

Abstract
Upadacitinib is a Janus kinase 1 inhibitor under development for the treatment of several inflammatory disorders including rheumatoid arthritis (RA). Upadacitinib was administered in the phase 2 RA trials primarily as twice‐daily regimens of an immediate‐release (IR) formulation. The upadacitinib extended‐release (ER) formulation was developed to enable once‐daily dosing. In the present study, upadacitinib pharmacokinetics were characterized after the administration of single and multiple once‐daily doses of the ER formulation in healthy subjects relative to single and multiple twice‐daily doses of the IR formulation. Increase in upadacitinib exposure was dose‐proportional over the evaluated 15‐ to 30‐mg ER dose range. Single 15‐ and 30‐mg ER doses provided equivalent AUC0–inf compared with single 12‐ and 24‐mg IR doses, respectively. A high‐fat breakfast increased upadacitinib ER Cmax and AUC0–inf by only 20% and 17%, respectively, relative to fasting conditions. The median time to peak plasma concentrations was 2 to 4 hours for the ER formulation, and steady state was achieved by day 4 of once‐daily dosing. Doses of 15 and 30 mg once daily using the ER formulation provided equivalent AUC0–24, comparable Cmax and Cmin, and a fluctuation index over a 24‐hour period at steady state similar to 6 and 12 mg twice daily, respectively, using the IR formulation. These results supported the use of upadacitinib 15‐ and 30‐mg doses of the ER formulation in the phase 3 trials in RA.
Keywords: upadacitinib, ABT‐494, pharmacokinetics, extended‐release formulation, rheumatoid arthritis, JAK1 inhibitors
Upadacitinib (previously known as ABT‐494; PubChem ICD: 58557659; https://pubchem.ncbi.nlm.nih.gov/) is a selective Janus kinase 1 (JAK1) inhibitor being developed for the treatment of inflammatory diseases, including rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, Crohn's disease, atopic dermatitis, and ankylosing spondylitis.1, 2, 3, 4, 5, 6 The JAK enzyme family consists of JAK1, JAK2, JAK3, and TYK2 enzymes, which are involved in cytokine signaling pathways involved in inflammatory disorders as well as several normal physiological functions (eg, erythropoiesis and immune function).7, 8 Selective inhibition of JAK1 more than other JAK isoforms has the potential to be efficacious in the treatment of inflammatory disorders while limiting the adverse effects on hemoglobin and immune function.9
The pharmacokinetics of upadacitinib were evaluated in phase 1 studies following the administration of immediate‐release (IR) capsules in single doses ranging from 1 to 48 mg to healthy subjects and multiple doses ranging from 3 to 24 mg twice daily to healthy subjects and 6 to 24 mg twice daily to subjects with rheumatoid arthritis.10 Following administration of the upadacitinib IR formulation, the maximum upadacitinib plasma concentration was achieved within 2 hours of dosing, followed by a biexponential decline in plasma levels. There was no significant accumulation of upadacitinib in plasma with twice‐daily dosing of the IR formulation, and approximately 20% of the upadacitinib dose was eliminated unchanged in urine.10 Upadacitinib is a nonsensitive substrate for cytochrome P450 (CYP) 3A with a minor contribution of CYP2D6 in vitro. Administration of the strong CYP3A inhibitor ketoconazole resulted in a 75% increase in upadacitinib AUC, whereas the CYP inducer rifampin decreased upadacitinib AUC by 60%.11 In a population pharmacokinetic analysis of phase 1 and 2 studies, the CYP2D6 metabolic phenotype showed no effect on the apparent upadacitinib oral clearance; mild and moderate renal impairment was estimated to result in 16% and 32% higher upadacitinib AUC compared with subjects with normal renal function.12 Intersubject variability in healthy subjects and subjects with rheumatoid arthritis (RA) was estimated to be 16% and 26%, respectively, for apparent upadacitinib oral clearance and 14% and 27%, respectively, for upadacitinib apparent central volume of distribution.12 Two dose‐ranging phase 2 studies evaluated the efficacy and safety of upadacitinib in subjects with RA who had inadequate response to methotrexate or anti‐tumor necrosis factor therapy.13, 14 Upadacitinib was administered primarily as twice‐daily regimens using the IR formulation in these phase 2 trials with doses ranging from 3 to 18 mg twice daily, in addition to 24 mg once daily in 1 of the 2 studies. The studies showed that doses of 6 and 12 mg twice daily using the IR formulation achieved the maximal efficacy in subjects with RA on background methotrexate treatment. Therefore, plasma exposures associated with 6 and 12 mg twice daily were selected as the target exposures to be evaluated in phase 3 studies in RA.
For patients with chronic diseases like RA, taking a drug twice daily can be cumbersome and may impact patients’ compliance with treatment compared with once‐daily dosing. To enhance patients’ compliance in the phase 3 studies and later in clinical settings, an extended‐release (ER) tablet formulation was developed with the objective of decreasing the peak‐to‐trough fluctuations in plasma concentrations with once‐daily dosing, therefore achieving daily AUC0–24 and minimum concentration (Cmin) comparable to IR doses of 6 and 12 mg twice daily. Another attribute of the target ER formulation was to avoid having a clinically relevant food effect to enable administration without regard to food intake. Several prototypes for the ER formulation were screened in vitro and in humans, and a formulation was selected that had the potential to meet the target pharmacokinetic profiles with 15‐ and 30‐mg strengths based on the initial pharmacokinetic assessment (data on file, AbbVie). The upadacitinib ER formulation uses an hydroxypropyl methylcellulose (HPMC) polymer that controls diffusion of the drug substance through a gel layer that forms during dissolution.
The current phase 1 study was conducted to characterize the pharmacokinetics of upadacitinib after single and multiple once‐daily doses of the ER formulation compared with single and multiple twice‐daily doses of the IR formulation used in phase 2 RA studies and to characterize the effect of a high‐fat meal on the ER formulation.
Methods
The study protocol was approved by the institutional review board (Vista Medical Center East Institutional Review Board, Waukegan, Illinois) of the study site (AbbVie Clinical Pharmacology Research Unit, Grayslake, Illinois), and all participants gave written informed consent before participation in the study. The study was conducted according to the International Conference on Harmonization Guidelines for Good Clinical Practice.
Study Design
The study was designed as a single‐center, 5‐part study in healthy subjects to compare the bioavailability of single and multiple doses of the upadacitinib ER formulation with the IR formulation under fasting conditions, to evaluate the pharmacokinetics, safety, and tolerability of multiple doses of the ER formulation, and to assess the effect of a high‐fat meal on the pharmacokinetics of the ER formulation. A separate cohort of subjects was enrolled in each study part.
The study design, which is summarized in Figure 1, was as follows:
In part 1, the bioavailability of single doses of upadacitinib 15‐mg ER tablets was compared with 12‐mg IR capsules in an open‐label, randomized, 2‐period, 2‐sequence crossover (between the 2 regimens) design in 11 subjects.
Part 2 was a single‐dose, open‐label, randomized, 3‐period, 2‐sequence crossover design comparing the bioavailability of upadacitinib 30‐mg ER tablets with 24‐mg IR capsules under fasting conditions. Part 2 also evaluated the effect of a high‐fat meal on the pharmacokinetics, safety, and tolerability of the 30‐mg ER formulation.
Parts 3 and 4 were multiple‐dose, randomized, open‐label, 2‐period, 2‐sequence crossover assessments under fasting conditions. Part 3 compared the bioavailability of upadacitinib 15 mg once daily using the ER tablets with 6 mg twice daily using IR capsules. Part 4 compared the bioavailability of 30 mg once daily using the ER tablets with 12 mg twice daily using IR capsules. Upadacitinib was administered in both parts 3 and 4 for 7 days under fasting conditions.
Part 5 was a multiple‐dose, randomized, double‐blind, placebo‐controlled evaluation in 3 parallel groups of subjects evaluating the pharmacokinetics, safety, and tolerability of multiple doses of upadacitinib 15‐ and 30‐mg ER tablets under nonfasting conditions. Subjects were randomized to receive upadacitinib or matching placebo for 7 days.
Figure 1.
Study design. A washout of 4 days was used between consecutive periods. IR, immediate release; ER, extended release; BID, twice daily; QD, once daily.
For the fasting regimens (study parts 1 through 4 except period 3 of part 2), subjects received the study drug after an overnight fast for the single‐dose regimens (parts 1 and 2) and the morning doses of the multiple‐dose fasting regimens (parts 3 and 4) and 3 hours after dinner for the evening dose of the multiple‐dose fasting IR regimens (parts 3 and 4). Subjects continued to fast for 4 hours after drug administration for all fasting regimens. For the high‐fat regimen (period 3 of part 2), the study drug was administered 30 minutes after a high‐calorie, high‐fat breakfast (750 to 1000 calories with fat constituting approximately 50% of the total caloric content).15 For the nonfasting regimens (part 5), the study drug was administered 30 minutes after a standardized breakfast.
Study Participants
Male and female subjects were eligible to participate in the study if they were between 18 and 55 years old; in general good health based on medical history, physical examination, laboratory profile, and 12‐lead electrocardiogram; and had a body mass index of 18 to 30 kg/m2. Subjects were excluded if they used any medications within 2 weeks prior to the first dose of study drug or if they used any known inhibitors (eg, amiodarone, clarithromycin, fluconazole, ciprofloxacin, itraconazole, ketoconazole, quinidine, fluoxetine, and paroxetine) or inducers (eg, carbamazepine, rifampin, phenobarbital, and phenytoin) of drug‐metabolizing enzymes within 30 days prior to the first dose of the study drug.
Pharmacokinetic Sampling
For single‐dose regimens, blood samples were collected prior to dosing and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, and 72 hours after dosing. For multiple‐dose twice‐daily regimens, blood samples were collected prior to dosing and 0.5, 1, 1.5, 2, 3, 4, 6, 9, and 12 hours after the morning and evening doses on days 1 and 7. For the multiple‐dose once‐daily regimens, blood samples were collected prior to dosing and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 hours after dosing on days 1 and 7. Additional blood samples were collected in the multiple‐dose regimens prior to dosing on days 3, 4, 5, and 6 as well as 36, 48, and 72 hours after the morning dose on day 7.
Plasma concentrations of upadacitinib were determined at AbbVie (North Chicago, Illinois) using a validated liquid chromatography method coupled with tandem mass spectrometric detection, as previously described.10 The lower limit of quantitation for upadacitinib in plasma was 0.05 ng/mL. The mean accuracy of the validated analytical method was 104.5%, and the coefficient of variation (%CV; as a measure for precision) was 7.4%. Samples quantified below the lowest standard (only 1.8% of postdose samples) were reported as zero.
Pharmacokinetic Analyses
Pharmacokinetic parameters of upadacitinib were determined using noncompartmental analyses with Phoenix software, version 7.0 (Certara, Princeton, New Jersey). Upadacitinib pharmacokinetic parameters included Cmax, time to Cmax (Tmax), terminal phase elimination half‐ life (t1/2), AUC from time 0 to the time of the last measurable concentration (AUCt) and to infinity (AUCinf) for single‐dose assessments or to 24 hours after the morning dose (AUC0–24) for multiple‐dose assessments, and the mean residence time after oral dosing (MRT). In addition, the minimum observed plasma concentration over a 24‐hour period on day 7 (Cmin), the fluctuation index on day 7 (calculated as [Cmax − Cmin]/Caverage), and day 7 accumulation ratio (Rac) for Cmax and AUC0‐24 were determined for multiple‐dose assessments.
Statistical Analyses
A linear mixed‐effects analysis was performed for the natural logarithms of Cmax, AUCt, and AUC∞ for the single‐dose relative bioavailability assessments in study parts 1 and 2 and for the natural logarithms of Cmax, AUC0–24, and Cmin on day 7 for the multiple‐dose relative bioavailability assessments in study parts 3 and 4. The model included effects for sequence, period, and regimen. Within the mixed‐effects modeling framework, the test regimen was compared with the reference regimen at a significance level of .05. The bioavailability of each test regimen relative to the corresponding reference regimen was estimated along with the 90% confidence interval obtained from the analyses of each parameter. To compare upadacitinib Cmax for the 15‐ and 30‐mg doses of the ER formulation to 6 and 12 mg, respectively, of the IR formulation (which were not studied in the same subjects as a crossover design following single dosing), the Cmax values for 6‐ and 12‐mg IR doses were calculated assuming linearity (as previously demonstrated10) from the 12‐ and 24‐mg doses, respectively.
In study part 5, dose proportionality for upadacitinib exposure was tested for the natural logarithms of dose‐normalized Cmax and AUC0–24 after the last dose on day 7. A repeated‐measures analysis was used for the pre–morning dose plasma concentration to assess the attainment of steady state using corresponding data from day 2 through day 7. The model included study day as the fixed effect and accounted for the correlation within the subject. Within the framework of the repeated‐measures analysis, trend analyses were performed on the contrasts in the study day effects to determine the earliest day after which there was no statistically significant change.
Safety and Tolerability
Safety and tolerability were evaluated during the study through adverse event monitoring, vital sign measurements, physical examinations, 12‐lead electrocardiograms (ECGs), and laboratory tests.
Results
Demographics and Subject Disposition
A total of 81 subjects were enrolled, of whom 76 completed the study. Among all 81 subjects, 71 subjects received upadacitinib and were included in the pharmacokinetic analyses, and 10 subjects received placebo. Data from all 81 subjects in the study were included in the safety analyses. The majority of subjects were male (71 of 81; 88%) and had a mean age of 36 years (range, 19 to 55 years) and mean body mass index of 26 kg/m2 (range, 18.5 to 29.8 kg/m2). Forty‐three of the 81 subjects were white (53%), 33 were black (41%), and 5 were of mixed race (6%).
Single‐Dose Pharmacokinetics and Bioavailability of Upadacitinib ER Formulation Relative to the IR Formulation and Effect of Food on the ER Formulation
Under fasting conditions, peak plasma concentrations of upadacitinib were reached within 2‐3 hours of administration of single 15‐ and 30‐mg doses of the ER tablet compared with 1 hour for 12‐ and 24‐mg doses of the IR capsule formulation (Figure 2 and Table 1).
Figure 2.

Upadacitinib mean ± SD plasma concentration‐versus‐time profiles following administration of single oral doses of upadacitinib IR and ER formulations to healthy subjects. The first 24 hours are shown on a larger scale in the inset. IR, immediate‐release; ER, extended‐release.
Table 1.
Upadacitinib Pharmacokinetic Parameters Following Administration of Single Doses of the IR and ER Formulations and Assessment of the Bioavailability of the ER Relative to the IR Formulations
| Parameter | 15 mg ER Fasting (n = 11) | 12 mg IR Fasting (n = 11) | Ratio of Central Values (ER to IR) (90%CI) | 30 mg ER Fasting (n = 12) | 24 mg IR Fasting (n = 12) | Ratio of Central Values (ER to IR) (90%CI) | 30 mg ER After High‐Fat Breakfast (n = 12) | Ratio of Central Values (30 mg ER After High‐Fat Breakfast to 30 mg ER Fasting) (90%CI) |
|---|---|---|---|---|---|---|---|---|
| Cmax (ng/mL) | 26.0 ± 9.7 | 64.6 ± 10.3 | 0.37 (0.31−0.45) | 63.7 ± 21.1 | 176 ± 65.6 | 0.37 (0.33−0.42) | 76.8 ± 29.6 | 1.20 (1.03−1.40) |
| AUCt (ng·h/mL) | 227 ± 60.0 | 231 ± 34.5 | 0.94 (0.87−1.01) | 477 ± 130 | 520 ± 130 | 0.91 (0.83−1.00) | 564 ± 145 | 1.18 (1.04−1.34) |
| AUC∞ (ng·h/mL) | 242 ± 63.6 | 234 ± 34.6 | 0.99 (0.91−1.08) | 491 ± 133 | 524 ± 133 | 0.93 (0.85−1.03) | 577 ± 157 | 1.17 (1.04−1.33) |
| Tmax (h) | 3.0 (1.0−4.0) | 1.0 (0.5−1.5) | — | 2.0 (1.5−4.0) | 0.5 (0.5−1.5) | — | 4.0 (1.5−8.0) | — |
| t1/2 (h) | 21.9 ± 23.1 | 18.3 ± 13.3 | — | 18.1 ± 14.5 | 15.3 ± 13.6 | — | 15.9 ± 11.0 | — |
IR, immediate release; ER, extended release; Cmax, maximum observed plasma concentration; AUCt, area under the plasma concentration‐time curve from time zero to time of last measurable concentration; AUC∞, area under the plasma concentration‐time curve from time zero to infinity; Tmax, time to Cmax; t1/2, terminal elimination half‐life.
Estimates are expressed as mean ± SD for all parameters except Tmax, which is presented as median (range).
Administration of single upadacitinib doses of 15 and 30 mg using the ER tablet formulation provided equivalent AUCt and AUCinf compared with single doses of 12 and 24 mg, respectively, using the IR capsule (90% confidence intervals for the ratio of central values were within the 0.8 to 1.25 equivalence bounds). Upadacitinib Cmax after single doses of 15 and 30 mg of the ER formulation were approximately 26% lower than the calculated Cmax values for 6 and 12 mg of the IR formulation, respectively, under fasting conditions. Mean upadacitinib MRT was 5 to 6 hours after administration of the IR capsule formulation and 13 to 19 hours following administration of the ER formulation.
Administration of a 30‐mg dose of the ER tablet after a high‐fat breakfast increased upadacitinib Cmax, AUCt, and AUCinf by only 20%, 18%, and 17%, respectively, and delayed Tmax by 2 hours relative to administration under fasting conditions (Table 1).
Multiple‐Dose Pharmacokinetics of Upadacitinib and Bioavailability of the ER Formulation Compared With the IR Formulation
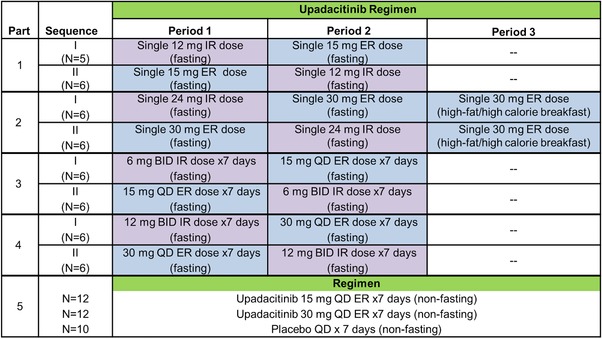
Multiple dosing with 15 and 30 mg daily of upadacitinib ER tablets provided equivalent AUC0‐24 compared with 6 and 12 mg twice daily, respectively of upadacitinib IR capsules under fasting conditions (Figure 3 and Table 2). The central values for upadacitinib Cmax and Cmin following administration of 15 and 30 mg once daily were within 13% of the respective central values following administration of 6 and 12 mg twice daily, respectively. The fluctuation index for once‐daily administration of the ER formulation was similar to that of twice‐daily administration of the IR formulation (Table 2).
Figure 3.
Upadacitinib mean ± SD steady‐state plasma concentration‐versus‐time profiles following administration of multiple doses of (A) upadacitinib 6 mg twice daily (IR formulation) and 15 mg once daily (ER formulation) under fasting conditions, (B) 12 mg twice daily (IR formulation) and 30 mg once daily (ER formulation) under fasting conditions, and (C) 15 mg once daily and 30 mg once daily (ER formulation) under nonfasting conditions. BID, twice daily; IR, immediate‐release; QD, once daily; ER, extended release.
Table 2.
Upadacitinib Steady‐State Pharmacokinetic Parameters Following Administration of Multiple Twice‐Daily Doses Using the IR Formulation and Multiple Once‐Daily Doses Using the ER Formulation
| Parameter | 15 mg Once‐Daily ER Fasting (n = 12) | 6 mg Twice‐Daily IR Fasting (n = 11) | Ratio of Central Values (90%CI) | 30 mg Once‐Daily ER Fasting (n = 11) | 12 mg Twice‐Daily IR Fasting (n = 11) | Ratio of Central Values (90%CI) | 15 mg Once‐Daily ER Nonfasting (n = 12) | 30 mg Once‐Daiy ER Nonfasting (n = 12) |
|---|---|---|---|---|---|---|---|---|
| Cmax (ng/mL) | 31.9 ± 11.2 | 33.9 ± 8.8 | 0.91 (0.74−1.12) | 68.2 ± 20.5 | 73.9 ± 14.2 | 0.90 (0.73−1.11) | 36.0 ± 8.8 | 79.5 ± 31.8 |
| AUC0–24 (ng·h/mL) | 279 ± 71.4 | 288 ± 63.5 | 0.94 (0.84−1.05) | 525 ±123 | 534 ± 97.8 | 0.97 (0.87−1.09) | 317 ± 68.1 | 582 ± 172 |
| Cmin | 3.1 ± 1.1 | 2.7 ± 0.6 | 1.09 (0.85−1.40) | 3.8 ± 1.6 | 3.8 ± 2.2 | 0.87 (0.75−1.02) | 2.8 ± 1.2 | 4.6 ± 1.8 |
| Fluctuation index | 2.5 ± 0.5 | 2.6 ± 0.3 | 2.9 ± 0.5 | 3.2 ± 0.4 | 2.5 ± 0.4 | 3.1 ± 0.5 | ||
| Tmax (h) | 2.5 (1.5−4.0) | 1.0 (0.5−14) | 3.0 (2.0−4.0) | 1.0 (0.5−1.5) | 4.0 (2.0−6.0) | 4.0 (1.5−6.0) | ||
| t1/2 (h) | 16.9 ± 12.0 | 24.5 ± 18.1 | 22.9 ± 21.1 | 11.5 ± 9.2 | 15.1 ± 10.1 | 13.6 ± 10.2 | ||
| Rac Cmax | 1.01 (0.65−3.01) | 0.97 (0.68−1.17) | 1.03 (0.40−1.82) | 0.98 (0.65−1.18) | 1.0 (0.84−1.3) | 1.02 (0.82−1.40) | ||
| Rac AUC0–24 | 1.11 (0.87−1.99) | 1.02 (0.88−1.09) | 1.11 (0.79−1.67) | 1.08 (0.97−1.18) | 1.0 (0.91−1.4) | 1.16 (0.92−1.31) |
IR, immediate release; ER, extended release; Cmax, maximum observed plasma concentration; AUC0–24, area under the plasma concentration‐time curve from time zero to time of last measurable concentration over a 24‐hour period; Cmin, minimum observed plasma concentration; fluctuation index was calculated as (Cmax ‐ Cmin)/Caverage, where Caverage is calculated as AUC0‐24/24, Tmax, time to Cmax; t1/2, terminal elimination half‐life; Rac, accumulation ratio.
Estimates are expressed as mean ± SD for all parameters except Tmax and Rac, which are presented as median (range).
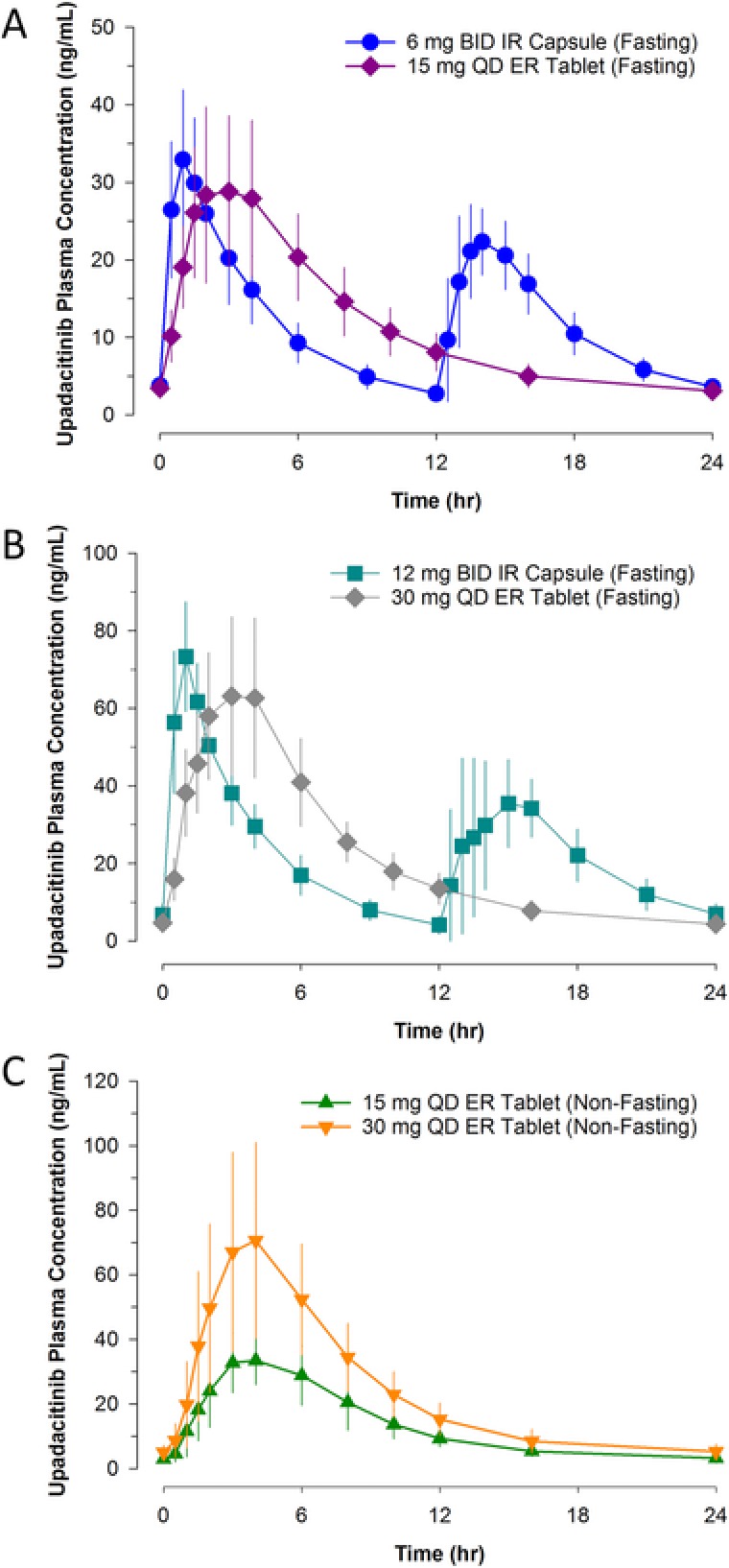
Upadacitinib steady‐state concentrations were achieved by day 4 of daily dosing, as the estimate of the mean dose‐normalized predose concentration did not significantly differ between day 4 and day 7 (Figure 4). No statistically significant differences were observed between 15 and 30 mg once daily in the terminal‐phase elimination rate constant or in the natural logarithms of dose‐normalized steady‐state Cmax or AUC0–24, indicating that upadacitinib exposures were dose‐proportional between 15‐ and 30‐mg once‐daily regimens. At steady state, the median Rac for Cmax and AUC0–24 was approximately 1, indicating lack of accumulation of upadacitinib in plasma with multiple once‐daily dosing using the ER formulation (Table 2).
Figure 4.
Upadacitinib mean ± SD predose plasma concentration by study day following administration of (A) 15 mg once daily and (B) 30 mg once daily using the ER formulation. SD, standard deviation; QD, once daily; ER, extended release.
Safety and Tolerability
Of the 81 subjects enrolled, 77 completed the study, and 4 discontinued because of adverse events of decreased neutrophil count (1 subject in the placebo group and 1 subject in the upadacitinib 30‐mg daily group), viral respiratory tract infection (1 subject in the upadacitinib 15‐mg daily group), and pain in extremity and rash (1 subject in the upadacitinib 6‐mg twice‐daily group). The events were assessed by the investigator as not related to upadacitinib. There was no pattern to the type or frequency of the adverse events that occurred. No serious adverse events or clinically significant changes in vital signs, ECG measurements, or laboratory measurements were observed during the course of the study.
Discussion
This study characterized the pharmacokinetics of upadacitinib following administration of the ER formulation and compared the bioavailability of upadacitinib from the ER formulation to that of the IR formulation, which was used in upadacitinib RA phase 2 trials. Single‐ and multiple‐dose evaluations were conducted for comprehensive assessment of the relative pharmacokinetic profiles. The study also characterized the effect of a high‐fat meal on upadacitinib exposure from the ER formulation. Results from this study, along with exposure‐response analyses of upadacitinib RA IR phase 2 trials, enabled advancing the ER formulation of upadacitinib directly in 6 large phase 3 trials in subjects with RA, without needing to evaluate the ER formulation in a phase 2 setting.
Evaluation of the efficacy and safety of upadacitinib in subjects with RA in 2 phase 2b dose‐ranging studies demonstrated that upadacitinib doses of 6 and 12 mg twice daily using the IR formulation maximized efficacy, measured as the percentage of subjects meeting American College of Rheumatology (ACR) 20%, 50%, and 70% improvement criteria (ACR20, ACR50, and ACR70, resepectively) at week 12,13, 14 and exposure‐response analyses confirmed these target exposures. Therefore, the objective was to develop an ER formulation that enables once‐daily dosing while having daily AUC and Cmin comparable to doses of 6 and 12 mg twice daily. The formulations did not necessarily have to be bioequivalent, given that these changes in the formulation were taking place prior to conducting the registrational phase 3 trials.
Early assessment of different prototypes for upadacitinib ER tablets indicated that the selected ER formulation for evaluation in this study had an oral bioavailability of 70% to 80% compared with the same dose administered using the IR formulation under fasting conditions (data not shown). Therefore, upadacitinib ER tablets were formulated to contain a slightly higher amount of upadacitinib compared with the total daily target dose of the IR formulation (eg, 15 mg ER compared with total daily dose of 12 mg IR) to account for the slightly lower relative bioavailability.
In this study, 15‐ and 30‐mg strengths of the ER formulation provided upadacitinib AUCinf equivalent to single doses of 12 and 24 mg, respectively, of the IR capsule formulation under fasting conditions. In addition, multiple dosing with 15 and 30 mg once daily using the ER tablets provided equivalent AUC0‐24 and comparable Cmax and Cmin compared with 6 and 12 mg twice daily of upadacitinib IR capsules under fasting conditions. The mean fluctuation index in upadacitinib plasma concentrations at steady state over a 24‐hour period was between 2.5 and 3.2 for the different regimens evaluated (Table 2). Therefore, the upadacitinib ER formulation provided a similar fluctuation index over a 24‐hour period with once‐daily dosing as opposed to needing twice‐daily dosing of the IR formulation (note that for dosing both ER and IR formulations at the same interval, the ER has a significantly lower fluctuation index than the IR, but with doubling the interdose interval for the ER relative to the IR, the fluctuation index becomes the same).
Administration of upadacitinib after a high‐fat meal had a limited effect (∼20% increase) on upadacitinib AUC and Cmax compared with administration under the fasting conditions. Based on population pharmacokinetic analysis, intersubject variability in apparent upadacitinib oral clearance is estimated to be 16% and 26% in healthy subjects and subjects with RA, respectively, and the residual error in upadacitinib concentrations is estimated to be 31%12; therefore, the small effect of a high‐fat meal on upadacitinib exposure is within the between‐subject variability in upadacitinib exposures and is not considered clinically relevant.
The effect of food on the pharmacokinetics of upadacitinib was assessed following administration of a high‐fat meal, as the worst‐case scenario for food effect, as recommended by the Food and Drug Administration.15 At steady state, upadacitinib AUC0–24, Cmax, and Cmin were also comparable when 30 mg once daily was administered under fasting conditions in part 4 and nonfasting conditions (after a standard breakfast) in part 5 (Table 2). Taken together, food intake has no clinically relevant effect on upadacitinib exposure when using the ER formulation; therefore, the upadacitinib ER formulation is being administered in ongoing clinical trials without regard to food. It is worth noting that food decreases the IR formulation Cmax by approximately 20%.11 Therefore, although the upadacitinib ER formulation Cmax is slightly lower than the IR formulation Cmax under fasting conditions, the upadacitinib Cmax is expected to be comparable in the 2 formulations when administered under nonfasting conditions because of the opposite directionality of the slight food effect on Cmax for each formulation.
In study parts 3 and 4, the upadacitinib evening IR dose was administered 3 hours after dinner and 4 hours prior to any additional food intake because it was not practical to have a longer fasting duration for that evening dose. This shorter fasting duration appears to have resulted in some lowering of upadacitinib evening Cmax and a slight increase in the following Ctrough for the IR formulation (manifested as some diurnal variability for the IR formulation in Figure 3). Given that food has no impact on upadacitinib AUC from the IR formulation,11 this shortened fasting condition does not impact the comparison between IR and ER formulation AUCs.
Following multiple once‐daily administration of the ER formulation, steady state was achieved by day 4, as demonstrated by the lack of a statistically significant difference in upadacitinib predose concentrations on days 4 and 7. Of note, mean upadacitinib predose trough concentration on day 7 was only 15% and 23% higher than upadacitinib predose concentration on day 2 for 15 and 30 mg once daily, respectively (Figure 4). In addition, there was no accumulation for upadacitinib in plasma with multiple once‐daily dosing of the ER formulation. Therefore, although steady state was statistically achieved by day 4, there was no clinically relevant difference in upadacitinib plasma concentrations after the first dose compared with steady state.
This assessment of the pharmacokinetics of upadacitinib ER formulation was conducted in healthy subjects. Based on population pharmacokinetic analyses of studies in healthy subjects as well as subjects with RA, apparent upadacitinib oral clearance is estimated to be 24% lower in RA patients compared with healthy subjects. Although this small difference in clearance is not expected to differ by formulation, confirmation of upadacitinib pharmacokinetics for the ER formulation in RA patients using population pharmacokinetic analyses of phase 3 trials is warranted.
Conclusions
The study demonstrated comparability of plasma exposure between the respective regimens of upadacitinib ER formulation and the IR formulation used in RA phase 2 studies, lack of a clinically relevant food effect on upadacitinib ER formulation, and favorable tolerability of the ER formulation in healthy subjects. These results show that the ER formulation of upadacitinib evaluated in this study achieves the target profile that enables once‐daily dosing in patients. Based on the results from this study, the ER formulation of upadacitinib was advanced to be used in all ongoing phase 3 studies in RA.
Declaration of Conflicting Interests
Mohamed‐Eslam F. Mohamed, Jiewei Zeng, Patrick J. Marroum, In‐Ho Song, and Ahmed A. Othman are employees of AbbVie Inc. and may hold AbbVie stock and/or stock options. Medical writing support was provided by Therese Stickler, a freelance writer under contract with AbbVie.
Funding
This study was supported by AbbVie Inc. AbbVie contributed to the study design, research, and interpretation of data and the writing, reviewing, and approving of the publication.
Ahmed A Othman, PhD, is a fellow of the American College of Clinical Pharmacology.
References
- 1. AbbVie . A study comparing ABT‐494 to placebo in subjects with rheumatoid arthritis on a stable dose of conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) who have an inadequate response to csDMARDs alone (SELECT‐NEXT) [ClinicalTrials.gov identifier NCT02675426]. Accessed July 13, 2017.
- 2. AbbVie . A multicenter, randomized, double‐blind, placebo‐controlled study of ABT‐494 for the induction of symptomatic and endoscopic remission in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to anti‐TNF therapy (Celest Study) [ClinicalTrials.gov identifier NCT02365649]. Accessed July 13, 2017.
- 3. AbbVie . A study to evaluate the safety and efficacy of ABT‐494 for induction and maintenance therapy in subjects with moderately to severely active ulcerative colitis [ClinicalTrials.gov identifier NCT02819635]. Accessed July 13, 2017.
- 4. AbbVie . A study to evaluate ABT‐494 in adult subjects with moderate to severe atopic dermatitis [ClinicalTrials.gov identifier NCT02925117]. Accessed July 13, 2017.
- 5. AbbVie . A study comparing ABT‐494 to placebo and to adalimumab in participants with psoriatic arthritis who have an inadequate response to at least one non‐biologic disease modifying anti‐rheumatic drug (SELECT ‐ PsA 1) [ClinicalTrials.gov identifier NCT03104400]. Accessed July 13, 2017.
- 6. AbbVie . A study evaluating the safety and efficacy of upadacitinib in subjects with active ankylosing spondylitis (SELECT Axis 1) [ClinicalTrials.gov Identifier NCT03178487]. Accessed July 13, 2017.
- 7. Leonard WJ. Role of Jak kinases and STATs in cytokine signal transduction. Int J Hematol. 2001;73(3):271–277. [DOI] [PubMed] [Google Scholar]
- 8. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune‐mediated disease. Immunity. 2012;36(4):542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs. 2014;23(8):1067–1077. [DOI] [PubMed] [Google Scholar]
- 10. Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet. 2016;55(12):1547–1558. [DOI] [PubMed] [Google Scholar]
- 11. Mohamed MF, Jungerwirth S, Asatryan A, Jiang P, Othman AA. Assessment of effect of CYP3A inhibition, CYP induction, OATP1B inhibition, and high‐fat meal on pharmacokinetics of the JAK1 inhibitor upadacitinib. Br J Clin Pharmacol. 2017;83(10):2242–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klünder B, Mohamed MF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analyses of phase I and II clinical trials. Clin Pharmacokinet. 2018;57:977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kremer JM, Emery P, Camp HS, et al. A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheumatol. 2016;68(12):2867–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Genovese MC, Smolen JS, Weinblatt ME, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2016;68(12):2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guidance for Industry . Food‐Effect Bioavailability and Fed Bioequivalence Studies. United States Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research; December 2002.