

Abstract
Autophagy is a crucial recycling process that is increasingly being recognized as an important factor in cancer initiation, cancer (stem) cell maintenance as well as the development of resistance to cancer therapy in both solid and hematological malignancies. Furthermore, it is being recognized that autophagy also plays a crucial and sometimes opposing role in the complex cancer microenvironment. For instance, autophagy in stromal cells such as fibroblasts contributes to tumorigenesis by generating and supplying nutrients to cancerous cells. Reversely, autophagy in immune cells appears to contribute to tumor‐localized immune responses and among others regulates antigen presentation to and by immune cells. Autophagy also directly regulates T and natural killer cell activity and is required for mounting T‐cell memory responses. Thus, within the tumor microenvironment autophagy has a multifaceted role that, depending on the context, may help drive tumorigenesis or may help to support anticancer immune responses. This multifaceted role should be taken into account when designing autophagy‐based cancer therapeutics. In this review, we provide an overview of the diverse facets of autophagy in cancer cells and nonmalignant cells in the cancer microenvironment. Second, we will attempt to integrate and provide a unified view of how these various aspects can be therapeutically exploited for cancer therapy.
Keywords: autophagy, cancer, immune cells, microenvironment, stroma, therapy
1. INTRODUCTION
Autophagy is an important homeostatic process in the human body that is responsible for the elimination of damaged and/or superfluous macromolecules such as proteins and lipids as well as the removal of damaged organelles like mitochondria. The successful execution of autophagy enables the recycling of nutrients, amino acids, and lipids and acts as a quality control mechanism to maintain organelle function.1, 2, 3, 4 The importance of autophagy is evidenced by the fact that a block in autophagic flux due to knockdown of core autophagy genes is detrimental during the early development in murine models.5, 6, 7, 8, 9, 10, 11 Perhaps not surprisingly, an increasing body of evidence highlights the important and multifaceted impact of autophagy in cancer. For instance, during tumor development, the autophagic process appears to function as a tumor suppressor and limits tumorigenesis.12, 13, 14, 15 In this respect, it is noteworthy that a single‐nucleotide polymorphism in the promoter region of the crucial autophagy‐related gene (ATG) ATG16L1, which putatively downregulates its expression level, associated with susceptibility to thyroid and colorectal cancer and has a significant negative impact on patient survival in local and advanced metastatic prostate cancer.16, 17, 18 Further, survival of patients with advanced lung adenocarcinoma upon epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor treatment is significantly impacted by functional genetic polymorphisms in core autophagy genes, thus highlighting the potential clinical impact of autophagic signaling on cancer development and response to therapy.19
In established cancers, autophagy activity is upregulated during treatment and associated with resistance to cancer therapy.20 Further, elevated autophagy maintains stemness in cancer stem cells (CSCs). Moreover, cancer cells appear to rely more on autophagy for continued survival than normal cellular counterparts. Consequently, the inhibition of autophagy is being explored for cancer therapy particularly in combination with other cytotoxic drugs to augment cytotoxicity.21, 22, 23 Autophagy occurring in the context of cancer therapy may, on the one hand, be a stress response that enables cancer cells to survive and evade apoptotic elimination.4 In this setting, inhibition of autophagy sensitizes cells to apoptotic cell death and may be of use to augment the efficacy of anticancer agents. On the contrary, autophagy may also be a driver of cytotoxic cell death and in this case inhibition of autophagy would inhibit cell death. This type of cell death has been termed autophagic cell death (ACD) and has been reported, eg, for radiation therapy.24, 25, 26, 27, 28 Thus, depending on the type of cell death inhibition of autophagy may be warranted for combination therapy.
It is evident that autophagy is more and more emerging as a potential target for cancer therapy. However, the complex microenvironment of an established tumor comprises many different cell types in addition to malignant cells that all to a different extent utilize and rely on the autophagic process. Indeed, as will be discussed in this review, autophagy not only clearly impacts on cancer (stem) cells, but also on stromal cells, endothelial cells, and (tumor‐infiltrated) innate and adaptive immune cells. Therefore, it is crucial to understand the impact of autophagy and its therapeutic targeting in the context of this diverse cellular composition of the tumor microenvironment.
In this review, we will first briefly detail the core autophagy machinery and regulatory pathways after which we will provide an overview of current thinking on the role of autophagy in cancer cells and the functioning of the diverse components within the tumor microenvironment (illustrated in Figure 1). Further, we will provide directions for incorporating the sometimes opposing effects of autophagy on tumor microenvironmental components for the future implementation of autophagy‐targeting drugs in cancer.
Figure 1.
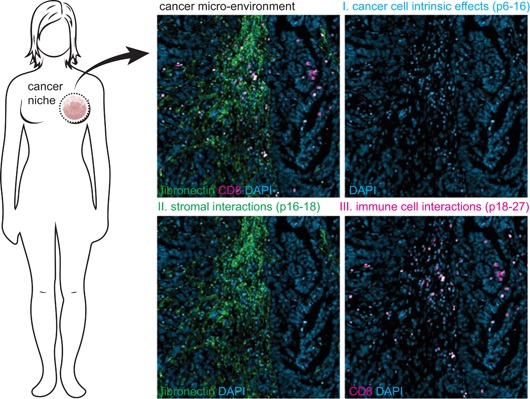
Review outline. This review highlights the impact of changes in autophagy within cancer cells, as well as in the context of the complex cancer microenvironment. Part I describes how aberrant autophagy can contribute to cancer initiation and maintenance as well as therapy resistance (pp. 6‐16). Part II describes the role of autophagy in different stromal cells within the tumor microenvironment, such as fibroblasts and mesenchymal stem cells (pp. 16‐18). Further, the impact of autophagy on anticancer immune responses is described (pp. 18‐27). Blue, 4′,6‐diamidino‐2‐phenylindole (DAPI) staining; green, fibronectin staining for stroma; red, CD8 staining for cytotoxic T‐cell [Color figure can be viewed at wileyonlinelibrary.com]
2. AUTOPHAGY SIGNALING AND REGULATORY PATHWAYS
The term autophagy defines a process that can occur in three different forms, with the most prominent form being macroautophagy, a form of autophagy that includes removal of proteins and/ or organelles. In the case of mitochondria, this process is called mitophagy. Second, when molecules that have to be degraded are directly invaginated by the lysosome, this process is called microautophagy. Third, proteins can be degraded via chaperone‐mediated autophagy (CMA). During CMA, proteins are targeted for degradation by heat shock protein hsc70 via their KFERQ‐like motif.29, 30 Unless specifically referred to, the term autophagy in this review describes macroautophagy. In the section below, we will detail basic autophagy pathways as well as highlight regulatory hubs that are important in cancer.
2.1. The core autophagy machinery
The execution of autophagy can be subdivided into the initiation phase, elongation phase, autophagosome maturation, autophagosome‐lysosome fusion, and degradation of content in autophagolysosomes (Figure 2A). The initiation of autophagy generally starts at the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1), the master regulator of autophagy, which under basal conditions represses the autophagy pathway by inhibiting the Unc‐51 like autophagy activating kinase 1 (ULK1) complex.31 However, upon increased nutrient demand or nutrient limiting conditions, mTORC1 is deactivated due to reduced upstream signaling from the phosphoinositide 3‐kinase (PI3K)/Akt and the mitogen‐activated protein kinase (MAPK) pathway, thereby enabling initiation of autophagy. In addition, the 5′‐adenosine monophosphate‐activated protein kinase (AMPK), a key kinase regulating cellular energy homeostasis, activates the ULK1 complex and inactivates mTORC1 when low energy levels are detected.19, 32 The activated ULK1 complex, together with the Beclin‐1‐VPS34 complex (a complex discussed in more detail in Section 2.2) initiates the formation of autophagosomes. The formation of autophagosomes can be inhibited by 3‐methyladenine (3‐MA), an inhibitor of VPS34. In contrast, rapamycin, an inhibitor of mTORC1, is generally used as an autophagy inducer. Of note, although mTOR and its complexes have many more functions besides regulating the autophagy pathway, eg, regulation of cell growth, proliferation, protein translation, and metabolism, the inhibition of mTOR is in this review generally used as an activator of autophagy.
Figure 2.
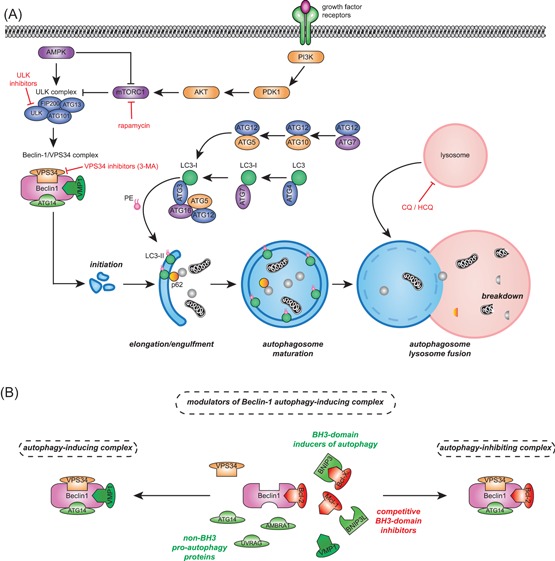
The autophagy pathway. A, The activation of autophagy is initiated by the reduced activity of the mechanistic target of rapamycin complex 1 (mTORC1) complex due to activated adenosine monophosphate‐activated protein kinase (AMPK) or decreased upstream growth signaling. mTORC1 is an inhibitor of the ULK complex, therefore reduced mTORC1 activity increases the activity of the ULK complex. The ULK complex together with the Beclin‐1/ VPS34 complex initiates the formation of autophagosomes. Dependent on the complex composition, Beclin‐1 can act as a molecular switch between autophagy and apoptosis (see B). The expansion and maturation of the autophagosomes is dependent on two ubiquitin‐like conjugation systems, which requires multiple autophagy proteins. First, ATG12‐ATG5 conjugate binds to ATG16, which stimulates LC3 lipidation. Second, LC3 is covalently conjugated to phosphatidylethanolamine (PE) generating LC3‐II, which is incorporated in the autophagosomal membrane. Incorporated LC3‐II is required for binding and internalization of adaptor proteins such as p62. Finally, the mature autophagosome fuses with lysosomes, after which its content is broken down by digestive enzymes. Indicated in red are pharmacological agents, chloroquine (CQ), hydroxychloroquine (HCQ), 3‐methyladenine (3‐MA), and ULK inhibitors, that inhibit autophagy. In addition, rapamycin activates autophagy by inhibiting mTORC1. B, Beclin‐1 is a core member of the VPS34/Beclin‐1 complex, which acts as a molecular switch in controlling autophagy downstream of the ULK1 complex. Depicted in red are the antiapoptotic members of the Bcl‐2 family BCL‐2, BCL‐XL, and MCL‐1 which can bind to Beclin‐1, through interaction with its BH3 domain, thereby inhibiting autophagy. Alternatively, Bcl‐2 interacting protein 3 (BNIP3) and Bcl‐2 interacting protein 3 like (BNIP3L; depicted in green) can competitively bind to antiapoptotic BLC‐2 members. Dissociation of antiapoptotic Bcl‐2 members from Beclin‐1, consequently activates autophagy. Other non‐BH3 proteins, also depicted in green, such as vacuole membrane protein 1 (VMP1), ATG14, UV radiation resistance‐associated gene (UVRAG), and activating molecule in Beclin‐1‐regulated autophagy protein 1 (AMBRA1) can also bind Beclin‐1, thereby activating autophagy. PDK1, pyruvate dehydrogenase kinase 1; PI3K, phosphoinositide 3‐kinase [Color figure can be viewed at wileyonlinelibrary.com]
Maturation of the autophagosome requires two ubiquitin‐like conjugation systems. First, ATG12 is covalently bound to ATG5, a process mediated by ATG7 and ATG10. The ATG12‐ATG5 conjugate is subsequently noncovalently connected to ATG16, which is required for the localization of ATG12 and ATG5 to the forming autophagosome.33 Second, LC3 is converted into LC3‐II, which starts with the proteolytic cleavage of LC3 by ATG4 to form LC3‐I. LC3‐I is then bound by ATG7, which transfers LC3‐I to ATG3.34, 35, 36 ATG3 subsequently catalyzes the conjugation of the lipid phosphatidylethanolamine (PE) to LC3‐I, thereby yielding LC3‐II. This lipidation step is enhanced by the ATG5/ATG12/ATG16 complex. Eventually, LC3‐II is inserted in the membrane of the elongating autophagosome. During the maturation of the autophagosome, proteins and organelles to be degraded are sequestered to the forming autophagosome by p62/sequestosome 1 (SQSTM1). For this purpose, p62 can directly interact with LC3.37 Finally, the mature autophagosome fuses with a lysosome to form the autolysosome. The lysosome‐associated membrane proteins (LAMP‐1 and LAMP‐2) are essential for this fusion and also maintain the integrity of lysosomal membranes.38 The macromolecules and organelles that have been entrapped in the autophagosomes are then degraded by the digestive enzymes of the lysosomes (eg, lipases, proteases, nucleases, sulfatases), which yields amino acids, fatty acids, and nucleotides for eventual reuse. The fusion of autophagosomes with lysosomes can be inhibited by chloroquine (CQ) or hydroxychloroquine (HCQ), both compounds that prevent acidification of the lysosomes.
Of note, the generation of LC3‐II is considered as a hallmark marker of autophagy induction, whereas its sustained accumulation is reflective of autophagy inhibition.39 In addition, p62 is degraded during the proper execution of autophagy, and its accumulation can be used as a marker for inhibition of autophagy.40
2.2. Bcl‐2 family members modulate Beclin‐1‐dependent autophagy
Beclin‐1 is an important regulatory hub to which proautophagic and antiautophagic proteins can bind (Figure 2B). First, the antiapoptotic proteins of the BCL‐2 family, eg, BCL‐2, BCL‐XL, and MCL‐1, can bind to the characteristic BH3 domain of Beclin‐1, which inhibits autophagy.41, 42, 43 Second, non‐Bcl‐2 family proteins like UV radiation resistance‐associated gene (UVRAG), activating molecule in Beclin‐1‐regulated autophagy protein 1 (AMBRA1), high mobility group box 1 (HMGB1), and vacuole membrane protein 1 (VMP1) can competitively bind to Beclin‐1 at the same domain, which can shift the balance to induction of autophagy.44, 45, 46, 47 In addition, the hypoxia‐inducible Bcl‐2 interacting protein 3 (BNIP3) and Bcl‐2 interacting protein 3 like (BNIP3L) proteins that also contain a BH3 domain can directly interact with BCL‐2 family members.48 This BNIP3‐Bcl‐2 interaction prevents Bcl‐2 binding to Beclin‐1 and, thereby, promotes autophagy. Alterations in the pool of Beclin‐1 interacting proteins can alter the balance of autophagy regulation. In line with this, gene silencing of Bcl‐2 using small interfering RNA (siRNA) in MCF‐7 cells triggered autophagy, whereas in neuron‐specific MCL‐1‐knockout mice autophagy was increased in neuronal cells.49, 50 Correspondingly, treatment of various cancer cell lines with BH3 mimetics that promote dissociation of Bcl‐2 or BCL‐XL from Beclin‐1‐activated autophagy.51, 52 Here, autophagy was inhibited by siRNA‐mediated knockdown of essential autophagy proteins.53 In a recent screen, three compounds were identified that specifically disrupt the binding between BCL‐2 and Beclin‐1.54 These compounds de‐repressed autophagy without causing any cytotoxicity.54 The induction of mitophagy can also be regulated by Bcl‐2 members. In brief, mitochondrial depolarization promoted Parkin and PTEN‐induced putative kinase 1 (PINK1)‐dependent induction of mitophagy, which was suppressed by transient overexpression of Bcl‐2 family members MCL‐1 and BLC‐XL.55, 56 In this case, inhibition of mitophagy was independent of Beclin‐1, but due to inhibition of Parkin translocation to depolarized mitochondria.55 Taken together, the elevated expression of members of the BCL‐2 family can reduce autophagy, including mitophagy.
3. PART I: THE ROLE OF AUTOPHAGY IN CANCER CELLS
Autophagy has a multifactorial impact on cancer and influences both cancer initiation and maintenance, as well as regulates cancer response to therapy. Alterations in autophagy levels due to mutations in key autophagy genes or aberrant activation of autophagy regulators have been associated with tumorigenesis (illustrated in Figure 3A). In this respect, cancer initiation is associated with reduced autophagy levels, which leads to the accumulation of oncogenes and reactive oxygen species (ROS). In contrast, during cancer maintenance, the activity of the autophagy pathway is often upregulated. This upregulation ensures sufficient energy supply and contributes to survival during stress, eg, hypoxia and metastasis (illustrated in Figure 3B). During anticancer therapy, autophagy is increased by which cancer cells survive and gain therapy resistance. In addition, CSCs appear to rely on autophagy to maintain stemness.
Figure 3.
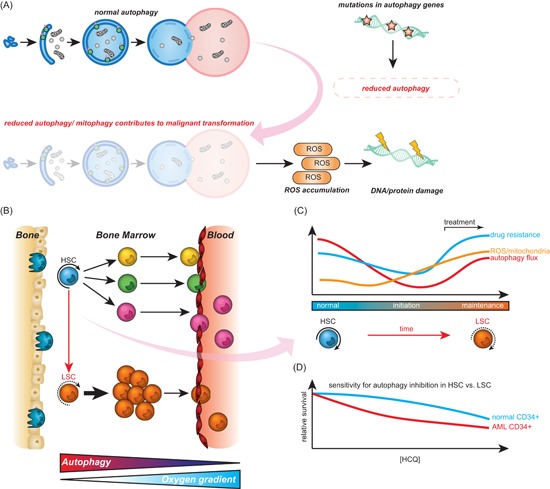
Autophagy during malignant transformation and cancer maintenance. A, Different pro‐oncogenic events such as mutation or monoallelic deletion of autophagy‐related genes can cause reduced autophagy activity. Reduced levels of autophagy/mitophagy can contribute to malignant transformation due to elevated levels of reactive oxygen species (ROS). B, Hematopoietic stem cells (HSCs) reside in specific bone marrow niches with low oxygen content and are characterized by high autophagy activity. During differentiation, the autophagy flux declines and mature cells leave the bone marrow (BM) environment and enter the blood‐stream. In leukemia, HSCs have acquired mutations which results in a block in differentiation and consequently accumulation of immature blasts in BM and peripheral blood of patients. C, Hypothetical model for changes in autophagy and ROS in HSCs during transformation. Normal HSCs have high autophagy flux, low mitochondrial activity, and ROS levels. During cancer initiation, autophagy is repressed (although not completely inhibited), causing accumulation of mitochondria and ROS, which in turn contributes to malignant transformation. During cancer maintenance, cancer cells re‐establish functional autophagy promoting tumor growth and survival. In addition, in response to drug treatment, autophagy is activated and acts as a survival mechanism for cancer cells. D, Both normal BM‐derived CD34+ and acute myeloid leukemia (AML) CD34+ cells need a certain level of autophagy to survive. Therefore, there is only a small therapeutic window of autophagy inhibition with autophagy inhibitors like hydroxychloroquine. LSC, leukemic stem cells [Color figure can be viewed at wileyonlinelibrary.com]
3.1. Impact of autophagy in early tumorigenesis
Autophagy is likely important for cancer initiation as mice with monoallelic deletion of the key autophagy regulator Beclin‐1 have an increased susceptibility to spontaneous tumor development.13 In line with this, monoallelic deletions of Beclin‐1 have been detected in human breast cancer, prostate, and ovarian cancer, whereas reduced expression of Beclin‐1 was detected in brain cancer.57, 58, 59, 60, 61 Similarly, monoallelic deletion of other essential autophagy genes such as ATG5, ATG7, or total loss of ATG4C have been associated with an increased risk of developing malignancies.14, 15 Based on this data autophagy appears to act as a tumor suppressor with reduced levels of autophagy associating with an accumulation of dysfunctional organelles and proteins that may contribute to malignant transformation. Of note, a low constitutive level of autophagy is required for cell survival, as evidenced by the fact that the knockout of ATG genes, Beclin‐1, or AMBRA1 is embryonically lethal in mice.13, 62 As described in more detail below, there are several mechanisms in cancer that can reduce autophagic flux, eg mutations in core autophagy genes that may trigger cancer development. These processes and their potential impact on cancer initiation are reviewed in more detail below.
3.1.1. Mutations in autophagy genes that affect autophagy levels during tumor development
Alterations in expression of various key autophagy genes have been reported for different types of cancer, including breast, lung, pancreatic, bladder cancer, and leukemia.63 As mentioned above, one of the common molecular aberrations is the loss of one of the alleles of the essential autophagy gene Beclin‐1. This aberration was detected in subsets of cancers, even in breast carcinoma cell lines that are often polyploid for the Beclin‐1 encoding chromosome 17.64, 65, 66 Interestingly, reduced autophagy due to allelic loss of Beclin‐1 in immortalized mouse kidney cells or mouse mammary epithelial cells led to a profound increase in DNA damage.67, 68 The increased DNA damage was associated with chromosomal abnormalities that are linked to cancer initiation, such as gene amplification and aneuploidy.67, 68 For example, in immortalized mouse kidney cells the chromosome number (normally 40) was increased to an average of 56 after allelic loss of Beclin‐1. 68 Moreover, mammary tissue in Beclin‐1+/− mice developed benign neoplasia with hyperproliferation, whereas reintroduction of Beclin‐1 expression in breast cancer MCF‐7 cells suppressed tumorigenesis.66, 69 However, mouse models with loss of Beclin‐1 or other essential autophagy proteins do not develop many different types of cancers.70 Also, Beclin‐1 is not specifically mutated or deleted in cancer, but rather lost due to deletions in chromosome 17Q21.70 So, it is not completely clear if the loss of Beclin‐1 directly contributes to cancer initiation. Similar to Beclin‐1, allelic loss of the autophagy component UVRAG or reduced expression of Bif‐1, both direct interactors with Beclin‐1, is also associated with cancer development, in this case, gastric and colon cancer.71, 72, 73 In brief, UVRAG forms a complex with Beclin‐1 to activate autophagy and loss of this protein resulted in impaired autophagy. Moreover, UVRAG prevented accumulation of abnormal chromosomes, although it is not clear whether this feature is autophagy dependent.74 Bif‐1 interacts with Beclin‐1 and UVRAG and also serves to activate autophagy.44 Consequently, loss of Bif‐1 expression reduces autophagy and in knockout mice resulted in an increased number of spontaneous tumors.44 Together with the above‐described data on Beclin‐1, these findings suggest that autophagy regulation by Beclin‐1 is an important hub that is deregulated in cancer. Further, disruption of Beclin‐1/UVRAG/BIF‐1 may cause genomic instability.75 In addition, GABARAPL1, an autophagy gene involved in of the initiation of autophagosome formation, was found to be downregulated in breast cancer, in this case, due to altered DNA methylation and histone deacetylation patterns.76 The functional outcome of downregulation of GABARAPL1 was a reduction in autophagic flux and increased tumorigenesis.77
In a recent screening approach, a more detailed picture of the mutational spectrum of 180 autophagy genes was obtained, using whole‐exome sequencing of 223 cases of myeloid neoplasm. Copy number alterations or missense mutations were detected in roughly 22% of autophagy‐associated genes and 14% of the studied cases.78 Interestingly, the majority of mutations were nonsynonymous substitutions that associated with adverse prognosis. Clonal hierarchy analysis indicated that these autophagy mutations were predominantly secondary events.78 In addition to mutations in core autophagy genes, mutations in the spliceosome that are linked to aberrant autophagy gene expression in myeloid malignancy were also found. For example, the splicing factor U2AF35, which is mutated in ~10% of patients with myelodysplastic syndrome, caused abnormal processing of ATG7 pre‐messenger RNA (pre‐mRNA) and consequently reduced the expression of ATG7.79 Interestingly, complete knockout of ATG7 in hematopoietic stem cells (HSCs) in mice causes severe anemia and in the long‐term triggered atypical myeloproliferation and accumulation of myeloid blasts in organs, all characteristics associated with myeloid malignancies.80, 81, 82 How autophagic flux is affected by these mutations remains to be functionally defined, but the likely outcome is a reduction in the level of autophagy. Indeed, the nonsynonymous substitutions observed in leukemia are often hypomorphic, that is mutations that cause reduced expression, suggesting that autophagy is repressed but not completely inhibited.78 In line with this, complete inhibition of autophagy due to, eg, bi‐allelic deletions or premature stop codons were not observed in any of the core autophagy genes in myeloid neoplasms.78 Further, in cross‐cancer unsupervised clustering analysis, autophagy‐associated transcript levels significantly correlated with overall survival in leukemia, kidney cancer, and endometrial cancer.83 Overall, these findings suggest that mutations in autophagy genes are relevant during tumorigenesis, with autophagy generally being downregulated but not lost.
3.1.2. Defective mitophagy causes accumulation of reactive oxygen species
Downregulation of mitophagy, the term used for the autophagic removal of dysfunctional mitochondria, can result in an increase in the formation of ROS.84, 85, 86 Disruption of mitophagy by knockout of essential autophagy genes such as ATG5, ATG7, ATG12, and FIP200 coincides with accumulation of defective mitochondria and increased ROS levels.7, 87, 88, 89 Such oxidative stress has been linked to cancer development and progression.90 For instance, persistent accumulation of ROS can damage proteins, fatty acids, and DNA, which may contribute to cancer development.90, 91, 92 Further, protein and lipid phosphatases can be inactivated upon oxidation of cysteine residues in the catalytic domain, causing changes in signaling pathways and affecting cell growth.93 Interestingly, the autophagy protein ATG4 is a cysteine protease that is overexpressed in several types of cancer and is highly sensitive to ROS.94, 95, 96 Redox modifications of cysteine residues in ATG4 prevent delipidation of LC3, thereby promoting sustained autophagy.96 In human adenocarcinoma cells, oxidative stress led to upregulation of ATG4 together with increased autophagy and increased invasion of cells through a Matrigel matrix.97 Another example of the interplay between ROS and autophagy is the accumulation of p62/SQSTM1, a scaffold protein for ubiquitinated cargo that is continuously cleared via basal autophagy.98 Accumulation of p62 aggregates due to crippling of autophagy causes oxidative stress and triggers the DNA damage response pathway.99 However, elevated ROS levels can also activate p53‐mediated apoptotic cell death.100 Of note, mutant p53 was shown to attenuate expression of ROS‐scavenging enzymes coinciding with high ROS levels, indicating that these cells are able to tolerate ROS levels to a higher degree.101 The exact interplay between autophagy and ROS in cancer development is highly complex, and it remains unclear how persistent elevation of ROS, due to defective autophagy can contribute to cancer development.
3.1.3. Autophagy prevents accumulation of oncoproteins
Reduced autophagy levels during tumorigenesis may also alter the intracellular levels of oncoproteins. Indeed, several oncoproteins have been shown to be a target for degradation via CMA. For example, BCR‐ABL, an oncoprotein formed by chromosomal translocation, was targeted to the autolysosome by CMA after treatment of chronic myeloid leukemia (CML) cell lines and primary CML patient–derived cells with the chemotherapeutic arsenic trioxide.102 In line with this data, inhibition of autophagy prevented arsenic trioxide–mediated suppression of BCR‐ABL expression.102 Defective autophagy was similarly associated with accumulation of the oncoprotein PML/RARA, the hallmark oncoprotein of acute promyelocytic leukemia.103 Moreover, treatment of acute myeloid leukemia (AML) cells with internal tandem duplications in fms‐like tyrosine kinase 3 (FLT3), referred to as FTL3‐ITD, with proteasome inhibitor bortezomib triggered autophagy‐dependent degradation of FLT3‐ITD and improved the overall survival in a xenografts.104 Further, the proto‐oncoprotein AF1Q, which is often overexpressed in AML and myelodysplastic syndrome and associated with an unfavorable prognosis, was targeted for breakdown by CMA.105, 106 Thus, autophagy and specifically CMA can clear various (proto)oncoproteins, and repression of this type of autophagy might contribute to tumorigenesis. Of note, autophagy can also aid the breakdown of tumor suppressor genes, like p53, as will be described below.
3.2. Autophagy in cancer maintenance
As evident from the preceding sections, autophagy can have a tumor suppressor function and is often downregulated in cancer. However, there is also clear evidence to suggest that autophagy is required for cancer (stem) cell maintenance. Indeed, increased autophagic flux or increased dependency on functional autophagy have been reported for various types of cancer, such as melanoma, CML, AML, and RAS‐driven cancers.107, 108, 109, 110, 111 For example, in solid cancers such as breast cancer and melanoma, increased LC3 puncta positively correlated with a more aggressive phenotype.110 Further, autophagic flux can aid cancer cell survival during cellular stress conditions, such as hypoxia and starvation.67, 112, 113 In addition, changes in autophagy can contribute to the maintenance of so‐called CSCs, a self‐renewing subpopulation of cancer cells with stem cell properties that for certain types of cancer, such as AML, is thought to drive the disease. The various roles of autophagy in cancer maintenance are detailed below (illustrated in Figure 3B).
3.2.1. Autophagy in the maintenance of CSCs function
CSCs are characterized by elevated levels of autophagy compared to more differentiated cancer cell populations, an observation confirmed in multiple cancer types, including urinary bladder and breast cancer.108, 114, 115 These CSCs expressed high levels of essential autophagy genes to maintain CSC properties and to remain dormant.114, 116 Further, elevated autophagy was required for the CSC‐mediated development of tumors in vivo in leukemia and breast cancer.115, 117, 118 However, the differentiation‐dependent level of autophagy is not specifically linked to malignantly transformed cells. Also normal hematopoietic, mesenchymal, and skin stem cells, have a higher level of autophagy as compared with more differentiated cells.119, 120 Thus, primitive cells have high autophagy levels in association with low ROS levels, which might be a protective mechanism for maintaining stem cell properties.119, 120 Correspondingly, the function of normal HSCs was lost in ATG7 and ATG12‐knockout mice. In the long term, this loss of function did coincide with the development of myeloproliferative syndrome, possibly a consequence of defective mitochondrial clearance in association with high ROS levels.82, 118, 121 Also, deletion of ATG5 or ATG7 in a mixed lineage leukemia murine AML model affected the survival and was associated with a decrease in a number of functional CSCs and a strong decrease in leukemic blasts in the peripheral blood indicating that autophagy has a critical function in leukemia maintenance.118 Similar findings were obtained with a bladder cancer cell line, and with breast cancer mammospheres, a model of CSCs with high levels of Beclin‐1 and an increase in autophagy.114 Thus, autophagy seems to be essential to preserve CSC function and to increase survivability.
3.2.2. Oncogenic mutations and autophagy
In established cancers, several oncogenes have been shown to induce autophagy and, thereby, contribute to cancer maintenance. For instance, oncogenic FLT3–ITD–positive AMLs cells are characterized by high levels of autophagy.122 Both pharmacological as well as genetic inhibition of autophagy in FLT3‐ITD in human AML cells markedly reduced cell proliferation and overcame acquired resistance to FLT3 inhibitors in mice. In addition, cancer driven by certain oncogenic RAS mutations as observed in a broad spectrum of tumors including colon, lung, and pancreatic cancers, appears to heavily depend on functional autophagy. For instance, basal levels of autophagy were increased in RAS‐transformed cancer cells even under nutrient‐rich conditions.112 Moreover, basal autophagy was strongly increased after overexpression of both mutant HRAS and KRAS in human mammary epithelial cells.123 The underlying mechanistic reason for mutant HRAS was found to be the activation of Beclin‐1 interacting partner NOXA, thereby upregulating autophagy.124 Genetic inhibition of autophagy in cells overexpressing mutant RAS attenuated glycolysis and inhibited proliferation.123 Similarly, ATG7 knockout in KRAS‐driven lung cancer cells increased ROS levels and triggered a striking depletion of the cellular nucleotide pool, which was rescued by supplementation with glutamine.125 In mouse models, the knockdown of ATG5 or ATG7 cells in RAS overexpressing cells triggered accumulation of dysfunctional mitochondria and reduced tumor growth.109, 126 Thus, RAS‐driven cancer cells exploit high levels of autophagy, which may position such cancers as targets for autophagy inhibition.
Further, oncogenic mutations in the tumor suppressor protein p53, a protein best known for its proapoptotic effect upon cellular stress, also clearly affect the autophagy pathway. For instance, elevated levels of autophagy were identified in mutant p53 expressing AML cells, whereas a reverse reduction in autophagy was detected in pancreas and breast cancer cell lines that expressed mutant p53.108, 127 These apparent contradictory data may be explained by the localization of p53, since p53 mutants that localized to the cytosol repressed autophagy, whereas p53 mutants localized to the nucleus did not.128 These clear differences in the effect of p53 mutants on autophagy may also impact on therapeutic response toward autophagy inhibition. Indeed, overexpression of mutant p53 in AML cells reduced the sensitivity toward HCQ treatment.108 Analogously, mutated p53 glioblastoma cells were less sensitive for CQ treatment.129 In contrast, CQ treatment impaired tumorigenesis in mutant KRAS pancreatic tumors with wildtype p53, but augmented tumorigenesis in the absence of p53.130 In this respect, it is important to note that wildtype p53 can differentially affect autophagy, with on the one hand inhibition of autophagy upon binding to proteins involved in autophagosome formation.128, 131, 132, 133 On the contrary, wildtype p53 can promote autophagy by inhibiting mTOR or by phosphorylation of Beclin‐1.134, 135, 136 Interestingly, the level of p53 itself is also regulated by autophagy. For instance, wildtype p53 is depleted via autophagy‐mediated degradation in renal cell carcinoma, which allows escaping from apoptotic cell death.137 In contrast, suppression of macroautophagy promotes the degradation of mutant p53 via CMA, which sensitizes various human cancer cell lines for cell death.138 Further, a truncated p53 isoform that inhibits wildtype p53 is degraded via autophagy.139
Thus, various known important oncogenic mutated proteins that are important in cancer maintenance are able to regulate autophagy, in most cases triggering elevated levels of autophagy that may aid in cancer cell survival.
3.2.3. Autophagy in cancer metabolism
Autophagy is a catabolic process whereby redundant organelles and proteins can re‐enter various metabolic pathways. Cancer cells typically metabolize glucose to lactate, even when sufficient oxygen is present to support oxidative phosphorylation, a phenomenon known as the Warburg effect.140 Of note, pyruvate kinase (PKM2) is the final enzyme in the glycolytic pathway that controls the glycolytic flux and is therefore important for preventing accumulation of glycolytic intermediates.141, 142 In cancer, PKM2 breakdown via CMA is increased, whereby reduced PKM2 associates with an accumulation of glycolytic intermediates that are rerouted toward branching biosynthetic pathways to support cancer growth.143 Likewise, the rate‐limiting enzyme hexokinase 2 (HK2) of the glycolytic pathway, was found to be selectively broken down via autophagy in liver cancer.144, 145 Together, this indicates that autophagy can control glycolysis at different levels and thus impacts on cancer metabolism. Indeed, glycolysis in MLL–ENL–driven leukemia is augmented by inhibition of autophagy, although the underlying mechanism remains to be determined.146 Of note, enhanced lactate secretion due to the Warburg effect can change the extracellular microenvironmental pH, which in turn can activate autophagy.147 For example, in breast carcinoma cells acute acidification led to an increase in LC3 puncta together with an increase in the expression of ATG5 and BNIP3.148 Thus, degradation of essential metabolic enzymes by autophagy may impact many aspects of central metabolism in cancer. Corroborating evidence hereof was obtained by labeling of wildtype or ATG7−/−KRAS driven lung cancer cells with heavy carbon and nitrogen isotopes, which in the autophagy‐deficient cells identified a significant depletion of amino acids linked to the tricarboxylic acid (TCA) cycle.149 Therefore, autophagy may provide cancer cells with a mechanism to efficiently redistribute metabolites enabling metabolic rewiring, which is required for malignant transformation.
3.2.4. Autophagy is upregulated in hypoxic tumor regions
Autophagy is also an important regulatory pathway during adaptation of cancer cells to hypoxic stress occurring in poorly oxygenated regions of the bone marrow due to AML infiltration or in hypoxic regions of solid cancers. Indeed, in xenograft models of human head and neck cancer, autophagy was associated with hypoxic tumor regions.113 Under hypoxic conditions, stabilization of hypoxia‐inducible factor 1α (HIF1α) was detected, leading to enhanced levels of Beclin‐1, increased LC3‐II/LC3‐I ratio and degradation of p62, eg, upon treatment of lung cancer cell lines with cisplatin.150 Likewise, in adenoid cystic carcinoma, the hypoxia mimetic CoCl2 stabilized HIF1α and induced autophagy.151 HIF1α activity among others upregulates expression of BNIP3 and BNIP3L, which can activate autophagy by shifting the balance of the regulatory Beclin‐1 hub toward autophagy induction (Figure 2B).48, 151, 152 In glioblastomas, increased expression of BNIP3 or ATG9A contributed to hypoxia‐associated growth, which could be blocked in vivo by HCQ.153, 154 Importantly, tumor cells in hypoxic regions proved to be particularly sensitive to HCQ treatment.113 In a panel of cancer cell lines, hypoxia‐induced cell death increased upon knockdown of Beclin‐1 or ATG7, with autophagy‐deficient cancer cells proliferating less in mouse xenograft models.155 Of note, xenografts of wildtype cell lines were characterized by increased LC3 and reduced p62 levels in hypoxic tumor regions, reflecting activation and execution of autophagy.155 Therefore, in a broad spectrum of cancers induction of autophagy contributes to survival in poorly oxygenated tumor areas.
3.2.5. Autophagy in anoikis and metastasis
Most cancer patients succumb to their disease due to metastatic spread of the original primary tumor, an event that can occur many years after initial seemingly successful treatment of the primary tumor. During metastatic spread, autophagy is thought to be crucial for cancer cell survival. First, cancer cells that spread to distal organs have to resist cell death due to loss of contact with the extracellular matrix (ECM), termed anoikis. Cells can resist anoikis partly through activation of autophagy as shown for metastatic hepatocellular carcinoma.156, 157 Similarly, transformed fibroblasts were characterized by a strong increase in autophagy after the loss of ECM contact. Further, anoikis was triggered upon inhibition of autophagy in cancer cell lines driven by either RAS or PI3K.123, 158, 159, 160 In an attachment‐free culture model system, tumor spheroids of various cancer cell lines depended on BNIP3‐associated autophagy for survival.161 Further, rapamycin‐mediated activation of autophagy improved spheroid growth, while autophagy inhibition induced apoptosis.161 Correspondingly, the levels of LC3B were significantly higher in metastases compared with primary tumors in breast cancer, liver cancer, and melanoma.110, 157, 162 Moreover, the incidence of metastases was reduced in metastatic liver cancer cells upon knockdown of Beclin‐1 or ATG5 in a mouse model, due to loss of resistance to anoikis.157 Thus, metastatic cells appear to be more dependent on functional autophagy to allow survival in the absence of ECM contact after which metastatic cells remain characterized by higher autophagy levels.
3.3. The role of autophagy in cytotoxic cancer therapy
Treatment of cancer cells with cytotoxic drugs inevitably leads to cellular stress. Consequently, activation of autophagy is widely described although, as detailed below, the impact of autophagy on cytotoxic therapy can differ depending on the type of cell death. Moreover, although the underlying cause of intrinsic and/or acquired drug resistance is likely multifactorial and often remains enigmatic, autophagy is increasingly recognized as being an important contributor to therapy resistance. In the sections below, the role of autophagy in cytotoxic cell death will be detailed, after which the role of autophagy in resistance to therapy is discussed.
3.3.1. Autophagy has a distinct impact depending on the type of cytotoxic cell death
Autophagy can be a stress response of cancer cells that enables cells to evade apoptotic elimination. An example hereof is the treatment of a triple‐negative breast cancer cell line with a plant‐derived anticancer drug that induced apoptosis and activated autophagy. Here, inhibition of autophagy with 3‐MA served to augment the level of apoptotic cell death.163 Similarly, in colorectal cancer cell lines, a proapoptotic polyamine analog simultaneously induced apoptosis and autophagy, with 3‐MA cotreatment enhancing induction of apoptotic cell death.164 In another breast cancer cell line model, the novel therapeutic drug NBT was found to induce autophagy and apoptosis, with apoptosis induction being increased upon CQ treatment.165 In CML cell lines, the antitumor agent asparaginase‐induced apoptosis and autophagy.166 Blockade of autophagy with three different autophagy inhibitors enhanced asparaginase‐induced cell death. Further, inhibition of autophagy in HeLa cells upregulated expression of p53 upregulated modulator of apoptosis (PUMA) via FOXO3a, which upon cotreatment with etoposide or doxorubicin upregulated apoptosis as defined by enhanced activation of effector caspase‐3/7.167, 168 This sensitizing effect of autophagy inhibition was abolished in cells lacking PUMA, indicating that FOXO3a‐dependent mechanism induction of PUMA contributes to drug resistance.167 Interestingly, an important regulator of initiator caspase‐8 activation, the antiapoptotic protein FLIP, also can regulate autophagy activity by competitive binding to ATG3 and preventing lipidation of LC3.169
Reversely, autophagy as part of ACD is required for cytotoxic cell death. An example hereof is cell death induced by a cardiac glycoside in non–small‐lung cancer cell lines, which was characterized by an increase in autophagic flux and was inhibited by 3‐MA.170 Treatment with this glycoside was accompanied by activation of the JNK signaling pathway, leading to a decrease in the level of Bcl‐2 and a concomitant shift toward Beclin‐1‐mediated induction of autophagy.170 Of note, although glycoside treatment elevated the level of intracellular ROS, antioxidant cotreatment did not prevent glycoside‐induced cell death indicating that ROS is a by‐product of ACD in this setting. In contrast, ROS was causal for ACD induction in triple‐negative breast cancer cells by the compound physagulide P purified from Chinese herbal medicine, with cotreatment with a ROS scavenger inhibiting ACD.28 Several other pathways can also be involved in therapy‐induced ACD. For instance, radiation treatment of breast cancer cell lines triggered ACD via activation of p53 and downstream p53 effector protein DRAM.25 In this case, cell viability was partially rescued upon treatment with 3‐MA or by knockdown of ATG5 or Beclin‐1.25 Further, treatment of breast cancer cells with a so‐called selective estrogen receptor modulator induced ACD via reducing ATP levels.26 Conversely, the addition of ATP restored cell viability, coinciding with a reduction in the LC3‐II/LC3‐I ratio, which indicates that ACD was averted.26 Furthermore, treatment with the glycan‐binding protein, Galectin‐9, triggered cell death in colon cancer cells, which was blocked by knockdown of Beclin‐1 or ATG5.171
In conclusion, autophagy during cytotoxic therapy can either be protective or can be instrumental for cell death induced by certain therapeutics. Thus, depending on the type of drug used in the treatment of cancer, the combination with autophagy inhibitors may be warranted or should be avoided.
3.3.2. The role of autophagy signaling in resistance to cancer therapy
As described above, autophagy during treatment may reduce sensitivity to cytotoxic therapy. Correspondingly, resistance to various types of therapy is characterized by enhanced basal levels of autophagy, as defined by increased conversion of LC3‐I to LC3‐II, increased numbers of LC3B puncta per cell, upregulated numbers of autophagolysosomes, and degradation of p62.172, 173, 174 For example, cisplatin‐resistant clones of ovarian cancer cell lines as well as an oral squamous cell carcinoma cell line were characterized by enhanced levels of autophagic flux.175 In radiotherapy‐resistant breast cancer cells, ionizing radiation also elevated basal autophagy levels, indicating a protective effect of autophagy against treatment.176 Similarly, treatment of pancreatic cancer, colorectal cancer, and AML cell lines with bortezomib was accompanied by elevated autophagic flux.172, 173 Importantly, in various cell lines and with different types of drugs, the cotreatment with autophagy inhibitors CQ or HCQ re‐sensitized cells to treatment.177, 178, 179, 180 For instance, in breast and esophageal squamous cancer cell lines, chemotherapy or radiotherapy induced an autophagy response accompanied by therapy resistance.180, 181 The cotreatment with CQ did not only reduce clonogenic survival of malignant cells in vitro, but also reduced tumor burden in murine models.180, 181 Of note, overexpression of multidrug resistance pumps, such as ABCG2, not only facilitates drug resistance by increasing drug efflux but also by increasing autophagic flux.182 In line with this, ABCG2‐mediated drug resistance was strongly inhibited by knockdown of either ATG5 or ATG7.182 In this respect, CSCs are also known to overexpress ABC transporters, which may upregulate autophagy and contribute to CSC resistance to chemotherapy.183 Further, in CSCs, autophagy was upregulated upon treatment with chemotherapy or photodynamic therapy, which contributed to CSCs survival and promoted therapy resistance.184, 185 Similarly, AML leukemic stem cells (LSCs) were characterized by elevated autophagic flux upon treatment with BET inhibitors, which contributed to resistance to therapy186 (Figure 3C). Of note, since both normal HSCs as well as LSCs need a certain amount of autophagy to survive, there is only a relatively small therapeutic window of autophagy inhibition with HCQ (Figure 3D).
Resistance toward antibody‐based therapy can also be regulated by autophagy, which has mainly been studied for cetuximab, an EGFR‐blocking antibody. For instance, cetuximab‐induced autophagy in various EGFR‐expressing cancer cell lines by downregulation of HIF1α and Bcl‐2, which promoted the association of Beclin‐1 with VPS34187 and dose dependently activated Beclin‐1‐mediated autophagy in colon carcinoma cell lines.188 Analogously, EGFR tyrosine kinase inhibitors activated autophagy by promoting Beclin‐1‐VPS34 complex formation.189 Importantly, chemical inhibition of autophagy or knockout of Beclin‐1 sensitized cancer cells for cetuximab‐induced apoptosis.187, 188 Interestingly, inactive EGFR is required for the induction of starvation‐induced autophagy.190 Together, this data clearly indicates that enhanced autophagy can associate with resistance to various types of cancer therapy. Thus, it is of clear relevance to gain insight into how autophagy facilitates resistance to therapy. In the following sections, the role of key autophagy‐regulating signaling pathways and cancer‐associated genetic mutations will be discussed in the context of resistance to therapy.
3.3.3. Key signaling pathways associated with autophagy‐dependent drug resistance
Many studies have focused on unraveling the mechanisms by which chemotherapy and radiation therapy induce resistance, with several key upstream signaling components being implicated. Most notably, deregulation of the upstream autophagy regulatory system AMPK, which can both activate ULK1 and repress mTOR signaling to promote autophagy, has been reported. For instance, treatment of a colorectal cancer cell line with the drug salidroside activated protective autophagy alone as well as in combination with other antitumor agents via activation of AMPK.191 When AMPK activity was blocked using a kinase inhibitor, autophagy was reduced as evidenced by a decrease in LC3‐II/LC3‐I ratio, which synergistically enhanced the cytotoxic effects of combined salidroside and chemotherapy treatment.191 In other studies, upregulation of autophagy was attributed to direct activation of ULK1. Specifically, AML LSCs that were resistant to treatment with BET inhibitor in vitro were characterized by ULK1 activation.186 In contrast, no ULK1 activation was detected in cells sensitive to BET inhibitor treatment. Interestingly, although ULK1 is supposed to be downstream of AMPK signaling, AMPK phosphorylation was detected in both BET inhibitor–sensitive and –resistant cells. Thus, resistance to treatment in these LSC appears to stem from ULK1 signaling that increases autophagic flux.186 In a follow‐up study, pharmacological inhibition of AMPK did induce apoptosis in BET‐resistant LSCs. AMPK and ULK1 were found to have a similar cytoprotective mechanism against chemotherapeutics in primary pancreatic cancer cells as well as pancreatic cell lines.192 Further, in a t(8;21) AML model, Kasumi‐1 cells survived short‐term treatment with histone deacetylase inhibitors by upregulation of autophagy.193 However, interactions between AMPK and mTOR were not investigated and long‐term resistance was not examined. Resistance to therapy due to upregulated autophagy can also be acquired through repression of the mTOR pathway as demonstrated for dexamethasone treatment in various leukemic cell lines.194 Similarly, activation of autophagy in an imatinib‐resistant CML line and in cisplatin‐resistant lung carcinoma cells was due to repression of mTOR signaling.195, 196 Altered signaling of upstream regulators of mTOR caused this repression of mTOR signaling, eg an increase in phosphorylation/activation of AMPK or a decrease in Akt signaling.194, 196 In targeted therapy, mTOR inhibitors as single agents did induce autophagy, but were ineffective anticancer therapeutics.197 However, when mTOR inhibitors were combined with autophagy inhibitors, prominent antileukemic effects were detected.197 In clonogenic assays, primary AML cells formed fewer colonies in combination therapy than single treatment. Similarly, knockdown of ULK1 in combination with mTOR inhibitor reduced the colony‐forming potential of primitive AML precursors.197
Another pathway involved in autophagy‐mediated resistance to therapy is the MAPK pathway, with chemotherapeutic treatment of hepatocellular carcinoma cell lines leading to increased MEK and ERK activity and induction of cytoprotective autophagy.198 This induction of autophagy was partly blocked by MEK inhibition.198 In cell lines carrying the oncogenic BRAF V600E mutation that have aberrant constitutive MAPK signaling, treatment with the specific V600E inhibitor vemurafenib resulted in AMPK–ULK1‐mediated autophagosome accumulation.199 Autophagy was similarly upregulated in BRAF‐mutated primary melanoma samples treated with BRAF inhibitor compared with baseline untreated samples. Interestingly, here induction of autophagy did not occur through AMPK‐ULK1 signaling, but was likely attributable to induction of endoplasmic reticulum (ER) stress response through CHOP, ATF4, and eIF2α.200 Similarly, in cutaneous BRAF‐mutated melanoma cell lines enhanced basal autophagy was observed.201 Oncogenic BRAF led to chronic ER‐stress, which in turn activated the JNK‐signaling cascade and contributed to autophagy induction, leading to therapy resistance.201 Of note, combined treatment of vemurafenib with autophagy inhibitor CQ almost completely blocked tumor growth in a xenograft mouse melanoma model, highlighting that cytoprotective autophagy was at least partially associated with resistance to vemurafenib. Thus, various types of chemotherapy as well as targeted drugs can trigger activation of autophagy that contributes to resistance to therapy.
3.3.4. HMGB1 positively regulates autophagy, contributing to therapy resistance
Recent evidence suggests that the nuclear protein HMGB1 is another critical regulator of autophagy that can mediate resistance during cancer treatment. Although normally in the nucleus, HMGB1 can translocate to the cytoplasm upon stress where it directly interacts with Beclin‐1 and displaces Bcl‐2. Consequently, cytoplasmic HMGB1 can activate autophagy. Many studies have linked increased HMGB1 protein levels to autophagy and therapy resistance.202, 203, 204, 205 For instance, upregulation of HMGB1 occurred during cisplatin treatment in non–small‐cell lung cancer cell lines, which associated with enhanced autophagy.206 Knockdown of HMGB1 reduced the levels of autophagy and increased cell death, with knockdown of HMGB1 being more efficient than treatment with well‐known autophagy inhibitor 3‐MA.206 Similarly, treatment with docetaxel upregulated HMGB1 protein, leading to enhanced autophagy levels.207 Upon continuous treatment with docetaxel, cells became resistant to therapy, with sensitivity being restored by knockdown of HMGB1 and reducing tumor growth in a xenograft model.207 In an analogous fashion, treatment of leukemic cell lines with different chemotherapeutic drugs upregulated expression of HMGB1. Upregulation of HMGB1 was associated with enhanced LC3‐II/LC3‐I ratios and protected from treatment‐induced cell death, which was prevented by knockdown of HMGB1.208 HMGB1 mediated resistance to chemotherapy via mTOR and Beclin‐1 was further reported in several different cancer cell lines.204, 207, 208 As discussed above, various other factors can induce mTOR, thereby, facilitating resistance to chemotherapy mediated by autophagy.
3.3.5. MicroRNAs in autophagy during treatment resistance
Several lines of evidence have emerged that indicate that microRNAs (miRNA), small noncoding RNAs that degrade mRNA and thereby reduce translation, may also play a regulatory role in autophagy signaling in therapy resistance. For instance, the reduced expression of miR‐23b in radiotherapy‐resistant pancreatic cancer cell lines enhanced the level of autophagy when compared with radiosensitive cell lines.209 MiR‐23b directly targeted and reduced ATG12 expression and overexpression of this miRNA in radiotherapy‐resistant cells blocked autophagy, as evidenced by reduced LC3‐II/LC3‐I ratio and reduced numbers of autophagosomes per cell, and re‐sensitized cells to radiation treatment.209 In epithelial ovarian cancer cell lines that were resistant to cisplatin treatment, a similar decrease in the level of miR‐429 was detected, which was associated with enhanced levels of autophagy.210 Correspondingly, overexpression of miR‐429 reduced autophagy via downregulation of ATG7 and increased cellular sensitivity to cisplatin treatment. Furthermore, doxycycline treatment reduced the expression of miR‐30a, a microRNA that directly targets Beclin‐1 mRNA, whereas the levels of miR140‐5p that targets IP3k2 mRNA were increased.211, 212 In both cases, induction of autophagy was enhanced and contributed to therapy resistance. In addition, treatment of colorectal cancer cells with cetuximab was associated with downregulation of another Beclin‐1 mRNA‐targeting miRNA, miR‐216b, again yielding elevated activation of autophagy and resistance to therapy.188
In conclusion, although still in early stages the available data collectively suggest that downregulation of various miRNAs can directly activate cytoprotective autophagy during therapy by upregulation of key components of the autophagy machinery. Thus, reduced miRNA expression appears to be causally related to autophagy‐mediated resistance to therapy.
3.3.6. Hypoxia as autophagy activating signal in therapy resistance
Several studies highlight that hypoxia‐induced autophagy contributes to resistance to therapy. For instance, in primary glioblastoma tissue samples, administration of the vascular endothelial growth factor neutralizing antibody bevacizumab increased tumor hypoxia. In turn, this hypoxia associated with upregulation of cytoprotective autophagy.154 Correspondingly, autophagy inhibition upon bevacizumab treatment of xenografts derived from glioblastoma multiforme patients resulted in increased survival.153 In a study conducted on breast cancer cell lines, hypoxia itself did not induce autophagy. However, upon taxol treatment in hypoxic conditions cancer cells did appear to activate cytoprotective autophagy through inhibition of the mTOR pathway.213 Similarly, chemotherapy resistance in triple‐negative breast CSCs was attributed to a combination of hypoxia and upregulation of autophagy occurring in xenograft models from these patients.214 Overall, this data implies that hypoxia‐mediated resistance to therapy is at least partly due to the induction of cytoprotective autophagy.
4. PART II: THE ROLE OF AUTOPHAGY IN THE TUMOR MICROENVIRONMENT
The tumor microenvironment is a specialized niche created during tumor development that plays an important role in terms of cancer progression, survival, and response to therapy. This microenvironment comprises of many different cell types, including fibroblasts, mesenchymal stem cells (MSCs), endothelial cells, and immune cells. All of these cell types to a different extent use autophagy in cellular functioning in cancer, with, eg, autophagy in stromal cells such as fibroblasts promoting tumorigenesis, whereas autophagy in immune cells such as cytotoxic T‐cell (CTLs) facilitates execution of anticancer immune responses. Thus, cells within the microenvironment may have opposing requirements for autophagy that may prove difficult to reconcile for autophagy‐targeting therapy in cancer. In this section, we will attempt to capture the role and importance of autophagy and the impact of potential therapeutic targeting of autophagy for several crucial tumor microenvironmental constituents, namely cancer‐associated fibroblasts (CAFs) and MSCs, endothelial cells, innate and adaptive immune cells.
4.1. Autophagy in the tumor microenvironment; stromal cells
4.1.1. Autophagy in stromal cells promotes cancer cell growth and survival
A positive influence of fibroblasts on cancer cell growth is well documented, with, eg, enhanced growth rates for both fibroblasts and colon cancer cell lines in cocultures, as well as enhanced growth rates of head and neck squamous cell carcinoma (HNSCC) cells and breast carcinoma cells.215, 216, 217 Similarly, primary patient‐derived AML cells survive and proliferate better in coculture with mouse stromal cells or human MSCs.218, 219, 220 In cocultures, fibroblasts were characterized by elevated levels of autophagy as, eg, evidenced by the accumulation of LC3‐positive vesicles.215, 216, 217 Importantly, inhibition of autophagy markedly attenuated the beneficial impact of fibroblast in such cocultures. Specifically, inhibition of autophagy using 3‐MA treatment reduced the growth rate of colon cancer cells, whereas treatment with CQ or knockdown of Beclin‐1 in fibroblasts prevented the increase in HNSCC proliferation in cocultures. Together these data indicate that cancer cells induce and exploit elevated levels of autophagy in stromal cells for their aberrant growth. In this respect, fibroblasts isolated from tumors indeed had higher autophagy activity than normal fibroblasts.216
In addition to promoting cancer cell proliferation, there are some clues that autophagy in stromal cells also helps to promote cancer cell survival and can protect against anticancer therapy. Specifically, in cocultures of cancer cells with fibroblasts the basal level of apoptosis in cancer cells decreased, a phenomenon reversed by inhibition of autophagy using CQ.217, 222, 223 Of note, this effect on basal apoptosis was significant, yet small with the basal level of apoptosis dropping from 5% in breast cancer monocultures to 1% in fibroblast cocultures. More importantly, fibroblasts protected breast cancer cells against treatment with tamoxifen, yielding 85% apoptosis in monocultures versus 45% in fibroblast cocultures.222 However, the relative importance of autophagy in this setting remains to be determined, as no autophagy inhibitors were applied to identify the impact of autophagy. Similarly, under serum deprivation conditions, MSCs were able to limit the induction of apoptosis in lung cancer cell lines through activation of autophagy.224 Interestingly, CAFs also resist stress better than normal fibroblasts, as fibroblasts isolated from ovarian cancer patients were more resistant to oxidative stress, with sensitivity being restored by Beclin‐1 or ATG5 knockout.221 Thus, autophagic signaling in stromal fibroblasts and MSCs can contribute to survival and growth of cancer cells.
4.1.2. Soluble factors secreted in stromal cell/cancer cocultures affect autophagic signaling
In many cases, the positive effect of fibroblasts on cancer cell growth was retained when cells were cultured in the absence of direct cell‐cell contact or when the conditioned medium of fibroblasts was used.215, 216, 225 In the latter case, the conditioned medium of CAFs outperformed that of normal fibroblasts.216, 225 Further, the supernatant of CAFs also protected melanoma and lung cancer cells from radiation‐induced cell death.226 This protumorigenic effect of secreted factors was due to autophagy signaling, as conditioned medium from CAFs pretreated with CQ failed to promote proliferation, migration, and invasion.216 Thus, CAFs secrete soluble factors through autophagy (called “secretory autophagy”) that are beneficial for cancer cells. Several secreted factors were identified, including various cytokines such as IGF1, IGF2, and CXCL12, all of which promoted survival of A375M melanoma and A549 lung cancer cells after radiation.226 Further, injection of CAFs at the site of tumors previously eradicated by radiation accelerated the subsequent development of tumor recurrence, which was abrogated by IGF2 knockout or 3‐MA treatment.226 This finding highlights the importance of this cytokine produced by CAFs under autophagy for cancer cell survival. Importantly, IGF2 produced by CAFs also induced autophagy in cancer cells, indicating a feed‐forward loop to promote autophagy in the tumor microenvironment. In a similar fashion, IL‐6 and IL‐8 secretion by CAFs was reduced upon knockdown of Beclin‐1, which decreased migration of HNSCC cells.216 Of note, direct addition of IL‐6 and IL‐8 to HNSCC cells promoted migration to a similar extent as coculture with CAFs, highlighting the importance of those cytokines for the autophagy‐mediated effect of fibroblasts. Cytokine production by fibroblasts was attributed to bFGF‐induced autophagy, with knockdown of bFGF in HNSCC cells reducing autophagy in fibroblast and reducing cytokine secretion. Similarly, TGF‐β secreted by breast cancer cells was shown to induce autophagy in CAFs.227 Thus, factors secreted by cancer cells can trigger activation of autophagy in CAFs, which concomitantly results in secretion of cytokines that elevate autophagy and have a protumorigenic effect on cancer cells. Hence, inhibiting autophagy in both cancer cells and cancer‐associated stromal cells likely outperforms inhibiting autophagy in cancer cells only. Indeed, simultaneous knockout of ATG7 in both MSCs and AML cells increased the sensitivity to cytarabine treatment compared with ATG7 knockout in AML cells alone.228
4.1.3. Cancer cells trigger metabolic reprogramming of cancer‐associated fibroblasts
In coculture experiments of fibroblasts and cancer cells, hypoxic stress was elevated in the fibroblast population, leading to the induction of autophagy and metabolic reprogramming. For instance, in coculture with breast cancer cells, HIF1α and NFκB signaling activated autophagy and, more specifically, mitophagy in CAFs.223 Similarly, coculture of fibroblast and colon cancer cells induced oxidative stress in fibroblasts and elevated the level of autophagy.215 Correspondingly, expression of constitutively active HIF1α in fibroblasts also induced autophagy/mitophagy, whereas treatment with HIF1α inhibitor echinomycin reduced levels of autophagy.229 Due to elevated mitophagy, the mitochondrial mass in fibroblasts was strongly reduced when cocultured with cancer cells.217 This resulted in a metabolic shift from the TCA cycle to the glycolytic pathway, also yielding increased production of ketones and lactate.215 A similar shift was detected in fibroblast engineered to overexpress the p53‐inducible autophagy inducer DRAM, leading to elevated autophagy, reduced mitochondrial mass, and an increase in secretion of ketones and lactate.230 In line with this, overexpression of ATG16L1 or BNIP3L, to induce autophagy, reduced fibroblast mitochondrial activity and increased glycolytic pathway activity.231 Interestingly, lactate and ketones produced by fibroblasts were utilized by cancer cells leading to increasing mitochondrial mass and mitochondrial oxidative metabolism of cancer cells in coculture with fibroblasts.217 Of note, HIF1α also directly activates the glycolysis pathway.232 Therefore, it is unclear whether elevated autophagy is the cause of glycolysis induction or that both pathways are simultaneously induced upon hypoxic stress. Taken together, cancer cells trigger hypoxic stress in fibroblasts leading to activation of autophagy and mitophagy and a metabolic switch from the TCA cycle to glycolysis. The metabolites produced by these fibroblasts are subsequently consumed by cancer cells and contribute to cancer cell growth and survival.226
Autophagy in fibroblasts has further been linked to reduced caveolin‐1 (cav‐1) expression in the stroma of breast cancer patients, a feature associated with poor survival.233, 234 Specifically, cav‐1 expression was downregulated in fibroblasts which were modulated to have elevated levels of autophagy.217, 223, 229, 231, 235, 236 Correspondingly, cav‐1 expression inversely correlated with autophagy and mitophagy in cell lines and patient‐derived human breast cancer samples.223 In mice, coinjection of breast cancer cells with fibroblasts yielded larger primary tumors and an increase in metastases, especially when fibroblasts were modulated for increased autophagic flux and reduced cav‐1 levels.217, 235, 236
Taken together, elevated levels of autophagy in CAFs promote cancer cell growth and survival, which among others is due to a metabolic switch of fibroblasts to glycolysis and the secretion of glycolytic by‐products.
4.1.4. Autophagy in endothelial cells modulates angiogenesis
Fast expanding tumors require sufficient angiogenesis. The importance of autophagy in this process is not yet thoroughly investigated, although some studies show that autophagy influences angiogenesis. For instance, the knockdown for ATG5 or treatment with 3‐MA of bovine aortic endothelial cells blocked angiogenesis (as determined by reduced tube length, migration, branching), whereas ATG5 overexpression promoted angiogenesis.237 Similarly, human dermal microvascular endothelial cells were blocked in angiogenesis upon 3‐MA or CQ treatment, whereas rapamycin promoted angiogenesis.238 In contrast, in the context of cancer cells, the inoculation of B16F10 melanoma cells in wildtype or heterogeneous Beclin‐1‐knockdown mice showed more angiogenesis in the Beclin‐1‐knockdown mice with concomitant bigger tumors and more lung metastasis.239 However, this effect was only seen under hypoxia and not normoxia. Also in vitro, Beclin‐1‐knockdown cells from lung epithelial cells and Beclin‐1 siRNA in wildtype cells yielded more angiogenesis under hypoxia but not under normoxia, which was regulated via HIF2α. Since cancer cells induce a state of hypoxia in adjacent fibroblast (as described above) it can be hypothesized that this also occurs in endothelial cells, leading to a reduction in angiogenesis when endothelial autophagy levels are high. In addition, modulating autophagy per se can suppress angiogenesis as has been shown in a model in chick eggs and human umbilical endothelial cells.240 Here, both rapamycin and 3‐MA disturbed normal blood vessel formation. Further, both downregulation of Atg7 or overexpression suppressed tube formation. However, rapamycin promoted endothelial cell migration. Thus, it is also possible that different processes in angiogenesis are differentially regulated by autophagy. Thus, it is currently unclear whether autophagy in endothelial cells should be inhibited or promoted to optimize anticancer effects.
4.2. Autophagy in cancer immunity; the cancer cell side of the coin
From the above paragraphs, it is clear that autophagy directly impacts on cancer proliferation and survival and thus is a target for inhibition in cancer cells. However, elevated autophagy levels in cancer cells can also have a diverse impact on anticancer T‐cell immunity. In brief, anticancer T‐cell immunity is a multilayered and intricately regulated process, which pivots on the recognition of antigenic peptides presented on the cancer cell surface in the so‐called major histocompatibility complex (MHC) class I to CTLs. Upon recognition of an MHC/peptide complex, the CTLs form an immunological synapse with the cancer cell and secrete cytotoxic and tumoricidal proteins, such as granzymes and perforins. Consequently, the cancer cell is eliminated by apoptosis.
Autophagy affects many aspects of this immune response and in cancer cells, for instance, inhibits proper formation of the immunological synapse and reduces the cytolytic potential of CTLs and natural killer (NK) cells (Figure 4). In addition, autophagy in cancer cells can have both a proimmune and an immune inhibitory effect by modulating the expression of immune checkpoints and steering the induction of so‐called immunogenic cell death (ICD). In this section of the review, the role of autophagy in anticancer immunity is discussed in detail.
Figure 4.
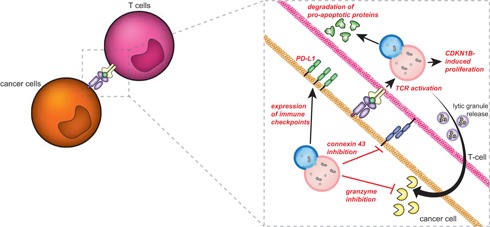
Autophagy in the tumor microenvironment impacts on anticancer immunity. Autophagy in cancer cells inhibits the anticancer immune response by reducing the efficacy of cytotoxic T‐cell and natural killer cell–mediated lysis by degrading granzyme B and connexin‐43. Further, autophagy is also required for T‐cell proliferation, survival, and induction of T‐cell memory by degrading proapoptotic proteins and maintaining mitochondrial homeostasis. Therefore, nonselective inhibition of autophagy in the tumor microenvironment will not only promote anticancer effects at the level of stroma and cancer cells, but will also dampen anticancer immune responses. CDKN1B, cyclin‐dependent kinase inhibitor 1B; PD‐L1, programmed cell death 1 and its ligand; TCR, T‐cell receptor [Color figure can be viewed at wileyonlinelibrary.com]
4.2.1. Autophagy in cancer cells inhibits anticancer immunity by reducing the sensitivity toward NK‐ and CTL‐mediated lysis
Cancer cells are lysed when they express MHC‐I molecules containing tumor‐derived antigenic peptides that are recognized by CTLs. To evade recognition and elimination by the immune system, cancer cells therefore often downregulate their MHC‐I expression.241, 242 Although this is mainly regulated by genetic mutations and epigenetic modifications, MHC‐I molecules are also degraded by autophagy. Indeed, inhibition of autophagy augmented cell surface expression of MHC‐I induced by treatment of melanoma cells with the immunostimulatory cytokine interferon γ (IFNγ).243 Importantly, the loss of MHC‐I expression (“missing self”) is also being recognized by NK cells, leading to the elimination of the cancer cell.241 However, several reports detail that high levels of autophagy in cancer cells reduces the efficacy of NK and CTL‐mediated cell lysis. For instance, autophagy in cancer cells affects the stability of the immunological synapse generated between the cytolytic immune cell and its target cell. Specifically, the formation of gap junctions, which requires connexin proteins such as connexin‐43, normally facilitates the exchange of small molecules between effector and target cell and is required for NK lysis.244 In melanoma cells, the accumulation of connexin‐43 at the immunological synapse was reduced under hypoxia, which was restored by inhibition of autophagy.245 Consequently, NK‐mediated cell lysis was restored. Interestingly, the gap junction protein connexin‐43 also transports active granzyme B, one of the main cytotoxic molecules of CTLs and NK cells, into the target cell.244 Thus, the degradation of connexin‐43 by autophagy may affect cytolysis in various ways. Interestingly, granzyme B is also a target of autophagy‐mediated breakdown, especially under hypoxic conditions. Correspondingly, granzyme B was predominantly detected in LC3 and Rab5‐positive fractions in hypoxic cells.246 Hence, autophagy in cancer cells contributed to resistance to lysis of lung cancer cell lines, breast cancer cells, and melanoma cells in hypoxic conditions by degrading granzyme B.246, 247, 248 A similar effect was detected upon hypoxia‐independent activation of HIFs, as seen in renal cancer with mutations in the von Hippel‐Lindau gene. In these cells, autophagy was upregulated, and cells were resistant toward NK‐mediated cell lysis.249 Autophagy not only affects CTL lysis in hypoxia, but also in normoxia with reduced sensitivity of melanoma cells toward CTL‐mediated lysis compared with healthy cells.250 In all cases, CTL‐ or NK cell–mediated lysis was restored by inhibition of autophagy.246, 249, 250 Of note, upregulation of autophagy also confers resistance to CTL lysis during the so‐called epithelial‐mesenchymal transition, a step necessary for cancer progression and metastasis, in breast carcinoma cells.251 Sensitivity to CTL‐mediated cell lysis in this setting was partly restored by Beclin‐1 knockdown. Thus, autophagy may affect cancer cell sensitivity to immune cell lysis at multiple levels.
Taken together, increased autophagy in cancer cells negatively affects sensitivity toward NK‐ and CTL‐mediated cell lysis through degradation of granzyme B and inhibition of the immunological synapse.
4.2.2. Autophagy in cancer cells regulates the expression of immune checkpoints
Immune checkpoints are coinhibitory receptor/ligand pairs that serve to dampen immune cell activity.252 A prominent example hereof is the receptor programmed cell death 1 (PD‐1) and its ligand (PD‐L1), which are expressed on activated T‐cell and antigen‐presenting cells (APCs), respectively. This checkpoint is a crucial inhibitor of anticancer T‐cell responses in the tumor microenvironment.253 Interestingly, activation of autophagy using mTOR inhibitor rapamycin decreased the expression of PD‐L1 in lung cancer cells in vitro and in vivo, whereas activation of mTOR increased expression of PD‐L1.254 Correspondingly, almost all human lung cancer patient samples (~90%) expressing PD‐L1 were characterized by increased mTOR signaling, whereas the majority (83%) of tumors negative for PD‐L1 also stained negative for active mTOR. Of note, signaling through PD‐L1 itself activated mTOR signaling in melanoma and ovarian cancer cells, with PD‐L1 blockade decreasing mTOR signaling in a mouse model of pancreatic cancer.255, 256 Thus, PD‐L1 activates mTOR and in a feed‐forward loop upregulates expression of PD‐L1, signaling that proceeded via mTORC1 and not mTORC2.254, 255 In line with the fact that mTORC1 is a major (negative) regulator of autophagy, melanoma, and ovarian cancer cells with low autophagic flux expressed higher levels of PD‐L1 than cells with high autophagic flux.255 Subsequent treatment with rapamycin to induce autophagy triggered a reduction in PD‐L1 expression and reactivated T‐cell–mediated anticancer immunity.254 Further, cotreatment with rapamycin and PD‐1 blocking antibodies more effectively reduced tumor growth than single treatment and was accompanied by reduced numbers of regulatory T‐cell and increased CD3+ T‐cell numbers. Thus, low levels of autophagy signaling associated with an increase in expression of PD‐L1.
4.2.3. Autophagy in cancer cells modulates the induction of immunogenic cell death
Autophagy can further impact on the process of ICD, a type of apoptosis that stimulates the development of anticancer T‐cell responses. ICD is induced by certain anticancer therapeutic strategies such as anthracyclines like mitoxantrone or doxorubicin, radiation therapy and photodynamic therapy (PDT).257 ICD requires the translocation of calreticulin to the cell surface and the release of several immune‐stimulating factors, among which HMGB1 and ATP.258, 259 During ICD, depletion of ATG5 or ATG7 in CT26 murine colon cancer cells or knockdown of Beclin‐1 reduced ATP‐release upon anthracycline treatment and inhibited in vivo anticancer immunity.258, 260 In contrast, ATG5 knockdown did not reduce ATP secretion in bladder cancer and melanoma cells after hypericin‐mediated PDT, although in this case knockdown of ATG5 did increase cell surface exposure of calreticulin.261 In contrast, calreticulin exposure was not affected upon anthracycline treatment of autophagy‐deficient and ‐competent CT26 cells or mouse embryonic fibroblasts (MEFs).258
The impact of autophagy inhibition on ICD also depends on which stage of autophagy is blocked, with calreticulin exposure being strongly reduced by blocking autophagy at early stages by silencing of ULK1, Beclin‐1, or ATG5.260 In contrast, calreticulin exposure was increased when autophagy was blocked at a late stage using vincristine, CQ, and bafilomycin A1. Of note, the induction of autophagy alone using rapamycin or mTOR siRNA was not enough to induce ATP secretion or calreticulin exposure.258, 260 Indeed, activation of the proapoptotic effector caspase‐8 is known to be pivotal for calreticulin exposure during ICD.262 Taken together, autophagy modulates the release of ATP and the cell surface exposure of calreticulin and thus contributes to ICD of cancer cells.
4.3. Autophagy in cancer immunity: the T‐cell side of the coin
Autophagy not only impacts on the immunogenicity of cancer cells, but is also pivotal for the correct functioning of APCs and T‐cell. For instance, the development of de novo T‐cell responses requires the presentation of antigenic peptides by professional APCs, most notably dendritic cells (DCs). DCs present the antigenic peptides to CTLs in the context of MHC‐I and further present peptides from endocytosed material to CD4+ helper T‐cell in the context of MHC class II. Activation of helper T‐cell licenses the DC to stimulate the clonal expansion of CTLs and is of great importance for anticancer T‐cell responses.263, 264, 265 Autophagy is critically involved in antigen presentation in both MHC‐I and MHC‐II in DCs. Finally, autophagy is also important in the functional activity of the immune cell itself, with T‐cell activity and generation of T‐cell memory requiring autophagy. Thus inhibition of autophagy in the context of cancer therapy also likely affects the activity of intratumoral T‐cell. Of note, the investigation of autophagy in the context of cancer immunity is in its infancy, but as detailed below, knowledge of core principles by which autophagy regulates T‐cell functioning has been gained among others in viral infection models.
4.3.1. Autophagy modulates surface MHC expression and alters the presentation of antigenic peptides
Anticancer T‐cell immunity is induced upon recognition of antigenic tumor peptides presented on the cell surface of professional APCs, such as DCs. However, the surface expression of the MHC‐I/peptide complex is directly affected by autophagy in DCs and macrophages (Figure 5A). For instance, expression of MHC‐I on murine macrophages and DCs was upregulated upon inhibition of autophagy using various chemical inhibitors or downregulation of core autophagy genes.243, 266 This upregulation was attributed to slower internalization of MHC‐I.266 Thus, in the absence of autophagy, MHC‐I molecules appear to be more stably expressed and less degraded.266 Correspondingly, DCs from VPS34‐deficient mice expressed more cell surface MHC‐I as well as MHC‐II.267 In contrast, surface expression of MHC‐II on macrophages was downregulated upon inhibition of autophagy using 3‐MA.166, 268
Figure 5.

Autophagy contributes to the formation of antigenic peptides in antigen‐presenting cells (APCs). Professional APCs, such as dendritic cells and macrophages, display antigenic peptides in the context of major histocompatibility complex class 1 (MHC‐I) or MHC‐II molecules to T‐cells, which will trigger an immune response. Autophagy reduces MHC‐I surface levels, which is converted upon autophagy inhibition. However, autophagy is also required for the generation of antigenic peptides. The inhibition of autophagy will therefore skew the peptidome, yielding less diversity In the antigens presented to T‐cells. Indeed autophagy, inhibition limits T‐cell activation by APCs [Color figure can be viewed at wileyonlinelibrary.com]
Thus, inhibition of autophagy upregulates surface expression of MHC‐I, although the impact on the surface expression of MHC‐II is less conclusively established. However, with one notable exception in an influenza model, functional studies highlight that despite an increase in surface MHC‐I expression the inhibition of autophagy actually weakens T‐cell responses.266 This weakening can likely be attributed to an alteration in the pool of immunogenic peptides presented in MHC, which has been best determined in the context of so‐called cross‐presentation in DCs. Cross‐presentation is a process which enables the loading of MHC‐I on DCs with extracellular antigens, which is important for activation of, eg, CTL responses in melanoma.269 DCs with cross‐presentation capacity are characterized by increased levels of autophagy compared with DCs that do not cross‐present, with inhibition of autophagy reducing MHC‐I‐mediated cross‐presentation of ovalbumin (OVA) and human cytomegalovirus (CMV) peptides.270, 271 Antigen presentation in MHC‐II was similarly altered upon inhibition of autophagy, with the reduced DC‐mediated processing of an immunodominant mycobacterial peptide, reduced presentation of herpes simplex virus (HSV) antigens, and vaccinia virus Ankara antigens.272, 273, 274 Consequently, antigen‐specific T‐cell responses were downregulated. Thus, autophagy inhibition modified the peptide pool presented in MHC and appeared to reduce the presentation of immunodominant epitopes.
In line with this role of autophagy in presenting appropriate antigenic peptides, knockout of the autophagy‐regulator VPS34 in DCs abrogated the induction of B16 melanoma‐specific CTLs in vivo, yielding a significantly higher incidence of lung metastases.275 In this setting, cross‐presentation of OVA peptides derived from apoptotic cells was strongly reduced, although VPS34‐deficient DCs did present higher levels of cytoplasmic OVA‐derived antigens.276 In addition, mice with autophagy‐deficient DCs failed to efficiently develop protective Th1 cell immunity and hence died faster upon challenge with a lethal dose of HSV.273 Further, starvation‐induced autophagy increased loading of intracellular and lysosomal‐derived peptides on MHC‐II molecules in human B‐lymphoblastoid cells.277
Taken together, most studies highlight that inhibition of autophagy negatively affects the MHC‐dependent presentation of tumor antigens by APCs and, thereby, negatively affects T‐cell immunity (Figure 5B).
4.3.2. Autophagy is required for T‐cell proliferation, survival, and generation of protective T‐cell memory
Autophagy plays a crucial in normal T‐cell functioning as evident from the severe reduction in basal peripheral T‐cell counts, especially CTL counts, in autophagy‐knockout mouse models.89, 267, 277, 278, 279, 280, 281, 282, 283, 284, 285, 286, 287 Naïve resting T‐cell have only minimal numbers of autophagosomes, but T‐cell receptor (TCR)‐mediated activation triggers a strong increase in autophagosome content in activated CD8+CD28+ CTLs and activated helper T‐cell subsets.278, 288, 289, 290, 291, 292 Induction of autophagy was augmented by T‐cell costimulation with the cytokine IL‐2 or by 4‐1BB signaling, which was accompanied by an increase in lysosomal content and colocalization of lysosomal LAMP staining with autophagosomal LC3 staining.278, 288, 290, 291, 292, 293 Moreover, in activated T‐cell transfected with an LC3‐GFP‐mCherry construct the LC3‐GFP signal was lost, indicative of fusion of autophagosomes with lysosomes. Thus, T‐cell activation increases autophagosome and lysosome content and increases autophagic flux.289
The importance of autophagy activation for T‐cell functioning became apparent upon knockout of various core autophagy genes and upstream autophagy regulators in T*cell. In all cases, this resulted in a poor proliferation of T‐cell upon TCR activation, which was not improved by addition of CD28 or IL‐2 costimulation.89, 267, 283, 284, 285, 286, 294, 295 Indeed, in T‐cell–specific ATG5 or ATG7‐knockout mouse models a strong decline in reactive CTLs was detected in models of lymphocytic choriomeningitis virus (MCMV), influenza virus or mouse CMV.280, 296 Notably, autophagy‐deficient T‐cell did express equal levels of the activation markers CD69 and CD25 upon TCR‐stimulation, suggesting that downstream TCR signaling does occur.89, 278, 283 Thus, it currently remains unclear whether the defect in the proliferating capacity of autophagy‐deficient T‐cell is on the level of the TCR or more downstream.
In addition to impaired cell proliferation, CTLs from ATG5‐knockout chimeras were ~50% more apoptotic than control T‐cell, and the viability of helper T‐cell from Beclin‐1‐knockout mice was also strongly reduced.278, 284 Further, spontaneous apoptosis or apoptosis after TCR activation in T‐cell was increased when core autophagy genes and regulators were knocked out.89, 267, 280, 281, 283, 285, 287 Indeed, ATG3 and ATG7‐knockout T‐cell contained more active caspase‐9 indicative of elevated levels of apoptosis, with viability being partly restored upon pan‐caspase inhibition.284, 287 In addition, upon TCR activation, autophagy‐deficient T‐cell secreted less proinflammatory cytokines, that is, IL‐2 and IFN‐γ, which may also negatively impact on their survival.282, 290 Taken together, inhibition of autophagy at multiple levels has a negative effect on T‐cell.
Importantly, autophagy is also pivotal for the development of T‐cell memory responses, with no development of protective T‐cell immunity in ATG5‐knockout mice upon rechallenge with influenza.279 Similarly, ATG7‐knockout mice vaccinated against MCMV failed to generate a T‐cell response upon re‐infection.280 Also the inhibition of CMA in T‐cell by knockdown of LAMP‐2A impaired the control of Listeria monocytogenes in mice upon rechallenge.293 Of note, residual peripheral T‐cell detected in autophagy‐deficient mouse models displayed a CD44highCD62low phenotype, a phenotype typically associated with effector and effector memory T‐cell.89, 267, 280, 287 Although this finding is in apparent contrast with the impaired induction of T‐cell memory, a similar “memory‐like” phenotype has been reported in lymphopenia in T‐cell–depleted mice.297 Similar to autophagy deficiency, such “memory‐like” T‐cell were, in fact, incapable of generating effective T‐cell immune responses. Thus, autophagy is pivotal for normal T‐cell function and, crucially, for the development of memory T‐cell that provide protective immunity. As a result, systemic, nontargeted application of autophagy inhibitors for cancer treatment likely also has a large negative effect on the function and activity of T‐cell, thereby, hampering the induction and/or execution of proper anticancer immune responses.
4.3.3. Autophagy‐dependent degradation of mitochondria and proapoptotic proteins maintain T‐cell homeostasis
The inhibition of autophagy negatively impacts on T‐cell, likely due to deregulated clearance of organelles and proteins, with transcriptional profiling of ATG5 wildtype vs knockout T‐cell among others suggesting a key involvement in mitochondrial homeostasis.89 In line with this analysis, knockout of ATG5 or other core autophagy genes was associated with an increase in mitochondrial mass compared to wildtype T‐cell, with a notable exception being Beclin‐1 knockout.267, 283, 284, 285, 286 Further, mitochondrial mass increased over time in inducible autophagy‐knockout models and was seen only in “aged” T‐cell in the periphery.89, 267, 283, 287 In autophagy‐deficient T‐cell, the increase in mitochondrial mass was accompanied by elevated levels of ROS, whereas activation of autophagy using autophagy‐inducer Torin‐1 reduced ROS levels and increased survival.280, 283, 285, 286, 287, 295 Correspondingly, treatment of hypoxic ATG5‐knockout T‐cell with ROS inhibitor N‐acetylcysteine restored T‐cell proliferation and prevented cell death.279
Autophagy also directly regulated expression of proapoptotic and antiapoptotic proteins in T‐cell. For instance, caspase‐8 and ‐3 protein levels were strongly increased in Beclin‐1‐knockout T‐cell, whereas no increase in mRNA levels was detected.284 Further, activation of autophagy using rapamycin triggered colocalization of LC3 and caspase‐3 and was accompanied by downregulation of caspase‐3 levels.284 Similarly, the level of several other apoptotic proteins like Bcl‐2, BIM, Bcl‐xl, Bax, cytochrome C, and AIF were increased upon interference with autophagy, although the impact of autophagy inhibition on these proteins varied among studies, possibly related to the timing of measurements, especially in inducible models.267, 281, 284, 287
Finally, autophagy also impacts on several key cell cycle regulators in T‐cell. For instance, TCR activation of T‐cell normally induces autophagy‐mediated degradation of cyclin‐dependent kinase (CDK) inhibitor 1B (CDKN1B), an inhibitor of cell cycle progression.294 Inhibition of autophagy prevented CDKN1B degradation upon TCR‐stimulation and, thereby, inhibited proliferation. Similarly, autophagy ensures degradation of Bcl‐10, a mediator of TCR‐to‐NF‐κB signaling, as well as Itch and Rcan1, two inhibitors of TCR signaling.293, 298
Taken together, the autophagy pathway is important for T‐cell survival and proliferation as it retains mitochondrial homeostasis and ensures the degradation of proapoptotic and antiproliferative proteins. An interesting exception to this rule was recently reported for a specific T‐cell subset, the so‐called Th9 T‐cell, which is reported to have potent anticancer immunity.299, 300 In this case, inhibition of autophagy prevented degradation of PU.1, the master transcription factor for Th9 cells.301 Hence, inhibition of autophagy enhanced differentiation of helper T‐cell to Th9 cells.
4.3.4. The role of autophagy in regulatory T‐cell functioning
Regulatory T (Treg) cells are a subpopulation of CD4+ T‐cell that inhibit effector T‐cell responses, with increased Treg infiltration in cancer associating with poor survival.302, 303 Compared to naïve CD4+ cells, Tregs contain more autophagosomes and have higher LC3‐II levels indicating the presence of increased levels of autophagy.304 Knockout of autophagy genes induced apoptosis of Treg cells and blocked Treg‐mediated suppression of effector T‐cell responses. This subsequently yielded higher percentages of tumor‐infiltrating CTLs and smaller tumors in a mouse model of colon adenocarcinoma.304 Further, animals with autophagy‐impaired Tregs were more prone to develop autoimmune diseases and adoptive transfer of Tregs from VPS34‐knockout mice failed to protect against colitis.267 Thus, autophagy is important for Treg immunosuppression. Autophagy inhibition may thus alleviate Treg immunosuppressive activity in cancer.
5. CONCLUSIONS AND PERSPECTIVES
Autophagy has a multifaceted impact on the cancer microenvironment and is an interesting target for cancer therapy. In established cancers, autophagy acts as a survival mechanism, eg, in conditions of elevated nutrient demand or low oxygen availability. Importantly, cancer cells elevate their autophagic flux during treatment to gain resistance toward (chemo)therapy. In addition, tumors activate autophagy in adjacent stromal cells to benefit from cytokines, growth factors and nutrients secreted upon induction of this pathway. Further, although the autophagy pathway is often repressed during early tumorigenesis, every cell requires a basal level of autophagy. Therefore, emerging cancer cells with downregulated levels of autophagy might be more reliant on remaining autophagy activity. Thus, therapeutic inhibition of autophagy may be well effective in both cancer cells with a high autophagic flux as well as cancer cells with a low autophagic flux.
Interestingly, many types of malignancies have a high prevalence in the aging population, while it has been suggested that a reduction in autophagy may be a contributor to the aging process. In line with this, autophagy was decreased in two‐third of HSCs of aged mice compared to young mice, with aged HSC with higher autophagy activity having better long‐term regenerative potential.87 Further, aging of the hematopoietic compartment is associated with myeloid malignancies, with a lineage skewing of HSCs due to upregulation of myeloid‐specific genes and downregulation of lymphoid genes.305, 306, 307, 308, 309 Intriguingly, similar myeloid skewing was detected upon monoallelic knockout of ATG7 or ATG12 in mice, which associated with the development of myeloproliferative syndrome.82, 87 Interestingly, treatment with autophagy inducer rapamycin also enhanced influenza‐specific CD8+ T‐cell responses in aged vaccinated mice, but not in aged mice with ATG7 knockout.280, 310, 311 Thus, reduced autophagy during ageing may increase susceptibility to tumorigenesis as well as negatively impact on T‐cell immunity. In this respect, activation of autophagy may even be a strategy for rejuvenation, with the treatment of mice with the autophagy inducer rapamycin extending their lifespan.312
An important process in ageing is cellular senescence, a stress response of damaged or aged cells characterized by permanent cell cycle arrest.313 Like senescence, autophagy is a pathway that is activated upon cellular stress, and both pathways appear connected. For instance, oncogenic HRAS triggered senescence in fibroblasts that coincided with increased autophagy.314 Similarly, overexpression of p21 or ULK3 triggered both senescence and autophagy in fibroblasts.314, 315 Further, the inhibition of autophagy suppressed irradiation‐induced senescence, collectively suggesting that autophagy positive regulates senescence.316 On the contrary, knockdown of ATG5 or ATG7 in human fibroblast cells or ATG7 in murine muscle stem cells also induced senescence.317, 318 Further, GATA4, a senescence regulator, was found to be selectively broken down via p62‐mediated autophagy, which was repressed when cells were undergoing senescence, suggesting that autophagy and senescence repress each other’s activity.319 However, future experiments will have to elucidate whether autophagy can directly influence senescence or whether it is a co‐occurring pathway and whether autophagy modulation may be used to regulate senescence.320
Based on the evidence reviewed here, the inhibition of autophagy in not only cancer cells but also in cancer‐supporting stromal cells may help increase cancer cell death, especially in combination with other therapeutic approaches. Indeed, several phase I/II clinical trials are currently being conducted in various cancers with established autophagy inhibitors, such as 3‐MA, CQ, or HCQ (Table 1). On the contrary, an effective anticancer immune response appears to require autophagy at multiple levels. For instance, autophagy promotes the generation of antigenic peptides to be presented in MHC‐I and MHC‐II molecules on APCs. Further, T‐cell rely on an active autophagy pathway for their proliferation and survival. Hence, systemic application of autophagy inhibitors would likely inhibit anticancer immune responses. Indeed, the in vitro treatment of T‐cell with CQ reduced T‐cell–dependent cell lysis, inhibited T‐cell proliferation and reduced cytokine secretion.321 Further, accumulation of autophagic vacuoles was observed in PBMCs of patients treated with HCQ, suggesting that inhibition of autophagy occurred in these immune cells.21, 22, 23, 322 In line with this, the inhibition of autophagy was associated with lymphopenia in 31% of the patients in the temsirolimus study, whereas autophagy activation using mTOR inhibitor everolimus negatively affected multiple immune cell subsets in renal carcinoma patients.21, 22 Thus, autophagy inhibition likely occurs with current therapeutics, but should be avoided in cancer‐associated immune cells.
Table 1.
Overview of phase I/II clinical trials in various cancers with established autophagy inhibitors. The table indicates the number of patients, treatment approach, patient outcome and the impact on autophagy activity
| Malignancies | Patient numbers | Treatment | Achievement/outcome | Autophagic response | References |
|---|---|---|---|---|---|
| • Non–small‐cell lung cancer | 8 | HCQ | Dose‐limiting toxicity: not determined Maximum dose tolerance: not determined Outcome: progressive disease median PFS 1.8 mo, median OS 9 mo |
Autophagic changes: not determined | Goldberg et al, A phase I study of erlotinib and HCQ in advanced non‐small cell lung cancer. J Thorac Oncol. 2012 |
| 19 | HCQ + erlotinib | Dose‐limiting toxicity: none Maximum dose tolerance: 1000 mg/d Recommended phase II dose: 1000 mg/d Outcome: PR, SD, median PFS 2 mo, median OS 10.6 mo |
Autophagic changes: not determined | ||
| • Colon rectal • Non–small‐lung cancer • Ovarian • Soft tissue Sarcoma • Renal • Breast • Melanoma • Carcinoid • Bladder • Prostate |
27 | Vorinostat (HDACi) + HCQ | Dose‐liming toxicity: 800 mg/d HCQ Maximum dose tolerance: 600 mg/d HCQ Outcome prolonged SD, PR |
Autophagic changes: no significant changes in AV accumulation at Day 49 IHC revealed increased MAP1LC3B at Day 49 |
Mahalingam et al, Combined autophagy and HDAC inhibition/Autophagy. 2014 |
| • Melanoma • Colorectal • Head and neck • Breast • Gastric/esophageal • Prostate • Pancreas • Non–small‐cell lung • Pheo/adrenocortical |
27 | Temsirolimus (mTOri) + HCQ | Dose‐limiting toxicity: 1200 mg/d HCQ Maximum dose tolerance: 1200 mg/d HCQ for 3 mo, subsequently lowered to 1000 mg/d HCQ Outcome: PFS of melanoma patients 3.5 mo, SD, no response |
Autophagic changes: with 1200 mg/d HCQ significant AV accumulation at 6 wk, increased AVs with temsirolimus + HCQ compared to single HCQ treatment | Rangwala et al, Combined MTOR and autophagy inhibition. Autophagy. 2014 |
| • Non–small‐cell lung cancer • Head and neck • Melanoma • Colon • Breast • Liposarcoma • Esophageal (SCC) • Brain metastasis |
37 | Temozolomide + HCQ | Dose‐limiting toxicity: none Maximum dose tolerance: not reached Recommended Phase II dose: 1200 mg/d Outcome: melanoma and breast PFS > 4 mo, PR, SD |
Autophagic changes: accumulation of AVs at 4 wk | Rangwala et al, Phase I trial of HCQ with dose‐intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014 |
| • Glioblastoma multiforme | 16 | Radiation therapy + temozolomide + HCQ | Dose‐limiting toxicity: 800 mg/d HCQ Maximum dose tolerance: 600 mg/d HCQ |
Autophagic changes: not determined | Rosenfeld et al, A phase I/II trial of HCQ in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastomas multiforme. Autophagy. 2014 |
| 76 | Radiation therapy + temozolomide + HCQ | Outcome: median OS 15.6 mo | Autophagic changes: increased AVs/cell and LC3‐II/I at 3 wk | ||
| • Relapsed/refractory myeloma | 25 | Bortezomib + HCQ | Dose‐limiting toxicity: none Maximum dose tolerance: 1200 mg/d HCQ Outcome: VGPR, MR, SD, and immediate progression |
Autophagic changes: increased AV and LC3‐II/I conversion (2 and 3 wk after treatment, respectively) | Vogl et al, Combined autophagy and proteasome inhibition. Autophagy. 2014 |
| • Metastatic pancreatic adenocarcinoma | 20 | HCQ | Dose‐limiting toxicity: not determined Maximum dose tolerance: not determined Outcome: Median PFS 45.6 d, overall survival 69 d |
Autophagy changes: inconsistent autophagy inhibition | Wolpin et al, Phase II and PD study of autophagy inhibition using HCQ in patients with metastatic pancreatic adenocarcinoma. The Oncologist. 2014 |
| • Pancreatic adenoma | 35 | HCQ + gemcitabine + surgery | Dose‐limiting toxicity: none Maximum dose tolerance: 1200 mg/d HCQ Outcome: median DFS 12.0 mo |
Autophagic changes: end of treatment 65% of patients showed increased LC3‐II staining, which correlated with improved DSF and OS | Boone et al, Safety and biologic response of preoperative autopagy inhibition in combination with Gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2015 |
| Sarcoma | Closed early | HCQ + sirolimus | Dose‐limiting toxicity: not determined Maximum dose tolerance: not determined Outcome: PR, SD, progressive disease, no reduction of tumor volume |
Autophagic changes: not determined | Chi et al, Double autophagy modulators reduce 2‐deoxyglucose uptake in sarcoma patients. Oncotarget. 2015 |
| • Advanced metastatic BRAF mutant melanoma | 11 | Debrafenib (BRAFi) + trametinib (MEKi) + HCQ | Dose‐limiting toxicity: not determined Maximum dose tolerance: not determined Outcome: low rate retinal toxicity |
Autophagic changes: not determined | Nti et al, Frequent subclinical macular changes in combined BRAF/MEK inhibition with high‐dose HCQ as treatment of advanced metastatic BRAF mutant melanoma. Retina. 2017 |
| Relapsed/refractory multiple myeloma | 6 | Rapamycin + cyclophosphamide + dexamethasone | Dose‐limiting toxicity: none Maximum dose tolerance: not determined Outcome: MR, SD |
Autophagic changes: not determined | Scott et al, Double autophagy stimulation using chemotherapy and mTOR inhibition combined with HCQ for autophagy modulation in patients with relapsed or refractory MM. Haematologica. 2017 |
| HCQ + cyclophosphamide + dexamethasone | Dose‐limiting toxicity: 1200 mg/d HCQ Maximum dose tolerance: not determined Outcome: PR, SD |
Autophagic changes: not determined | |||
| 18 | Rapamycin + HCQ + cyclophosphamide + dexamethasone | Dose‐limiting toxicity: 1200 mg/d HCQ Maximum dose tolerance: 800 mg/d HCQ Outcome: VGPR, PR, MR, SD, immediate progression, median PFS 8.6 mo, median OS 11.3 mo |
Autophagic changes: increased AV counts in 600‐1200 mg/d HCQ |
Abbreviations: 3‐MA, 3‐Methyladenine; CQ, chloroquine; HCQ, hydroxychloroquine; AV, autophagic vacuoles; DFS, disease‐free survival; IHC, immunohistochemistry; MR, minor response; OS, overall survival; PFS, progression‐free survival; PR, partial response; SD, stable disease; VGPR, very good partial response.
Of note, in a phase II study in glioblastoma patients, 600 mg/d HCQ was found to be the maximum tolerated dose, a dose at which autophagy inhibition was not consistently achieved in the tumor.323 Doses used in other early clinical cancer studies range from 800 to 1200 mg/d (Table 1). Thus, the therapeutic window for the clinical use of CQ seems to be quite small. Indeed, as also recently reported by us, there is only a small therapeutic window of autophagy inhibition with HCQ between CD34+ AML cells and healthy normal bone marrow‐derived CD34+ cells (Figure 3D).108 Moreover, current autophagy inhibitors such as CQ have a poor biodistribution profile, with levels of CQ in the blood being much higher than in the tumor.324
Taken together, it is clear that development of therapeutic strategies that inhibit autophagy more selectively in cancer cells appears warranted. Hereto, the development of novel autophagy inhibitors that have an increased activity profile in vivo, with limited cytotoxicity may help improve the therapeutic window for autophagy inhibition. For instance Lys05, a bisaminoquinoline and synthetic derivative of CQ was 10‐fold more potent than HCQ through better accumulation and deacidification of lysosomes and was effective in inhibiting autophagy in xenograft studies.325 Another inhibitor of interest is ARN5187, which blocks the final step of autophagolysosome maturation, with superior cytotoxic activity over HCQ in various cancer cell lines.326 Several other approaches to inhibit autophagy that target upstream regulatory events are currently under investigation in leukemia and solid cancer treatment. These include the PI3K inhibitor buparlisib, which selectively targets VPS34.327 First promising results in phase I single‐agent studies were observed in solid cancers as well as advanced leukemias, although toxicity was detected.328, 329 Other upstream targets being investigated include ULK.330, 331, 332 One of the more potent inhibitors of ULK1, SBI‐0206965, had prominent anticancer activity in human non–small‐cell lung cancer and adenocarcinoma.332, 333 All of these new autophagy‐targeting drugs are in early stages, with future clinical trials being awaited to gain insight into toxic effects and potential anticancer efficacy.
Further, increasing the tumor selectivity of autophagy drugs can be pursued, eg by use of liposomal formulations. Such formulations have been widely used to enhance drug retention and alter biodistribution by passive or active targeting. In this respect, encapsulation of chemotherapeutic drugs such as doxorubicin in liposomes has yielded more effective formulations with less side effects.334 Such formulations can be further optimized by using antibody‐conjugated and tumor‐targeted liposomes.335 Of note, CQ liposomes have already been generated in the context of CQ as a malaria drug and were also suitable to simultaneously deliver CQ and a tumoricidal drug.336, 337, 338, 339, 340, 341 The codelivery of CQ and doxorubicin improved the anticancer activity compared with liposomal doxorubicin.341 Alternatively, drugs such as CQ may be directly targeted to cancer cells using antibody‐drug conjugates.342
Future studies will have to determine whether selective targeting of cancer cells can prevent the adverse effect of inhibition of anticancer immunity. Concerning in this respect is the reported upregulation of PD‐L1 upon inhibition of autophagy, which results in dampening of immune responses. If further proven, combined treatment with autophagy and PD‐1/PD‐L1 checkpoint inhibitors may prove a straight‐forward approach to circumvent this issue. Nevertheless, inhibition of autophagy also alters the composition of peptides presented in MHC and thus may still impact on cancer immunity. Studies that elucidate this potential effect of autophagy inhibition in more detail are urgently needed to steer design of future clinical studies.
As it is evident that autophagy can steer the pool of antigenic peptides that are presented in MHC‐I and MHC‐II on the cell surface, attempts have been made to exploit autophagy for the purpose of therapeutic vaccination. For instance, fusion of NY‐ESO‐1, a cancer‐testis antigen frequently overexpressed in melanoma, to LC3 resulted in autophagosome targeting and augmented NY‐ESO‐1‐specific antimelanoma helper T‐cell responses.343 An additional approach to exploit autophagy for induction of immunity is through autophagosome‐based vaccination. Here, autophagosomes are isolated from cancer cells treated with a proteasome inhibitor. Such autophagosomes contain tumor‐associated antigens and on the surface express CLEC9A ligands that facilitate endocytosis by APCs.344 DCs pulsed with such autophagosomes were more efficient in inducing OVA‐specific T‐cell responses compared with soluble protein.345, 346 This autophagosome vaccination approach reduced B16F10 melanoma cell growth, eliminated 3LL Lewis lung tumors, and protected mice from a rechallenge with sarcoma.346, 347 Of note, tumor cells may also release autophagosomes themselves and modulation of autophagy could, therefore, trigger the release of autophagosomes from tumor cells and positively impact on anticancer T‐cell immune responses.280, 348
Of note, an important issue that needs to be addressed for any new type of therapy to enter clinical practice is the identification of appropriate patient stratification criteria. In this respect, current clinical trials do not have inclusion/exclusion criteria that take autophagic activity in the tumor into account. Further, appropriate testing of the efficacy of autophagy inhibition in cancer cells of patients will have to be developed, with current trials mainly monitoring autophagy in peripheral blood mononuclear cells (PBMCs) as a surrogate marker of response or in tumor biopsies. However, the level of autophagy in PBMCs does not seem to correlate with autophagy inhibition in the tumor microenvironment.23 Therefore, positron emission tomography/computed tomography and magnetic resonance imaging probes for ATG activity are currently being developed. Thus, research in the upcoming years should focus on not only identifying optimal inhibitors of autophagy in patients, but also on the identification of appropriate patient selection criteria and monitoring tools to position autophagy targeting for clinical use.
In conclusion, a host of evidence has emerged on the importance of autophagy in cancer cells and its validity as a target for this disease. However, the various cell types in the tumor microenvironment differ in their reliance on autophagy, making it hard to predict the exact outcome of autophagy inhibition in cancer. Further detailed investigations into the specific impact in this complex milieu are needed to steer rational design of therapeutic targeting of autophagy in specific cancer subtypes and combination strategies.
ACKNOWLEDGMENTS
The authors acknowledge Dr Marco de Bruyn for his help with the figures.
Biographies
H Folkerts, MSc, studied applied medical biology and medical biology at the Saxion University and the University of Amsterdam (UVA), The Netherlands. During several internships at the University of Amsterdam, he worked on genetic metabolic diseases, in the lab of Prof R.J.A. Wanders. His master thesis focused on cellular metabolism in the context of cancer. In 2012 he joined the lab of hematology at the University Medical Center Groningen (UMCG), where he worked for 5 years on the role of autophagy in the normal hematopoiesis and leukemia, supervised by Prof Dr E Vellenga and Prof Dr J.J. Schuringa. Currently, he is working as a postdoctoral researcher in the Department of Hematology, supervised by Prof Dr E. Vellenga. His current work is focused on understanding the role of autophagy in leukemia and characterizing leukemic stem cells based on their cellular metabolic status.
S Hilgendorf, MSc, acquired a bachelor degree in health science in 2009, and a master degree in molecular life sciences in 2011 at the Maastricht University (UM), the Netherlands. Susan worked on hypermethylated tumor suppressor genes during an internship at the UM and investigated biomarkers for tumor hypoxia during an internship at the Ontario Cancer Institute, Canada. From 2011‐2016 she performed her PhD research at the Department of Hematology of the University Medical Center Groningen (UMCG)/University of Groningen, supervised by Prof Dr E. Vellenga and Prof Dr J.J. Schuringa. Here Susan investigated the role of mutated genes in leukemia. From 2017‐2018 Susan worked as a postdoctoral researcher at the Department of Hematology of the UMCG on a project involving the activation of anticancer immune responses.
Prof Dr E Vellenga, hematologist, is during many years involved in clinical and translational studies in acute myeloid leukemia (AML). Fields of interest are the development of multihit models by using cord blood cells and oncogenes of interest thereby mimicking the transforming events during tumor initiation. In addition, the signaling pathways involved have been studied including NF‐κB and STAT signaling in normal and leukemic cells. These investigations are closely connected to clinical studies in AML patients as a member of Leukemia working party of the HOVON. In the past, clinical co‐operative studies have been performed on the application of growth factor during induction chemotherapy in newly diagnosed patients with AML and the importance of autologous stem cell transplantation as consolidation course for intermediate risk AML patients. Currently, Edo is the HOVON coordinator for high‐risk acute promyelocytic leukemia patients in the European APOLLO consortium.
Prof Dr E Bremer is a tenure‐track professor of immunohematology at the Department of Hematology of the UMCG. The central aim of his research is the development of new cancer (immuno)therapeutics that can (re) activate innate and adaptive anticancer immunity or trigger tumor‐selective cell death. In addition, his research has uncovered an autophagy‐dependent cell death pathway induced by the glycan‐binding protein Galectin‐9 in cancer cells, which is being further elucidated for its potential application in cancer therapy. Finally, research in his group is focusing on the role of autophagy in anticancer immune responses and the therapeutic targeting of autophagy in cancer.
Dr VR Wiersma, acquired a bachelor degree in biotechnology in 2008, and a master degree in Medical Pharmaceutical Sciences in 2010 at the University of Groningen, the Netherlands. From 2010 to 2014 she performed her PhD research at the Department of Surgery, Surgical Research Laboratory, of the University edical Center Groningen (UMCG)/University of Groningen, supervised by Prof Dr W. Helfrich and Prof Dr E. Bremer. Her PhD research was focused on the preclinical evaluation of the anticancer activity of TRAIL‐fusion proteins and the glycan‐binding protein, Galectin‐9. For Galectin‐9 she uncovered a cancer‐specific toxicity pathway that involved autophagy. From 2014‐2016 she worked as Postdoctoral Researcher at the Department of Gynecology of the UMCG where she preclinically evaluated the activity of antibodies for cancer immunotherapy. In 2016, Valerie started at the Department of Hematology where she is currently the project leader of the research involving the therapeutic potential of Galectin‐9 in AML and other cancers. Main scientific interests of Valerie comprise: oncology, immunology, autophagy, cell death, and therapeutic targeting.
Folkerts H, Hilgendorf S, Vellenga E, Bremer E, Wiersma VR. The multifaceted role of autophagy in cancer and the microenvironment. Med Res Rev. 2019;39:517‐560. 10.1002/med.21531
Contributor Information
Edwin Bremer, Email: e.bremer@umcg.nl.
Valerie R. Wiersma, Email: v.wiersma@umcg.nl.
References
REFERENCES
- 1. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111(9):1208‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding W‐X, Yin X‐M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allen GFG, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin‐independent mitophagy. EMBO Rep. 2013;14:1127‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheong H, Wu J, Gonzales LK, Guttentag SH, Thompson CB, Lindsten T. Analysis of a lung defect in autophagy‐deficient mouse strains. Autophagy. 2014;10:45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol. 2005;169:425‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032‐1036. [DOI] [PubMed] [Google Scholar]
- 9. Malhotra R, Warne JP, Salas E, Xu AW, Debnath J. Loss of Atg12, but not Atg5, in pro‐opiomelanocortin neurons exacerbates diet‐induced obesity. Autophagy. 2015;11(1):145‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature. 2008;456:264‐268. [DOI] [PubMed] [Google Scholar]
- 11. Yoshii SR, Kuma A, Akashi T, et al. Systemic analysis of Atg5‐null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev Cell. 2016;39:116‐130. [DOI] [PubMed] [Google Scholar]
- 12. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23(16):2891‐2906. [DOI] [PubMed] [Google Scholar]
- 13. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077‐15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takamura A, Komatsu M, Hara T, et al. Autophagy‐deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mariño G, Salvador‐Montoliu N, Fueyo A, Knecht E, Mizushima N, López‐Otín C. Tissue‐specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin‐3. J Biol Chem. 2007;282:18573‐18583. [DOI] [PubMed] [Google Scholar]
- 16. Huijbers A, Plantinga TS, Joosten LAB, et al. The effect of the ATG16L1 Thr300Ala polymorphism on susceptibility and outcome of patients with epithelial cell‐derived thyroid carcinoma. Endocr Relat Cancer. 2012;19(3):L15‐L18. [DOI] [PubMed] [Google Scholar]
- 17. Nicoli ER, Dumitrescu T, Uscatu CD, et al. Determination of autophagy gene ATG16L1 polymorphism in human colorectal cancer. Rom J Morphol Embryol. 2014;55(1):57‐62. [PubMed] [Google Scholar]
- 18. Huang C‐Y, Huang S‐P, Lin VC, et al. Genetic variants of the autophagy pathway as prognostic indicators for prostate cancer. Sci Rep. 2015;5:14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan J, Zhang N, Yin L, et al. Clinical implications of the autophagy core gene variations in advanced lung adenocarcinoma treated with gefitinib. Sci Rep. 2017;7:17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1391‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vogl DT, Stadtmauer EA, Tan K‐S, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10(8):1380‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahalingam D, Mita M, Sarantopoulos J, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10(8):1403‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Z, Wang B, Yu F, et al. The roles of mitochondria in radiation‐induced autophagic cell death in cervical cancer cells. Tumour Biol. 2016;37(3):4083‐4091. [DOI] [PubMed] [Google Scholar]
- 25. Cui L, Song Z, Liang B, Jia L, Ma S, Liu X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol Rep. 2016;35(6):3639‐3647. [DOI] [PubMed] [Google Scholar]
- 26. Kim DE, Kim Y, Cho D‐H, et al. Raloxifene induces autophagy‐dependent cell death in breast cancer cells via the activation of AMP‐activated protein kinase. Mol Cells. 2015;38(2):138‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peng P, Kuo W‐H, Tseng H‐C, Chou F‐P. Synergistic tumor‐killing effect of radiation and berberine combined treatment in lung cancer: the contribution of autophagic cell death. Int J Radiat Oncol Biol Phys. 2008;70(2):529‐542. [DOI] [PubMed] [Google Scholar]
- 28. Yu P, Zhang C, Gao C‐Y, et al. Anti‐proliferation of triple‐negative breast cancer cells with physagulide P: ROS/JNK signaling pathway induces apoptosis and autophagic cell death. Oncotarget. 2017;8(38):64032‐64049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaushik S, Cuervo AM. Chaperone‐mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koga H, Martinez‐Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone‐mediated autophagy. Nat Commun. 2011;2:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wong P‐M, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walczak M, Martens S. Dissecting the role of the Atg12‐Atg5‐Atg16 complex during autophagosome formation. Autophagy. 2013;9(3):424‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861‐2873. [DOI] [PubMed] [Google Scholar]
- 35. Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395‐398. [DOI] [PubMed] [Google Scholar]
- 36. Tanida I, Tanida‐Miyake E, Ueno T, Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a protein‐activating enzyme for multiple substrates including human Apg12p, GATE‐16, GABARAP, and MAP‐LC3. J Biol Chem. 2001;276:1701‐1706. [DOI] [PubMed] [Google Scholar]
- 37. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131‐24145. [DOI] [PubMed] [Google Scholar]
- 38. Huynh KK, Eskelinen E‐L, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26(2):313‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542‐545. [DOI] [PubMed] [Google Scholar]
- 40. Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181‐197. [DOI] [PubMed] [Google Scholar]
- 41. Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl‐xL’s target recognition versatility revealed by the structure of Bcl‐xL in complex with the BH3 domain of Beclin‐1. J Mol Biol. 2007;372:223‐235. [DOI] [PubMed] [Google Scholar]
- 42. Pattingre S, Tassa A, Qu X, et al. Bcl‐2 antiapoptotic proteins inhibit Beclin 1‐dependent autophagy. Cell. 2005;122:122927‐122939. [DOI] [PubMed] [Google Scholar]
- 43. Tai WT, Shiau CW, Chen HL, et al. Mcl‐1‐dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC‐59 in hepatocellular carcinoma cells. Cell Death Dis. 2013;4:e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi Y, Coppola D, Matsushita N, et al. Bif‐1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Strappazzon F, Vietri‐Rudan M, Campello S, et al. Mitochondrial BCL‐2 inhibits AMBRA1‐induced autophagy. EMBO J. 2011;30(7):1195‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Molejon MI, Ropolo A, Re AL, Boggio V, Vaccaro MI. The VMP1‐Beclin 1 interaction regulates autophagy induction. Sci Rep. 2013;3:1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bellot G, Garcia‐Medina R, Gounon P, et al. Hypoxia‐induced autophagy is mediated through hypoxia‐inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570‐2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akar U, Chaves‐Reyez A, Barria M, et al. Silencing of Bcl‐2 expression by small interfering RNA induces autophagic cell death in MCF‐7 breast cancer cells. Autophagy. 2008;4(5):669‐679. [DOI] [PubMed] [Google Scholar]
- 50. Germain M, Nguyen AP, Le Grand JN, et al. MCL‐1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011;30:395‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Malik SA, Orhon I, Morselli E, et al. BH3 mimetics activate multiple pro‐autophagic pathways. Oncogene. 2011;30:3918‐3929. [DOI] [PubMed] [Google Scholar]
- 52. Pedro JMBS, Wei Y, Sica V, et al. BAX and BAK1 are dispensable for ABT‐737‐induced dissociation of the BCL2‐BECN1 complex and autophagy. Autophagy. 2015;11:452‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl‐X(L) and a BH3‐like domain in Beclin‐1. EMBO J. 2007;26:2527‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chiang W‐C, Wei Y, Kuo Y‐C, et al. High‐throughput screens to identify autophagy inducers that function by disrupting Beclin 1/Bcl‐2 binding. ACS Chem Biol. 2018;13:2247‐2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl‐2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1‐dependent mitophagy. Mol Cell. 2014;55:451‐466. [DOI] [PubMed] [Google Scholar]
- 56. Vives‐Bauza C, Zhou C, Huang Y, et al. PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eccles DM, Russell SE, Haites NE, et al. Early loss of heterozygosity on 17q in ovarian cancer. The Abe Ovarian Cancer Genetics Group. Oncogene. 1992;7:2069‐2072. [PubMed] [Google Scholar]
- 58. Gao X, Zacharek A, Salkowski A, et al. Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 1995;55:1002‐1005. [PubMed] [Google Scholar]
- 59. Miracco C, Cosci E, Oliveri G, et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30:429‐436. [PubMed] [Google Scholar]
- 60. Pickford F, Masliah E, Britschgi M, et al. The autophagy‐related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118:2190‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Saito H, Inazawa J, Saito S, et al. Detailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2‐cM region on 17q21.3 often and commonly deleted in tumors. Cancer Res. 1993;53:3382‐3385. [PubMed] [Google Scholar]
- 62. Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121‐1125. [DOI] [PubMed] [Google Scholar]
- 63. Eissa S, Matboli M, Awad N, Kotb Y. Identification and validation of a novel autophagy gene expression signature for human bladder cancer patients. Tumour Biol. 2017;39:1010428317698360. [DOI] [PubMed] [Google Scholar]
- 64. Aita VM, Liang XH, Murty VVVS, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59‐65. [DOI] [PubMed] [Google Scholar]
- 65. Jiang ZF, Shao LJ, Wang WM, Yan XB, Liu RY. Decreased expression of Beclin‐1 and LC3 in human lung cancer. Mol Biol Rep. 2012;39:259‐267. [DOI] [PubMed] [Google Scholar]
- 66. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809‐1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karantza‐Wadsworth V, Patel S, Kravchuk O, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367‐1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672‐676. [DOI] [PubMed] [Google Scholar]
- 70. Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res. 2014;12(4):485‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene. 2004;23:639‐645. [DOI] [PubMed] [Google Scholar]
- 72. Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy‐related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39:1059‐1063. [DOI] [PubMed] [Google Scholar]
- 73. Lee JW, Jeong EG, Soung YH, et al. Decreased expression of tumour suppressor Bax‐interacting factor‐1 (Bif‐1), a Bax activator, in gastric carcinomas. Pathology. 2006;38:312‐315. [DOI] [PubMed] [Google Scholar]
- 74. Zhao Z, Oh S, Li D, et al. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev Cell. 2012;22(5):1001‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 76. Hervouet E, Claude‐Taupin A, Gauthier T, et al. The autophagy GABARAPL1 gene is epigenetically regulated in breast cancer models. BMC Cancer. 2015;15:15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Boyer‐Guittaut M, Poillet L, Liang Q, et al. The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA‐MB‐436 breast cancer cells. Autophagy. 2014;10:986‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Visconte V, Przychodzen B, Han Y, et al. Complete mutational spectrum of the autophagy interactome: a novel class of tumor suppressor genes in myeloid neoplasms. Leukemia. 2017;31:505‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Park SM, Ou J, Chamberlain L, et al. U2AF35(S34F) promotes transformation by directing aberrant ATG7 pre‐mRNA 3′ end formation. Mol Cell. 2016;62:479‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu F, Lee JY, Wei H, et al. FIP200 is required for the cell‐autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116:4806‐4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mortensen M, Ferguson DJP, Edelmann M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A. 2010;107:832‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mortensen M, Soilleux EJ, Djordjevic G, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208(3):455‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lebovitz CB, Robertson AG, Goya R, et al. Cross‐cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11(9):1668‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Frank M, Duvezin‐Caubet S, Koob S, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297‐2310. [DOI] [PubMed] [Google Scholar]
- 85. Klionsky DJ, Cuervo AM, Dunn WA, Jr. , Levine B, van der Klei IJ, Seglen PO. How shall I eat thee? Autophagy. 2007;3:413‐416. [DOI] [PubMed] [Google Scholar]
- 86. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3‐5. [DOI] [PubMed] [Google Scholar]
- 87. Ho TT, Warr MR, Adelman ER, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543(7644):205‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liang CC, Wang C, Peng X, Gan B, Guan JL. Neural‐specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285:3499‐3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Stephenson LM, Miller BC, Ng A, et al. Identification of Atg5‐dependent transcriptional changes and increases in mitochondrial mass in Atg5‐deficient T lymphocytes. Autophagy. 2009;5(5):625‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stanicka J, Russell EG, Woolley JF, Cotter TG. NADPH oxidase‐generated hydrogen peroxide induces DNA damage in mutant FLT3‐expressing leukemia cells. J Biol Chem. 2015;290:9348‐9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Van Remmen H, Ikeno Y, Hamilton M, et al. Life‐long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29‐37. [DOI] [PubMed] [Google Scholar]
- 93. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336‐20342. [DOI] [PubMed] [Google Scholar]
- 94. Liu PF, Leung CM, Chang YH, et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy. 2014;10:1454‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rothe K, Lin H, Lin KBL, et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123:3622‐3634. [DOI] [PubMed] [Google Scholar]
- 96. Scherz‐Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Barry EL, Mosher DF. Factor XIIIa‐mediated cross‐linking of fibronectin in fibroblast cell layers. Cross‐linking of cellular and plasma fibronectin and of amino‐terminal fibronectin fragments. J Biol Chem. 1989;264(7):4179‐4185. [PubMed] [Google Scholar]
- 98. Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986‐1990. [DOI] [PubMed] [Google Scholar]
- 99. Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Perri F, Pisconti S, Della Vittoria Scarpati G. p53 mutations and cancer: a tight linkage. Ann Transl Med. 2016;4(24):522‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kalo E, Kogan‐Sakin I, Solomon H, et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J Cell Sci. 2012;125(Pt 22):5578‐5586. [DOI] [PubMed] [Google Scholar]
- 102. Goussetis DJ, Gounaris E, Wu EJ, et al. Autophagic degradation of the BCR‐ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120:3555‐3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy‐induced degradation of the PML/RARA oncoprotein. Blood. 2010;116:2324‐2331. [DOI] [PubMed] [Google Scholar]
- 104. Larrue C, Saland E, Boutzen H, et al. Proteasome inhibitors induce FLT3‐ITD degradation through autophagy in AML cells. Blood. 2016;127:882‐892. [DOI] [PubMed] [Google Scholar]
- 105. Li P, Ji M, Lu F, et al. Degradation of AF1Q by chaperone‐mediated autophagy. Exp Cell Res. 2014;327(1):48‐56. [DOI] [PubMed] [Google Scholar]
- 106. Tse W, Deeg HJ, Stirewalt D, Appelbaum FR, Radich J, Gooley T. Increased AF1q gene expression in high‐risk myelodysplastic syndrome. Br J Haematol. 2005;128(2):218‐220. [DOI] [PubMed] [Google Scholar]
- 107. Altman BJ, Jacobs SR, Mason EF, et al. Autophagy is essential to suppress cell stress and to allow BCR‐Abl‐mediated leukemogenesis. Oncogene. 2011;30:1855‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Folkerts H, Hilgendorf S, Wierenga ATJ, et al. Inhibition of autophagy as a treatment strategy for p53 wild‐type acute myeloid leukemia. Cell Death Dis. 2017;8(7):e2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang Y, Wang XD, Lapi E, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A. 2012;109:13325‐13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rouschop KMA, van den Beucken T, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120(1):127‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ojha R, Jha V, Singh SK, Bhattacharyya S. Autophagy inhibition suppresses the tumorigenic potential of cancer stem cell enriched side population in bladder cancer. Biochim Biophys Acta. 2014;1842:2073‐2086. [DOI] [PubMed] [Google Scholar]
- 115. Gong C, Bauvy C, Tonelli G, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem‐like/progenitor cells. Oncogene. 2013;32:2261‐2272. 2272e.1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chaterjee M, van Golen KL. Breast cancer stem cells survive periods of farnesyl‐transferase inhibitor‐induced dormancy by undergoing autophagy. Bone Marrow Res. 2011;2011:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liu Q, Chen L, Atkinson JM, Claxton DF, Wang HG. Atg5‐dependent autophagy contributes to the development of acute myeloid leukemia in an MLL‐AF9‐driven mouse model. Cell Death Dis. 2016;7:e2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sumitomo Y, Koya J, Nakazaki K, et al. Cytoprotective autophagy maintains leukemia‐initiating cells in murine myeloid leukemia. Blood. 2016;128(12):1614‐1624. [DOI] [PubMed] [Google Scholar]
- 119. Gómez‐Puerto MC, Verhagen LP, Braat AK, Lam EWF, Coffer PJ, Lorenowicz MJ. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy. 2016;12:1804‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gomez‐Puerto MC, Folkerts H, Wierenga ATJ, et al. Autophagy proteins ATG5 and ATG7 are essential for the maintenance of human CD34(+) hematopoietic stem‐progenitor cells. Stem Cells. 2016;34(6):1651‐1663. [DOI] [PubMed] [Google Scholar]
- 121. Mohrin M, Bourke E, Alexander D, et al. Hematopoietic stem cell quiescence promotes error‐prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Heydt Q, Larrue C, Saland E, et al. Oncogenic FLT3‐ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. 2018;37(6):787‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras‐mediated oncogenic transformation. Mol Biol Cell. 2011;22:165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras‐induced expression of Noxa and Beclin‐1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23‐35. [DOI] [PubMed] [Google Scholar]
- 125. Guo JY, Teng X, Laddha SV, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras‐driven lung cancer cells. Genes Dev. 2016;30:1704‐1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Guo JY, Karsli‐Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K‐ras‐induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cordani M, Oppici E, Dando I, et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol Oncol. 2016;10(7):1008‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Morselli E, Tasdemir E, Maiuri MC, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7(19):3056‐3061. [DOI] [PubMed] [Google Scholar]
- 129. Kim EL, Wustenberg R, Rubsam A, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol. 2010;12(4):389‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296‐300. [DOI] [PubMed] [Google Scholar]
- 131. Morselli E, Shen S, Ruckenstuhl C, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10(16):2763‐2769. [DOI] [PubMed] [Google Scholar]
- 132. Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Scherz‐Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53‐dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A. 2010;107(43):18511‐18516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Duan L, Perez RE, Davaadelger B, Dedkova EN, Blatter LA, Maki CG. p53‐regulated autophagy is controlled by glycolysis and determines cell fate. Oncotarget. 2015;6:23135‐23156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Feng Z, Hu W, de Stanchina E, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF‐1‐AKT‐mTOR pathways. Cancer Res. 2007;67:3043‐3053. [DOI] [PubMed] [Google Scholar]
- 136. Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP‐kinase promotes autophagy by weakening its interactions with Bcl‐2 and Bcl‐XL. Autophagy. 2009;5(5):720‐722. [DOI] [PubMed] [Google Scholar]
- 137. Kang JH, Lee J‐S, Hong D, et al. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2‐chaperoned autophagy. Cell Death Dis. 2016;7:e2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Vakifahmetoglu‐Norberg H, Kim M, Xia H, et al. Chaperone‐mediated autophagy degrades mutant p53. Genes Dev. 2013;27:1718‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Horikawa I, Fujita K, Jenkins LMM, et al. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53‐mediated senescence. Nat Commun. 2014;5:4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Liberti MV, Locasale JW. The Warburg Effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lv L, Li D, Zhao D, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone‐mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Xie J, Dai C, Hu X. Evidence that does not support pyruvate kinase M2 (PKM2)‐catalyzed reaction as a rate‐limiting step in cancer cell glycolysis. J Biol Chem. 2016;291:8987‐8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zhu H, Luo H, Zhu X, Hu X, Zheng L, Zhu X. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: a meta‐analysis. Oncotarget. 2017;8:1628‐1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Jiao L, Zhang HL, Li DD, et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy. 2017;14:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Xia HG, Najafov A, Geng J, et al. Degradation of HK2 by chaperone‐mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. 2015;210:705‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Watson A, Riffelmacher T, Stranks A, et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015;1:15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wojtkowiak JW, Rothberg JM, Kumar V, et al. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012;72:3938‐3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Guo JY, White E. Autophagy, metabolism, and cancer. Cold Spring Harb Symp Quant Biol. 2016;81:73‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wu H‐M, Jiang Z‐F, Ding P‐S, Shao L‐J, Liu R‐Y. Hypoxia‐induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015;5:12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wu H, Huang S, Chen Z, Liu W, Zhou X, Zhang D. Hypoxia‐induced autophagy contributes to the invasion of salivary adenoid cystic carcinoma through the HIF‐1α/BNIP3 signaling pathway. Mol Med Rep. 2015;12(5):6467‐6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mazure NM, Pouysségur J. Atypical BH3‐domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy. 2009;5:868‐869. [DOI] [PubMed] [Google Scholar]
- 153. Abdul Rahim SA, Dirkse A, Oudin A, et al. Regulation of hypoxia‐induced autophagy in glioblastoma involves ATG9A. Br J Cancer. 2017;117(6):813‐825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Hu Y‐L, DeLay M, Jahangiri A, et al. Hypoxia‐induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tan Q, Wang M, Yu M, et al. Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia. 2016;18:347‐355. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 156. Fu XT, Shi YH, Zhou J, et al. MicroRNA‐30a suppresses autophagy‐mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 2018;412:108‐117. [DOI] [PubMed] [Google Scholar]
- 157. Peng YF, Shi YH, Ding ZB, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9:2056‐2068. [DOI] [PubMed] [Google Scholar]
- 158. Avivar‐Valderas A, Salas E, Bobrovnikova‐Marjon E, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616‐3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chen N, Eritja N, Lock R, Debnath J. Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3‐kinase in three‐dimensional culture. Oncogene. 2013;32:2543‐2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Palorini R, Votta G, Pirola Y, et al. Protein kinase A activation promotes cancer cell resistance to glucose starvation and anoikis. PLOS Genet. 2016;12:e1005931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Chen JL, David J, Cook‐Spaeth D, et al. Autophagy induction results in enhanced anoikis resistance in models of peritoneal disease. Mol Cancer Res. 2017;15:26‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Peng YF, Shi YH, Shen YH, et al. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLOS One. 2013;8:e74407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Tian S, Chen Y, Yang B, et al. F1012‐2 inhibits the growth of triple negative breast cancer through induction of cell cycle arrest, apoptosis, and autophagy. Phytother Res. 2018;32:908‐922. [DOI] [PubMed] [Google Scholar]
- 164. Gurkan AC, Arisan ED, Yerlikaya PO, Ilhan H, Unsal NP. Inhibition of autophagy enhances DENSpm‐induced apoptosis in human colon cancer cells in a p53 independent manner (2018;41:297‐317. [DOI] [PubMed] [Google Scholar]
- 165. Yan X, Yang L, Feng G, et al. Lup‐20(29)‐en‐3β,28‐di‐yl‐nitrooxy acetate affects MCF‐7 proliferation through the crosstalk between apoptosis and autophagy in mitochondria. Cell Death Dis. 2018;9(2):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Song P, Ye L, Fan J, et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget. 2015;6(6):3861‐3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Fitzwalter BE, Towers CG, Sullivan KD, et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev Cell. 2018;44(5):555‐565. e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Thorburn J, Andrysik Z, Staskiewicz L, et al. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7(1):45‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Lee J‐S, Li Q, Lee J‐Y, et al. FLIP‐mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11(11):1355‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Trenti A, Grumati P, Cusinato F, Orso G, Bonaldo P, Trevisi L. Cardiac glycoside ouabain induces autophagic cell death in non‐small cell lung cancer cells via a JNK‐dependent decrease of Bcl‐2. Biochem Pharmacol. 2014;89(2):197‐209. [DOI] [PubMed] [Google Scholar]
- 171. Wiersma VR, de Bruyn M, Wei Y, et al. The epithelial polarity regulator LGALS9/galectin‐9 induces fatal frustrated autophagy in KRAS mutant colon carcinoma that depends on elevated basal autophagic flux. Autophagy. 2015;11(8):1373‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Fang J, Rhyasen G, Bolanos L, et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy‐mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood. 2012;120(4):858‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Min H, Xu M, Chen Z, et al. Bortezomib induces protective autophagy through AMP‐activated protein kinase activation in cultured pancreatic and colorectal cancer cells. Cancer Chemother Pharmacol. 2014;74(1):167‐176. [DOI] [PubMed] [Google Scholar]
- 174. Naik PP, Mukhopadhyay S, Panda PK, et al. Autophagy regulates cisplatin‐induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018;51(1):e12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Qiu S, Sun L, Jin Y, An Q, Weng C, Zheng J. Silencing of BAG3 promotes the sensitivity of ovarian cancer cells to cisplatin via inhibition of autophagy. Oncol Rep. 2017;38(1):309‐316. [DOI] [PubMed] [Google Scholar]
- 176. Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99(3):287‐292. [DOI] [PubMed] [Google Scholar]
- 177. Cook KL, Warri A, Soto‐Pantoja DR, et al. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+breast cancer. Clin Cancer Res. 2014;20(12):3222‐3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Degtyarev M, De Mazière A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN‐null tumors to lysosomotropic agents. J Cell Biol. 2008;183(1):101‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Ratikan JA, Sayre JW, Schaue D. Chloroquine engages the immune system to eradicate irradiated breast tumors in mice. Int J Radiat Oncol Biol Phys. 2013;87(4):761‐768. [DOI] [PubMed] [Google Scholar]
- 180. Wiedmer T, Blank A, Pantasis S, et al. Autophagy inhibition improves sunitinib efficacy in pancreatic neuroendocrine tumors via a lysosome‐dependent mechanism. Mol Cancer Ther. 2017;16:2502‐2515. [DOI] [PubMed] [Google Scholar]
- 181. Lu C, Xie C. Radiation‐induced autophagy promotes esophageal squamous cell carcinoma cell survival via the LKB1 pathway. Oncol Rep. 2016;35(6):3559‐3565. [DOI] [PubMed] [Google Scholar]
- 182. Ding R, Jin S, Pabon K, Scotto KW. A role for ABCG2 beyond drug transport: Regulation of autophagy. Autophagy. 2016;12(5):737‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Begicevic R‐R, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. 2017;18(11):2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wei M‐F, Chen M‐W, Chen K‐C, et al. Autophagy promotes resistance to photodynamic therapy‐induced apoptosis selectively in colorectal cancer stem‐like cells. Autophagy. 2014;10(7):1179‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ojha R, Jha V, Singh SK. Gemcitabine and mitomycin induced autophagy regulates cancer stem cell pool in urothelial carcinoma cells. Biochim Biophys Acta. 2016;1863(2):347‐359. [DOI] [PubMed] [Google Scholar]
- 186. Jang JE, Eom J‐I, Jeung H‐K, et al. AMPK‐ULK1‐mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res. 2017;23(11):2781‐2794. [DOI] [PubMed] [Google Scholar]
- 187. Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF‐1alpha and Bcl‐2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010;70(14):5942‐5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Chen Z, Gao S, Wang D, Song D, Feng Y. Colorectal cancer cells are resistant to anti‐EGFR monoclonal antibody through adapted autophagy. Am J Transl Res. 2016;8(2):1190‐1196. [PMC free article] [PubMed] [Google Scholar]
- 189. Wei Y, Zou Z, Becker N, et al. EGFR‐mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Tan X, Thapa N, Sun Y, Anderson RA. A kinase‐independent role for EGF receptor in autophagy initiation. Cell. 2015;160(1–2):145‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Li H, Chen C. Inhibition of autophagy enhances synergistic effects of Salidroside and anti‐tumor agents against colorectal cancer. BMC Complement Altern Med. 2017;17(1):538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Jang JE, Eom J‐I, Jeung H‐K, et al. Targeting AMPK‐ULK1‐mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy. 2017;13(4):761‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Torgersen ML, Engedal N, Boe S‐O, Hokland P, Simonsen A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood. 2013;122(14):2467‐2476. [DOI] [PubMed] [Google Scholar]
- 194. Jiang L, Xu L, Xie J, et al. Inhibition of autophagy overcomes glucocorticoid resistance in lymphoid malignant cells. Cancer Biol Ther. 2015;16(3):466‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Cao L, Wang J, Ma D, Wang P, Zhang Y, Fang Q. Heme oxygenase‐1 contributes to imatinib resistance by promoting autophagy in chronic myeloid leukemia through disrupting the mTOR signaling pathway. Biomed Pharmacother. 2016;78:30‐38. [DOI] [PubMed] [Google Scholar]
- 196. Wu T, Wang M‐C, Jing L, et al. Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des Devel Ther. 2015;9:6421‐6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Altman JK, Szilard A, Goussetis DJ, et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014;20(9):2400‐2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Tong Y, Huang H, Pan H. Inhibition of MEK/ERK activation attenuates autophagy and potentiates pemetrexed‐induced activity against HepG2 hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2015;456(1):86‐91. [DOI] [PubMed] [Google Scholar]
- 199. Sanduja S, Feng Y, Mathis RA, et al. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene. 2016;35(40):5295‐5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Ma X‐H, Piao S‐F, Dey S, et al. Stress‐induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124(3):1406‐1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Corazzari M, Rapino F, Ciccosanti F, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015;22(6):946‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Xia J, Yu X, Song X, Li G, Mao X, Zhang Y. Inhibiting the cytoplasmic location of HMGB1 reverses cisplatin resistance in human cervical cancer cells. Mol Med Rep. 2017;15(1):488‐494. [DOI] [PubMed] [Google Scholar]
- 203. Shrivastava S, Mansure JJ, Almajed W, et al. The role of HMGB1 in radioresistance of bladder cancer. Mol Cancer Ther. 2016;15(3):471‐479. [DOI] [PubMed] [Google Scholar]
- 204. Wang L, Zhang H, Sun M, Yin Z, Qian J. High mobility group box 1‐mediated autophagy promotes neuroblastoma cell chemoresistance. Oncol Rep. 2015;34(6):2969‐2976. [DOI] [PubMed] [Google Scholar]
- 205. Zhang Y‐X, Yuan Y‐Q, Zhang X‐Q, Huang D‐L, Wei Y‐Y, Yang J‐G. HMGB1‐mediated autophagy confers resistance to gemcitabine in hormone‐independent prostate cancer cells. Oncol Lett. 2017;14(5):6285‐6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Zhang R, Li Y, Wang Z, Chen L, Dong X, Nie X. Interference with HMGB1 increases the sensitivity to chemotherapy drugs by inhibiting HMGB1‐mediated cell autophagy and inducing cell apoptosis. Tumour Biol. 2015;36(11):8585‐8592. [DOI] [PubMed] [Google Scholar]
- 207. Pan B, Chen D, Huang J, et al. HMGB1‐mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol Cancer. 2014;13:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Yang L, Yu Y, Kang R, et al. Up‐regulated autophagy by endogenous high mobility group box‐1 promotes chemoresistance in leukemia cells. Leuk Lymphoma. 2012;53(2):315‐322. [DOI] [PubMed] [Google Scholar]
- 209. Wang P, Zhang J, Zhang L, et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology. 2013;145(5):1133‐1143. e12 [DOI] [PubMed] [Google Scholar]
- 210. Zou J, Liu L, Wang Q, et al. Downregulation of miR‐429 contributes to the development of drug resistance in epithelial ovarian cancer by targeting ZEB1. Am J Transl Res. 2017;9(3):1357‐1368. [PMC free article] [PubMed] [Google Scholar]
- 211. Xu R, Liu S, Chen H, Lao L. MicroRNA‐30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin‐1‐mediated autophagy. Oncol Rep. 2016;35(3):1757‐1763. [DOI] [PubMed] [Google Scholar]
- 212. Wei R, Cao G, Deng Z, Su J, Cai L. MiR‐140‐5p attenuates chemotherapeutic drug‐induced cell death by regulating autophagy through inositol 1,4,5‐trisphosphate kinase 2 (IP3k2) in human osteosarcoma cells. Biosci Rep. 2016;36(5):e00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Notte A, Ninane N, Arnould T, Michiels C. Hypoxia counteracts taxol‐induced apoptosis in MDA‐MB‐231 breast cancer cells: role of autophagy and JNK activation. Cell Death Dis. 2013;4:e638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Bousquet G, Bouchtaoui ME, Sophie T, et al. Targeting autophagic cancer stem‐cells to reverse chemoresistance in human triple negative breast cancer. Oncotarget. 2017;8(21):35205‐35221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Zhou W, Xu G, Wang Y, et al. Oxidative stress induced autophagy in cancer associated fibroblast enhances proliferation and metabolism of colorectal cancer cells. Cell Cycle. 2017;16(1):73‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. New J, Arnold L, Ananth M, et al. Secretory autophagy in cancer‐associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017;77(23):6679‐6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Martinez‐Outschoorn UE, Balliet RM, Rivadeneira D, et al. Oxidative stress in cancer associated fibroblasts drives tumor‐stroma co‐evolution: a new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9(16):3256‐3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. van Gosliga D, Schepers H, Rizo A, van der Kolk D, Vellenga E, Schuringa JJ. Establishing long‐term cultures with self‐renewing acute myeloid leukemia stem/progenitor cells. Exp Hematol. 2007;35(10):1538‐1549. [DOI] [PubMed] [Google Scholar]
- 219. Schuringa JJ, Schepers H. Ex vivo assays to study self‐renewal and long‐term expansion of genetically modified primary human acute myeloid leukemia stem cells. Methods Mol Biol. 2009;538:287‐300. [DOI] [PubMed] [Google Scholar]
- 220. Carretta M, de Boer B, Jaques J, et al. Genetically engineered mesenchymal stromal cells produce IL‐3 and TPO to further improve human scaffold‐based xenograft models. Exp Hematol. 2017;51:36‐46. [DOI] [PubMed] [Google Scholar]
- 221. Wang Q, Xue L, Zhang X, Bu S, Zhu X, Lai D. Autophagy protects ovarian cancer‐associated fibroblasts against oxidative stress. Cell Cycle. 2016;15(10):1376‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Ko Y‐H, Lin Z, Flomenberg N, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biol Ther. 2011;12(12):1085‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Martinez‐Outschoorn UE, Trimmer C, Lin Z, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9(17):3515‐3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Zhang M‐H, Hu Y‐D, Xu Y, et al. Human mesenchymal stem cells enhance autophagy of lung carcinoma cells against apoptosis during serum deprivation. Int J Oncol. 2013;42(4):1390‐1398. [DOI] [PubMed] [Google Scholar]
- 225. Wheeler SE, Shi H, Lin F, et al. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor‐associated fibroblasts in preclinical models. Head Neck. 2014;36(3):385‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang Y, Gan G, Wang B, et al. Cancer‐associated fibroblasts promote irradiated cancer cell recovery through autophagy. EBioMedicine. 2017;17:45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Karakas HE, Kim J, Park J, et al. A microfluidic chip for screening individual cancer cells via eavesdropping on autophagy‐inducing crosstalk in the stroma niche. Sci Rep. 2017;7(1):2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Piya S, Kornblau SM, Ruvolo VR, et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma‐mediated chemoresistance in acute myeloid leukemia. Blood. 2016;128(9):1260‐1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Chiavarina B, Whitaker‐Menezes D, Migneco G, et al. HIF1‐alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment‐specific oncogenesis. Cell Cycle. 2010;9(17):3534‐3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Salem AF, Whitaker‐Menezes D, Lin Z, et al. Two‐compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012;11(13):2545‐2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Capparelli C, Guido C, Whitaker‐Menezes D, et al. Autophagy and senescence in cancer‐associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11(12):2285‐2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co‐selecting interconnected physiological pathways. Cancer Metab. 2014;2(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Sloan EK, Ciocca DR, Pouliot N, et al. Stromal cell expression of caveolin‐1 predicts outcome in breast cancer. Am J Pathol. 2009;174(6):2035‐2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Ma X, Liu L, Nie W, et al. Prognostic role of caveolin in breast cancer: a meta‐analysis. Breast. 2013;22(4):462‐469. [DOI] [PubMed] [Google Scholar]
- 235. Capparelli C, Whitaker‐Menezes D, Guido C, et al. CTGF drives autophagy, glycolysis and senescence in cancer‐associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11(12):2272‐2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Castello‐Cros R, Bonuccelli G, Molchansky A, et al. Matrix remodeling stimulates stromal autophagy, “fueling” cancer cell mitochondrial metabolism and metastasis. Cell Cycle. 2011;10(12):2021‐2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Du J, Teng R‐J, Guan T, et al. Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol Cell Physiol. 2012;302(2):C383‐C391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Sachdev U, Cui X, Hong G, et al. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J Vasc Surg. 2012;55(1):180‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Lee S‐J, Kim H‐P, Jin Y, Choi AMK, Ryter SW. Beclin 1 deficiency is associated with increased hypoxia‐induced angiogenesis. Autophagy. 2011;7(8):829‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lu W‐H, Shi Y‐X, Ma Z‐L, et al. Proper autophagy is indispensable for angiogenesis during chick embryo development. Cell Cycle. 2016;15(13):1742‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Fruci D, Benevolo M, Cifaldi L, et al. Major histocompatibility complex class i and tumour immuno‐evasion: how to fool T‐cells and natural killer cells at one time. Curr Oncol. 2012;19(1):39‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Li B, Lei Z, Lichty BD, et al. Autophagy facilitates major histocompatibility complex class I expression induced by IFN‐γ in B16 melanoma cells. Cancer Immunol Immunother. 2010;59(2):313‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Tittarelli A, Mendoza‐Naranjo A, Farías M, et al. Gap junction intercellular communications regulate NK cell activation and modulate NK cytotoxic capacity. J Immunol. 2014;192(3):1313‐1319. [DOI] [PubMed] [Google Scholar]
- 245. Tittarelli A, Janji B, Van Moer K, Noman MZ, Chouaib S. The selective degradation of synaptic connexin 43 protein by hypoxia‐induced autophagy impairs natural killer cell‐mediated tumor cell killing. J Biol Chem. 2015;290(39):23670‐23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Baginska J, Viry E, Berchem G, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer‐mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110(43):17450‐17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Noman MZ, Janji B, Kaminska B, et al. Blocking hypoxia‐induced autophagy in tumors restores cytotoxic T‐cell activity and promotes regression. Cancer Res. 2011;71(18):5976‐5986. [DOI] [PubMed] [Google Scholar]
- 248. Noman MZ, Buart S, Van Pelt J, et al. The cooperative induction of hypoxia‐inducible factor‐1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL‐mediated cell lysis. J Immunol. 2009;182(6):3510‐3521. [DOI] [PubMed] [Google Scholar]
- 249. Messai Y, Noman MZ, Hasmim M, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014;74(23):6820‐6832. [DOI] [PubMed] [Google Scholar]
- 250. Khazen R, Müller S, Gaudenzio N, Espinosa E, Puissegur M‐P, Valitutti S. Melanoma cell lysosome secretory burst neutralizes the CTL‐mediated cytotoxicity at the lytic synapse. Nat Commun. 2016;7:10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Akalay I, Janji B, Hasmim M, et al. Epithelial‐to‐mesenchymal transition and autophagy induction in breast carcinoma promote escape from T‐cell‐mediated lysis. Cancer Res. 2013;73(8):2418‐2427. [DOI] [PubMed] [Google Scholar]
- 252. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Lastwika KJ, Wilson W, Li QK, et al. Control of PD‐L1 expression by oncogenic activation of the AKT‐mTOR pathway in non‐small cell lung cancer. Cancer Res. 2016;76(2):227‐238. [DOI] [PubMed] [Google Scholar]
- 255. Clark CA, Gupta HB, Sareddy G, et al. Tumor‐intrinsic PD‐L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 2016;76(23):6964‐6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Zhao L, Li C, Liu F, et al. A blockade of PD‐L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2017; 10:2115‐2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54‐61. [DOI] [PubMed] [Google Scholar]
- 258. Michaud M, Martins I, Sukkurwala AQ, et al. Autophagy‐dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573‐1577. [DOI] [PubMed] [Google Scholar]
- 259. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll‐like receptor 4‐dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050‐1059. [DOI] [PubMed] [Google Scholar]
- 260. Li D‐D, Xie B, Wu X‐J, et al. Late‐stage inhibition of autophagy enhances calreticulin surface exposure. Oncotarget. 2016;7(49):80842‐80854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Garg AD, Dudek AM, Ferreira GB, et al. ROS‐induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy. 2013;9(9):1292‐1307. [DOI] [PubMed] [Google Scholar]
- 262. Panaretakis T, Kepp O, Brockmeier U, et al. Mechanisms of pre‐apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Thibodeau J, Bourgeois‐Daigneault M‐C, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology. 2012;1(6):908‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Accolla RS, Lombardo L, Abdallah R, Raval G, Forlani G, Tosi G. Boosting the MHC Class II‐restricted tumor antigen presentation to CD4+T helper cells: a critical issue for triggering protective immunity and re‐orienting the tumor microenvironment toward an anti‐tumor state. Front Oncol. 2014;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Dickgreber N, Stoitzner P, Bai Y, et al. Targeting antigen to MHC class II molecules promotes efficient cross‐presentation and enhances immunotherapy. J Immunol. 2009;182(3):1260‐1269. [DOI] [PubMed] [Google Scholar]
- 266. Loi M, Müller A, Steinbach K, et al. Macroautophagy proteins control MHC Class I levels on dendritic cells and shape anti‐viral CD8(+) T‐cells responses. Cell Rep. 2016;15(5):1076‐1087. [DOI] [PubMed] [Google Scholar]
- 267. Parekh VV, Wu L, Boyd KL, et al. Impaired autophagy, defective T‐cells homeostasis, and a wasting syndrome in mice with a T‐cells‐specific deletion of Vps34. J Immunol. 2013;190(10):5086‐5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Choi S‐H, Gonen A, Diehl CJ, et al. SYK regulates macrophage MHC‐II expression via activation of autophagy in response to oxidized LDL. Autophagy. 2015;11(5):785‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Khan N, Vidyarthi A, Pahari S, et al. Signaling through NOD‐2 and TLR‐4 bolsters the T‐cells priming capability of dendritic cells by inducing autophagy. Sci Rep. 2016;6:19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Mintern JD, Macri C, Chin WJ, et al. Differential use of autophagy by primary dendritic cells specialized in cross‐presentation. Autophagy. 2015;11(6):906‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Tey S‐K, Khanna R. Autophagy mediates transporter associated with antigen processing‐independent presentation of viral epitopes through MHC class I pathway. Blood. 2012;120(5):994‐1004. [DOI] [PubMed] [Google Scholar]
- 272. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15(3):267‐276. [DOI] [PubMed] [Google Scholar]
- 273. Lee HK, Mattei LM, Steinberg BE, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32(2):227‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Thiele F, Tao S, Zhang Y, et al. Modified vaccinia virus Ankara‐infected dendritic cells present CD4+T‐cell epitopes by endogenous major histocompatibility complex class II presentation pathways. J Virol. 2015;89(5):2698‐2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Parekh VV, Pabbisetty SK, Wu L, et al. Autophagy‐related protein Vps34 controls the homeostasis and function of antigen cross‐presenting CD8α+dendritic cells. Proc Natl Acad Sci U S A. 2017;114(31):E6371‐E6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T‐cells immunity in melanoma. Cancer Cell. 2016;30(2):324‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102(22):7922‐7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He Y‐W. A critical role for the autophagy gene Atg5 in T‐cells survival and proliferation. J Exp Med. 2007;204(1):25‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Schlie K, Westerback A, DeVorkin L, et al. Survival of effector CD8+T‐cells during influenza infection is dependent on autophagy. J Immunol. 2015;194(9):4277‐4286. [DOI] [PubMed] [Google Scholar]
- 280. Puleston DJ, Zhang H, Powell TJ, et al. Autophagy is a critical regulator of memory CD8(+) T‐cell formation. ELife. 2014;3:e0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Oami T, Watanabe E, Hatano M, et al. Suppression of T‐cell autophagy results in decreased viability and function of T‐cells through accelerated apoptosis in a murine sepsis model. Crit Care Med. 2017;45(1):e77‐e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Lin C‐W, Lo S, Hsu C, et al. T‐cell autophagy deficiency increases mortality and suppresses immune responses after sepsis. PLOS One. 2014;9(7):e102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Jia W, He Y‐W. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. 2011;186(9):5313‐5322. [DOI] [PubMed] [Google Scholar]
- 284. Kovacs JR, Li C, Yang Q, et al. Autophagy promotes T‐cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012;19(1):144‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide‐3 kinase Vps34 is required for naive T‐cell homeostasis. Proc Natl Acad Sci U S A. 2012;109(22):8670‐8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Roy SG, Stevens MW, So L, Edinger AL. Reciprocal effects of rab7 deletion in activated and neglected T‐cells. Autophagy. 2013;9(7):1009‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Pua HH, Guo J, Komatsu M, He Y‐W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182(7):4046‐4055. [DOI] [PubMed] [Google Scholar]
- 288. Li C, Capan E, Zhao Y, et al. Autophagy is induced in CD4+T‐cells and important for the growth factor‐withdrawal cell death. J Immunol. 2006;177(8):5163‐5168. [DOI] [PubMed] [Google Scholar]
- 289. Watanabe R, Fujii H, Shirai T, Saito S, Ishii T, Harigae H. Autophagy plays a protective role as an anti‐oxidant system in human T‐cells and represents a novel strategy for induction of T‐cell apoptosis. Eur J Immunol. 2014;44(8):2508‐2520. [DOI] [PubMed] [Google Scholar]
- 290. Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T‐cell activation. J Immunol. 2010;185(12):7349‐7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Arnold CR, Pritz T, Brunner S, et al. T‐cell receptor‐mediated activation is a potent inducer of macroautophagy in human CD8(+)CD28(+) T‐cells but not in CD8(+)CD28(‐) T‐cells. Exp Gerontol. 2014;54:75‐83. [DOI] [PubMed] [Google Scholar]
- 292. Botbol Y, Patel B, Macian F. Common γ‐chain cytokine signaling is required for macroautophagy induction during CD4+T‐cell activation. Autophagy. 2015;11(10):1864‐1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Valdor R, Mocholi E, Botbol Y, et al. Chaperone‐mediated autophagy regulates T‐cell responses through targeted degradation of negative regulators of T‐cell activation. Nat Immunol. 2014;15(11):1046‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Jia W, He M‐X, McLeod IX, Guo J, Ji D, He Y‐W. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell‐cycle inhibitor CDKN1B/p27Kip1. Autophagy. 2015;11(12):2335‐2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Matsuzawa Y, Oshima S, Takahara M, et al. TNFAIP3 promotes survival of CD4 T‐cells by restricting MTOR and promoting autophagy. Autophagy. 2015;11(7):1052‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Xu X, Araki K, Li S, et al. Autophagy is essential for effector CD8(+) T‐cell survival and memory formation. Nat Immunol. 2014;15(12):1152‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Voehringer D, Liang H‐E, Locksley RM. Homeostasis and effector function of lymphopenia‐induced “memory‐like” T‐cells in constitutively T‐cell‐depleted mice. J Immunol. 2008;180(7):4742‐4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Paul S, Kashyap AK, Jia W, He Y‐W, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T‐cell receptor activation of NF‐κB. Immunity. 2012;36(6):947‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Vegran F, Apetoh L, Ghiringhelli F. Th9 cells: a novel CD4 T‐cell subset in the immune war against cancer. Cancer Res. 2015;75(3):475‐479. [DOI] [PubMed] [Google Scholar]
- 300. Lu Y, Hong S, Li H, et al. Th9 cells promote antitumor immune responses in vivo. J Clin Invest. 2012;122(11):4160‐4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Rivera Vargas T, Cai Z, Shen Y, et al. Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nat Commun. 2017;8(1):559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Zhou Y, Shao N, Aierken N, et al. Prognostic value of tumor‐infiltrating Foxp3+regulatory T‐cells in patients with breast cancer: a meta‐analysis. J Cancer. 2017;8(19):4098‐4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. de Jong RA, Leffers N, Boezen HM, et al. Presence of tumor‐infiltrating lymphocytes is an independent prognostic factor in type I and II endometrial cancer. Gynecol Oncol. 2009;114(1):105‐110. [DOI] [PubMed] [Google Scholar]
- 304. Wei J, Long L, Yang K, et al. Autophagy enforces functional integrity of regulatory T‐cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016;17(3):277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D, Pronk CJ. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLOS One. 2016;11(7):e0158369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Cho RH, Sieburg HB, Muller‐Sieburg CE. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood. 2008;111(12):5553‐5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208(13):2691‐2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Beerman I, Bhattacharya D, Zandi S, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107(12):5465‐5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Rossi DJ, Bryder D, Zahn JM, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194‐9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Zhang H, Puleston DJ, Simon AK. Autophagy and immune senescence. Trends Mol Med. 2016;22(8):671‐686. [DOI] [PubMed] [Google Scholar]
- 311. Phadwal K, Alegre‐Abarrategui J, Watson AS, et al. A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T‐cells. Autophagy. 2012;8(4):677‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Kang C, Elledge SJ. How autophagy both activates and inhibits cellular senescence. Autophagy. 2016;12(5):898‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Young ARJ, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Capparelli C, Chiavarina B, Whitaker‐Menezes D, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer‐associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo‐angiogenesis. Cell Cycle. 2012;11(19):3599‐3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Huang Y‐H, Yang P‐M, Chuah Q‐Y, et al. Autophagy promotes radiation‐induced senescence but inhibits bystander effects in human breast cancer cells. Autophagy. 2014;10(7):1212‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLOS One. 2011;6(8):e23367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. García‐Prat L, Martínez‐Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37‐42. [DOI] [PubMed] [Google Scholar]
- 319. Kang C, Xu Q, Martin TD, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Kwon Y, Kim JW, Jeoung JA, Kim M‐S, Kang C. Autophagy is pro‐senescence when seen in close‐up, but anti‐senescence in long‐shot. Mol Cells. 2017;40(9):607‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Schmidt RLJ, Jutz S, Goldhahn K, et al. Chloroquine inhibits human CD4+T‐cell activation by AP‐1 signaling modulation. Sci Rep. 2017;7:42191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Rangwala R, Leone R, Chang YC, et al. Phase I trial of hydroxychloroquine with dose‐intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1369‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Rosenfeld MR, Ye X, Supko JG, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10(8):1359‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. McAfee Q, Zhang Z, Samanta A, et al. Autophagy inhibitor Lys05 has single‐agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109(21):8253‐8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. De Mei C, Ercolani L, Parodi C, et al. Dual inhibition of REV‐ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene. 2015;34(20):2597‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Galluzzi L, Bravo‐San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16(7):487‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Ragon BK, Kantarjian H, Jabbour E, et al. Buparlisib, a PI3K inhibitor, demonstrates acceptable tolerability and preliminary activity in a phase I trial of patients with advanced leukemias. Am J Hematol. 2017;92(1):7‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Rodon J, Braña I, Siu LL, et al. Phase I dose‐escalation and ‐expansion study of buparlisib (BKM120), an oral pan‐Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32(4):670‐681. [DOI] [PubMed] [Google Scholar]
- 330. Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol. 2015;10(1):257‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Petherick KJ, Conway OJL, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)‐dependent autophagy. J Biol Chem. 2015;290(18):11376‐11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Egan DF, Chun MGH, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59(2):285‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Tang F, Hu P, Yang Z, et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non‐small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol Rep. 2017;37(6):3449‐3458. [DOI] [PubMed] [Google Scholar]
- 334. Barenholz Y. Doxil®—the first FDA‐approved nano‐drug: lessons learned. J Control Release. 2012;160(2):117‐134. [DOI] [PubMed] [Google Scholar]
- 335. Sofou S, Sgouros G. Antibody‐targeted liposomes in cancer therapy and imaging. Expert Opin Drug Deliv. 2008;5(2):189‐204. [DOI] [PubMed] [Google Scholar]
- 336. Khan MA, Jabeen R, Nasti TH, Mohammad O. Enhanced anticryptococcal activity of chloroquine in phosphatidylserine‐containing liposomes in a murine model. J Antimicrob Chemother. 2005;55(2):223‐228. [DOI] [PubMed] [Google Scholar]
- 337. Qiu L, Jing N, Jin Y. Preparation and in vitro evaluation of liposomal chloroquine diphosphate loaded by a transmembrane pH‐gradient method. Int J Pharm. 2008;361(1–2):56‐63. [DOI] [PubMed] [Google Scholar]
- 338. Ibrahim S, Tagami T, Ozeki T. Effective‐loading of platinum‐chloroquine into PEGylated neutral and cationic liposomes as a drug delivery system for resistant malaria parasites. Biol Pharm Bull. 2017;40(6):815‐823. [DOI] [PubMed] [Google Scholar]
- 339. Xie F, Zhang S, Liu J, et al. Codelivery of salinomycin and chloroquine by liposomes enables synergistic antitumor activity in vitro. Nanomed. 2016;11(14):1831‐1846. [DOI] [PubMed] [Google Scholar]
- 340. Qiu L, Yao M, Gao M, Zhao Q. Doxorubicin and chloroquine coencapsulated liposomes: preparation and improved cytotoxicity on human breast cancer cells. J Liposome Res. 2012;22(3):245‐253. [DOI] [PubMed] [Google Scholar]
- 341. Gao M, Xu Y, Qiu L. Sensitization of multidrug‐resistant malignant cells by liposomes co‐encapsulating doxorubicin and chloroquine through autophagic inhibition. J Liposome Res. 2017;27(2):151‐160. [DOI] [PubMed] [Google Scholar]
- 342. Hendriks D, Choi G, de Bruyn M, Wiersma VR, Bremer E. Antibody‐based cancer therapy: successful agents and novel approaches. Int Rev Cell Mol Biol. 2017;331:289‐383. [DOI] [PubMed] [Google Scholar]
- 343. Fonteneau JF, Brilot F, Münz C, Gannagé M. The tumor antigen NY‐ESO‐1 mediates direct recognition of melanoma cells by CD4+T‐cells after intercellular antigen transfer. J Immunol. 2016;196(1):64‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Page DB, Hulett TW, Hilton TL, Hu H‐M, Urba WJ, Fox BA. Glimpse into the future: harnessing autophagy to promote anti‐tumor immunity with the DRibbles vaccine. J Immunother Cancer. 2016;4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Li Y, Wang L‐X, Yang G, Hao F, Urba WJ, Hu H‐M. Efficient cross‐presentation depends on autophagy in tumor cells. Cancer Res. 2008;68(17):6889‐6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Li Y, Wang L‐X, Pang P, et al. Tumor‐derived autophagosome vaccine: mechanism of cross‐presentation and therapeutic efficacy. Clin Cancer Res. 2011;17(22):7047‐7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Twitty CG, Jensen SM, Hu H‐M, Fox BA. Tumor‐derived autophagosome vaccine: induction of cross‐protective immune responses against short‐lived proteins through a p62‐dependent mechanism. Clin Cancer Res. 2011;17(20):6467‐6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Zhou M, Wen Z, Cheng F, et al. Tumor‐released autophagosomes induce IL‐10‐producing B cells with suppressive activity on T lymphocytes via TLR2‐MyD88‐NF‐κB signal pathway. Oncoimmunology. 2016;5(7):e1180485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Goldberg SB, Supko JG, Neal JW, et al. A phase I study of erlotinib and hydroxychloroquine in advanced non‐small‐cell lung cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. 2012;7(10):1602‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. The Oncologist. 2014;19(6):637‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Boone BA, Bahary N, Zureikat AH, et al. Safety and biologic response of pre‐operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2015;22(13):4402‐4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Chi M‐S, Lee C‐Y, Huang S‐C, et al. Double autophagy modulators reduce 2‐deoxyglucose uptake in sarcoma patients. Oncotarget. 2015;6(30):29808‐29817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Nti AA, Serrano LW, Sandhu HS, et al. Frequent subclinical macular changes in combined BRAF/MEK inhibition with high‐dose hydroxychloroquine as treatment for advanced metastatic BRAF mutant melanoma: Preliminary results from a phase I/II clinical treatment trial. Retina Phila Pa 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Scott EC, Maziarz RT, Spurgeon SE, et al. Double autophagy stimulation using chemotherapy and mTOR inhibition combined with hydroxychloroquine for autophagy modulation in patients with relapsed or refractory multiple myeloma. Haematologica. 2017;102(7):e261‐e265. [DOI] [PMC free article] [PubMed] [Google Scholar]