

Abstract
We report physiologically based pharmacokinetic‐modeling analyses to determine olaparib (tablet or capsule) drug–drug interactions (DDIs). Verified DDI simulations provided dose recommendations for olaparib coadministration with clinically relevant CYP3A4 modulators to eliminate potential risk to patient safety or olaparib efficacy. When olaparib is given with strong/moderate CYP3A inhibitors, the dose should be reduced to 100/150 mg b.i.d. (tablet), and 150/200 mg b.i.d. (capsule). Olaparib administration is not recommended with strong/moderate CYP3A inducers. No dose reductions are required with weak CYP3A inhibitors/inducers. Olaparib was shown to be a weak inhibitor of CYP3A (1.6‐fold increase in exposure of a sensitive CYP3A probe) and to have no effect on P‐glycoprotein or UGT1A1 substrates. Finally, this model was used to simulate exposure in scenarios where clinical data of olaparib are lacking, such as severe renal or hepatic impairment populations, and provided initial dosing recommendations in pediatric patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ In vitro ADME studies suggested that olaparib is a substrate of CYP3A, a reversible/time‐dependent inhibitor of CYP3A, an inhibitor of UGT1A1, P‐gp, and an inducer of CYP3A.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What DDIs occur when olaparib tablets/capsules are coadministered with mild/moderate CYP3A inhibitors/inducers or CYP3A, P‐gp, or UGT1A1 probe substrates? Is olaparib tablet exposure affected in mild/moderate/severe hepatically/renally impaired patients? What dosing recommendations are for pediatric patients?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The developed PBPK model was used to simulate exposure in scenarios where clinical data of olaparib are lacking, such as DDIs with CYP modulators, severe renal or hepatic impairment populations, or in pediatric patients. This analysis supported the olaparib drug label as follows: when given with strong/moderate CYP3A inhibitors, olaparib tablet dose should be reduced to 100/150 mg b.i.d., and capsule dose to 150/200 mg b.i.d.: olaparib is not recommended with strong/moderate CYP3A inducers: no dose reductions are required with weak CYP3A inhibitors/inducers: little change in the exposure of olaparib in patients with mild/moderate hepatic impairment was observed.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Given the challenges of conducting DDIs, or special population (organ impairment and pediatric) studies in oncology, our model‐based approach demonstrates that it is beneficial to utilize verified PBPK models to simulate changes in drug exposure in various scenarios to guide dosing information when clinical trials are not possible.
Olaparib (Lynparza) is a potent PARP inhibitor that has demonstrated antitumor activity.1, 2, 3, 4 To receive the original capsule‐formulation dose (400 mg twice daily (b.i.d.)), patients are required to take 8 × 50‐mg large capsules b.i.d. (16 units/day). A tablet formulation was developed to facilitate delivery of olaparib doses in fewer, smaller units (300 mg b.i.d.; 2 × 150‐mg tablets b.i.d.; 4 units/day). Both capsule and tablet formulations are approved.5, 6 Phase III trials of olaparib monotherapy using the tablet (300 mg b.i.d.) in patients with platinum‐sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2, NCT01874353), or human epidermal growth factor receptor 2 (HER2)‐negative metastatic breast cancer and a BRCA1/2 mutation (OlympiAD, NCT02000622), demonstrated a clinically meaningful and statistically significant difference in progression‐free survival in patients receiving olaparib compared with placebo or physician's choice chemotherapy, respectively.4, 7
Comparison of the capsule and tablet formulations using a single aqueous dissolution medium and standard USP dissolution apparatus showed a slower rate of release for the capsule formulation compared to the tablet formulation (AstraZeneca unpublished data). The in vitro dissolution profile of the olaparib tablet and capsule formulations is discussed further in the Supplementary Supporting Information. Clinical investigation of the relative bioavailability of the tablet vs. capsule formulation indicated that at doses up to 100 mg, the two formulations had similar area under the plasma concentration–time curve (AUC) values but the maximum plasma concentration (Cmax) values exhibited by the tablet formulation were significantly higher. At doses >100 mg, exposure to olaparib capsule formulation increased less than proportionally with dose, whereas exposure to olaparib tablets increased approximately proportionally with dose; thus, they are not bioequivalent.8 During formulation development, consideration was given to the low solubility of olaparib and the potential impact upon exposure, particularly at higher doses. Both capsule and tablet formulations have been developed as enabled formulations rather than conventional immediate‐release formulations; however, different mechanisms are employed to enable enhanced performance. The capsule consists of a dispersion of crystalline olaparib within a semisolid lauroyl macrogol‐32 glycerides (LMG) matrix, which has been shown to enhance olaparib solubility. Exposure to olaparib increases less than proportionally with the capsule formulation for doses ≥100 mg owing to solubility limitations. Olaparib solubility in the hot‐melt‐extruded tablet formulation is enhanced by presenting olaparib in amorphous form. The tablet formulation also has a higher drug‐loading capacity (Table S1).
Olaparib clearance is primarily through metabolism by oxidation via cytochrome P450 (CYP)3A4/5 (84% of total clearance), and to a lesser extent by renal elimination (16% of total clearance; Figure S1). The olaparib mass balance absorption, distribution, metabolism, and excretion (ADME) study9 results are described in the Supplementary Supporting Information and shown in Figure S1. Patients with breast, prostate, or ovarian cancer may suffer from varying degrees of hepatic impairment.10, 11, 12 The effect of mild/moderate hepatic or renal impairment on the pharmacokinetics of single‐dose olaparib tablet in oncology patients has been investigated (NCT01894243, NCT01894256).13 Following a single oral 300 mg dose of olaparib tablet formulation to patients with mild renal impairment, AUC and Cmax increased by 24% and 15%, respectively, and to patients with moderate renal impairment AUC and Cmax increased by 44% and 26%, respectively, compared to patients with normal renal function. Mild and moderate hepatic impairment had a minimal effect on exposure (change in AUC and Cmax <15% compared to patients with normal hepatic function).
Drug–drug interactions (DDIs) are one of the major causes of drug withdrawal from the market.14 Oncology patients often have multiple comorbidities and therefore take multiple drugs; it is difficult to determine whether adverse events are the result of side effects from a single drug, interactions between two or more drugs, or exacerbations of the patient's underlying disease(s).15 Given the challenges in conducting pharmacokinetic (PK) studies in patients with hepatic or renal impairment, as well as numerous DDI studies of anticancer drugs, it can be beneficial to utilize physiologically based PK (PBPK) models to predict changes in drug exposure in various scenarios to guide dosing where clinical trials have not been conducted. Validated PBPK models can also provide initial dosing recommendations for pediatric development plans.16, 17, 18, 19
In vitro enzymology data have suggested that olaparib metabolism is predominantly CYP3A‐mediated and olaparib has the potential to cause CYP3A reversible and time‐dependent inhibition (TDI) and induction.20 The DDI potential of olaparib is therefore multivariate, and predicting the net effect on CYP3A in vivo is challenging. The DDI of olaparib tablet with a CYP3A inhibitor (itraconazole) and inducer (rifampicin) has been studied in clinical trials of patients with solid tumors21; however, equivalent studies have not been performed with the capsule. In vitro data have also indicated that olaparib shows inhibition of P‐glycoprotein (P‐gp) and UGT1A1 with measured IC50 values of 76 and 96.7 μM, respectively.5, 6
The objectives of the studies reported here were to:
Develop a mechanistic PBPK model of olaparib tablet and capsule formulations using Simcyp (Sheffield, UK) with olaparib (capsule and tablet) physiological parameters, human in vitro data, and human in vivo data to simulate olaparib exposure.
Utilize observed clinical data21 to verify model predictions of the effect of coadministration of olaparib with a CYP3A4 inhibitor (itraconazole) or inducer (rifampicin).
Bridge the DDI outcome of the tablet to the capsule to provide dose‐adjustment information for the approved capsule formulation by predicting the effect of weak to potent CYP3A inhibitors and inducers on exposure to olaparib capsule using the PBPK model. In addition, provide dose‐adjustment recommendations for the tablet formulation given with weak to moderate CYP3A inhibitors or inducers.
Simulate the DDI risk of olaparib as a perpetrator drug using the probe CYP3A substrates midazolam and simvastatin, the P‐gp substrate digoxin, and the UGT1A1 substrate raltegravir.
Utilize the PBPK models developed based on single‐dose olaparib clinical trial DDIs to simulate DDI outcomes following multiple‐dose administration.
Apply clinical data from olaparib tablet studies in patients with mild/moderate renal or hepatic impairment to the PBPK models to predict changes in olaparib exposure when given to patients with mild, moderate, or severe renal or hepatic impairment after multiple dosing.
Use the PBPK models to predict olaparib exposure in pediatric patients to recommend dosing regimens for clinical pediatric studies.
Results
PBPK simulations of olaparib tablet‐ and capsule‐monotherapy exposure
Figure 1 a shows the mean concentration–time profile in plasma simulated by the PBPK tablet model for a single tablet dose of olaparib (300 mg) in the fasted state, accounting for CYP3A and P‐gp inhibition and induction parameters; Figure 1 b shows that for multiple tablet doses. The predicted PK parameters (AUC and Cmax) were within 1.5‐fold of the PK parameters observed for olaparib tablet monotherapy in the clinical trials21, 22 following single or multiple oral tablet doses (Table 1).23 For multiple tablet dosing, there was a predicted ∼24% decrease in olaparib clearance and therefore an increase in AUC at steady state (AUCss) of 1.35‐fold over day 1 predicted AUC; this was in agreement with observed clinical data (NCT01921140).22 A comparison of the observed and predicted clearance and distribution at steady state is shown in Table S7.
Figure 1.
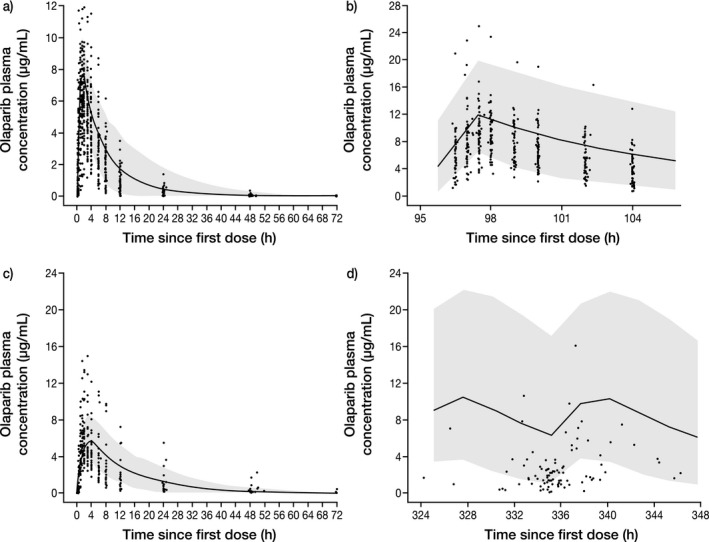
Simulated plasma concentration–time profile for (a) single‐dose olaparib tablet (300 mg), (b) multiple‐dose olaparib tablet (300 mg b.i.d.), (c) single‐dose olaparib capsule (400 mg), and (d) multiple‐dose olaparib capsule (400 mg b.i.d.), compared with observed olaparib clinical data. The continuous line represents the median prediction using the PBPK model; the shaded area represents the 95% prediction intervals. Closed circles are observed data points from olaparib clinical trials NCT01921140 (a,b) and NCT01851265 (c), as well as pooled clinical trial data from NCT00572364, NCT00516373, NCT00494234, NCT00494442, NCT00628251, and NCT00777582 (d).
Table 1.
Mean AUC and Cmax from PBPK model simulations compared with observed clinical data of olaparib exposure following single or multiple doses (with and without CYP3A4 TDI parameters) and olaparib tablet (300 mg) or capsule (400 mg) monotherapy
| Parameters from observed dataa, 22 | Parameters from simulation | Mean ratio of simulated/observed (plus or minus ratio values SD)23 | |||||
|---|---|---|---|---|---|---|---|
| Olaparib single dose | Olaparib formulation | AUCt, h·µg/mL(range) | Cmax, µg/mL(range) | AUCt, h·µg/mL(range) | Cmax, µg/mL(range) | AUC | Cmax |
| 300 mg | Tablet | 49.4 (15.7–144) | 7.41 (2.6–19.1) | 65.4 (24.2–191) | 8.11 (5.47–14.2) | 1.32 (0.94) | 1.09 (0.44) |
| 400 mg | Capsule | 56.2 (21.9–299) | 4.44 (3–15) | 90.7 (24.4–276) | 5.90 (2.80–12.8) | 1.61 (1.23) | 1.32 (0.72) |
| Olaparib multiple dose | Olaparib formulation | AUCss, h·µg/mL(range) | Cmax, µg/mL(range) | AUCss, h·µg/mL(range) | Cmax, µg/mL(range) | AUC | Cmax |
| 300 mg bid | Tablet | 63.8 (25.5–189) | 9.71 (4.8–24.9) | 93 (9.82–240.8) | 12.3 (3.22–24.3) | 1.46 (1.01) | 1.26 (0.64) |
| 400 mg bida | Capsule | 55.2 (8.57–154) | 6.25 (1.57–14.2) | 105.4 (20.1–319) | 10.7 (2.71–29.6) | 1.91 (1.61) | 1.71 (1.20) |
Data are expressed as arithmetic mean. Ranges are shown as minimum–maximum values.
For the capsule, multiple‐dosing pooled PK analysis from NCT00572364, NCT00516373, NCT00494234, NCT00494442, NCT00628251, and NCT00777582 were used (data on file). AUCt, area under the plasma concentration–time curve from time zero to the last measurable timepoint; SD, standard deviation.
The predicted median plasma concentration–time profile of a single olaparib capsule dose (400 mg) was comparable to that observed for olaparib monotherapy from pooled clinical trials (Figure 1 c). Given the closeness to the mean of the observed data (Table 1), it was considered appropriate to use the model to predict systemic concentrations of olaparib following single oral doses of the capsule 400 mg (Figure 1) to fasting patients and to perform DDI predictions with olaparib and CYP3A inhibitors. For multiple capsule dosing, there was a predicted ∼9% decrease in olaparib clearance and therefore an increase in AUCss of 1.08‐fold over day 1 predicted AUC. The observed PK data were overpredicted by 1.9‐fold (Figure 1 d). The model used to predict the DDIs of olaparib with CYP3A and P‐gp substrates incorporated both TDI and induction of CYP3A4 to reflect the net effect of olaparib as a perpetrator.
PBPK simulations of olaparib tablet and capsule DDIs
Predicted and clinically observed changes for olaparib tablet and capsule exposure when dosed with CYP3A inhibitors, inducers, and probe substrates, as well as P‐gp and UGT1A1 substrates, are shown in Figure 2, Figure S3, Table 2, and Table S3.
Figure 2.
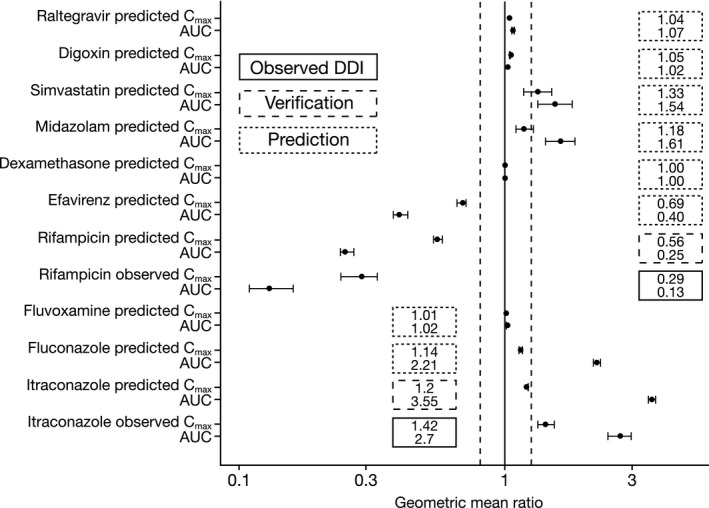
DDI predictions using the olaparib tablet model (AUC and Cmax ratios with 95% CI*) and observed olaparib tablet clinical data for interactions with itraconazole and rifampicin. *90% CIs are shown for observed clinical data for olaparib tablet monotherapy in clinical trials “21, 22” following single or multiple oral tablet doses, respectively.22 CI, confidence interval. Vertical dotted lines represent the (80% and 125%) interval.
Table 2.
Mean AUC and Cmax ratios and model‐driven olaparib tablet or capsule dose optimization determined* from PBPK model simulations investigating the effect of CYP3A inhibitors and inducers on the exposure of olaparib tablet and capsule, and of olaparib tablet and capsule on the exposure of the CYP3A probe substrates midazolam and simvastatin, the P‐gp substrate digoxin, and the UGT1A1 substrate raltegravir
| Olaparib as a victim | ||||||
|---|---|---|---|---|---|---|
| Olaparib formulation and dose | CYP3A inhibitor/inducers schedule and dose | Parameters from observed data21 | Parameters from simulation | Olaparib prescribing information5, 6 | ||
| AUC ratio (95% CI) | Cmax ratio (95% CI) | AUC ratio (95% CI) | Cmax ratio (95% CI) | |||
| CYP3A inhibitors | ||||||
| Tablet, 100 mg single dose given 96 hours after initial itraconazole dose | Itraconazole, 0 mg vs. itraconazole, 200 mg qd for 7 days | 2.7(2.44–2.97) | 1.42(1.33–1.52) | 3.55(3.46–3.65) | 1.20 (1.2–1.21) | Reduce olaparib dose to 100 mg bid |
| Capsule, 100 mg single dose given 96 hours after initial itraconazole dose | Itraconazole, 0 mg vs. itraconazole, 200 mg qd for 7 days | — | — | 2.52 (2.39–2.67) | 1.33 (1.30–1.37) | Reduce olaparib dose to 150 mg bid |
| Tablet, 100 mg single dose given 96 hours after initial fluconazole dose | Fluconazole, 0 mg vs. fluconazole, 200 mg qd for 7 days | — | — | 2.21 (2.14–2.28) | 1.14 (1.13–1.16) | Reduce olaparib dose to 150 mg bid |
| Capsule, 100 mg single dose given 96 hours after initial fluconazole dose | Fluconazole, 0 mg vs. fluconazole, 200 mg qd for 7 days | — | — | 1.98 (1.92–2.05) | 1.17 (1.15–1.19) | Reduce olaparib dose to 200 mg bid |
| Tablet, 100 mg single dose given 96 hours after initial fluvoxamine dose | Fluvoxamine, 0 mg vs. fluvoxamine, 50 mg qd for 7 days | — | — | 1.02 (1.01–1.02) | 1.01 (1.01–1.01) | Permitted with olaparib |
| Capsule, 100 mg single dose given 96 hours after initial fluvoxamine dose | Fluvoxamine, 0 mg vs. fluvoxamine, 50 mg qd for 7 days | — | — | 1.01 (1.01–1.01) | 1.01 (1.01–1.01) | Permitted with olaparib |
| CYP3A inducers | ||||||
| Tablet, 300 mg single dose given 216 hours after initial rifampicin dose | Rifampicin, 0 mg vs. rifampicin, 600 mg qd for 13 days | 0.13 (0.11–0.16) | 0.29 (0.24–0.33) | 0.25 (0.24–0.27) | 0.56 (0.54–0.58) | Not recommended |
| Capsule, 300 mg single dose given 216 hours after initial rifampicin dose | Rifampicin, 0 mg vs. rifampicin, 600 mg qd for 13 days | — | — | 0.29 (0.27–0.31) | 0.55 (0.52–0.58) | Not recommended |
| Tablet, 300 mg single dose given 216 hours after initial efavirenz dose | Efavirenz, 0 mg vs. efavirenz, 600 mg qd for 13 days | — | — | 0.40 (0.38–0.43) | 0.69 (0.66–0.71) | Not recommended |
| Capsule, 400 mg single dose given 216 hours after initial efavirenz dose | Efavirenz, 0 mg vs. efavirenz, 600 mg qd for 13 days | — | — | 0.47 (0.44–0.50) | 0.66 (0.63–0.69) | Not recommended |
| Tablet, 300 mg single dose given 216 hours after initial dexamethasone dose | Dexamethasone, 0 mg vs. dexamethasone, 8 mg qd for 13 days | — | — | 1.00 (0.99–1.00) | 1.00 (1.00–1.00) | Permitted with olaparib |
| Capsule, 400 mg single dose given 216 hours after initial dexamethasone dose | — | — | 1.00 (0.99–1.00) | 1.00 (1.00–1.00) | Permitted with olaparib | |
| Olaparib as a perpetrator | ||||||
| Probe substrate and dose | Olaparib formulation, schedule and dose | Substrate parameters from simulation | Dosing recommendations | |
|---|---|---|---|---|
| AUC ratio (95% CI) | Cmax ratio (95% CI) | |||
| Midazolam 5 mg single dose given 96 hours after initial olaparib dose | Tablet, 0 mg vs. tablet, 300 mg bid for 7 days | 1.61 (1.42–1.83) | 1.18 (1.10–1.27) | EMA: Caution should be exercised when substrates which are sensitive or with a narrow therapeutic margin are combined with olaparib FDA: None stated |
| Capsule, 0 mg vs. capsule, 400 mg bid for 7 days | 1.45 (1.27–1.65) | 1.11 (1.04–1.19) | EMA: Caution should be exercised when sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic margin are combined with olaparib FDA: None stated | |
| Simvastatin 40 mg single dose given 96 hours after initial olaparib dose | Tablet, 0 mg vs. tablet, 300 mg bid for 7 days | 1.54 (1.33–1.78) | 1.33 (1.18–1.49) | EMA: Caution should be exercised when substrates which are sensitive or with a narrow therapeutic margin are combined with olaparib FDA: None stated |
| Capsule, 0 mg vs. capsule, 400 mg bid for 7 days | 1.47 (1.27–1.71) | 1.27 (1.13–1.43) | EMA: Caution should be exercised when sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic margin are combined with olaparib FDA: None stated | |
| Digoxin 0.5 mg (0.25 mg in capsule study) single dose given 96 hours after initial olaparib dose | Tablet, 0 mg vs. tablet, 300 mg bid for 7 days | 1.02 (1.02–1.02) | 1.05 (1.04–1.05) |
In vitro, olaparib inhibits the efflux transporter P‐gp (IC50 = 76 μM), therefore it cannot be excluded that olaparib may cause clinically relevant drug interactions with substrates of P‐gp (e.g. simvastatin, pravastatin, dabigatran, digoxin and colchicine). Appropriate clinical monitoring is recommended for patients receiving this type of medicinal product concomitantly. FDA: None stated |
| Capsule, 0 mg vs. capsule, 400 mg bid for 7 days | 1.02 (1.02–1.02) | 1.03 (1.03–1.03) |
EMA: In vitro, olaparib inhibits the efflux transporter P‐gp (IC50 = 76 μM), therefore it cannot be excluded that olaparib may cause clinically relevant drug interactions with substrates of P‐gp (e.g. simvastatin, pravastatin, dabigatran, digoxin and colchicine). Appropriate clinical monitoring is recommended for patients receiving this type of medicinal product concomitantly. FDA: None stated |
|
| Raltegravir 400 mg single dose given 96 hours after initial olaparib dose | Tablet, 0 mg vs. tablet, 300 mg bid for 8 days | 1.07 (1.06–1.08) | 1.04 (1.04–1.04) | EMA: Olaparib inhibited UGT1A1 in vitro, however, PBPK simulations suggest this is not of clinical importance. FDA: None stated |
| Capsule, 0 mg vs. capsule, 400 mg bid for 7 days | 1.04 (1.03–1.05) | 1.02 (1.01–1.03) | Olaparib inhibited UGT1A1 in vitro, however, PBPK simulations suggest this is not of clinical importance | |
Study simulation parameters required for dose adjustment recommendation: the simulated olaparib/CYP3A modulator exposure as monotherapy should be <2‐fold that of the coadministered combination. Data are expressed as geometric mean. qd, once daily.
When PBPK model simulations were performed, matching clinical studies of olaparib tablets in combination with the CYP3A inhibitor itraconazole or inducer rifampicin, the ratio of the predicted to observed olaparib exposure was within the predefined (2‐fold; widely acceptable clinically insignificant criteria for wider therapeutic drugs)24 range of the observed exposure (Table 2, Figure S4, Table S3).
PBPK model simulations of olaparib tablet or capsule coadministered with CYP3A inhibitors provided a consistent predicted increase in olaparib AUC and Cmax, with higher fold changes occurring with strong CYP3A inhibition, reducing with moderate and mild inhibition. Conversely, when coadministered with CYP3A inducers, olaparib AUC and Cmax were decreased (dexamethasone coadministration: no effect).
When olaparib was coadministered with the CYP3A probe substrate midazolam, the predicted increases in AUC and Cmax of midazolam were 1.6‐ and 1.2‐fold (tablet) and 1.5‐ and 1.1‐fold (capsule), respectively. Sensitivity analysis of the CYP3A inhibition parameters Ki (reversible inhibition) and KI (TDI) suggested that an AUC ratio for midazolam of >2‐fold was observed only when these parameter values (corrected for unbound fraction) were <10 and <40 μmol/L, respectively (Figure S10; shown for TDI). The amount of active CYP3A enzyme in the liver was decreased by 24% (tablet) and 20% (capsule), whereas it was increased via induction in the gut (Figure S13). Coadministration of the CYP3A substrate simvastatin with olaparib (tablet and capsule) resulted in a predicted increase in simvastatin AUC and Cmax of 1.5‐ and 1.3‐fold (tablet), respectively. When olaparib was combined with the P‐gp substrate digoxin (capsule and tablet) or the UGT1A1 substrate raltegravir, no effect on exposure to either was observed.
PBPK simulations for recommendations for olaparib tablet and capsule dose adjustment when coadministering olaparib with CYP3A inhibitors, CYP3A inducers, or probe substrates of CYP3A, P‐gp, or UGT1A1
Simulations for olaparib tablet or capsule DDIs were performed to guide dose‐adjustment recommendations for patients receiving olaparib coadministered with other drugs. Table 4 shows the predicted exposure ratios and dose‐adjustment recommendations for olaparib tablet or capsule in combination with CYP3A inhibitors, CYP3A inducers, and probe substrates of CYP3A, P‐gp, or UGT1A1.
Table 4.
Actual olaparib tablet exposure observed in adult patients and predicted olaparib tablet exposure in different pediatric age groups (geometric mean with 95% confidence interval)
| Age group | Olaparib dose, mg bid | Geometric mean AUC, h·µg/mL (95% CI) | Geometric mean Cmax, μg/mL (95% CI) |
|---|---|---|---|
| Adults SOLO2 (observed) | 300 | 40.7 (37.7–44.0) | 7.12 (6.75–7.51) |
| Adults OlympiAD (observed) | 300 | 41.2 (36.1‐47.0) | 6.41 (5.64‐7.29) |
| Adults | 300 | 40.3 (36.7–44.2) | 6.99 (6.58–7.41) |
| 12–17 years | 300 | 39.9 (36.5–43.6) | 7.74 (7.24–8.28) |
| 6–12 years |
200 150 |
47.7 (43.9–51.8) 35.8 (33.0–38.9) |
10.2 (9.55–10.8) 7.62 (7.17–8.11) |
| 2–6 years |
100 75 |
42.6 (39.2–46.3) 32.0 (29.4–34.7) |
8.94 (8.43–9.48) 6.71 (6.33–7.11) |
| 1–2 years |
60 50 |
40.1 (36.9–43.5) 33.4 (30.8–36.3) |
8.18 (7.75–8.63) 6.82 (6.46–7.20) |
| 0.5–1 years |
40 30 |
43.8 (40.3–47.5) 32.8 (30.2–35.6) |
8.19 (7.75–8.66) 6.14 (5.81–6.50) |
| 3–6 months |
20 15 |
42.8 (39.6–46.3) 32.1 (29.7–34.7) |
6.80 (6.44–7.18) 5.10 (4.83–5.39) |
| 1–3 months |
10 7.5 |
43.6 (40.3–47.2) 32.7 (30.2–35.4) |
5.84 (5.50–6.20) 4.38 (4.13–4.65) |
As reported previously,21 exposure to olaparib (tablet) was significantly increased when coadministered with the potent CYP3A inhibitor, itraconazole. Both olaparib tablet (100 mg) and capsule (150 mg) single dose administered in combination with itraconazole were predicted to give an exposure range that matched (within 2‐fold) monotherapy predicted exposures of olaparib tablet (300 mg) and capsule (400 mg). For moderate CYP3A inhibitors, such as fluconazole, coadministration with olaparib tablet (150 mg) or capsule (200 mg) resulted in an exposure range and mean exposure closely matched (within 2‐fold) with the observed and predicted monotherapy exposure of the olaparib tablet and capsule approved dose. There was no change in the predicted exposure of olaparib (tablet or capsule) when given with a weak CYP3A inhibitor such as fluvoxamine.
There was a reduction in exposure of olaparib (tablet or capsule) when coadministered with a strong or moderate inducer of CYP3A, such as rifampicin or efavirenz, respectively, whereas no change was observed for either formulation when given with a weak CYP3A inducer such as dexamethasone.
When olaparib was coadministered with CYP3A, P‐gp, or UGT1A1 probe substrates, no clinically significant effect was observed for P‐gp and UGT1A1 substrates, with a small increase in exposure to CYP3A substrates observed.
PBPK simulations for olaparib tablet and capsule given to patients with renal or hepatic impairment
The PBPK model reasonably predicted renal‐impairment effects within 0.67–1.5‐fold of observed olaparib tablet exposure from matched clinical studies (NCT01894256, NCT01894243; Table S4).13 For moderate hepatic impairment, the initial PBPK model overpredicted olaparib tablet exposure by >2‐fold. After the model was revised to include observed clinical trial data (see Methods), it predicted observed single‐dose clinical data in patients with hepatic impairment within ∼1.25‐fold. Table 3 shows the predicted exposure ratios for olaparib tablet or capsule in patients with mild, moderate, or severe renal or hepatic impairment. Table S8 shows the simulations of multiple confounding factors which otherwise are impossible to be studied clinically (e.g., moderate renal impairment patients with a strong CYP inhibitor as a comedication).
Table 3.
Mean olaparib tablet or capsule AUC and Cmax ratios from PBPK model simulations investigating the effect of hepatic or renal impairment
| Type and severity of impairment | Study simulation parameters required for dose adjustment recommendation | Observed exposure ratiod, 13 | Predicted exposure ratio | ||||
|---|---|---|---|---|---|---|---|
| AUC ratio (90% CI) | Cmax ratio (90% CI) | AUC ratio (90% CI) | Cmax ratio (90% CI) | ||||
| Mild renal impairment (creatinine clearance 51–80 mL/min) | Single olaparib tablet 300 mg dose using matched observed patient demographics and matched renal clearance from patients with mild or moderate renal impairment vs. patients with normal renal impairment from study NCT0189425613 | 1.24 (1.06–1.47) | 1.15 (1.04–1.27) | 1.40 (1.39–1.40) | 1.04 (1.03–1.04) | ||
| Moderate renal impairment (creatinine clearance 31–50 mL/min) | 1.44 (1.10‐1.89) | 1.38 (1.06‐1.48) | 1.89 (1.89–1.90) | 1.09 (1.07–1.10) | |||
| Severe renal impairment and endstage renal disease (creatinine clearance ≤ 30 mL/min) | — | — | 2.21 (2.19–2.22) | 1.11 (1.10–1.12) | |||
| Mild hepatic impairmenta (Child–Pugh A) | Single olaparib tablet 300 mg dose using matched observed patient demographics of patients with mild or moderate hepatic impairment vs. patients with normal renal impairment from study NCT0189424313 | 1.15 (0.77–1.72) | 1.12 (0.82–1.55) | 1.38 (1.27–1.50)b | 1.26 (1.26–1.28)c | 1.10 (1.06–1.14)b | 1.06 (1.05–1.07)c |
| Moderate hepatic impairmenta (Child–Pugh B) | 1.08 (0.66–1.74) | 0.87 (0.63–1.22) | 2.50 (2.49–3.19)b | 1.26 (1.15–1.32)c | 1.53 (1.48–1.59)b | 0.78 (0.77–0.80)c | |
| Severe hepatic impairmenta (Child–Pugh C) | — | — | 3.88 (3.74–4.02)b | 1.06 (1.03–1.08)c | 2.23 (2.18–2.27)b | 0.59 (0.58–0.59)c | |
| Capsule formulation | |||||||
| Mild renal impairment (creatinine clearance 51–80 mL/min) | — | — | 1.48 (1.44–1.52) | 1.21 (1.19–1.24) | |||
| Moderate renal impairment (creatinine clearance 31–50 mL/min) | — | — | 1.95 (1.92–1.98) | 1.28 (1.26–1.31) | |||
| Severe renal impairment and end‐stage renal disease (creatinine clearance ≤ 30 mL/min) | — | — | 2.27 (2.25–2.29) | 1.31 (1.28–1.33) | |||
| Mild hepatic impairmenta (Child–Pugh A) | — | — | 0.95 (0.94–0.97)c | 1.16 (1.15–1.16)c | |||
| Moderate hepatic impairmenta (Child–Pugh B) | — | — | 1.54 (1.52–1.56)c | 1.27 (1.26–1.28)c | |||
| Severe hepatic impairmenta (Child–Pugh C) | — | — | 2.20 (2.13–2.28)c | 1.04 (1.03–1.06)c | |||
Modified PBPK model with fa reduced by 34% and minimal PBPK, assuming normal GFR for predicted simulations.
Full/ADAM PBPK model.
Minimal/FO PBPK model.
GLS mean ratio. ADAM, advanced dissolution, absorption and metabolism; fa, fraction of absorption; FO, first order absorption; GFR, glomerular filtration rate; IC50, 50% inhibitory concentration.
PBPK simulations of olaparib tablets in pediatric oncology patients
The updated PBPK model reasonably captured adult olaparib‐exposure profiles, so the model was applied to determine the olaparib tablet doses that resulted in equivalent exposure range to olaparib in pediatric patients of different ages by considering the ontogeny of the CYP enzymes (Table 4). Body weight normalized clearance values were compared among different age groups, increasing gradually from 1–3 months to 6 months to 1 year age groups, and were generally consistent from 1 year and older age groups (Figure S15). As olaparib is mainly metabolized by CYP3A enzymes, this increase of clearance in the younger age groups reflects the maturation of the CYP3A enzymes.
Exposure–response relationship for both safety & efficacy
Based on a PKPD analysis of data from a recent phase III study (SOLO2; NCT01874353), no exposure–response relationships were found for efficacy or safety endpoints, except for with hemoglobin count and fatigue. The PKPD relationship for fatigue was flat, and for hemoglobin count the relationship was more significant. A 50% increase in exposure to olaparib is predicted to result in an ∼17.5% drop in hemoglobin level, which was considered to be significant. The decrease of hemoglobin count is expected to be reasonably managed in the clinical setting.
Discussion
We report here a series of PBPK‐based model experiments used to determine the DDIs for olaparib (tablet or capsule formulation) as a victim when coadministered with CYP3A inhibitors and CYP3A inducers and as a perpetrator when coadministered with CYP3A, P‐gp, or UGTA1A probe substrates. Additionally, the models were utilized to determine the pharmacokinetics of olaparib tablet or capsule in patients with mild, moderate, or severe renal or hepatic impairment, and in pediatric subjects (tablet only).
The PBPK models developed were robustly defined using a mechanistic approach. The capsule and tablet formulations are not bioequivalent; the capsule exhibits solubility‐limited absorption and nonlinear pharmacokinetics, and the PBPK model accurately predicted exposure to olaparib for both formulations. The in vivo plasma concentration–time profile for olaparib tablet monotherapy was predicted to be within ∼1.5‐fold of the observed monotherapy clinical data. Clinical DDI studies with a strong CYP3A inhibitor, itraconazole, and a strong CYP3A inducer,21 rifampicin, were used to verify the olaparib tablet PBPK model and to bridge the tablet DDI study results to DDI predictions for the capsule, taking into account differences in absorption between the two formulations. The in vivo plasma concentration–time profile for olaparib capsule monotherapy was predicted to be within 1.9‐fold of the observed monotherapy clinical data.
The PBPK capsule model used to predict drug interactions incorporated both TDI and induction parameters of CYP3A. Although the observed olaparib PK data were overpredicted by 1.9‐fold for the capsule formulation when CYP3A TDI parameters were included, the predicted AUC change for the CYP3A probe substrates midazolam and simvastatin and the P‐gp substrate digoxin reflected the worst‐case scenario, as ∼1.9‐fold overprediction of portal‐vein concentrations of olaparib is likely to be the driving force for the DDIs.
The tablet and capsule models were applied to inform drug labeling of olaparib by predicting olaparib multiple‐dose DDI (Table 2). The magnitude of the change following olaparib tablet dosing to patients with mild renal or mild/moderate hepatic impairment was low.13, 25 The PBPK model overpredicted moderate hepatic impairment by >2‐fold. This is likely to be because of several factors, including: physiological parameters related to blood flow away from the liver26; variation in drug absorption (decrease in exposure due to congestion and decreased blood flow in intestinal mucosa)27; displacement of drugs bound to albumin (protein‐binding changes can be important but cannot be predicted without quantifying human serum albumin and α‐acidic glycoprotein contributions)28; quantification of the reductions in CYP and transporter activity (the contribution of CYP enzymes/transporters to drug clearance and volume distribution is required to progress to more accurate predictions)28; reduced renal metabolism (e.g., morphine glucuronidation); or reduced glomerular filtration and/or tubular secretion.28 In addition, in patients with moderate hepatic impairment, cirrhosis may decrease gastrointestinal absorption because of congestion and decreased blood flow in the intestinal mucosa.29 After reducing the fraction absorbed value to 0.60 from the original value of 0.94, based on literature observations, and switching to minimal PBPK from full PBPK, predictions of single‐dose exposure to olaparib in patients with mild/moderate hepatic impairment were within ∼1.25‐fold of observed clinical data (NCT01894243). Additional information on the modified physiological parameters used for the renal impaired patients are shown in Table S5. The decrease in olaparib absorption observed appeared to counteract the effect of metabolism decrease, resulting in little change in the exposure of olaparib in patients with moderate hepatic impairment. The reduced CYP3A abundance in patients with hepatic impairment is supported by clinical studies investigating the pharmacokinetics of CYP3A4 substrates (alprazolam, alfentanil, midazolam, rivaroxaban), which included plasma protein‐binding measurements and cohorts of patients with hepatic impairment compared with healthy age‐matched subjects.30, 31, 32 There are several drugs predominantly metabolized by CYP3A that have little change in drug exposure when given to patients with hepatic impairment (e.g., doravirinin, bosutinib, ombitasvir, ritonavir)33; the studies reported here show that olaparib appears to behave in a similar manner.
Finally, the tablet model was used to predict the exposure to olaparib when given to pediatric subjects of different ages by accounting for the ontogeny of the CYP enzymes. Simulations for adults using the approved tablet 300 mg b.i.d. dose were comparable to those observed in vivo (NCT01921140). Olaparib tablet doses resulting in an olaparib exposure range equivalent to those observed in adult patients for a range of pediatric ages (Table 4). The dosing recommendations will be verified and revised if necessary when clinical data are available in patients <18 years old and when an appropriate formulation for younger children is available.
There are several limitations to this study. First, the effect of olaparib tablet or capsule, when coadministered with a CYP3A or P‐gp probe substrate, has not been tested in an in vivo clinical study; therefore, the developed model has not been verified against any clinical study data. However, olaparib is itself a CYP3A and P‐gp substrate that relies heavily on CYP3A for clearance, and the single‐ and multiple‐dose plasma concentration–time profiles have been adequately defined: within 1.5‐fold of the observed data for the tablet25 and within 1.9‐fold for the capsule. Furthermore, the model predicted only a minimal magnitude of effect of olaparib capsule on CYP3A and Pg‐p. A second challenge of this PBPK approach is the prediction of simultaneous inhibition and induction of CYP3A enzymes. This may not be robust with mechanistic static models.34 However, we have shown improved agreement with observed clinical data (predictions were within 1.9‐fold of observed olaparib monotherapy clinical data) when the PBPK model incorporated all necessary and adequately characterized system and drug parameters. Recent studies have also used robust PBPK‐modeling approaches to successfully predict the clinical drug interactions for mixed CYP3A inhibitors and inducers,35, 36 further supporting our mechanistic PBPK‐modeling approach to quantify the net effect of olaparib drug interactions.
In conclusion, in vitro studies suggested that metabolism of olaparib in humans is primarily mediated by CYP3A. The PBPK tablet model developed here adequately predicted the observed clinical tablet monotherapy and DDI data with potent CYP3A modulators and allowed simulation of the effect of other CYP3A modulators on olaparib tablet pharmacokinetics; it has also adequately predicted data following administration in patients with hepatic or renal impairment. Similarly, the PBPK capsule model adequately predicted the observed clinical capsule monotherapy pharmacokinetics and has provided DDI data with CYP3A modulators. The models developed have provided important information for olaparib tablet or capsule dose recommendations for coadministering olaparib with CYP3A4 modulators to eliminate the potential risk to patient safety or olaparib efficacy. Olaparib was shown to be a weak inhibitor of CYP3A and to have no effect on P‐gp or UGT1A1 substrates. Finally, this model allowed prediction of initial dosing recommendations in pediatric subjects.
Methods
PBPK model development
Olaparib tablet PBPK model
PBPK‐modeling software (Simcyp, v. 16.1) was used to build PBPK models for simulating the human exposure of olaparib tablet or capsule (Table 5).37, 38, 39, 40
Table 5.
Olaparib tablet and capsule model input parameters used for PBPK model development
| Olaparib formulation | |||
|---|---|---|---|
| Parameters and models | Tablet (source) | Capsule (source) | |
| Physiochemical properties | MW | 434.5 (measured data) | 434.5 (measured data) |
| Log P | 1.55 (measured data) | 1.55 (measured data) | |
| pKa | Neutral | Neutral | |
| B/P ratio | 0.7 (measured data) | 0.7 (measured data) | |
| fu,plasma | 0.181 (measured data) | 0.181 (measured data) | |
| Dosage form | Film‐coated tablets, immediate release | Gelucire 44/14 capsule | |
| Absorption | Absorption model | ADAM | ADAM |
| fu,gut | 0.259 (derived using ratio of fu,plasma to B/P) | 0.259 (derived using ratio of fu,plasma to B/P) | |
| Peff,man | 36.23 × 10−4 cm/s (estimated based on clinical data) | 6.44 × 10−4 cm/s (estimated based on clinical data) | |
| Distribution | Distribution model | Full PBPK | Full PBPK |
| Vss (L/kg) | 0.378 (predicted in Simcyp from physiochemical data)a | 0.378 (predicted in Simcyp from physiochemical data)a | |
| Elimination | Clearance type | Enzyme kinetics | Enzyme kinetics |
| CLpo (L/h) | 7.6 (estimated based on clinical data) | 6.07 (estimated based on clinical data) | |
| CYP3A CLint (µL/min/pmol of isoform) | 0.057 (retrograde approach) | 0.0412 (retrograde approach) | |
| Additional HLM CLint | 0.218 (retrograde approach) | 0.156 (retrograde approach) | |
| Renal clearance (L/h) | 1.48 (based on clinical data: NCT01894256) | 1.48 (based on clinical data: NCT01894256) | |
| Interaction | CYP3A4 reversible inhibition Ki (Ki=IC50/2) (μM) | 59.5 (measured data) | 59.5 (measured data) |
| CYP3A4 TDI KI (μM) | 72.2 (measured data) | 72.2 (measured data) | |
| CYP3A4 TDI Kinact (/h) | 4.05 (measured data) | 4.05 (measured data) | |
| CYP3A4 induction EC50 (μM) | 17.7 (measured data) | 17.7 (measured data) | |
| CYP3A4 induction Emax (fold) | 48.1 (measured data) | 48.1 (measured data) | |
| fu,mic/fu,inc | 0.892 (Simcyp predicted) | 0.892 (Simcyp predicted) | |
| Pg‐P inhibition (μM) | 73.9 (measured data; Ki calculated using Simcyp calculator) | 73.9 (measured data; Ki calculated using Simcyp calculator) | |
| UGT1A1 (Ki=IC50/2) (μM) | 48.4 (measured data) | 48.4 (measured data) | |
| Population | All simulations based on oncology patients39 except for the pediatric simulations, which are based on a healthy population40 | ||
| Clinical data |
D0816C00004 (NCT01921140): Tablet monotherapy data D0816C00024 (NCT00777582): Tablet monotherapy data at steady state D0816C00007 (NCT01900028): Tablet DDI study with itraconazole D0816C00008 (NCT01929603): Tablet DDI study with rifampicin D0816C00006 (NCT01894256): Tablet renal impairment study D081AC00001 (NCT01851265), D0810C00001 (NCT00572364): Capsule monotherapy data D0810C00002 (NCT00516373), D0810C00008 (NCT00494234), D0810C00009 (NCT00494442), D0810C00010 (NCT00633269), D0810C00012 (NCT00628251), D0810C00024 (NCT00777582), Capsule monotherapy data at steady state |
||
No in vitro data are available to estimate Vss in patients, therefore Vss of olaparib was estimated to be 0.378 L/kg using the Rodgers and Rowland 2007 method37; this value is in a reasonable agreement with apparent volume distribution (0.438 L/kg) estimated using a population pharmacokinetic approach.38
For in vitro dissolution information on the tablet and capsule formulations, please see the Supplementary Materials.
B/P, blood‐to‐plasma ratio; CLint, intrinsic clearance; CLpo, apparent oral clearance; EC50, half‐maximal effective concentration; Emax, maximum achievable response; fu,gut, fraction unbound in gut enterocytes; fu,inc, fraction unbound in the incubation; fu,mic, fraction unbound in microsomes; fu,plasma, fraction unbound in plasma; HLM, human liver microsomes; KI, inhibitory constant for time‐dependent inhibition; Ki, inhibitory constant for reversible inhibition; Kinact, rate of enzyme inactivation; MW, molecular weight; Peff,man, human intestinal effective permeability; pKa, acid dissociation constant (logarithmic scale); Vss, volume of distribution at steady state.
Based on olaparib drug disposition and DDI mechanisms, the PBPK tablet model was built in three stages. First, the model was developed using in vitro and physical‐chemistry data with a derived value of in vivo human intestinal effective permeability (Peff,human) parameter to drive first‐order absorption from an oral dose (Supplementary Supporting Information). An in vivo derived estimate of apparent oral clearance (CLpo) via a population PK model was used to predict the intrinsic clearance of olaparib (CLint). Second, the Peff,human and CLpo values were revised using the parameter‐estimation option within Simcyp to fit Peff,human and CLpo using emerging NCT01921140 clinical data (remaining parameters were fixed). Finally, the model was checked for consistency against clinical PK data and the in vivo clearance was converted into enzyme kinetic clearance using the retrograde model. To determine if the olaparib tablet model could replicate the plasma concentration–time profile observed in patients following single (300 mg) or multiple 300 mg b.i.d.) dosing, simulations were compared with matched clinical trial data (NCT01921140).22 Metabolic routes of clearance were incorporated into the simulation of the plasma concentration–time curves using in vitro data, to predict the effects of the CYP3A inducer, rifampicin, and the CYP3A inhibitor, itraconazole, on olaparib tablet exposure. Additionally, in vivo monotherapy data from clinical studies (NCT01929603, NCT01900028, NCT01921140),21 were used to verify the models. The final PBPK model used for simulation was based on a “middle‐out” approach.41
Olaparib capsule PBPK model development and bridging the formulations
The olaparib capsule PBPK model was developed in line with the tablet model, observed apart from differences in absorption between the two formulations, as well as CYP3A and P‐gp inhibition and induction parameters (Table 5); the capsule model used refitted parameters (from NCT01851265) for extent of CLpo and rate of Peff,human, and segmental solubility was used instead of intrinsic solubility. To determine replication of the plasma concentration–time profile of olaparib observed in patients following single (400 mg) or multiple capsule (400 mg b.i.d.) dosing, simulations were compared with clinical trial data (single dosing: NCT01851265)25; multiple dosing was compared with pooled data from NCT00572364, NCT00516373, NCT00494234, NCT00494442, NCT00628251, NCT00777582, and D0810C00007. Predefined acceptance criteria to assess the predictive performance of the PBPK model was set to be between 0.5 and 2‐fold, which was based on clinical relevance criteria.24
Sensitivity analyses to optimize the PBPK models
The impact of model assumptions and uncertainties in the model parameters on the predictive performance of the olaparib PBPK model was investigated using sensitivity analyses (Supplementary Supporting Information; Figures S8–S13, Figure S17).
Verification of DDIs using oral olaparib tablet in oncology patients
Clinical trial DDI data of olaparib tablet dosed alone or in combination with itraconazole or rifampicin were used to verify the tablet PBPK model simulations. Simulations comprised 100 patients (10 trials of 10 patients each), except for itraconazole simulations, which used 140 patients (5 trials of 28 patients each). Simulations used a representative oncology patient population39 and were performed with patients in a fasted state matching observed clinical data.
Clinical DDI studies with the strong CYP3A inhibitor itraconazole and inducer rifampicin were used to verify the olaparib tablet DDI model and to bridge the tablet DDI study results with capsule DDI simulations. Verified PBPK models were used to perform prospective prediction of DDIs where no clinical data were available (Figure [Link]), as described below. Input parameters for all drugs other than olaparib, dexamethasone, and raltegravir used reference values from the Simcyp compound library (Table 5, Table S2). Simulated PK profiles after oral administration of multiple doses of itraconazole, OH‐itraconazole, rifampicin, efavirenz, fluvoxamine, or single doses of midazolam, simvastatin, and digoxin were consistent with the observed literature data from clinical studies42, 43, 44, 45, 46, 47, 48 and verification summary reports can be found in the Simcyp members area (https://members.simcyp.com/account/libraryFiles/). Verification of dexamethasone and raltegravir are detailed in Pilla Reddy et al., 201849 and Hartman et al., 2013.50 For both tablets and capsules predefined acceptance criteria to assess the predictive performance of the PBPK model was set to be between 0.5‐ and 2‐fold, based on clinical relevance criteria.24
Prospective DDI predictions for olaparib tablet and capsule
Olaparib tablet and capsule simulations are shown in Figure S2 Briefly, olaparib tablet/capsule exposure was simulated in the presence and absence of multiple doses of the CYP3A4 inhibitors itraconazole (200 mg replicating NCT01900028 (tablet study)),21 fluconazole (200 mg), and fluvoxamine (50 mg) and CYP3A4 inducers rifampicin (600 mg replicating NCT01929603 (tablet study)),21 efavirenz (600 mg), and dexamethasone (8 mg). Exposures of the CYP3A substrates midazolam (5 mg) and simvastatin (40 mg), and the P‐gp substrate digoxin (0.5 mg and 0.25 mg with olaparib tablet and capsule formulations, respectively) and UGT1A1 substrate raltegravir (400 mg) were simulated following a single dose in the presence and absence of daily dosing of olaparib tablets/capsules.
Simulating olaparib tablet exposure when dosed to patients with hepatic or renal impairment
Simulations were conducted to predict changes in olaparib tablet plasma exposure in patients with hepatic or renal impairment at steady state using predefined Simcyp mild, moderate, and severe hepatically/renally impaired populations,39 except for the Simcyp values for creatinine clearance in hepatically impaired patients, which used clinical values (NCT01894256; Table S6),25 which were in the normal range. The methodology for these simulated study designs is described further in the Supplementary Supporting Information.
The methodology for simulating olaparib tablet exposure when dosed to pediatric patients is also described in the Supplementary Supporting Information.
Funding
This study was sponsored by AstraZeneca. Medical writing assistance was provided by Martin Goulding, PhD, from Mudskipper Business Ltd, funded by AstraZeneca.
Conflict of interest
The authors are all employees of AstraZeneca and own stock in AstraZeneca.
Author contributions
V.P.R. wrote the article; V.P.R., K.B., G.S., D.Z., and M.L. designed the study; V.P.R., K.B., G.S., D.Z., and M.L. performed the research; V.P.R., K.B., G.S., D.Z., and M.L. analyzed the data; V.P.R., K.B., G.S., D.Z., and M.L. contributed new reagents/analytical tools.
Supporting information
Acknowledgments
The authors thank the patients, their families, and the clinical teams who worked on the studies. The authors thank Alex McCormick, Mike Walker (both formally of AstraZeneca, Macclesfield, UK), and Owen Jones (AstraZeneca, Cambridge, UK) for assistance in carrying out the study procedures described. Medical writing assistance was provided by Martin Goulding, PhD, from Mudskipper Business Ltd, funded by AstraZeneca.
V.P.R. and M.L. are joint first authors.
References
- 1. Kaufman, B. et al Olaparib monotherapy in patients with advanced cancer and a germ‐line BRCA1/2 mutation. J. Clin. Oncol. 33, 244–250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ledermann, J . et al Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 15, 852–861 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Tutt, A . et al Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet. 376, 235–244 (2010). [DOI] [PubMed] [Google Scholar]
- 4. Pujade‐Lauraine, E . et al Olaparib tablets as maintenance therapy in patients with platinum‐sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT‐Ov21): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 18, 1274–1284 (2017). [DOI] [PubMed] [Google Scholar]
- 5. European Medicines Agency . Lynparza (olaparib); EPAR. <http://www.ema.europa.eu/ema/index.jsp?curl5/pages/medicines/human/medicines/003726/human_med_001831.jsp> (2015).
- 6. FDA . Lynparza prescribing information. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208558s000lbl.pdf> (2017).
- 7. Robson, M . et al Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 377, 523–533 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Molife, R . et al A phase I study to determine the comparative bioavailability of two different oral formulations of the PARP inhibitor, olaparib (AZD2281), in patients with advanced solid tumors. J. Clin. Oncol. 28(7S), abst 2599 (2010). [Google Scholar]
- 9. Center for Drug Evaluation and Research . Clinical Pharmacology and Biopharmaceutics Reviews; Olaparib. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/206162Orig1s000ClinPharmR.pdf> (2016).
- 10. Thadhani, R. , Pascual, M. & Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 334, 1448–1460 (1996). [DOI] [PubMed] [Google Scholar]
- 11. Wyld, L . et al Prognostic factors for patients with hepatic metastases from breast cancer. Br. J. Cancer. 89, 284–290 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bacalbasa, N . et al Liver resection for ovarian cancer liver metastases as part of cytoreductive surgery is safe and may bring survival benefit. World J. Surg. Oncol. 13, 235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rolfo, C . et al Pharmacokinetics and safety of olaparib in patients with advanced solid tumours and hepatic or renal impairment In 18th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics. Washington, DC, 13–18 March abst PII–121 (2017). [Google Scholar]
- 14. Huang, S.M . et al New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 48, 662–670 (2008). [DOI] [PubMed] [Google Scholar]
- 15. Percha, B. & Altman, R.B. Informatics confronts drug‐drug interactions. Trends Pharmacol. Sci. 34, 178–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thai, H.T. , Mazuir, F. , Cartot‐Cotton, S. & Veyrat‐Follet, C. Optimizing pharmacokinetic bridging studies in paediatric oncology using physiologically‐based pharmacokinetic modelling: application to docetaxel. Br. J. Clin. Pharmacol. 80, 534–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walsh, C . et al Development of a physiologically based pharmacokinetic model of actinomycin D in children with cancer. Br. J. Clin. Pharmacol. 81, 989–998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Hasselt, J.G. , van Eijkelenburg, N.K. , Beijnen, J.H. , Schellens, J.H. & Huitema, A.D. Optimizing drug development of anti‐cancer drugs in children using modelling and simulation. Br. J. Clin. Pharmacol. 76, 30–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khalil, F. & Laer, S. Physiologically based pharmacokinetic modeling: methodology, applications, and limitations with a focus on its role in pediatric drug development. J. Biomed. Biotechnol. 2011, 907461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCormick, A. , Swaisland, H. , Reddy, V.P. , Learoyd, M. & Scarfe, G. In vitro evaluation of the inhibition and induction potential of olaparib, a potent poly(ADP‐ribose) polymerase inhibitor, on cytochrome P450. Xenobiotica. 48, 555‐564 (2017). [DOI] [PubMed] [Google Scholar]
- 21. Dirix, L . et al Effect of itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: results of two phase I open‐label studies. Clin. Ther. 38, 2286–2299 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Plummer, R . et al Olaparib tablet formulation: effect of food on the pharmacokinetics after oral dosing in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 76, 723–729 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Jiang, X.L. , Zhao, P. , Barrett, J.S. , Lesko, L.J. & Schmidt, S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacometrics Syst. Pharmacol. 2, e80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner, C. , Pan, Y. , Hsu, V. , Sinha, V. & Zhao, P. Predicting the effect of CYP3A inducers on the pharmacokinetics of substrate drugs using physiologically based pharmacokinetic (PBPK) modeling: an analysis of PBPK submissions to the US FDA. Clin. Pharmacokinet. 55, 475–483 (2016). [DOI] [PubMed] [Google Scholar]
- 25. Rolfo, C. et al Effect of food on the pharmacokinetics of olaparib after oral dosing of the capsule formulation in patients with advanced solid tumors. Adv. Ther. 32, 510–522 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Foti, R.S. Strategies and retrospective data analysis in hepatic impairment studies. In 19th North American Regional ISSX Meeting. San Francisco, CA, 19–23 October abst SC4.4 (2014).
- 27. Rodighiero, V. Effects of liver disease on pharmacokinetics. An update. Clin. Pharmacokinet. 37, 399–431 (1999). [DOI] [PubMed] [Google Scholar]
- 28. Palatini, P. & De Martin, S. Pharmacokinetic drug interactions in liver disease: an update. World J. Gastroenterol. 22, 1260–1278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ono, C. , Hsyu P.H., Abbas, R. , Loi, C.M. & Yamazaki, S. Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug‐drug and drug‐disease interactions. Drug Metab. Dispos. 45, 390–398 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Juhl, R.P. , Van Thiel, D.H. , Dittert, L.W. & Smith, R.B. Alprazolam pharmacokinetics in alcoholic liver disease. J. Clin. Pharmacol. 24, 113–119 (1984). [DOI] [PubMed] [Google Scholar]
- 31. Mueck, W. , Kubitza, D. & Becka, M. Co‐administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br. J. Clin. Pharmacol. 76, 455–466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baririan, N. et al Alfentanil‐induced miosis as a surrogate measure of alfentanil pharmacokinetics in patients with mild and moderate liver cirrhosis. Clin. Pharmacokinet. 46, 261–270 (2007). [DOI] [PubMed] [Google Scholar]
- 33. University of Washington . The University of Washington Drug Interaction Database Program. <https://www.druginteractioninfo.org/> (2018).
- 34. Prueksaritanont, T. et al Drug‐drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 15, 629–645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu, Y. , Loi, C.M. , Hoffman, J. & Wang, D. Physiologically based pharmacokinetic modeling of palbociclib. J. Clin. Pharmacol. 57, 173–184 (2017). [DOI] [PubMed] [Google Scholar]
- 36. Yamazaki, S. , Johnson, T.R. & Smith, B.J. Prediction of drug‐drug interactions with crizotinib as the CYP3A substrate using a physiologically based pharmacokinetic model. Drug Metab. Dispos. 43, 1417–1429 (2015). [DOI] [PubMed] [Google Scholar]
- 37. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 38. Peer, C.J. et al Population pharmacokinetic analyses of the effect of carboplatin pretreatment on olaparib in recurrent or refractory women's cancers. Cancer Chemother. Pharmacol. 80, 165–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheeti, S. , Budha, N.R. , Rajan, S. , Dresser, M.J. & Jin, J.Y. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm. Drug Dispos. 34, 141–154 (2013). [DOI] [PubMed] [Google Scholar]
- 40. Jamei, M. et al The Simcyp population‐based ADME simulator. Expert Opin. Drug Metab. Toxicol. 5, 211–223 (2009). [DOI] [PubMed] [Google Scholar]
- 41. Shebley, M . et al Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin. Pharmacol. Ther. [Epub ahead of print] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barone, J.A. et al Food interaction and steady‐state pharmacokinetics of itraconazole capsules in healthy male volunteers. Antimicrob. Agents Chemother. 37, 778–784 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prueksaritanont, T. et al Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ke, A. , Barter, Z. , Rowland‐Yeo, K. & Almond, L. Towards a best practice approach in PBPK modeling: case example of developing a unified efavirenz model accounting for induction of CYPs 3A4 and 2B6. CPT Pharmacometrics Syst. Pharmacol. 5, 367–376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Vries, M.H. , Van Harten, J. , Van Bemmel, P. & Raghoebar, M. Pharmacokinetics of fluvoxamine maleate after increasing single oral doses in healthy subjects. Biopharm. Drug Dispos. 14, 291–296 (1993). [DOI] [PubMed] [Google Scholar]
- 46. Kyrklund, C. et al Rifampin greatly reduces plasma simvastatin and simvastatin acid concentrations. Clin. Pharmacol. Ther. 68, 592–597 (2000). [DOI] [PubMed] [Google Scholar]
- 47. Eckermann, G. , Lahu, G. , Nassr, N. & Bethke, T.D. Absence of pharmacokinetic interaction between roflumilast and digoxin in healthy adults. J. Clin. Pharmacol. 52, 251–257 (2012). [DOI] [PubMed] [Google Scholar]
- 48. Abel, S. , Russell, D. , Whitlock, L.A. , Ridgway, C.E. & Muirhead, G.J. Effect of maraviroc on the pharmacokinetics of midazolam, lamivudine/zidovudine, and ethinyloestradiol/levonorgestrel in healthy volunteers. Br. J. Clin. Pharmacol. 65(Suppl 1), 19–26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pilla Reddy, V. , Walker, M. , Sharma, P. , Ballard, P. & Vishwanathan, K. Development, verification, and prediction of osimertinib drug‐drug interactions using PBPK modelling approach to inform drug label. CPT Pharmacometrics Syst. Pharmacol. [Epub ahead of print] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hartmann, G. , Gibson, C. , Rizk, M. & Xu, Y. Retrospective analysis of raltegravir (Isentress™) oral pharmacokinetics in healthy volunteers using the SimCYP PBPK model. Simcyp Consortium Meeting 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials