

Abstract
Medicines Adaptive Pathways to Patients (MAPPs) seeks to foster access to novel beneficial treatments for the right patient groups at the earliest appropriate time in the product life‐span, in a sustainable fashion. We summarize the MAPPs engagement process and critical questions to be asked at each milestone of the product life‐span. These considerations are of relevance for regulatory and access pathways that strive to address the “evidence vs. access” conundrum.
The mapps concept
MAPPs is a concept that “seeks to foster access to [novel] beneficial treatments for the right patient groups at the earliest appropriate time in the product life‐span, in a sustainable fashion.”1 It is also commonly referred to as adaptive pathways.
MAPPs seek to address the “evidence vs. access” conundrum faced by patients, healthcare professionals, healthcare decision‐makers, and pharmaceutical innovators. The conundrum refers to the delicate trade‐offs between ensuring rapid access to promising treatments for patients in urgent need, on the one hand, and ensuring that patients, healthcare professionals, and other decision‐makers possess adequate information on the benefits and risks at the time of launch on the other. This is at the heart of “Why” stakeholders wish to engage in MAPPs. For an in‐depth description of this conundrum and of the principles and goals underpinning the MAPPs approach, please refer to previous work.1, 2, 3, 4, 5
MAPPs is not an official designation and is not intended to create new regulatory or legal frameworks. Instead, MAPPs is a concept that aims to make better use of various existing regulatory pathways (e.g., conditional marketing authorization in the EU) and procedures for medicines development, marketing authorization, reimbursement (e.g., preauthorization for payer coverage), on‐market clinical use and monitoring of benefits and harms, depending on jurisdiction and agencies. In order to successfully couple swift access for patients with swift reduction of remaining uncertainties, MAPPs relies on a number of elements (or “building blocks”) briefly described in Figure 1. Conceptually, MAPPs could be described as the preplanned and smartly designed combination of these building blocks, achieved through coordinated and repeated dialog of the principal stakeholders—a conceptual shift from the traditional, sequential model of interactions.
Figure 1.

Building blocks of MAPPs. HTA, Health Technology Assessment; P&R, Pricing and Reimbursement; RCT, Randomized Clinical Trial. Republished with permission from www.adaptsmart.eu.
MAPPs has garnered interest but it became clear that many aspects still need to be addressed and aligned between stakeholders (including patients, healthcare professionals, regulators, health technology assessment (HTA) bodies, payers, developers, and EU and national policy makers) before it can become a reality. Moreover, the MAPPs concept was discussed controversially. While some stakeholders embraced it or were at least willing to explore further, others voiced a range of concerns. Concerns raised included the lowering of regulatory evidence standards, putting patients at risk of unsafe treatments, and forcing healthcare systems to pay for unproven drugs.6, 7, 8
Against this background, the European Innovative Medicines Initiative 2 (IMI2) established and funded the multistakeholder ADAPT SMART consortium (Accelerated Development of Appropriate Patient Therapies: a Sustainable, Multistakeholder Approach from Research to Treatment‐outcomes). The objective of ADAPT SMART is to provide an enabling platform and engage in a dialog with all concerned stakeholders for the coordination of MAPPs‐related activities, focusing primarily but not exclusively on the EU healthcare systems.
The consortium comprises all relevant stakeholders in the healthcare system: patients' organizations, researchers, healthcare providers, research‐based industry, regulators, and HTA bodies (HTABs). Payers are not formal partners, but some EU payer organizations are willing to engage in constructive dialog with the consortium and have constructively participated in meetings and other forms of discussions.
The consortium does not conduct primary research; it performs gap analysis and consensus building to inform future research priorities and other activities—all with a view to facilitate a common understanding and accelerate the availability of MAPPs.
A wide gap between stakeholders' views became apparent early on in the course of the ADAPT SMART project, namely: to what kind of novel medicines and clinical conditions should a MAPPs approach be applied? Some stakeholders advocated that MAPPs become the default pathway for most new products. Others argued it should be reserved for a small and well‐defined number of exceptional medicines in development. Another gap was noticed between the standpoints of different actors in the healthcare environment about when and how to engage with each other to enable a seamless process across the lifecycle of a product for timely patient access.
In an attempt to bridge these gaps, the consortium has produced two technical documents: “Discussion paper on Engagement Criteria for MAPPs”9 is essentially about prioritization (“why to engage”), while “Seamless Process and Decision Points of an Adaptive Pathway”10 is process focused (“when and how to engage”).
In this article we describe the background and divergent starting points of the discussions and synthesize the broad lines of consensus delineated in the documents. Other elements of the MAPPs concept, such as the pros and cons of MAPPs for individual stakeholder groups or methodological considerations for knowledge generation, managed entry agreements, or on‐market monitoring and ensuring appropriate utilization, have been addressed by the consortium1 but are outside the scope of this article.
Why, when, and how to engage in mapps
There was broad agreement among stakeholders that the overarching goal of MAPPs—to allow timely access to beneficial treatments for patients who need them—is worth pursuing and will likely require more coordination among decision‐makers to make better use of existing processes and tools than exists today. However, two road blocks were identified early on in the discussions: first, MAPPs requires a high level of interactions and resource capacity and capability from all stakeholders involved. The resources, skill sets, and processes may not be readily mobilized in many organizations that have not been designed to enable such interactions, e.g., some public healthcare payer organizations, patient organizations, or even some pharmaceutical companies. It was pointed out that MAPPs will have to be limited to selected products and indications of high unmet medical need where it is worthwhile to engage in the process and genuinely speed up the time to patient access.
Second, it became apparent that some stakeholders and decision‐makers were worried not only about the scientific uncertainty associated with early availability—which is at the heart of what MAPPs seeks to address proactively—but also about behavioral uncertainties. Behavioral uncertainty is unrelated to scientific knowledge about a product's benefits and harms, but addresses risks associated with human or organizational behavior in situations of mutual interdependence. For example: Will companies deliver the studies they promised to perform at the time of launch? Will payers allow a price to go up in case the product demonstrates higher than expected value? Will patient groups accept the withdrawal of reimbursement in case early expectations of benefits are not met? Will prescribers conform to the agreed‐on label use? Will regulators come up with unfeasible postmarketing requirements?
Stakeholder groups and especially payers insisted that such uncertainties need to be mitigated if they are to engage in the MAPPs process. Variants of some of these uncertainties apply today in select circumstances outside of the MAPPs construct, e.g., conditional marketing authorizations (CMA) and implementation of risk management plan measures. Therefore, the consortium also assessed experience‐based knowledge to gain insights for MAPPs.
With this shared understanding, the consortium developed a set of “engagement criteria for MAPPs”.9 These criteria are question‐based and are designed to guide stakeholder groups in their consideration of using a MAPPs process for a given product (Table 1). They are elaborated below, in the context of the proposed adaptive pathway moments.
Table 1.
Framework of questions to be addressed by stakeholders when considering MAPPs for a given medicinal product
|
Republished with permission from ADAPT SMART discussion paper on engagement criteria for MAPPS.9
The MAPPs concept is predicated on coordinated and iterative dialog among the stakeholders. The aim of these interactions is to jointly chart a course of actions for drug development, authorization, HTA, pricing and reimbursement (P&R), utilization, and monitoring across the entire product life‐span. While some current drug developments programs have “adaptive elements” such as targeted development to achieve early approval, MAPPs is different from the traditional, sequential approach with limited concerted multistakeholder interactions and only partly overlapping processes. It aims to address the information needs of all decision‐makers in a single, efficient evidence generation plan and to enable rapid action by sequential decision‐makers (e.g., regulators, HTA bodies, payers).
The consortium's reflections on how to best manage multistakeholder interactions, and how they differ from the current process, are described as a seamless process with defined consultations milestones, termed “adaptive pathway moments.”10 Each of the initial consultation moments may be followed by one or more reassessment moments where new data and information is reviewed and decisions refined based on the collected data. A pictographic representation of the proposed medicine life‐span, process steps, and core “adaptive pathway moments” is shown in Figure 2.
Figure 2.
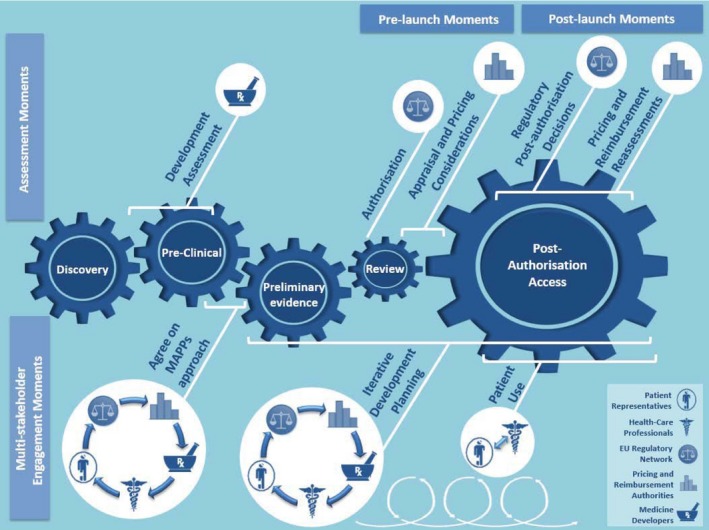
Proposal for the core multistakeholder engagement and assessment moments enabling an adaptive pathway. Within the diagram, each product life‐span phase is symbolized by a blue cog. The separate cogs are comparatively sized to represent the characteristic duration of the phase during the lifespan of a typical medicine. The top half of the figure includes the assessment or decision moments by the stakeholder represented. Although these moments may be informed by multistakeholder inputs, in the end the designated stakeholder makes an assessment and ultimate decision based on their respective remits. Within the bottom portion of the diagram, moments of multistakeholder engagement are illustrated. Each “moment” represented may actually be a series of engagement moments. For further explanation, please refer to Ref. 10. Modified with permission from Seamless process and Decision Points of an Adaptive Pathway.10
The proposed adaptive pathway moments introduce a number of process modifications compared to typical development approaches—all intended to enhance the stakeholder interactions in order to gain insights on the relevance of data generation and facilitate timely patient access. The complexities of logistical coordination, nuanced remits and responsibilities, and differing processes of the stakeholders will likely necessitate the assignment of a project manager to coordinate the logistics and the enhanced, iterative engagement moments and to serve as a consistent point of contact for all stakeholders.10
In the following we describe the individual proposed adaptive pathway moments along the product life‐span and how they relate to the key questions summarized in Table 1.
Preclinical adaptive pathways moment
Initially, around the time of preclinical development, the medicine developers should conduct their preparatory work and assess the suitability of a new product for MAPPs. The assessment should be guided by the questions in Table 1 and the medicine developer should seek input from the perspectives of at least patient representatives, regulators, HTA bodies, or P&R authorities. Such initial input should ideally be provided in an environment without formal commitments by stakeholders, for example, in the format of the EMA's adaptive pathways pilots.11
The essential question at this early development stage is the anticipated willingness of each stakeholder in contributing resources to provide meaningful advice on the evidence generation plan. In turn, their willingness is likely driven by the answer to the first question in Table 1: Does the product have sufficient promise to address a (high) unmet medical need?
Stakeholders in the consortium agreed that the MAPPs focus should be on disease transformative medicines, targeting well‐defined patient populations with a high unmet medical need, i.e., life‐threatening or severely debilitating conditions for which no treatment or no satisfactory treatment exists. Hence, early, noncommittal interaction between the developer and other stakeholders may be about the disease's main features, epidemiology, natural history, and evolution with available treatment or standard of care options. If relevant, the unmet medical need will be described separately for well‐defined subpopulations to select patients who could benefit best from the medicine.12
In order to justify the benefits of early availability on the market, a product developed within MAPPs is expected to provide clinically relevant improvements in patient‐relevant outcome(s). This implies 1) a high probability that the initial promise of the product will be confirmed once more data come in and that there is a reasonable expectation of confirming added value of the product in the clinical setting, and 2) that the effect size will likely be sufficient to improve the patient's daily life and/or life expectancy in a meaningful way.
It may not be useful to engage in detailed discussions of the clinical evidence generation plan at this early stage. However, recent experience has shown for some “complex” products (e.g., cell‐based or gene‐based medicines) that product quality—rather than clinical data—may become the bottleneck for early availability on the market. We here refer to Question 6 of the engagement criteria, about Chemistry, Manufacturing, and Controls (CMC) aspects of pharmaceutical development (Table 1). For such products, it may be advisable to seek a very early dialog between the developers and the authorities concerned, in order to facilitate an effective lifespan management of the CMC documentation, and agreement on how the CMC development strategy will be implemented. An agreed strategy should ensure that the quality of the product or the validity of the clinical trial data will not be compromised by an early access.
Preliminary evidence adaptive pathways moment
As more evidence about the product is accruing, the MAPPs interactions are expected to intensify. Once initial clinical data become available, all of the questions listed in Table 1 become relevant. Importantly, does the promise of truly addressing the unmet need still hold? Are stakeholders still committed to dedicate the necessary resources to continue the process? Is there institutional support?
At this juncture, the key deliverable from the process will be a mutually acceptable iterative development plan. An “iterative development plan” is a development plan initiated by the developer, which integrates the feedback from the stakeholders and outlines the development of the medicine during its lifespan, including the phase after initial authorization and P&R; it supports an adaptive pathway approach from beginning to end. As described above, it is envisaged that development planning discussions likely begin as interactions without committments11 and progress to more formal interactions such as, e.g., parallel consultation procedure with EMA and HTABs.
The development plan should clarify what evidence package must be available before product launch. The initial evidence package would support the regulatory authorization (i.e., provide evidence of a positive benefit–risk profile according to current scientific standards) but at the same time support HTA and enable the initial P&R negotiations and design of suitable managed entry agreements (MEAs; or alternative P&R approaches) across the different healthcare systems.
The development plan should also lay out what data and information will have to be provided after the initial marketing authorization and reimbursement decisions. It would be expected to consider many aspects of product development and evidence generation such as biomarkers, surrogate and composite endpoints, subpopulations, CMC issues, adaptive trial design, patient reported outcomes (PROs), and key milestones to review postauthorization evidence requirements. It should be clarified whether there is adequate infrastructure available for postlaunch monitoring of the product (e.g., existing disease registries) or whether new infrastructure needs to be put in place.
At this stage, payers, and perhaps other stakeholders, will likely wish to start considering issues of appropriate product utilization and workable strategies for payers with clearly defined decision points in case the product underperforms (Table 1 and below).
Given that clinical development from the preliminary evidence adaptive pathways moment up to initial authorization may take several years, unexpected findings or environmental changes may necessitate one or more reassessment moments or loops (Figure 2).
Prelaunch adaptive pathways moment
Once the initial, agreed‐upon evidence package has matured and provided the product has performed as expected with regard to benefits and harms, the developer will trigger the prelaunch adaptive pathways moment.
A key goal of this adaptive pathway moment would be to reach final agreement among all stakeholders on the treatment‐eligible population. Definition of the treatment‐eligible population will likely be driven by the preagreed development plan (i.e., patient selection criteria in the clinical trials) but also by the product's (under/over)‐performance as judged at the time of launch.
It is anticipated that marketing authorization (MA) may be conditional (i.e., CMA), unless a full MA in a well‐defined (sub‐)population is agreed upon based on evidence generated. Under MAPPs, local HTA recommendations and reimbursement decisions will hopefully rapidly follow the regulatory decision since HTA bodies and some payers would have been involved in the early and iterative adaptive pathways moments. If so, the MAPPs goal of timely access for patients with a high unmet need would have been achieved.
However, it bears reminding that the initial product launch is only one (albeit important) milestone in the MAPPs concept. The critical outstanding issues of continued knowledge generation and on‐market management of the product will have been discussed and planned earlier, but it is at the prelaunch state that they need to be conclusively agreed on by all stakeholders.
Under the MAPPs concept, continuous knowledge generation throughout the on‐market lifespan of a medicine aims to 1) achieve rapid reduction of remaining uncertainties around efficacy/effectiveness and safety; 2) allow for broadening (or narrowing) of the treatment‐eligible population where justified; and 3) inform payers about the use and effectiveness in a consistent way in order to enable flexible (adaptive) pricing and reimbursement schemes.
The type of postlaunch studies (randomized controlled trials (RCTs), single‐arm trial, observational studies, etc.) as well as available data collection infrastructure to use (e.g., patient registries) will be finalized and agreed on. For regulators, this will most likely be laid out in the risk management plan (RMP13); for HTA bodies and payers, additional postlaunch evidence requirements should align with the RMP and may lead to an integrated research plan that include HTA/payers requirements and the RMP. More work is needed to align the requirements of the regulators, the HTA bodies, and the payer organizations to make the system more agile and efficient.
Developing a pathway for continued knowledge generation is a necessary but not sufficient part of the MAPPs concept during the on‐market phase of the product life‐span. Two additional issues need consideration; they relate to on‐market utilization of the product and payer strategies and are addressed by Questions 3 and 4 in Table 1.
In the attempt to have a MAPPs product reaching the patient at an early timepoint—for the benefit of a specific patient (sub‐)population—MAPPs places much emphasis on the appropriate treatment eligible population in routine healthcare. The emphasis is motivated by the notion that the degree of acceptable uncertainty about a product's benefits and harms must be proportional to the degree of unmet patient need. This general principle has been reflected in regulatory practice for decades and is in line with ethical principles on trading‐off patients potential benefits and harms.14 Including the patients' voice in these deliberations is key for MAPPs. Assuming that different (sub‐)populations face different degrees of unmet need and will benefit differently from a given treatment, on‐market utilization should be in line with the target population agreed by the stakeholders. Normally, this will mean a "smaller" well‐defined and targeted population initially, with new subpopulations added as more information about the product becomes available from real‐world data (RWD) and/or new RCTs. Involvement of healthcare providers and patients in these deliberations is key to a successful implementation.
Unmanaged off‐label use may compromise sound medical practice, harm patients, and undermine the regulators' mission of protecting patients as well as the payers' mission to make best use of limited healthcare funds.15 While off‐label prescribing may be detrimental to patients in many treatment scenarios, it is even more of a concern in the MAPPs scenario.
An initial assessment of the potential for off‐label use of the product and of the opportunities to mitigate such use must be discussed at the time of MAPPs selection (at the latest during the Preliminary evidence adaptive pathways moment). At the time of launch an action plan will need to be agreed to ensure appropriate product utilization.
Such a plan could include, e.g., assessment of the potential volume of off‐label use, educational programs, and materials for physicians and patients (i.e., guidelines and patient information leaflets), use restrictions, empowerment of patient representatives, etc., and would need to be agreed with all stakeholders. Where appropriate (components of) the utilization plan could be embedded in the risk mitigation part of the regulatory RMP.
Early on in the discussions, payers highlighted their unease with a particular aspect of MAPPs (captured in Question 4 of the MAPPs engagement criteria; see Table 1): Are there workable “strategies” for payers in case the product underperforms?
The MAPPs concept relies on continuous knowledge generation and iterative assessment of benefit‐risk and value of a product. It is possible that one of two undesirable situations may arise after an initial marketing authorization and reimbursement decision had been granted: 1) new data show that the product's performance, and therefore its value, is less than expected (e.g., the effect size, duration of response, or percentage of responders are lower than the original estimate); or 2) post‐MA commitments are not satisfied and the presumed benefit–risk and value of the product cannot be confirmed.
Payers are worried they would then “be locked into paying too high a price for an underperforming or poorly documented product.” This situation would arise if the company refuses to lower the price and payers were unable to terminate reimbursement for political reasons—as patients may resist having a treatment option taken away. (Note that this payer concern would not apply in situations where the benefit–risk changed to outright negative; the regulator would be expected to withdraw the marketing authorization.)
To mitigate this concern, and allow payers to commit to MAPPS, an appropriate strategy (sometimes referred to by payers as disengagement or “exit” strategy) needs to be worked out. At the time before launch, the strategy should be negotiated by way of a transparent commitment and agreement between the company and individual healthcare payers. In practice, this may take the form of managed entry agreements (MEA); these are formal arrangements between a developer and a payer or healthcare provider that enables access to (coverage/reimbursement of) a health technology such as a medicinal product subject to specified conditions. Several different MEA formats have already been implemented, but they are not yet widely used.16, 17
In a MAPPs scenario, the terms of the MEAs may specify the consequences on price and reimbursement conditions of successful and unsuccessful confirmation of the value proposition. This is why an adaptive pathway approach requires an adaptive pricing approach with price and conditions negotiated by each payer and revisited at set milestones in light of the results of the evidence available postauthorization. Conceptually, the MEA should allow price points to move up or down, in line with forthcoming information about the product's demonstrated performance.
Postlaunch adaptive pathways moment
Postlaunch adaptive pathway moments could always be triggered ad hoc by any of the stakeholders involved (e.g., when unexpected, important information becomes available) but in the MAPPs scenario, it is expected that stakeholders have agreed on preset milestones when to reevaluate the product. This could take the form of set dates (e.g., after 2 years) or a timepoint related to patient treatment experiences (e.g., once 1,000 patients had on‐market exposure to the product, or after 3,000 patient‐years).
Stakeholders will review the data and information accumulated postlaunch and seek to address the relevant MAPPs questions: Have new data come in as promised? If so, are the new data credible and informative? Is there still reason to believe the product addresses an unmet need? Is the product's initial promise (effect size, safety profile, clinical value) fulfilled, based on the new information? Is the product utilized as foreseen? Should the treatment‐eligible population be enlarged or narrowed?
At this juncture, stakeholders will need to agree whether to maintain or modify the original on‐market evidence generation plan.
In line with legal obligations, regulators will determine whether the new information warrants a change in the label. In case the product had been conditionally authorized in the first place, a decision needs to be made whether data are sufficient to justify conversion to a regular marketing authorization.
Payers and the company will review whether the new evidence triggers any changes in the P&R as per the terms of the MEA, or they may wish to renegotiate P&R conditions.
As on‐market experience accumulates, uncertainty is progressively reduced. The product “matures” and it is expected that the intervals between the reassessment moments will gradually widen (unless regulated by law), and—barring any surprises—the MAPPs‐related evidence generation plan will be gradually phased out and the degree of utilization oversight relaxed.
Conclusions
We have given a high‐level description of the MAPPs engagement process, attempting to synthesize the Why, When, and How of the process steps and the critical questions to be asked at each milestone. In doing so, we acknowledge that many questions remain unanswered,10 including, for example, the modalities of patient and healthcare professionals' contributions to or patient responsibility within the overall MAPPs process and the resourcing of MAPPs activities. One size does not fit all products or all clinical conditions in all countries and it is expected that these issues will gradually be resolved as initial pilots and other collaborative efforts generate more collaborative experience within the stakeholder communities. The gap analysis currently performed by the ADAPT SMART consortium will help identify research projects and other MAPPs enablers that should even more support products entering MAPPs.
Discussions held by the ADAPT SMART consortium revealed the need for some nontangible preconditions to be in place in order to enable the MAPPs interactions: Some stakeholders may feel more comfortable contributing to a MAPPs process if they were assured that their early involvement would be nonbinding; hence, an initial informal environment might encourage early MAPPS cases.
MAPPs is a scientific concept, not a legal framework. Hence, stakeholders have the (reassuring) option to opt out or to stop the MAPPs approach if they feel it ceases to add value. In such cases, the drug developer may still decide to continue development “as usual.”
The nature of the early informal, and later formal multistakeholder interactions, will require a delicate balance between maintaining appropriate levels of transparency vs. confidentiality, being respectful of the political mandate of each stakeholder's involvement, and ensuring mutual trust and confidence.
There remains another challenge not be underestimated: MAPPs is a life‐span approach to learning about and utilizing a drug product. This will necessarily require all stakeholders to not only adopt the concept but also adopt a long‐term view to achieve “collective and cumulative learning”18 along the journey.
What impact has the MAPPs concept made to date? While there are products currently in a “MAPPS‐like” development, MAPPs is too novel for a product to have already gone through successfully and product‐specific information is not yet in the public domain. However, there is at least one product in preauthorization development for which MAPPs interactions have been publicly released by the sponsor19 and for which MAPPs may help achieve timely access for patients—provided the results from ongoing studies meet expectations.
While this may be a hoped‐for outcome in the future, we argue that MAPPs has even today stimulated progress on the level of public debate and stakeholder perceptions. Some of the building blocks of MAPPs (Figure 1) have become mainstream: a focus on patient groups with high unmet need (as opposed to the blockbuster approach), the need for iterative development and assessment throughout the product life‐span, and the need for multistakeholder collaboration along the entire product life‐span are now widely accepted in principle, and partly implemented (consider, for example, the joint early regulator‐HTA dialogs in the EU).20 Other building blocks will take longer to be fully accepted and implemented; for example, the appropriate use of real‐world data, frameworks to allow for adaptive pricing and reimbursement, or ways to ensure appropriate on‐market utilization. We remain confident that these outstanding issues will gradually be resolved and MAPPs can be applied in the best interest of patients.
Disclaimer
The work leading to these results was conducted as part of the ADAPT SMART consortium (Accelerated Development of Appropriate Patient Therapies: a Sustainable, Multi‐stakeholder Approach from Research to Treatment‐outcomes). For further information please refer to www.adaptsmart.eu. This article only reflects the views of the stated authors and shall not be understood or quoted as being made on behalf of, or reflecting the position of, any participating organization or stakeholder, public or private. It is not intended to replace or complement official guidelines that may be in place or in development. The article is merely intended to inform and drive future discussions on MAPPs, both within the ADAPT SMART consortium and in the wider scientific and healthcare communities.
Funding
This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115890. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA.
Conflict of interest
Solange Corriol‐Rohou is an employee and shareholder of Astra Zeneca, Sue Forda is an employee and shareholder of Eli Lilly and Company, Angelika Joos is an employee and shareholder of MSD, Mark Mayer is an employee and shareholder of Eli Lilly and Company, Vinciane Pirard is an employee and shareholder of Sanofi.
Acknowledgments
The authors acknowledge the contributions to this article of the following experts: Richard Barker (CASMI), Magda Chlebus (EFPIA), Isabelle Clamou (Amgen), Elisabeth Cobbs (MSD), Pauline Evers (EGAN), Christine Fletcher (Amgen), Edith Frénoy (EFPIA), Alicia Granados (Sanofi), Birthe Holm, (EURORDIS), Patrick Hopkinson (BMS), Yilin Huo (Lilly), Craig Johnson (GSK), Alexandre Joyeux (Novartis), Anne‐Sophie Lapointe (EURORDIS), Susan Longman (Novartis), Christine Mayer‐Nicolai (Merck KGaA, Germany), Brian Mayhew (Novartis), Thomas Metcalfe (Roche), Lise Murphy (EURORDIS), Hans Ovelgönne (EMA/CBG‐MEB), Samantha Parker (Lysogene), Ralf Rischke (Boehringer‐Ingelheim), Alexander Roediger (MSD), Claudine Sapede (Roche), Isabelle Stoeckert (Bayer), Nanguneri Sumathi (Merck KgaA, Germany), Karin van Baelen (Johnson & Johnson), Elizabeth Vroom (United Parent Projects Muscular Dystrophy), Richard West (Behcet's Society UK, EURORDIS).
References
- 1.ADAPT SMART website. <http://adaptsmart.eu/home/>. Accessed 28 March 2018.
- 2. Baird, L.G. et al Accelerated access to innovative medicines for patients in need. Clin. Pharmacol. Ther. 96, 559–571 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Eichler, H.G. et al Adaptive licensing: taking the next step in the evolution of drug approval. Clin. Pharmacol. Ther. 91, 426–437 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Schulthess, D. et al Medicines Adaptive Pathways to Patients (MAPPs) A Story of International Collaboration Leading to Implementation. TIRS Volume: 50 issue: 3, 347–354 (2015). [DOI] [PubMed] [Google Scholar]
- 5. Eichler, H.G. et al From adaptive licensing to adaptive pathways: delivering a flexible life‐span approach to bring new drugs to patients. Clin. Pharmacol. Ther. 97, 234–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis, C. , Lexchin, J. , Jefferson, T. , Gøtzsche, P. & McKee, M. “Adaptive pathways” to drug authorisation: adapting to industry? BMJ 354, i4437 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Ermisch, M. et al Payers' views of the changes arising through the possible adoption of adaptive pathways. Front. Pharmacol. 7, 305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. HAI — “Adaptive licensing” or “adaptive pathways”: Deregulation under the guise of earlier access. (Oct 2015).
- 9. ADAPT SMART website. Discussion paper on Engagement Criteria for MAPPs. <http://adaptsmart.eu/wp-content/uploads/2016/02/ADAPT-SMART-Engagement-Criteria-Final1.pdf>. Accessed 28 March 2018.
- 10. ADAPT SMART website. Seamless Process and Decision Points of an Adaptive Pathway. <http://adaptsmart.eu/wp-content/uploads/2017/06/ADAPT-SMART-seamless-pathway_final_website.pdf>. Accessed 28 March 2018.
- 11. European Medicines Agency website. Adaptive Pathways. <http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000601.jsp&mid=WC0b01ac05807d58ce>. Accessed 28 March 2018.
- 12. ADAPT SMART website. Early Access: Is it worth it? <http://adaptsmart.eu/early-access-is-it-worth-it/>. Accessed 28 March 2018.
- 13. European Medicines Agency website. Risk management plan (RMP): questions and answers. <http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q_and_a/q_and_a_detail_000171.jsp&mid=WC0b01ac0580a53f5f>. Accessed 28 March 2018.
- 14. Eichler, H.G. , Pignatti, F. , Flamion, B. , Leufkens, H. & Breckenridge, A. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat. Rev. Drug Discov. 7, 818–826 (2008). [DOI] [PubMed] [Google Scholar]
- 15. Jonville‐Béra, A.P. , Béra, F. & Autret‐Leca, E. Are incorrectly used drugs more frequently involved in adverse drug reactions? A prospective study. Eur. J. Clin. Pharmacol. 61, 231–236 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Ferrario, A. & Kanavos, P. Managed Entry Agreements for Pharmaceuticals: The European Experience (EMiNet, Brussels, Belgium, 2013). [Google Scholar]
- 17. Bouvy, J.C. , Sapede, C. & Garner, S. Managed entry agreements for pharmaceuticals in the context of adaptive pathways in Europe. Front. Pharmacol < 10.3389/fphar.2018.00280> <https://www.frontiersin.org/articles/10.3389/fphar.2018.00280/full>. Accessed 28 March 2018. [DOI] [PMC free article] [PubMed]
- 18.Lazonick, et al US Pharma's Financialized Business Model. Working Paper No. 60. July 13, 2017 (revised September 8, 2017). Lazonick et al. US Pharma's financialized business model. 2017. Accessed 28 March 2018.
- 19. FierceBiotech website. Phil Taylor. <https://www.fiercebiotech.com/biotech/bluebird-bio-sees-europe-as-first-market-for-its-gene-therapies>. Accessed 28 March 2018.
- 20. European Medicines Agency website. Parallel consultation with regulators and health technology assessment bodies. <http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001857.jsp&mid=WC0b01ac0580a11c96>. Accessed 28 March 2018.