in worldwide product labels.
Figure 3.
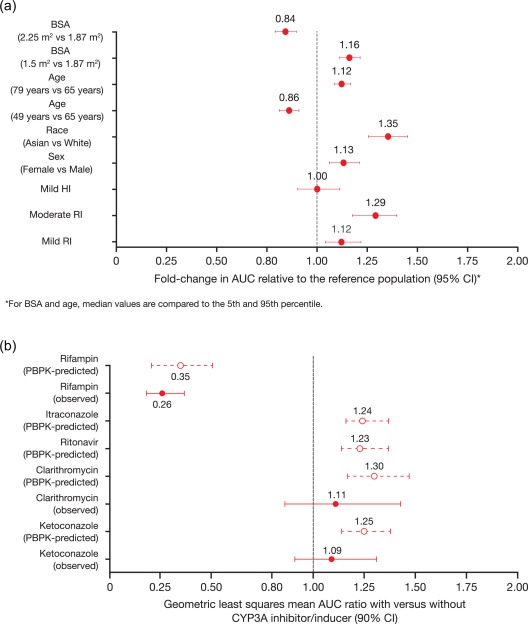
(a) Fold change in ixazomib AUC according to baseline covariates (test vs. reference), and (b) PBPK model‐predicted and observed geometric least squares mean AUC ratios for ixazomib with and without various strong CYP3A inhibitors and strong CYP3A inducers.18 For predicted data, error bars represent the 5th and 95th percentile. Panel b reproduced from Gupta, N. et al. J. Clin. Pharmacol. 58, 180–192. doi:10.1002/jcph.988 (2017)18 under the Creative Commons license: https://creativecommons.org/licenses/by-nc/4.0/legalcode
APPLICATION OF A PBPK MODEL TO FACILITATE REGULATORY REVIEW
Population PBPK modeling has emerged as an important tool within the field of drug development over the past 10 years,48 and is being increasingly applied across the development continuum. Notably, increased use of PBPK modeling in FDA submissions for assessing the potential for DDIs has been reported, with models demonstrating close concordance with observed effects of cytochrome P450 (CYP) enzyme inhibitors and inducers.49, 50 PBPK modeling was used for this purpose in the development of ixazomib,18 in order to provide scientific plausibility in explaining the observed totality of DDI results with ixazomib as the object drug and to facilitate regulatory review of the DDI potential.1
Metabolism by multiple CYP enzymes and non‐CYP proteins is expected to be the major clearance mechanism for ixazomib. At clinically relevant ixazomib concentrations, in vitro studies using human cDNA‐expressed cytochrome P450 isozymes showed that no specific CYP isozyme predominantly contributes to ixazomib metabolism. At higher than clinical concentrations, ixazomib was metabolized by multiple CYP isoforms, with 42% relative contribution of CYP3A4 estimated based on the results of in vitro studies using human cDNA‐expressed CYP isozymes.1 In clinical DDI studies, ixazomib AUC was not meaningfully altered upon coadministration of strong CYP3A inhibitors (ketoconazole and clarithromycin), indicating a minor role for CYP3A in ixazomib clearance.18 However, rifampin, a prototypic strong inducer of PXR‐inducible enzymes, decreased ixazomib systemic exposure by 74%,18 raising questions during regulatory review regarding the explanation for this apparent disconnect and the level of risk for DDIs with other strong CYP3A inhibitors.
In order to address this issue, a PBPK model was developed to examine if a minor contribution of CYP3A to ixazomib clearance was consistent with the observed totality of DDI study results with CYP3A inhibitors and inducers. Through the incorporation of a minor contribution of CYP3A to overall ixazomib clearance and by quantitatively considering the strength of induction of CYP3A and intestinal P‐glycoprotein by rifampin, the PBPK model was able to reconcile well the clinical DDI study results, with model‐predicted geometric mean AUC ratios falling within the 90% CI for the observed ratios from each DDI study.18 Based on this verification of the model, the final model was also used to simulate additional clinical DDI studies to forecast the expected magnitude of unstudied interactions. The effects of coadministration of the strong CYP3A inhibitors ritonavir and itraconazole on the PK of ixazomib were investigated, and findings showed similar geometric least‐squares mean AUC ratios to those simulated for ketoconazole and clarithromycin (Figure 3 b). These simulation results provided further support for the lack of a clinically meaningful CYP3A inhibitor effect on ixazomib PK, and were used during regulatory review to explain the clinically significant effect of rifampin despite the lack of a strong CYP3A inhibitor effect on ixazomib PK.1
MODEL‐BASED ANALYSES TO SUPPORT FAVORABLE BENEFIT–RISK OF PROPOSED DOSE
As cancer becomes a chronic disease, a key question in oncology drug development is regarding the appropriateness of the selected dose and schedule to optimize benefit–risk.51, 52 A recent analysis of oncology new molecular entity submissions to the FDA indicated that almost a quarter of submissions from 2011 to 2017 were issued with a postmarketing requirement/commitment to conduct ER analyses for dose‐optimization or to evaluate alternative dose regimens.7
The approved dose and schedule of ixazomib, as employed in TOURMALINE‐MM1 in combination with Rd,3 is 4 mg weekly in 28‐day cycles.1 Initial analysis of two phase I single‐agent studies in RRMM demonstrated a favorable efficacy/safety profile with weekly vs. twice‐weekly dosing, and a phase I/II study of ixazomib‐Rd in newly diagnosed MM (NDMM) determined that utilizing a 4 mg weekly dose in this combination resulted in an acceptable efficacy/safety profile.21 The favorable benefit–risk profile of this dose was quantitatively supported by ER analyses of efficacy and safety data in the pivotal trial.21
The first key finding of these analyses was that ixazomib exposure (in terms of time‐averaged systemic exposure – AUC/day) was not identified as a significant predictor of PFS (P = 0.26), with a longer median PFS seen with ixazomib‐Rd across all four ixazomib exposure quartiles vs. with placebo‐Rd (Figure 4 a).21 Similarly, logistic regression analyses showed that the probability of achieving a complete response (CR), a very good partial response or better (≥VGPR), or a partial response or better (≥PR) was not associated with ixazomib exposure. However, in contrast, statistically significant relationships were identified between ixazomib exposure and the probability of treatment‐emergent adverse events (TEAEs) of clinical interest, notably the probabilities of grade ≥3 thrombocytopenia, and grade ≥2 rash, fatigue, and diarrhea (Figure 4 b–e).21 Based on the dose‐linear PK of ixazomib and the observed exposure–safety relationships, it was estimated that the odds of grade ≥3 thrombocytopenia and anemia, and grade ≥2 rash, fatigue, diarrhea, nausea, and peripheral neuropathy will be reduced by ∼20–50% with a dose reduction from 4 mg to 3 mg.21 These findings thus support the established dose‐reduction guidelines for ixazomib, which recommend this decrement as the first dose decrease required for specific toxicities during treatment.1
Figure 4.
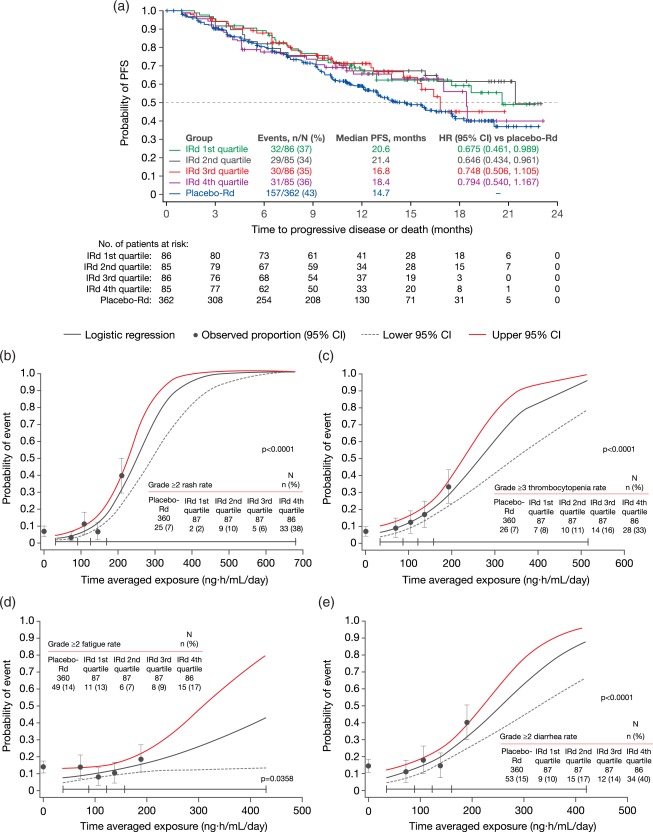
(a) Ixazomib exposure–PFS analysis in the ixazomib‐Rd and placebo‐Rd arms of TOURMALINE‐MM1.21 Kaplan–Meier curves show PFS distributions in the placebo‐Rd arm and in the ixazomib‐Rd (IRd) arm by quartiles of ixazomib exposure. (b–e) Observed incidence and predicted probability of (b) grade ≥2 rash, (c) grade ≥3 thrombocytopenia, (d) grade ≥2 fatigue, and (e) grade ≥2 diarrhea as a function of ixazomib exposure using a logistic regression model.21 Black circles and error bars show the event probabilities plus 95% CI in the placebo‐Rd arm and in the IRd arm within each ixazomib exposure quartile. Reproduced from Gupta, N. et al. Target. Oncol. 12, 643–654. https://doi.org/10.1007/s11523-017-0524-3 (2017)21 under the Creative Commons license: https://creativecommons.org/licenses/by-nc/4.0/legalcode
Furthermore, an exposure–lenalidomide relative dose intensity (RDI) analysis established that a lenalidomide RDI of ≥60% was less likely to be achieved with increasing ixazomib exposure in TOURMALINE‐MM1,21 as outlined in Figure 5 a. Together with the exposure–safety analyses, these findings suggest that increasing the weekly ixazomib dose beyond 4 mg in combination with Rd could result in higher rates of TEAEs, which could require lenalidomide dose reductions and thereby negatively impact the RDI of lenalidomide. As a consequence, patients would receive a lower dose intensity of lenalidomide, which could counteract the potential positive effects of a higher ixazomib dose on the overall efficacy of the ixazomib‐Rd regimen (Figure 5 b).21 These conclusions from the ER analyses were important in the development of ixazomib, as they were used to support the dose‐reduction guidelines in a Japan phase II bridging study (NCT02917941) to maximize the benefit–risk profile for this population, given the modestly higher exposures of ixazomib in this population.21
Figure 5.
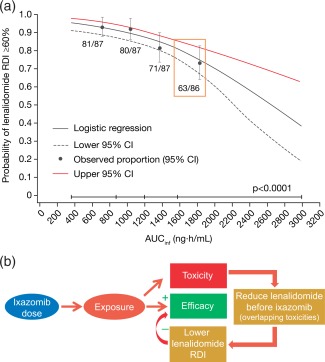
(a) Analysis of lenalidomide RDI according to ixazomib exposure (AUCinf) in the ixazomib‐Rd arm of TOURMALINE‐MM1, showing probability of lenalidomide RDI ≥60%.21 Black circles and error bars show the event probabilities plus 95% CI in the ixazomib‐Rd arm within each ixazomib exposure quartile. (b) Schematic illustrating the relationship between ixazomib dose, systemic exposure, and the RDI of lenalidomide in the ixazomib‐Rd regimen.21 Reproduced from Gupta, N. et al. Target. Oncol. 12, 643–654. https://doi.org/10.1007/s11523-017-0524-3 (2017) 21 under the Creative Commons license: https://creativecommons.org/licenses/by-nc/4.0/legalcode
These findings from ER analyses are also important in the context of suggestions that a more intensive regimen or higher dose may be feasible in some patients,53 due to the activity reported in other clinical studies of higher or twice‐weekly ixazomib dosing and the relatively limited additional toxicities seen with ixazomib‐Rd compared to placebo‐Rd in TOURMALINE‐MM1. For example, higher weekly dosing of ixazomib (5.5 mg) was investigated in a phase II study in RRMM in combination with dexamethasone, demonstrating a higher response rate; however, greater toxicity was also seen compared to the 4 mg dose.54 Additionally, twice‐weekly dosing of ixazomib 3 mg has been investigated in combination with Rd in a phase I/II study in NDMM, demonstrating notable activity but with potentially greater toxicity than seen in the similar phase I/II study utilizing weekly dosing.55
EXPOSURE–RESPONSE ANALYSES IN SUPPORT OF TITRATION REGIMENS
Dose titration represents an important potential approach for achieving optimal dosing of oncology drugs,56 with a recent proposal that an individualized dose‐titration algorithm should supersede the concept of a single maximum tolerated dose,57 and a number of agents recently receiving approvals incorporating the potential for upward dose titration.7 Such an approach may help account for interindividual variability in PK and susceptibility to key toxicities. It may also enable clinicians to adapt their use of a particular agent to different treatment settings without requiring development of therapeutic drug‐monitoring algorithms,58, 59 which have faced challenges with respect to qualification as promising tools for translation to clinical use in practice settings.60, 61, 62, 63 With ixazomib, the 4 mg starting dose used in the TOURMALINE‐MM1 phase III study in patients with RRMM was determined based on early‐phase study data for single‐agent ixazomib in heavily pretreated patients with RRMM and for ixazomib‐Rd in NDMM.4, 5, 55, 64 As demonstrated by the ER analyses21 conducted using data from TOURMALINE‐MM1 and by the safety and tolerability profile of ixazomib‐Rd in that study,3 a 4 mg dose of ixazomib provided a favorable benefit–risk balance in the treatment of RRMM. However, the development of ixazomib also encompasses its investigation in the maintenance setting, both posttransplantation and following initial induction therapy in transplant‐ineligible NDMM patients, and in this treatment setting the benefit–risk considerations differ somewhat compared to RRMM or initial therapy for NDMM.52, 65 With patients typically having already achieved a PR or better, development of ixazomib in the maintenance setting needed to proceed with a focus on ensuring long‐term tolerability while permitting durable disease control.
In this context of ixazomib development, additional ER analyses were conducted on phase I data for single‐agent ixazomib in RRMM4 with the aim of informing the appropriate ixazomib dose for use in phase III maintenance studies, including the potential for a dose‐titration approach.20 Significant relationships were identified between ixazomib exposure (in terms of time‐averaged systemic exposure – AUC/day) and the rate of clinical benefit, as well as the rates of grade ≥3 thrombocytopenia and neutropenia, and grade ≥2 rash, fatigue, and diarrhea, and these analyses were then used to determine the probabilities of clinical benefit and TEAEs in relation to dose.20 At a 3 mg dose of ixazomib, it was predicted that the probabilities of TEAEs would be reduced compared to the 4 mg dose. Furthermore, the 3 mg dose was expected to be within the clinically active range for ixazomib based on exposure–clinical benefit rate analyses (Figure 6). Accordingly, to appropriately balance benefit vs. risk in the long‐term maintenance setting, a starting dose of 3 mg with escalation to 4 mg, if tolerated, is being used in the two ongoing maintenance trials of ixazomib, specifically TOURMALINE‐MM3 (NCT02181413), a phase III study of ixazomib vs. placebo as maintenance therapy posttransplant in MM patients with posttransplant response (≥PR), and TOURMALINE‐MM4 (NCT02312258), a phase III study of ixazomib vs. placebo as maintenance therapy in MM patients not eligible for transplant achieving ≥PR after 6–12 months of initial therapy.66 Furthermore, this example more broadly illustrates the value of characterizing exposure–efficacy and exposure–safety relationships in early clinical development for tailoring dosing strategies in pivotal clinical development guided by differential context‐specific benefit–risk considerations. For example, while dose selection in advanced disease may be guided by the need for more aggressive pharmacotherapy to rapidly reduce disease burden and associated symptoms, supported by medical management of treatment‐related toxicities with close monitoring, the goal of long‐term treatment in the maintenance setting is to achieve durable disease control at the selected dose with a safety/tolerability profile that will maximize adherence and decrease risk of poor compliance, which in turn could promote development of resistance and disease recurrence.
Figure 6.
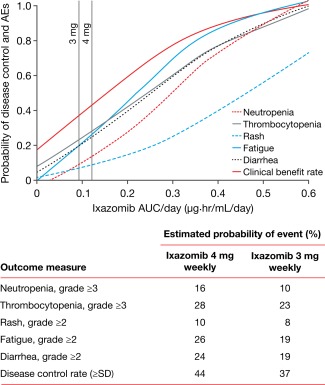
Relationship between ixazomib exposure and TEAEs or clinical benefit rate.20 Reproduced from Gupta, N. et al. Invest. New Drugs 34, 338–346, https://doi.org/10.1007/s10637-016-0346-7 (2016) under the Creative Commons license: https://creativecommons.org/licenses/by-nc/4.0/legalcode
MODEL‐BASED META‐ANALYSIS FOR GO/NO‐GO DECISION‐MAKING
Despite steady progress in the research and development of cancer therapeutics, the efficiency of oncology drug development remains suboptimal, with an estimated probability of success of 5% from phase I and 33% from phase III over the 2006–2015 clinical development timeframe.67 Late‐phase failures consume valuable patient resources in large and expensive confirmatory trials when the level of evidence for overall proof‐of‐concept and positive benefit–risk balance may simply not have been there to justify their conduct in the first place.68 While decisions to initiate pivotal development in oncology are often based on promising response rates observed in early clinical development (e.g., in phase I expansion cohorts or phase II signal‐seeking trials), a principled approach to decision‐making needs to consider prior disease‐specific knowledge of the performance characteristics of response rate as a decision‐enabling endpoint to forecast the likelihood of desired survival outcomes in phase III. This is of particular importance in MM, in which randomized controlled trials are expected for regulatory approval and the typical size of confirmatory phase III trials is ∼250–500 patients per arm.22 Accordingly, it was of strategic importance to maximize the probability of success for phase III trials of ixazomib‐based combinations in lifecycle management and to prioritize combinations most likely to win from phase II ORR data.
MBMA approaches are becoming established as an important analytical method in drug development that can help streamline the development process through the modeling of head‐to‐head trials and aspects of drug pharmacology, such as dose–response relationships within a drug class, considering potency and PK differences.69, 70, 71, 72 Furthermore, utilizing response rates from early‐phase studies, MBMA can be used for predicting PFS and overall survival (OS) based on an enhanced quantitative knowledge management framework, provided a relationship across these endpoints is qualified. For example, an MBMA in glioblastoma demonstrated the lack of meaningful correlation between ORR and OS, whereas a strong correlation between PFS and OS could be demonstrated.73 This example illustrates the importance of quantitative interrogation of relationships between ORR and survival outcomes within a disease to inform principled decisions on ORR data from early‐phase studies. In the first example of such an approach being utilized in RRMM, data from TOURMALINE‐MM1 and six other phase III clinical trials of other regimens studied in this setting were included in an MBMA that was developed using linear regression to establish a relationship between ORR data in phase II studies and PFS in phase III studies.22 A strongly correlated linear relationship was established using the model, the predictive ability of which was demonstrated by comparing predicted to observed PFS with ixazomib‐Rd. MBMA predicted a PFS of 20 months based on an ORR of 78% for ixazomib‐Rd (Figure 7),22 consistent with results of TOURMALINE‐MM1.3 This model has provided a useful quantitative framework to help prioritize ixazomib‐based combinations for further clinical development based on Bayesian inference of expected PFS and probability of technical success from ORR observed in phase I/II studies. MBMA approaches such as this can thus help estimate the probability of technical success of a phase III trial and thereby decrease the failure rate and speed up the implementation of such studies based on early data from phase II studies without necessarily waiting for mature PFS data, which would otherwise require longer and larger early‐phase trials. Predicting the likelihood of achieving the gold‐standard efficacy targets that have been established in the target product profile for a novel agent can subsequently inform “Go/No‐Go” decisions regarding its development and, indeed, the appropriate relative prioritization of development of novel pipeline agents within the same indication when such approaches are deployed at the portfolio level.
Figure 7.
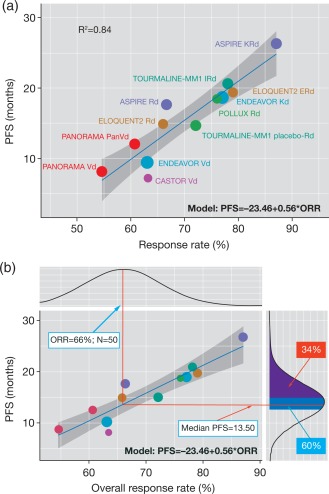
(a) Relationship between ORR and median PFS using data from seven phase III studies. The blue line represents the linear regression line and the gray band represents the 95% CI. (b) An illustrative example of predicting PFS using ORR. The probability of achieving the target product profile (PFS 15 months) is 34% (purple area) and the probability of achieving the minimum detectable PFS is 60% (blue area). Reproduced from Teng, Z. et al. Clin. Transl. Sci. e‐pub ahead of print (2017) 22 under the Creative Commons license: https://creativecommons.org/licenses/by-nc/4.0/legalcode
CONCLUSION
This review has discussed the pivotal role played by MIDD in the development of ixazomib. When viewed from a broader perspective, the following instructive themes emerge from this example that are widely applicable to oncology drug development:
Challenge the conventional wisdom of BSA‐based dosing.
Conserve valuable patient resources and gain efficiency in drug development by eliminating the need for dedicated QT studies and leveraging the power of model‐based analyses.
Minimize redundant clinical investigation by complementing DDI/special population data with in silico population pharmacology analyses.
Strive to design more inclusive clinical trials with model‐informed posology to optimize benefit–risk for special patient populations.
Consider titration‐based dosing approaches to optimize benefit–risk.
Commit to principled decision‐making bolstered by quantitative knowledge management of historical data to characterize the therapeutic potential.
The findings of the analyses summarized here impacted internal development decisions and the regulatory review process, and informed product labeling for ixazomib. MIDD leveraged a number of valuable pharmacometric tools and strategies in the comprehensive pharmacologic characterization of ixazomib and, in doing so, contributed to the rapid development of ixazomib in RRMM, whereby initial FDA approval was granted in November 2015.1
In conclusion, MIDD approaches are vital to modern oncology drug development for optimizing benefit–risk across contexts of clinical use, addressing anticipated regulatory review requirements, minimizing redundant clinical investigation, enabling complete prescribing guidance at first approval, and promoting a culture of principled decision‐making in drug development, with the ultimate goal of making novel anticancer therapies available to patients with a strong sense of urgency.
FUNDING
The research was funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
CONFLICT OF INTEREST/DISCLOSURES
N.G., M.J.H., H.Y., Z.T., R.L., D.B., C.P., G.L., H.vdV., and K.V. are employees of Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. P.M.D. is an employee of Certara Strategic Consulting, Breda, The Netherlands, which conducted analytical work for Millennium Pharmaceuticals, Inc. A.K. is an employee of Certara USA, Inc., Princeton, NJ, USA, which conducted analytical work for Millennium Pharmaceuticals, Inc. As an Associate Editor for Clinical Pharmacology & Therapeutics, Karthik Venkatakrishnan was not involved in the review or decision process for this article.
AUTHOR CONTRIBUTIONS
N.G. and K.V. wrote the article; N.G., M.J.H., H.Y., Z.T., R.L., D.B., C.P., G.L., H.vdV., and K.V. designed the component research described in the article; N.G., M.J.H., P.M.D., H.Y., A.K., Z.T., R.L., D.B., C.P., G.L., H.vdV., and K.V. performed the component research described in the article; all authors reviewed and revised the article, and approved the final version for submission.
ACKNOWLEDGMENTS
The authors thank all the patients and their families, the investigators, and the ixazomib team members who took part in the studies reported herein. The authors acknowledge the valuable contribution of Yeamin Huh, of Ann Arbor Pharmacometrics Group, Ann Arbor, MI, USA, in conducting the analytical work reported herein. The authors also acknowledge editorial support from Steve Hill of FireKite, an Ashfield company, part of UDG Healthcare plc, during the development of this article, which was funded by Millennium Pharmaceuticals, Inc., and complied with Good Publication Practice 3 ethical guidelines (Battisti et al., Ann. Intern. Med. 163, 461–464 (2015)).
References
- 1. Millennium Pharmaceuticals Inc . NINLARO® (ixazomib) capsules, for oral use. United States Prescribing Information. 2016.
- 2. Takeda Pharma A/S . NINLARO: European Product Assessment Report (EPAR) — Product Information. Annex I — Summary of Product Characteristics. 2017.
- 3. Moreau, P. et al Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 374, 1621–1634 (2016). [DOI] [PubMed] [Google Scholar]
- 4. Kumar, S.K. et al Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood 124, 1047–1055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richardson, P.G. et al Phase 1 study of twice‐weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood 124, 1038–1046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta, N. et al Pharmacokinetics and safety of ixazomib plus lenalidomide‐dexamethasone in Asian patients with relapsed/refractory myeloma: a phase 1 study. J. Hematol. Oncol. 8, 103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faucette, S. , Wagh, S. , Trivedi, A. , Venkatakrishnan, K. & Gupta, N. Reverse translation of US Food and Drug Administration reviews of oncology new molecular entities approved in 2011‐2017: lessons learned for anticancer drug development. Clin. Transl. Sci. e‐pub ahead of print; doi: 10.1111/cts.12527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Efpia Mid Workgroup et al Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venkatakrishnan, K. et al Optimizing oncology therapeutics through quantitative translational and clinical pharmacology: challenges and opportunities. Clin. Pharmacol. Ther. 97, 37–54 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Visser, S.A. , de Alwis, D.P. , Kerbusch, T. , Stone, J.A. & Allerheiligen, S.R. Implementation of quantitative and systems pharmacology in large pharma. CPT Pharmacometrics Syst. Pharmacol. 3, e142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellanti, F. , van Wijk, R.C. , Danhof, M. & Della Pasqua, O. Integration of PKPD relationships into benefit‐risk analysis. Br. J. Clin. Pharmacol. 80, 979–991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rowland, M. , Lesko, L.J. & Rostami‐Hodjegan, A. Physiologically based pharmacokinetics is impacting drug development and regulatory decision making. CPT Pharmacometrics Syst. Pharmacol. 4, 313–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friedrich, C.M. A model qualification method for mechanistic physiological QSP models to support model‐informed drug development. CPT Pharmacometrics Syst. Pharmacol. 5, 43–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Standing, J.F. Understanding and applying pharmacometric modelling and simulation in clinical practice and research. Br. J. Clin. Pharmacol. 83, 247–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkins, J.J. et al Thoughtflow: standards and tools for provenance capture and workflow definition to support model‐informed drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 6, 285–292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gupta, N. et al Population pharmacokinetic analysis of ixazomib, an oral proteasome inhibitor, including data from the phase III TOURMALINE‐MM1 study to inform labelling. Clin. Pharmacokinet. 56, 1355–1368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupta, N. , Zhao, Y. , Hui, A.M. , Esseltine, D.L. & Venkatakrishnan, K. Switching from body surface area‐based to fixed dosing for the investigational proteasome inhibitor ixazomib: a population pharmacokinetic analysis. Br. J. Clin. Pharmacol. 79, 789–800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta, N. et al Effects of strong CYP3A inhibition and induction on the pharmacokinetics of ixazomib, an oral proteasome inhibitor: results of drug‐drug interaction studies in patients with advanced solid tumors or lymphoma and a physiologically based pharmacokinetic analysis. J. Clin. Pharmacol. 58, 180–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta, N. , Huh, Y. , Hutmacher, M.M. , Ottinger, S. , Hui, A.M. & Venkatakrishnan, K. Integrated nonclinical and clinical risk assessment of the investigational proteasome inhibitor ixazomib on the QTc interval in cancer patients. Cancer Chemother. Pharmacol. 76, 507–516 (2015). [DOI] [PubMed] [Google Scholar]
- 20. Gupta, N. , Labotka, R. , Liu, G. , Hui, A.M. & Venkatakrishnan, K. Exposure‐safety‐efficacy analysis of single‐agent ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma: dose selection for a phase 3 maintenance study. Invest. New Drugs 34, 338–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gupta, N. et al Dose and schedule selection of the oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma: clinical and model‐based analyses. Target. Oncol. (2017) [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Teng, Z. et al Model‐based meta‐analysis for multiple myeloma: a quantitative drug‐independent framework for efficient decisions in oncology drug development. Clin. Transl. Sci. 12, 643–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathijssen, R.H. , de Jong, F.A. , Loos, W.J. , van der Bol, J.M. , Verweij, J. & Sparreboom, A. Flat‐fixed dosing versus body surface area based dosing of anticancer drugs in adults: does it make a difference? Oncologist 12, 913–923 (2007). [DOI] [PubMed] [Google Scholar]
- 24. Millennium Pharmaceuticals Inc . VELCADE® (bortezomib) for injection, for subcutaneous or intravenous use. 2017.
- 25. Onyx Pharmaceuticals Inc . KYPROLIS® (carfilzomib) for injection, for intravenous use. 2017.
- 26. Beumer, J.H. , Chu, E. & Salamone, S.J. Body‐surface area‐based chemotherapy dosing: appropriate in the 21st century? J. Clin. Oncol. 30, 3896–3897 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Gupta, N. et al A pharmacokinetics and safety phase 1/1b study of oral ixazomib in patients with multiple myeloma and severe renal impairment or end‐stage renal disease requiring haemodialysis. Br. J. Haematol. 174, 748–759 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gupta, N. et al Pharmacokinetics of ixazomib, an oral proteasome inhibitor, in solid tumour patients with moderate or severe hepatic impairment. Br. J. Clin. Pharmacol. 82, 728–738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sparreboom, A. & Verweij, J. Advances in cancer therapeutics. Clin. Pharmacol. Ther. 85, 113–117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elez, M.E. et al First‐in‐human phase I study of lurbinectedin (PM01183) in patients with advanced solid tumors. Clin. Cancer Res. 20, 2205–2214 (2014). [DOI] [PubMed] [Google Scholar]
- 31. Miller, A.A. , Rosner, G.L. , Egorin, M.J. , Hollis, D. , Lichtman, S.M. & Ratain, M.J. Prospective evaluation of body surface area as a determinant of paclitaxel pharmacokinetics and pharmacodynamics in women with solid tumors: Cancer and Leukemia Group B Study 9763. Clin. Cancer Res. 10, 8325–8331 (2004). [DOI] [PubMed] [Google Scholar]
- 32. Rudek, M.A. et al Fixed‐dose capecitabine is feasible: results from a pharmacokinetic and pharmacogenetic study in metastatic breast cancer. Breast Cancer Res. Treat. 139, 135–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sawyer, M. & Ratain, M.J. Body surface area as a determinant of pharmacokinetics and drug dosing. Invest. New Drugs 19, 171–177 (2001). [DOI] [PubMed] [Google Scholar]
- 34. Bins, S. , Ratain, M.J. & Mathijssen, R.H. Conventional dosing of anticancer agents: precisely wrong or just inaccurate? Clin. Pharmacol. Ther. 95, 361–364 (2014). [DOI] [PubMed] [Google Scholar]
- 35. US FDA Center for Drug Evaluation and Research . Clinical pharmacology and biopharmaceutics reviews, application number 203585Orig1s000: Omacetaxine mepesuccinate. 2012.
- 36. Rosshandler, Y. , Shen, A.Q. , Cortes, J. & Khoury, H.J. Omacetaxine mepesuccinate for chronic myeloid leukemia. Expert Rev. Hematol. 9, 419–424 (2016). [DOI] [PubMed] [Google Scholar]
- 37. Darpo, B. , Nebout, T. & Sager, P.T. Clinical evaluation of QT/QTc prolongation and proarrhythmic potential for nonantiarrhythmic drugs: the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use E14 guideline. J. Clin. Pharmacol. 46, 498–507 (2006). [DOI] [PubMed] [Google Scholar]
- 38. Bockorny, M. , Chakravarty, S. , Schulman, P. , Bockorny, B. & Bona, R. Severe heart failure after bortezomib treatment in a patient with multiple myeloma: a case report and review of the literature. Acta Haematol. 128, 244–247 (2012). [DOI] [PubMed] [Google Scholar]
- 39. Danhof, S. , Schreder, M. , Rasche, L. , Strifler, S. , Einsele, H. & Knop, S. ‘Real‐life’ experience of preapproval carfilzomib‐based therapy in myeloma — analysis of cardiac toxicity and predisposing factors. Eur. J. Haematol. 97, 25–32 (2016). [DOI] [PubMed] [Google Scholar]
- 40. Grandin, E.W. , Ky, B. , Cornell, R.F. , Carver, J. & Lenihan, D.J. Patterns of cardiac toxicity associated with irreversible proteasome inhibition in the treatment of multiple myeloma. J. Card. Fail. 21, 138–144 (2015). [DOI] [PubMed] [Google Scholar]
- 41. Garnett, C.E. et al Concentration‐QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J. Clin. Pharmacol. 48, 13–18 (2008). [DOI] [PubMed] [Google Scholar]
- 42. Darpo, B. et al Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin. Pharmacol. Ther. 97, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 43. Darpo, B. et al The IQ‐CSRC prospective clinical Phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?” Ann. Noninvasive Electrocardiol. 19, 70–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sarapa, N. , & Britto, M.R. Challenges of characterizing proarrhythmic risk due to QTc prolongation induced by nonadjuvant anticancer agents. Expert Opin. Drug Saf. 7, 305–318 (2008). [DOI] [PubMed] [Google Scholar]
- 45. Huh, Y. & Hutmacher, M.M. Evaluating the use of linear mixed‐effect models for inference of the concentration‐QTc slope estimate as a surrogate for a biological QTc model. CPT Pharmacometrics Syst. Pharmacol. 4, e00014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. United States Food and Drug Administration . E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs — Questions and Answers (R3) Guidance for Industry. 2017.
- 47. Australian Government — Department of Health Therapeutic Goods Administration . Guidance 15: Biopharmaceutic studies. 2015.
- 48. Zhuang, X. & Lu, C. PBPK modeling and simulation in drug research and development. Acta Pharm. Sin. B 6, 430–440 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wagner, C. et al Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 54, 117–127 (2015). [DOI] [PubMed] [Google Scholar]
- 50. Wagner, C. , Pan, Y. , Hsu, V. , Sinha, V. & Zhao, P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin. Pharmacokinet. 55, 475–483 (2016). [DOI] [PubMed] [Google Scholar]
- 51. Lu, D. et al A survey of new oncology drug approvals in the USA from 2010 to 2015: a focus on optimal dose and related postmarketing activities. Cancer Chemother. Pharmacol. 77, 459–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Minasian, L. , Rosen, O. , Auclair, D. , Rahman, A. , Pazdur, R. & Schilsky, R.L. Optimizing dosing of oncology drugs. Clin. Pharmacol. Ther. 96, 572–579 (2014). [DOI] [PubMed] [Google Scholar]
- 53. Richardson, P.G. et al The investigational proteasome inhibitor ixazomib for the treatment of multiple myeloma. Future Oncol. 11, 1153–1168 (2015). [DOI] [PubMed] [Google Scholar]
- 54. Kumar, S.K. et al Randomized phase 2 trial of ixazomib and dexamethasone in relapsed multiple myeloma not refractory to bortezomib. Blood 128, 2415–2422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Richardson, P. et al Twice‐weekly ixazomib plus lenalidomide‐dexamethasone in patients with newly diagnosed multiple myeloma: long‐term follow‐up data for patients who did not undergo stem cell transplantation. Haematologica 102, 317–318 (2017). [Google Scholar]
- 56. Sachs, J.R. , Mayawala, K. , Gadamsetty, S. , Kang, S.P. & de Alwis, D.P. Optimal dosing for targeted therapies in oncology: drug development cases leading by example. Clin. Cancer Res. 22, 1318–1324 (2016). [DOI] [PubMed] [Google Scholar]
- 57. Norris, D.C. Dose Titration Algorithm Tuning (DTAT) should supersede ‘the’ Maximum Tolerated Dose (MTD) in oncology dose‐finding trials. F1000Res 6, 112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lankheet, N.A.G. et al Optimizing the dose in cancer patients treated with imatinib, sunitinib and pazopanib. Br. J. Clin. Pharmacol. 83, 2195–2204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang, Y. , Chia, Y.L. , Nedelman, J. , Schran, H. , Mahon, F.X. & Molimard, M. A therapeutic drug monitoring algorithm for refining the imatinib trough level obtained at different sampling times. Ther. Drug Monit. 31, 579–584 (2009). [DOI] [PubMed] [Google Scholar]
- 60. Widmer, N. et al Review of therapeutic drug monitoring of anticancer drugs part two—targeted therapies. Eur. J. Cancer 50, 2020–2036 (2014). [DOI] [PubMed] [Google Scholar]
- 61. Decosterd, L.A. , Widmer, N. , Zaman, K. , Cardoso, E. , Buclin, T. & Csajka, C. Therapeutic drug monitoring of targeted anticancer therapy. Biomark. Med. 9, 887–893 (2015). [DOI] [PubMed] [Google Scholar]
- 62. Bardin, C. et al Therapeutic drug monitoring in cancer—are we missing a trick?, Eur. J. Cancer 50, 2005–2009 (2014). [DOI] [PubMed] [Google Scholar]
- 63. Paci, A. et al Review of therapeutic drug monitoring of anticancer drugs part 1—cytotoxics. Eur. J. Cancer 50, 2010–2019 (2014). [DOI] [PubMed] [Google Scholar]
- 64. Kumar, S.K. et al Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open‐label phase 1/2 study. Lancet Oncol. 15, 1503–1512 (2014). [DOI] [PubMed] [Google Scholar]
- 65. Postel‐Vinay, S. et al Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents—dose‐limiting Toxicity and Toxicity Assessment Recommendation Group for Early Trials of Targeted therapies, an European Organisation for Research and Treatment of Cancer‐led study. Eur. J. Cancer 50, 2040–2049 (2014). [DOI] [PubMed] [Google Scholar]
- 66. Palumbo, A. et al Two phase 3 studies of the oral proteasome inhibitor (PI) ixazomib for multiple myeloma (MM) in the maintenance setting: TOURMALINE‐MM3, and ‐MM4. J. Clin. Oncol. 34, TPS8068 (2016). [Google Scholar]
- 67. Thomas, D.W. , Burns, J. , Audette, J. , Carroll, A. , Dow‐Hygelund, C. & Hay, M. Clinical development success rates 2006‐2015. BIO Biomedtracker Amplion (2016) [Epub ahead of print]. [Google Scholar]
- 68. Seruga, B. , Ocana, A. , Amir, E. & Tannock, I.F. Failures in phase III: causes and consequences. Clin. Cancer Res. 21, 4552–4560 (2015). [DOI] [PubMed] [Google Scholar]
- 69. Mould, D.R. Model‐based meta‐analysis: an important tool for making quantitative decisions during drug development. Clin. Pharmacol. Ther. 92, 283–286 (2012). [DOI] [PubMed] [Google Scholar]
- 70. Mandema, J.W. , Gibbs, M. , Boyd, R.A. , Wada, D.R. & Pfister, M. Model‐based meta‐analysis for comparative efficacy and safety: application in drug development and beyond. Clin. Pharmacol. Ther. 90, 766–769 (2011). [DOI] [PubMed] [Google Scholar]
- 71. Lu, D. et al Model‐based meta‐analysis for quantifying Paclitaxel dose response in cancer patients. CPT Pharmacometrics Syst. Pharmacol. 3, e115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Perez‐Pitarch, A. , Guglieri‐Lopez, B. , Ferriols‐Lisart, R. & Merino‐Sanjuan, M. A model‐based meta‐analysis of sofosbuvir‐based treatments in chronic hepatitis C patients. Int. J. Antimicrob. Agents 47, 184–194 (2016). [DOI] [PubMed] [Google Scholar]
- 73. Han, K. et al Progression‐free survival as a surrogate endpoint for overall survival in glioblastoma: a literature‐based meta‐analysis from 91 trials. Neuro. Oncol. 16, 696–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]