

Abstract
Background & Aims
Assessment of hepatic steatosis by transient elastography (TE)‐based controlled attenuation parameter (CAP) might predict hepatic decompensation. Therefore, we aimed to evaluate the prognostic value of CAP in patients with compensated advanced chronic liver disease (cACLD) and decompensated cirrhosis (DC).
Methods
A total of 430 patients who underwent TE (liver stiffness ≥10 kPa) and CAP measurements were included in this retrospective analysis. Half of patients (n = 189) underwent simultaneous HVPG measurement. In cACLD patients, first hepatic decompensation was defined by new onset of ascites, hepatic encephalopathy or variceal bleeding. In patients with DC, the following events were considered as further hepatic decompensation: requirement of paracentesis, admission for/development of grade 3/4 hepatic encephalopathy, variceal (re‐)bleeding or liver‐related death.
Results
First hepatic decompensation occurred in 25 of 292 (9%) cACLD patients, while 46 of 138 (33%) DC patients developed further hepatic decompensation during a median follow‐up of 22 and 12 months respectively. CAP was not predictive of first (cACLD; per 10 dB/m; hazard ratio [HR]: 0.97, 95% confidence interval [95% CI]: 0.91‐1.03, P = 0.321) or further hepatic decompensation (DC; HR: 0.99, 95% CI: 0.94‐1.03, P = 0.554) in adjusted analysis. Using the well‐established CAP cut‐off of ≥248 dB/m for hepatic steatosis, the incidence of first (cACLD; P = 0.065) and further hepatic decompensation (DC; P = 0.578) was similar in patients with hepatic steatosis or without. Serum albumin levels (per mg/dL; HR: 0.83, 95% CI: 0.77‐0.89, P < 0.001) and MELD‐Na (per point; HR: 1.15, 95% CI: 1.04‐1.28, P = 0.006) in cACLD and MELD‐Na (per point; HR: 1.12, 95% CI: 1.05‐1.19, P < 0.0001) in DC patients were the only parameters independently associated with first and further hepatic decompensation, respectively.
Conclusion
Controlled attenuation parameter does not predict the development of first (cACLD)/further (DC) hepatic decompensation, while serum albumin levels and MELD‐Na are of prognostic value.
Keywords: compensated advanced chronic liver disease, controlled attenuation parameter, decompensated cirrhosis, hepatic decompensation
Abbreviations
- 95%CI
95% confidence interval
- ACLF
acute‐on‐chronic liver failure
- ACLD
advanced chronic liver disease
- AUROC
area under the receiver operating characteristic curve
- cACLD
compensated advanced chronic liver disease
- CAP
controlled attenuation parameter
- CSPH
clinically significant portal hypertension
- DC
decompensated cirrhosis
- HBV
hepatitis B virus
- HR
hazard ratio
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HVPG
hepatic venous pressure gradient
- IQR
interquartile range
- LRE
liver‐related events
- MELD‐Na
model for end‐stage liver disease including sodium
- NSBB
non‐selective beta‐blocker
- SD
standard deviation
- SVR
sustained virologic response
- TE
transient elastography
- TIPS
transjugular intrahepatic portosystemic shunt
Key points.
CAP did not predict the occurrence of first or further hepatic decompensation in patients with compensated advanced chronic liver disease or decompensated cirrhosis, respectively.
Serum albumin levels and MELD‐Na yield prognostic information.
1. INTRODUCTION
Portal hypertension, as assessed by hepatic venous pressure gradient (HVPG), drives the development of complications (ie, hepatic decompensation) in patients with advanced chronic liver disease (ACLD).1, 2, 3, 4
Animal studies have shown that diet‐induced steatohepatitis is associated with the development of portal hypertension in the absence of fibrosis5 by inducing sinusoidal endothelial dysfunction and thereby increasing intrahepatic vascular resistance.6 These results were confirmed in a small series of patients with noncirrhotic nonalcoholic fatty liver disease (NAFLD) reporting an association between steatosis and elevated portal pressure.7
Transient elastography (TE) has been shown to correlate with HVPG8, 9, 10, 11 and the development of complications of portal hypertension.12, 13 TE‐based controlled attenuation parameter (CAP) measurement is a novel tool for the quantification of hepatic steatosis,14 and robust cut‐offs for different grades of hepatic steatosis have previously been established by individual patient‐based meta‐analysis.15 Recently, CAP has been linked to the incidence of liver‐related events (LRE) in patients with compensated ACLD (cACLD).16 Importantly, this study did not provide information on HVPG, which is a well‐established predictor of hepatic decompensation.13, 17 Thus, it is unclear, whether CAP is an independent predictor of LRE.
Another recent study did not observe an association between CAP and hepatic decompensation in patients with liver disease.13 However, the majority of patients included in the latter study did not have significant liver fibrosis, as reflected by low median liver stiffness values. Accordingly, only a very small proportion of patients developed hepatic decompensation, which substantially limits the generalizability of the findings to patients with ACLD,13 who are at considerable risk of LRE.18 In addition, the role of CAP in predicting further hepatic decompensation in patients with decompensated cirrhosis (DC) remains unknown.
Therefore, we aimed to assess the prognostic value of CAP for first (cACLD) and further (DC) hepatic decompensation in a thoroughly characterized cohort of ACLD patients undergoing TE with CAP including a large subgroup of patients who also underwent simultaneous HVPG measurement.
2. PATIENTS AND METHODS
2.1. Study design
Patients undergoing TE and CAP measurement19 between 01 January 2014 to 31 December 2016 at the Medical University of Vienna were included in this retrospective analysis. Clinical follow‐up was evaluated by checking the patients’ electronic reports until 31 September 2017.
2.2. Patients and definitions
Patients were excluded if they had (a) invalid TE measurements (nonfasting, <10 valid measurements, IQR/median >0.320); (b) history of hepatocellular carcinoma (HCC), hepatitis C virus (HCV) treatment,21, 22, 23, 24 transjugular intrahepatic portosystemic shunt (TIPS) or liver transplantation; (c) missing data/lack of clinical follow‐up, (d) cardiac cirrhosis; or (e) evidence of severe hepatitis/hepatic inflammation or acute liver failure. Furthermore, patients with baseline liver stiffness value <10 kPa were excluded as these patients are unlikely to have ACLD.25 Patient characteristics and important laboratory parameters at baseline as well as clinical follow‐up were evaluated by two authors separately (B.S. and L.S.) by chart review. Patients were grouped according to the presence (decompensated cirrhosis, DC) or absence (compensated advanced chronic liver disease, cACLD) of previous hepatic decompensation.26 In patients with DC, the following events were considered as further hepatic decompensation: requirement of paracentesis, admission for grade 3/4 hepatic encephalopathy, variceal (re‐)bleeding or liver‐related death. In cACLD patients, first hepatic decompensation was defined by the new onset of ascites, hepatic encephalopathy or variceal bleeding. Even though new onset of jaundice is commonly referred to as a decompensating event, we did not incorporate jaundice, since this term is poorly defined.17 This is in line with previous studies investigating risk factors for hepatic decompensation.17, 27
Acute‐on‐chronic liver failure (ACLF) was diagnosed according to the EASL‐CLIF definition.28, 29
2.3. Clinically significant portal hypertension
Clinically significant portal hypertension (CSPH) was defined as a HVPG ≥10 mm Hg. The Vienna Hepatic Hemodynamic Lab at the Medical University of Vienna performed the measurements according to a standardized operating procedure.19
2.4. Noninvasive measurements and cut‐offs for hepatic steatosis
Hepatic steatosis was assessed by TE‐based CAP using a FibroScan® 502 Touch (Echosens, Paris, France), applying previously described reliability criteria for TE.20 The following cut‐offs for steatosis grades were derived from a recently published meta‐analysis15: any steatosis/≥S1 (lipid accumulation in >5% of hepatocytes): ≥248 dB/m2, ≥S2 (>33% of hepatocytes): ≥268 dB/m2 and ≥S3 (>66% of hepatocytes): ≥280 dB/m2.
It is not possible to obtain valid liver stiffness/CAP values by TE in most patients with pronounced perihepatic ascites. Moreover, based on our experience, the presence of severe ascites makes catheterization of the hepatic veins more difficult. Therefore, patients with severe ascites commonly undergo paracentesis prior to HVPG measurement. Importantly, there is evidence from both experimental and clinical studies that TE provides reliable measurements in phantoms/patients with a thin lamella of water/ascites.30 However, if we were unable to obtain liver stiffness measurement by TE (eg, due to pronounced perihepatic ascites), patients were excluded.
2.5. Statistics
Statistical analyses were performed using IBM SPSS Statistics 23 (SPSS Inc., Armonk, NY, USA) and GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Continuous variables were reported as mean ± standard deviation (SD) or median (IQR), and categorical variables were shown as numbers (n) and proportions (%) of patients. Comparisons of continuous variables were performed using Student's t test or Mann‐Whitney U test, as applicable. The incidence of first/further hepatic decompensation was assessed by the Kaplan‐Meier method and compared between the two groups using the log‐rank test. Multivariate Cox regression analysis with stepwise backward selection was used to determine prognostic factors independently associated with hepatic decompensation. Patients entered the Kaplan‐Meier analysis and Cox models at the time of CAP measurement. Time to first/further hepatic decompensation was defined as time to clinical decompensation or liver‐related death. Patients were censored at the time of liver transplantation, end of follow‐up or non‐liver‐related death. Cox regression included variables that showed differences between patients with and without first/further hepatic decompensation or those which we considered highly relevant based on the previous literature (eg, HVPG). Area under the receiver operating characteristic curve (AUROC) analysis was performed for determining the optimal CAP cut‐off for predicting hepatic decompensation. A two‐sided P‐value ≤0.05 was considered as statistically significant.
2.6. Ethics
This study was approved by the ethics committee of the Medical University of Vienna (EK No.: 2013/2016 and 1124/2017). Since this is a retrospective analysis, the requirement of a written informed consent was waived by the ethics committee of the Medical University of Vienna.
3. RESULTS
3.1. Patients
In total, 3601 patients underwent simultaneous TE and CAP measurements within the inclusion period (Figure 1). As depicted in Figure 1, 2714 patients had to be excluded. Finally, 430 patients were included for further analysis (Figure 1). A total of 292 patients were assigned to the cACLD and 138 to the DC group. Half of patients (n = 86 in the cACLD and n = 103 in the DC group) also underwent simultaneous HVPG measurement.
Figure 1.
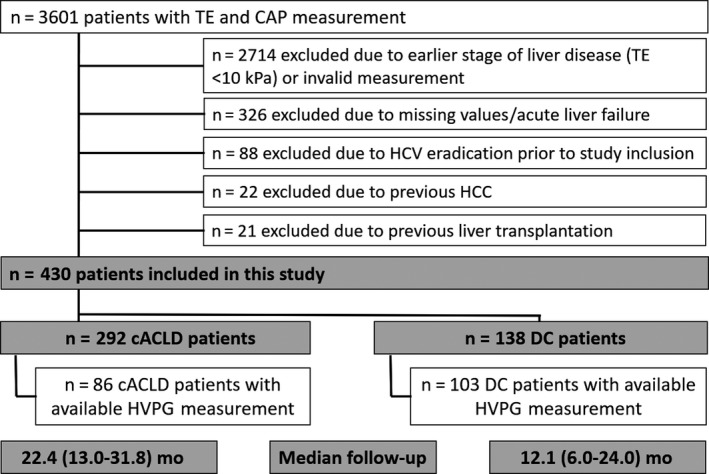
Patient flow chart showing the number of included and excluded patients as well as the number of patients assigned to the group of compensated advanced chronic liver disease and decompensated cirrhosis. CAP, controlled attenuation parameter; cACLD, compensated advanced chronic liver disease; DC, decompensated cirrhosis; HVPG, hepatic venous pressure gradient; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; TE, transient elastography
3.2. Patient characteristics (Tables 1 and 2)
Table 1.
Comparison of baseline characteristics of patients with compensated advanced chronic liver disease (cACLD) with vs without hepatic decompensation during follow‐up
| cACLD patients, n = 292 | Without first hepatic decompensation, n = 267 | With first hepatic decompensation, n = 25 | P‐value | |
|---|---|---|---|---|
| Sex, male/female (% male) | 190/102 (65%) | 176/91 (66%) | 14/11 (56%) | 0.320 |
| Age, yr | 54 ± 12 | 54 ± 12 | 57 ± 12 | 0.197 |
| BMI, kg/m2 | 27 ± 6 | 28 ± 6 | 26 ± 5 | 0.177 |
| Aetiology | ||||
| Viral hepatitis, n (%) | 195 (67) | 181 (68) | 14 (56) | 0.097 |
| (N)AFLD, n (%) | 43 (15) | 40 (15) | 3 (12) | |
| Other, n (%) | 38 (13) | 34 (13) | 4 (16) | |
| Cryptogenic, n (%) | 16 (5) | 12 (4) | 4 (16) | |
| Diabetes, n (%) | 59 (20) | 51 (19) | 8 (32) | 0.125 |
| Fasting glucose, mg/dL | 107 ± 23 | 106 ± 22 | 112 ± 32 | 0.611 |
| Triglycerides, mg/dL | 115 ± 71 | 116 ± 71 | 99 ± 72 | 0.245 |
| Total cholesterol, mg/dL | 162 ± 52 | 164 ± 52 | 144 ± 40 | 0.066 |
| Statin treatment, n (%) | 26 (9) | 22 (8) | 4 (16) | 0.383 |
| INR | 1.2 ± 0.3 | 1.2 ± 0.3 | 1.3 ± 0.3 | 0.095 |
| Albumin, g/L | 42 ± 4 | 42 ± 4 | 37 ± 5 | <0.001 |
| MELD‐Na score, points | 9.4 ± 3.3 | 9.1 ± 3.2 | 12.2 ± 3.9 | 0.001 |
| Liver stiffness, median kPa (range) | 18 (12‐28) | 17 (12‐26) | 26 (21‐47) | 0.001 |
| CAP, dB/m | 265 ± 63 | 266 ± 64 | 246 ± 59 | 0.126 |
| HVPG, median mm Hg (range)a | 14 (9‐19) | 13 (8‐18) | 17 (11‐22) | 0.156 |
| CSPH, n (%)a | 60 (70) | 50 (68) | 10 (77) | 0.746 |
BMI, body mass index; CAP, controlled attenuation parameter; cACLD, compensated advanced chronic liver disease; CSPH, clinically significant portal hypertension; HVPG, hepatic venous pressure gradient; INR, international normalized ratio; MELD‐Na, model for end‐stage liver disease including sodium; (N)AFLD, (non)alcoholic fatty liver disease.
Statistically significant P‐values are shown in bold.
Data were available in 86 patients (n = 73 without decompensation and n = 13 with decompensation).
Table 2.
Comparison of baseline characteristics of patients with decompensated cirrhosis with vs without further decompensation
| Decompensated cirrhosis patients, n = 138 | No further hepatic decompensation, n = 92 | Further hepatic decompensation, n = 46 | P‐value | |
|---|---|---|---|---|
| Sex, male/female (% male) | 86/54 (62) | 57/35 (62) | 27/19 (59) | 0.711 |
| Age, yr | 54 ± 12 | 53 ± 12 | 54 ± 13 | 0.808 |
| BMI, kg/m2 | 25 ± 5 | 25 ± 5 | 24 ± 5 | 0.771 |
| Aetiology | ||||
| Virus hepatitis, n (%) | 33 (24) | 20 (22) | 13 (28) | 0.738 |
| (N)AFLD, n (%) | 72 (52) | 51 (55) | 21 (46) | |
| Other, n (%) | 17 (12) | 11 (12) | 6 (13) | |
| Cryptogenic, n (%) | 16 (12) | 10 (11) | 6 (13) | |
| Diabetes, n (%) | 23 (17) | 16 (17) | 7 (15) | 0.747 |
| Fasting glucose, mg/dL | 101 ± 21 | 99 ± 18 | 122 ± 53 | 0.662 |
| Triglycerides, mg/dL | 91 ± 49 | 90 ± 48 | 94 ± 52 | 0.667 |
| Total cholesterol mg/dL | 151 ± 54 | 151 ± 52 | 150 ± 57 | 0.870 |
| Statin treatment, n (%) | 8 (6) | 5 (5) | 3 (7) | 0.350 |
| History of variceal bleeding, n (%) | 43 (31) | 24 (26) | 19 (41) | 0.069 |
| History of or current ascites | ||||
| No ascites | 29 (21) | 22 (24) | 7 (15) | 0.422 |
| Mild/moderate (%) | 87 (63) | 57 (62) | 30 (65) | |
| Severe, n (%) | 22 (16) | 13 (14) | 9 (20) | |
| History of or current hepatic encephalopathy, n (%) | 45 (33) | 24 (26) | 21 (46) | 0.021 |
| INR | 1.4 ± 0.3 | 1.3 ± 0.3 | 1.4 ± 0.2 | 0.298 |
| Albumin, g/L | 35 ± 6 | 35 ± 4 | 35 ± 6 | 0.624 |
| MELD‐Na score, points | 15 ± 5 | 14 ± 5 | 16 ± 5 | 0.004 |
| Liver stiffness, kPa | 46 (26‐69) | 46 (26‐70) | 46 (27‐69) | 0.895 |
| CAP, dB/m | 235 ± 66 | 238 ± 66 | 231 ± 68 | 0.575 |
| HVPG, mm Hga | 19 (15‐23) | 19 (15‐22) | 21 (17‐25) | 0.023 |
BMI, body mass index; CAP, controlled attenuation parameter; cACLD, compensated advanced chronic liver disease; HVPG, hepatic venous pressure gradient; INR, international normalized ratio; MELD‐Na, model for end‐stage liver disease including sodium; (N)AFLD, (non)alcoholic fatty liver disease.
Statistically significant P‐values are shown in bold.
Data were available in 103 patients (n = 70 without decompensation and n = 33 with decompensation).
Patient characteristics are shown separately for cACLD (Table 1) and DC patients (Table 2). The majority of cACLD patients were male (65%) with a mean age of 54 ± 12 years. The cACLD cohort comprised 173 patients with chronic HCV infection, 30 with NAFLD, 23 with chronic hepatitis B virus (HBV) infection, 12 with alcoholic liver disease, 11 with cholestatic liver disease, eight with autoimmune hepatitis and 35 patients had other aetiologies of liver disease. Patients had a high median liver stiffness of 18 (12‐28) kPa, and the mean CAP value was in the range of mild steatosis (256 ± 59 dB/m) resulting in a prevalence of any hepatic steatosis (≥248 dB/m) of 60%. In the subgroup of patients with information on HVPG (n = 86), median HVPG was 14 (9‐19) mm Hg with more than two‐thirds of patients presenting with CSPH (69.8%).
Similarly, more than half of DC patients were male (62%) with a mean age of 54 ± 12 years. The majority of patients (n = 68) had alcoholic liver disease, followed by HCV infection (n = 30), cholestatic liver disease (n = 6), NAFLD (n = 4), HBV infection (n = 3) and other aetiologies of liver disease (n = 27). The most common previous decompensating event was ascites (79%), followed by hepatic encephalopathy (33.0%) and variceal bleeding (31%). The mean CAP value was 235 ± 66 dB/m, and 38% had any hepatic steatosis (≥248 dB/m).
The subgroup of DC patients with available HVPG data (n = 103) presented with pronounced portal hypertension as indicated by a median HVPG value of 19 (15‐23) mm Hg.
3.3. Development of first/further hepatic decompensation and ACLF (Figure 2)
Figure 2.
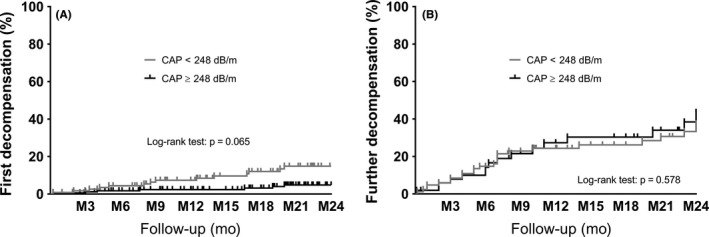
Incidence of first/further hepatic decompensation according to baseline CAP </≥248 dB/m in (A) patients with compensated advanced chronic liver disease and (B) patients with decompensated cirrhosis. CAP, controlled attenuation parameter; M, months
During a median follow‐up of 22 (13‐32) months, 25 of 292 (9%) cACLD patients developed first hepatic decompensation (Figure 2A). In the DC group, 46/138 (33%) patients experienced further hepatic decompensation during a median follow‐up period of 12.1 (6‐24) months (Figure 2B). The most common decompensating event in the cACLD group was new onset of ascites, which occurred in 15 (5%) patients, followed by de novo hepatic encephalopathy (n = 5, 2%), and first variceal bleeding (n = 5, 2%). In the DC group, the major event leading to further hepatic decompensation was requirement of paracentesis (n = 19, 14%), followed by hospital admission for hepatic encephalopathy grade 3/4 (n = 13, 9%), and variceal (re‐)bleeding (n = 9, 6%), and five additional patients (4%) died from a liver‐related cause.
In total, hepatic decompensation was associated with ACLF in four patients in the cACLD (<248 dB/m: 1/118 [1%] vs ≥248 dB/m: 3/174 [2%]) and 11 patients in the DC (<248 dB/m: 9/85 [11%] vs ≥248 dB/m: 2/53 [4%]) group.
3.4. Comparison of baseline characteristics between patients with and without first/further hepatic decompensation during follow‐up (Tables 1 and 2)
Patients with first hepatic decompensation (cACLD group) during follow‐up had significantly lower baseline serum albumin levels (37 ± 5 vs 42 ± 4 g/L; P < 0.001), significantly higher MELD‐Na score (12 ± 4 vs 9 ± 3 points; P = 0.001) as well as significantly higher liver stiffness (26 [21‐47] vs 17 [12‐26] kPa; P = 0.001). Apart from these three variables, baseline characteristics including CAP values (266 ± 64 vs 246 ± 59 dB/m; P = 0.126) were comparable between cACLD patients with and without first hepatic decompensation (Table 1).
MELD‐Na score (16 ± 5 vs 14 ± 5 points; P = 0.004), proportion of patients with previous or current hepatic encephalopathy (46% vs 26%; P = 0.021) and baseline HVPG (21 [11‐31] vs 19 [3‐29] mm Hg; P = 0.023; in patients with available data) were significantly higher in patients presenting with further hepatic decompensation (DC group). Again, CAP values (238 ± 66 vs 231 ± 68 dB/m; P = 0.575) as well as the other evaluated baseline characteristics were comparable between DC patients with and without further hepatic decompensation (Table 2).
3.5. Predictors of the development of first/further hepatic decompensation (Table 3)
Table 3.
Predictors of the development of (further) hepatic decompensation in (A) patients with compensated advanced chronic liver disease and (B) decompensated cirrhosis
| Patient characteristics | Multivariate, first step | Multivariate, final step | ||||
|---|---|---|---|---|---|---|
| HR | 95%CI | P‐value | HR | 95%CI | P‐value | |
| (A) | ||||||
| Albumin, per g/L | 0.84 | 0.77–0.92 | <0.001 | 0.83 | 0.77–0.89 | <0.001 |
| MELD‐Na, per point | 1.14 | 1.02–1.27 | 0.022 | 1.15 | 1.04–1.28 | 0.006 |
| CAP, per 10 dB/m | 0.97 | 0.91–1.03 | 0.321 | ‐ | ||
| TE, per kPa | 1.02 | 1.00–1.04 | 0.108 | ‐ | ||
| (B) | ||||||
| History of or current hepatic encephalopathy | 1.81 | 1.00–3.28 | 0.052 | 1.68 | 0.94–3.01 | 0.082 |
| History of or current ascites | 1.64 | 0.72–3.75 | 0.237 | ‐ | ||
| MELD‐Na, per point | 1.11 | 1.05–1.19 | 0.001 | 1.12 | 1.05–1.19 | <0.001 |
| CAP, per 10 dB/m | 0.99 | 0.94–1.03 | 0.554 | ‐ | ||
95%CI, 95% confidence interval; CAP, controlled attenuation parameter; HR, hazard ratio; MELD‐Na, model for end‐stage liver disease including sodium.
Statistically significant P‐values are shown in bold.
Multivariate Cox regression analysis evaluating predictors of first hepatic decompensation in patients with cACLD showed that lower serum albumin level (per g/L; hazard ratio (HR): 0.83 [95% CI: 0.77‐0.89]; P < 0.001) and higher MELD‐Na score (per point; HR: 1.15 [95% CI: 1.04‐1.28]; P = 0.006) were independent predictors of hepatic decompensation (Table 3A). In contrast, neither CAP nor liver stiffness was associated with first hepatic decompensation.
In patients with DC, multivariate Cox regression analysis showed that higher MELD‐Na score (per point; HR: 1.12 [95% CI: 1.05‐1.19]; P < 0.001) was the only independent predictor for the development of further hepatic decompensation (Table 3B). Moreover, history of or current hepatic encephalopathy tended to increase the risk of further hepatic decompensation (HR: 1.68 [95% CI: 0.94‐3.01]; P = 0.082). However, neither CAP nor a history of or current ascites was independently predictive of further hepatic decompensation.
3.6. Predictors of the development of first/further hepatic decompensation in patients with information on HVPG (Table S3)
Multivariate Cox regression analysis evaluating predictors of first hepatic decompensation in patients with cACLD showed that lower serum albumin level (per g/L; HR: 0.850 [95% CI: 0.745‐0.969]; P = 0.015) and higher MELD‐Na score (per point; HR: 1.251 [95% CI: 1.020‐1.533]; P = 0.032) were independent predictors (Table S3A).
In patients with DC, higher HVPG (per mm Hg; HR: 1.079 [95% CI: 1.001‐1.164]; P = 0.047) and higher MELD‐Na score (per point; HR: 1.132 [95% CI: 1.046‐1.225]; P = 0.002) were independent predictors for the development of further hepatic decompensation in the final step of backward selection (Table S3B).
3.7. First/further hepatic decompensation according to the presence/absence of hepatic steatosis diagnosed by CAP (Tables S1 and S2, Figure 2, Figures S1 and S2)
Baseline characteristics of patients presenting with a CAP value in the steatotic vs nonsteatotic range were comparable, apart from a higher BMI (cACLD: 29 ± 7 vs 25 ± 4 kg/m2, P < 0.001; DC: 26 ± 6 vs 24 ± 4 kg/m2, P = 0.005), a higher albumin level (cACLD: 43 ± 4 vs 41 ± 5 g/L, P = 0.007), higher triglyceride levels (DC: 102 ± 56 vs 85 ± 43 md/dL, P = 0.045), as well as a lower MELD‐Na score (cACLD: 9 ± 3 vs 10 ± 4 points, P = 0.001) in the CAP ≥248 dB/m subgroups (Table S1). Furthermore and not surprisingly, the proportion of patients with (N)AFLD was significantly higher in the CAP ≥248 dB/m subgroup (cACLD: 20% vs 8%, P = 0.037; Table S2). Importantly, in the subgroup of patients who underwent HVPG measurement, HVPG was comparable between patients with and without any hepatic steatosis (CAP <248 dB/m: 18 (13‐22) vs CAP ≥248 dB/m: 16 (11‐21); P = 0.296).
We used Kaplan‐Meier analysis and log‐rank test to compare the incidence of first/further hepatic decompensation. As shown in Figure 2, there was no difference in the incidence of first/further hepatic decompensation, neither in the cACLD (log‐rank test: P = 0.065), nor in the DC group (log‐rank test: P = 0.578). When using a CAP cut‐off ≥220 dB/m, CAP even had a protective effect for the development of first hepatic decompensation as shown in Figure S2 (log‐rank test: P = 0.021).
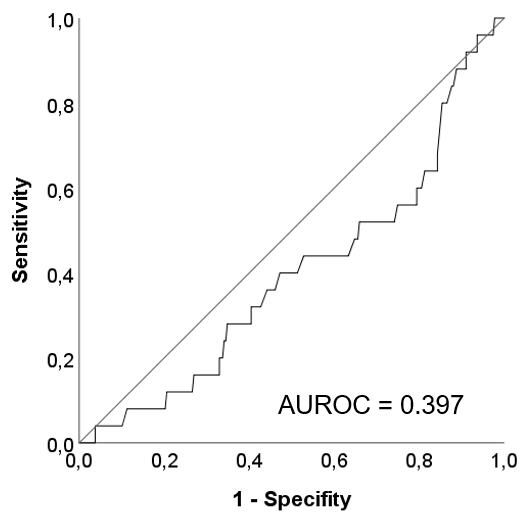
Finally, AUROC analysis was performed to determine whether CAP predicts clinical decompensation, if analysed as a dichotomous variable (Figure S1). The AUROC values were 0.397 (95% CI: 0.282‐0.513; P = 0.090) in the cACLD group and 0.472 (95% CI: 0.368‐0.577; P = 0.599) in the DC group. Therefore, CAP had no predictive value for first/further hepatic decompensation, independently of the cut‐off.
3.8. Impact of aetiological and supportive treatment (Table S4A‐D)
Furthermore, as aetiological and supportive treatments might have impacted our results, the number of patients receiving vs not receiving anti‐HCV and non‐selective beta‐blocker (NSBB) therapy during follow‐up was compared (Table S4A‐D). In the cACLD group, 80% (n = 64) of HCV‐infected patients with a CAP <248 dB/m and 88% (n = 82) of HCV‐infected patients with a CAP ≥248 dB/m (P = 0.140) achieved sustained virologic response (SVR) to anti‐HCV therapy during the study period. In the DC group, results were comparable, n = 14 (70%) vs n = 6 (60%; P = 0.690) had SVR.
Additionally, NSBB treatment was equally distributed between patients with a CAP <248 dB/m vs patients with a CAP ≥248 dB/m (cACLD group: 40 (34%) vs 53 (31%), P = 0.536; DC group: 55 (65%) vs 35 (66%), P = 0.873).
4. DISCUSSION
The development of noninvasive tools for the diagnosis of portal hypertension and prediction of complications is of great clinical relevance. TE has been shown to be a useful method for predicting CSPH25, 31 and liver‐related events.12, 13 Besides liver stiffness, CAP has recently been proposed as an independent predictor of clinical decompensation in patients with cACLD.16 The results of Margini and co‐workers indicate that CAP is particularly useful for subclassifying patients with liver stiffness values suggestive of CSPH (ie, ≥21 kPa).16 In their study, a CAP value ≥220 dB/m seemed to be an excellent predictor of first hepatic decompensation,16 which was explained by the potential role of hepatic steatosis for liver disease progression.
In our cohort, we studied the prognostic impact of CAP using a cut‐off of ≥248 dB/m. This value was chosen as it has recently been shown to be the optimal cut‐off for diagnosing hepatic steatosis in an individual patient data meta‐analysis.15 Interestingly, using this cut‐off, CAP had no impact on the incidence of first hepatic decompensation in patients with cACLD, even after adjusting for other important prognostic parameters (eg, MELD‐Na, serum albumin levels, as well as HVPG in a subgroup of patients). Furthermore, HVPG values were comparable between patients with and without hepatic steatosis, suggesting that hepatic steatosis does not aggravate portal hypertension.
Since aetiological treatment (eg, HCV therapy24) as well as NSBB use32 could potentially have influenced our results, the proportions of patients achieving SVR or receiving NSBB treatment were compared. Importantly, these treatments were equally distributed between patients with CAP </≥248 dB/m, and thus, it is unlikely that they had an impact on our results.
Interestingly, we even observed a trend towards a lower incidence of first hepatic decompensation among patients with hepatic steatosis. Histological steatosis frequently regresses with liver fibrosis progression, and thus, patients with hepatic steatosis had less severe liver disease at baseline, as indicated by higher serum albumin levels and lower MELD‐Na.33, 34, 35 Importantly, after adjusting for these and other potentially relevant baseline characteristics, we observed no independent association between CAP and first hepatic decompensation.
In contrast to the study by Margini et al,16 our study comprised a subgroup of patients with information on HVPG, which is, based on the literature, a highly relevant predictor of hepatic decompensation17 and liver‐related mortality.26, 36 Although the authors tried to account for the severity of portal hypertension by adjusting for liver stiffness,16 the well‐established impact of high HVPG values was neglected in their study, since the correlation with liver stiffness becomes weaker in patients with HVPG ≥10‐12 mm Hg.21, 37 Our findings are in line with Liu et al,13 who also did not report an association between CAP values and the incidence of hepatic decompensation in a large cohort of patients at different stages of liver disease. However, the majority of patients included in the latter study did not have significant liver fibrosis. Thus, both malignancies other than HCC and cardiovascular events were more common than LRE. Unfortunately, the authors did not perform a subgroup analysis in patients with cACLD who are at considerable risk for developing hepatic decompensation in the short term.13
Compensated and DC are two pathophysiologically distinct stages of liver disease.26, 29, 38, 39 This is the first study to evaluate the prognostic impact of CAP in DC by investigating the determinants of further hepatic decompensation in a separate analysis. However, similar to patients with cACLD, CAP ≥248 dB/m was not predictive for further hepatic decompensation.
While we could not find any impact of CAP on hepatic decompensation, we were able to confirm the results of previous studies on predictors of first/further hepatic decompensation in patients with compensated and DC: Low serum albumin level and high MELD‐Na were independently predictive for first hepatic decompensation in the setting of cACLD, which is in line with previous reports.26, 40 Furthermore, MELD‐Na and HPVG (in the subgroup of patients with available HVPG measurement) were independent predictors of further hepatic decompensation in patients with DC.2
We have to acknowledge several limitations of our study. First, a considerable number of patients were excluded from analysis. As a consequence, sample size was limited. However, the absence of an association between CAP and hepatic decompensation cannot be explained by this limitation, since the rates of hepatic decompensation were even numerically (and statistically significantly) lower in cACLD patients with CAP ≥248 dB/m or CAP ≥220 dB/m. Second, only a low number of patients with (N)AFLD aetiology were included; therefore, we cannot exclude that CAP has a prognostic impact on hepatic decompensation in these patients. Lastly, our study is limited by its retrospective single‐centre design. However, clinical events were reviewed by two authors separately. Moreover, the incidence rates of first/further hepatic decompensation was in line with the literature,17, 26, 41, 42 with ascites being the most common decompensation event.39
In conclusion, CAP does not predict first/further hepatic decompensation in cACLD or in patients with DC. Serum albumin level and MELD‐Na score are important independent predictors of first hepatic decompensation in cACLD, while MELD‐Na and HVPG predicted further hepatic decompensation in patients with DC.
AUTHOR CONTRIBUTIONS
B.S., T.R. and M.M. involved in concept of study. P.S., T.B., R.P., A.F., T.R. and M.M. carried out transient elastography (TE) and hepatic venous pressure gradient (HVPG) measurements. B.S., L.S., G.S. and L.W.U. collected the data. B.S. and M.M. performed the statistical analysis. B.S. and M.M. wrote the manuscript. All authors involved in revision for important intellectual content and approval of the final version of the manuscript.
CONFLICT OF INTEREST
The authors have nothing to disclose regarding this study.
Supporting information
{kind=link}
{kind=link}
Scheiner B, Steininger L, Semmler G, et al. Controlled attenuation parameter does not predict hepatic decompensation in patients with advanced chronic liver disease. Liver Int. 2019;39:127–135. 10.1111/liv.13943
Handling Editor: Janus Ong
REFERENCES
- 1. Villanueva C, Albillos A, Genesca J, et al. Development of hyperdynamic circulation and response to eta‐blockers in compensated cirrhosis with portal hypertension. Hepatology. 2016;63(1):197‐206. [DOI] [PubMed] [Google Scholar]
- 2. Reiberger T, Mandorfer M. Beta adrenergic blockade and decompensated cirrhosis. J Hepatol. 2017;66(4):849‐859. [DOI] [PubMed] [Google Scholar]
- 3. Scheiner B, Parada‐Rodriguez D, Bucsics T, et al. Non‐selective beta‐blocker treatment does not impact on kidney function in cirrhotic patients with varices. Scand J Gastroenterol. 2017;52(9):1008‐1015. [DOI] [PubMed] [Google Scholar]
- 4. Reiberger T, Puspok A, Schoder M, et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien Klin Wochenschr. 2017;129(Suppl 3):135‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Francque S, Wamutu S, Chatterjee S, et al. Non‐alcoholic steatohepatitis induces non‐fibrosis‐related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010;30(3):365‐375. [DOI] [PubMed] [Google Scholar]
- 6. Pasarin M, La Mura V, Gracia‐Sancho J, et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One. 2012;7(4):e32785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Francque S, Verrijken A, Mertens I, et al. Noncirrhotic human nonalcoholic fatty liver disease induces portal hypertension in relation to the histological degree of steatosis. Eur J Gastroenterol Hepatol. 2010;22(12):1449‐1457. [DOI] [PubMed] [Google Scholar]
- 8. Reiberger T, Ferlitsch A, Payer BA, et al. Noninvasive screening for liver fibrosis and portal hypertension by transient elastography–a large single center experience. Wien Klin Wochenschr. 2012;124(11–12):395‐402. [DOI] [PubMed] [Google Scholar]
- 9. Reiberger T, Ferlitsch A, Payer BA, et al. Non‐selective beta‐blockers improve the correlation of liver stiffness and portal pressure in advanced cirrhosis. J Gastroenterol. 2012;47(5):561‐568. [DOI] [PubMed] [Google Scholar]
- 10. Shi KQ, Fan YC, Pan ZZ, et al. Transient elastography: a meta‐analysis of diagnostic accuracy in evaluation of portal hypertension in chronic liver disease. Liver Int. 2013;33(1):62‐71. [DOI] [PubMed] [Google Scholar]
- 11. You MW, Kim KW, Pyo J, et al. A meta‐analysis for the diagnostic performance of transient elastography for clinically significant portal hypertension. Ultrasound Med Biol. 2017;43(1):59‐68. [DOI] [PubMed] [Google Scholar]
- 12. Robic MA, Procopet B, Metivier S, et al. Liver stiffness accurately predicts portal hypertension related complications in patients with chronic liver disease: a prospective study. J Hepatol. 2011;55(5):1017‐1024. [DOI] [PubMed] [Google Scholar]
- 13. Liu K, Wong VW, Lau K, et al. Prognostic value of controlled attenuation parameter by transient elastography. Am J Gastroenterol. 2017;112:1812‐1823. [DOI] [PubMed] [Google Scholar]
- 14. Sasso M, Beaugrand M, de Ledinghen V, et al. Controlled attenuation parameter (CAP): a novel VCTE guided ultrasonic attenuation measurement for the evaluation of hepatic steatosis: preliminary study and validation in a cohort of patients with chronic liver disease from various causes. Ultrasound Med Biol. 2010;36(11):1825‐1835. [DOI] [PubMed] [Google Scholar]
- 15. Karlas T, Petroff D, Sasso M, et al. Individual patient data meta‐analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol. 2017;66(5):1022‐1030. [DOI] [PubMed] [Google Scholar]
- 16. Margini C, Murgia G, Stirnimann G, et al. Controlled attenuation parameter as an additional tool for the non‐invasive prediction of first clinical decompensation in compensated advanced chronic liver disease. J Hepatol. 2017;66(1):S91‐S92. [Google Scholar]
- 17. Ripoll C, Groszmann R, Garcia‐Tsao G, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133(2):481‐488. [DOI] [PubMed] [Google Scholar]
- 18. D'Amico G, Pasta L, Morabito A, et al. Competing risks and prognostic stages of cirrhosis: a 25‐year inception cohort study of 494 patients. Aliment Pharmacol Ther. 2014;39(10):1180‐1193. [DOI] [PubMed] [Google Scholar]
- 19. Ferlitsch A, Bota S, Paternostro R, et al. Evaluation of a new balloon occlusion catheter specifically designed for measurement of hepatic venous pressure gradient. Liver Int. 2015;35(9):2115‐2120. [DOI] [PubMed] [Google Scholar]
- 20. Schwabl P, Bota S, Salzl P, et al. New reliability criteria for transient elastography increase the number of accurate measurements for screening of cirrhosis and portal hypertension. Liver Int. 2015;35(2):381‐390. [DOI] [PubMed] [Google Scholar]
- 21. Mandorfer M, Kozbial K, Schwabl P, et al. Sustained virologic response to interferon‐free therapies ameliorates HCV‐induced portal hypertension. J Hepatol. 2016;65(4):692‐699. [DOI] [PubMed] [Google Scholar]
- 22. Schwabl P, Mandorfer M, Steiner S, et al. Interferon‐free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment Pharmacol Ther. 2017;45(1):139‐149. [DOI] [PubMed] [Google Scholar]
- 23. Lens S, Alvarado‐Tapias E, Marino Z, et al. Effects of all‐oral anti‐viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus‐associated cirrhosis. Gastroenterology. 2017;153(5):1273‐1283 e1271. [DOI] [PubMed] [Google Scholar]
- 24. Mandorfer M, Reiberger T, Peck‐Radosavljevic M. Monitoring the evolution of portal hypertension after sustained virologic response. Gastroenterology. 2018;154(5):1550‐1551. [DOI] [PubMed] [Google Scholar]
- 25. Berzigotti A. Non‐invasive evaluation of portal hypertension using ultrasound elastography. J Hepatol. 2017;67(2):399‐411. [DOI] [PubMed] [Google Scholar]
- 26. D'Amico G, Garcia‐Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217‐231. [DOI] [PubMed] [Google Scholar]
- 27. Berzigotti A, Garcia‐Tsao G, Bosch J, et al. Obesity is an independent risk factor for clinical decompensation in patients with cirrhosis. Hepatology. 2011;54(2):555‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moreau R, Jalan R, Gines P, et al. Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144(7):1426‐1437, 1437 e1421‐1429. [DOI] [PubMed] [Google Scholar]
- 29. European Association for the Study of the Liver , Electronic address easloffice@easloffice.eu, European Association for the Study of the Liver . EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406‐460. [DOI] [PubMed] [Google Scholar]
- 30. Kohlhaas A, Durango E, Millonig G, et al. Transient elastography with the XL probe rapidly identifies patients with nonhepatic ascites. Hepat Med. 2012;4:11‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abraldes JG, Bureau C, Stefanescu H, et al. Noninvasive tools and risk of clinically significant portal hypertension and varices in compensated cirrhosis: the “Anticipate” study. Hepatology. 2016;64(6):2173‐2184. [DOI] [PubMed] [Google Scholar]
- 32. Mandorfer M, Reiberger T. Beta blockers and cirrhosis, 2016. Dig Liver Dis. 2017;49(1):3‐10. [DOI] [PubMed] [Google Scholar]
- 33. Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow‐up study of forty‐two patients for up to 21 years. Hepatology. 1990;11(1):74‐80. [DOI] [PubMed] [Google Scholar]
- 34. Abdelmalek M, Ludwig J, Lindor KD. Two cases from the spectrum of nonalcoholic steatohepatitis. J Clin Gastroenterol. 1995;20(2):127‐130. [DOI] [PubMed] [Google Scholar]
- 35. Charlton M, Kasparova P, Weston S, et al. Frequency of nonalcoholic steatohepatitis as a cause of advanced liver disease. Liver Transpl. 2001;7(7):608‐614. [DOI] [PubMed] [Google Scholar]
- 36. Ripoll C, Banares R, Rincon D, et al. Influence of hepatic venous pressure gradient on the prediction of survival of patients with cirrhosis in the MELD Era. Hepatology. 2005;42(4):793‐801. [DOI] [PubMed] [Google Scholar]
- 37. Vizzutti F, Arena U, Romanelli RG, et al. Liver stiffness measurement predicts severe portal hypertension in patients with HCV‐related cirrhosis. Hepatology. 2007;45(5):1290‐1297. [DOI] [PubMed] [Google Scholar]
- 38. Zipprich A, Garcia‐Tsao G, Rogowski S, Fleig WE, Seufferlein T, Dollinger MM. Prognostic indicators of survival in patients with compensated and decompensated cirrhosis. Liver Int. 2012;32(9):1407‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. D'Amico G, Morabito A, Pagliaro L, Marubini E. Survival and prognostic indicators in compensated and decompensated cirrhosis. Dig Dis Sci. 1986;31(5):468‐475. [DOI] [PubMed] [Google Scholar]
- 40. Nahon P, Ganne‐Carrie N, Degos F, et al. Serum albumin and platelet count but not portal pressure are predictive of death in patients with Child‐Pugh A hepatitis C virus‐related cirrhosis. Gastroenterol Clin Biol. 2005;29(4):347‐352. [DOI] [PubMed] [Google Scholar]
- 41. Tanaka R, Itoshima T, Nagashima H. Follow‐up study of 582 liver cirrhosis patients for 26 years in Japan. Liver. 1987;7(6):316‐324. [DOI] [PubMed] [Google Scholar]
- 42. Fattovich G, Giustina G, Degos F, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow‐up study of 384 patients. Gastroenterology. 1997;112(2):463‐472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials