

Abstract
The liver bears unique immune properties that support both immune tolerance and immunity, but the mechanisms responsible for clearance versus persistence of virus‐infected hepatocytes remain unclear. Here, we dissect the factors determining the outcome of antiviral immunity using recombinant adenoviruses that reflect the hepatropism and hepatrophism of hepatitis viruses. We generated replication‐deficient adenoviruses with equimolar expression of ovalbumin, luciferase, and green fluorescent protein driven by a strong ubiquitous cytomegalovirus (CMV) promoter (Ad‐CMV‐GOL) or by 100‐fold weaker, yet hepatocyte‐specific, transthyretin (TTR) promoter (Ad‐TTR‐GOL). Using in vivo bioluminescence to quantitatively and dynamically image luciferase activity, we demonstrated that Ad‐TTR‐GOL infection always persists, whereas Ad‐CMV‐GOL infection is always cleared, independent of the number of infected hepatocytes. Failure to clear Ad‐TTR‐GOL infection involved mechanisms acting during initiation as well as execution of antigen‐specific immunity. First, hepatocyte‐restricted antigen expression led to delayed and curtailed T‐cell expansion—10,000‐fold after Ad‐CMV‐GOL versus 150‐fold after Ad‐TTR‐GOL‐infection. Second, CD8 T‐cells primed toward antigens selectively expressed by hepatocytes showed high PD‐1/Tim‐3/LAG‐3/CTLA‐4/CD160 expression levels similar to that seen in chronic hepatitis B. Third, Ad‐TTR‐GOL but not Ad‐CMV‐GOL‐infected hepatocytes escaped being killed by effector T‐cells while still inducing high PD‐1/Tim‐3/LAG‐3/CTLA‐4/CD160 expression, indicating different thresholds of T‐cell receptor signaling relevant for triggering effector functions compared with exhaustion. Conclusion: Our study identifies deficits in the generation of CD8 T‐cell immunity toward hepatocyte‐expressed antigens and escape of infected hepatocytes expressing low viral antigen levels from effector T‐cell killing as independent factors promoting viral persistence. This highlights the importance of addressing both the restauration of CD8 T‐cell dysfunction and overcoming local hurdles of effector T‐cell function to eliminate virus‐infected hepatocytes.
Abbreviations
- CFSE
carboxyfluorescein succinimidyl ester
- CMV
cytomegalovirus
- DC
dendritic cell
- EGFP
enhanced green fluorescent protein
- GFP
green fluorescent protein
- HBV
hepatitis B virus
- IFN
interferon
- IL
interleukin
- LSEC
liver sinusoidal endothelial cell
- MHC
major histocompatibility complex
- moDC
monocyte‐derived dendritic cell
- PFU
plaque‐forming units
- p.i.
post injection
- sALT
serum alanine aminotransferase
- TCR
T‐cell receptor
- TTR
transthyretin
The liver bears particular immune competence through its unique microenvironment together with liver‐resident, antigen‐presenting cell populations that regulate both local and systemic immune responses.1, 2, 3 The incidence of infection with hepatitis viruses A to E is high and more than 300 million persons suffer from chronic viral hepatitis B, C, and D.4, 5 The factors leading to the failure of the immune response clearing virus‐infected hepatocytes are still not entirely understood. Exploring the mechanistic basis of local regulation of antiviral immunity in the liver that determines clearance or persistence is important to understand disease pathogenesis and to develop future successful immune therapies.
It is well recognized that CD8 T‐cell immunity against virus‐infected hepatocytes is controlled at various levels, such as inefficient intrahepatic priming of CD8 T‐cells by antigen‐presenting hepatocytes resulting in clonal deletion,6, 7 by control of CD8 T‐cell effector function either through regulatory T‐cells8 or myeloid‐derived suppressor cells,9 through natural killer cells killing activated virus‐specific CD8 T‐cells,10 or induction of CD8 T‐cell dysfunction.11, 12 Local antigen presentation appears to play a major role during regulation of CD8 T‐cell immunity in the liver2, 3: (1) Tolerogenic intrahepatic dendritic cells (DCs) contribute to transplantation tolerance13; (2) liver sinusoidal endothelial cells (LSECs) cross‐prime CD8 T‐cells to generate memory T‐cells with proliferative potential that relocate to lymphoid tissue; and (3) cross‐presenting LSECs activate circulating effector CD8 T‐cells, triggering a noncanonical tumor necrosis factor (TNF)‐mediated T‐cell effector function that eliminates virus‐infected hepatocytes.14, 15 High levels of circulating viral antigens are associated with the induction of CD8 T‐cell nonresponsiveness in experimental viral infection models such as lymphocytic choriomeningitis virus (LCMV) and in hepatitis virus infection.16 In particular, hepatitis B virus (HBV) infection shows liver tropism and hepatocyte‐restricted expression of viral antigens together with no measurable induction of innate immunity.17, 18 This combination of liver targeting, hepatocyte‐restricted antigen expression and release of soluble antigen with low innate immune activation poses a challenge for the immune system to mount protective immunity. Although certain antigen‐presenting cell populations in lymphoid tissues are specialized for viral gene expression to induce CD8 T‐cell immunity, cross‐priming by specialized DCs in the presence of type I interferon signaling allows for induction of protective CD8 T‐cell immunity.19 However, infection with hepatitis viruses, in particular HBV, escapes both of these mechanisms through hepatocyte‐restricted gene expression and absence of interferon induction.
We generated recombinant adenoviruses in which expression of secreted antigen is driven by the hepatocyte‐specific transthyretin (TTR) promoter linked through 2A sites to luciferase and enhanced GFP (EGFP) expression (Ad‐TTR‐GOL). Luciferase allows for sensitive detection of infected hepatocytes by in vivo bioluminescence measurement. We discovered that infection with Ad‐TTR‐GOL, which was characterized by hepatocyte‐selective, but low antigen expression, was not eliminated, whereas infection with the same virus, but cytomegalovirus (CMV) promoter–driven antigen expression (Ad‐CMV‐GOL), was cleared within 18 days. Co‐infection with both viruses revealed that effector CD8 T‐cells were not able to clear Ad‐TTR‐GOL infection, indicating a so‐far unappreciated role of low‐level antigen expression in infected hepatocytes for immune escape.
Materials and Methods
Mice and Patient Samples
C57Bl/6 mice were purchased from Charles River (Sulzfeld, Germany). H‐2Kb‐restricted T‐cell receptor (TCR) transgenic CD45.1+ OT‐1 mice were bred and maintained under specific pathogen‐free conditions in the central animal facility of the Klinikum Rechts der Isar according to the guidelines of the Federation of Laboratory Animal Science Association. Male mice between the ages of 6 and 10 weeks were used. Cell depletion was injected intravenously with 30 µg antimouse CD8α (clone 2.43, BioXCell), 300 µg antimouse CD4 (GK1.5, BioXCell), or 300 µg antimouse NK1.1 (PK136, BioXCell).
Patients with the diagnosis of chronic HBV infection who presented at the University Medical Center Freiburg outpatient liver center were recruited in the study after obtaining written informed consent from each patient and approval by the Ethics Committee of the Albert Ludwigs University of Freiburg (Germany). Intrahepatic lymphocytes, obtained during diagnostic liver biopsy, were compared with peripheral immune responses and analyzed for HBV‐specific immune responses and expression of inhibitory receptors. All investigations were conducted according to the principles expressed in the Declaration of Helsinki.
IN VIVO Bioluminescence Imaging
Mice anesthetized with 2.5% Isoflurane and D‐Luciferin‐K‐Salt (PJK GmbH, Germany) were injected intraperitoneally at 100 mg/kg body weight 5 minutes before bioluminescence imaging. In vivo bioluminescence imaging was performed with an IVIS Lumina LT‐Series III instrument (PerkinElmer LAS, Germany).
IN VITRO CD8 T‐Cell Activation and Proliferation Assays
CD44low naïve carboxyfluorescein succinimidyl ester (CFSE) labeled (2 µM) OT‐1 CD8 T‐cells were isolated by MACS sorting (Milteny Biotec, Germany) and incubated with splenic CD11c+ DCs isolated from the spleen after 6 hours of infection with Ad‐CMV‐GOL or Ad‐TTR‐GOL. After 1 and 3 days of coculture, CD8 T‐cells were harvested and analyzed by flow cytometry for dilution of CFSE as a marker for proliferation.
CD8 T‐Cell Cytotoxicity Assay
Freshly isolated primary murine hepatocytes were seeded on collagenR‐coated 96‐well E‐plates (ACEA Biosciences) in William’s E (WE) medium (PAN Biotech, Germany) containing 200 mM Glutamine, 1 M Hepes pH 7.4, 10,000 U/mL Pen‐Strep, 50 mg/mL gentamycin, 0.005 ng/mL insulin, 1.6% DMSO, and 10% fetal bovine serum (FBS). After seeding, the medium was exchanged to a medium containing 1% FBS. Twenty hours after seeding, cells were washed with Hank’s balanced salt solution and infected with Ad‐CMV‐GOL or Ad‐TTR‐GOL at a multiplicity of infection (moi) of 5. Forty‐eight hours after infection, the medium was exchanged and in vitro–activated OT‐1 CD8 T‐cells were added at an effector‐to‐target ratio of 10:1. For activation of OT‐1 CD8 T‐cells, splenocytes from OT‐1 transgenic mice were cultured for 72 hours with 100 µg/mL ovalbumin. Hepatocyte cell death was measured in the impedance‐based Xcelligence RTCA MP (ACEA Biosciences) device.
Results
Generation of Recombinant Adenoviruses for Hepatocyte‐Specific Gene Expression
To characterize antiviral CD8 T‐cell immunity against infected hepatocytes, we generated recombinant replication‐deficient adenoviruses with known hepatotropism20 coding for an antigen together with reporter molecules. We used the immunologically well‐characterized antigen ovalbumin that is secreted by infected cells.21 We cloned ovalbumin with EGFP and luciferase and/or iRFP720, as markers to quantitatively detect infection, into an expression cassette linking the genes through 2A sites. This ensures that the luciferase signal detected by in vivo bioluminescence directly correlates with the amount of expressed ovalbumin.22 We used a CMV promoter to drive strong gene expression in every infected cell (Ad‐CMV‐GOL and Ad‐CMV‐GiRO). For selective gene expression in hepatocytes, we generated an adenovirus with gene expression controlled by the hepatocyte‐restricted TTR promoter (Ad‐TTR‐GOL), thus combining adenoviral hepatotropism with hepatocyte‐specific gene expression (Supporting Fig. S1A). Infection with these recombinant adenoviruses led to clear reporter signals in the liver (Fig. 1A). Consistent with the known adenoviral hepatotropism, we detected bioluminescence or iRFP720 fluorescence for both viruses in isolated livers, but not lung, kidneys, gut, or heart (Fig. 1B). Importantly, we found a weaker bioluminescence signal after Ad‐TTR‐GOL, compared with Ad‐CMV‐GOL infection. We therefore performed a dose titration of Ad‐TTR‐GOL and Ad‐CMV‐GOL, revealing a direct correlation between infectious dose and bioluminescence signal for both viruses (Fig. 1C). Comparing infection with similar numbers of Ad‐CMV‐GOL or Ad‐TTR‐GOL showed that TTR promoter–driven luciferase expression was about 100‐fold weaker (Fig. 1C). Because reporter and ovalbumin genes are linked by 2A sites, we assume that ovalbumin expression was also 100‐fold lower after Ad‐TTR‐GOL infection. Thus, we generated identical recombinant adenoviruses that differed only in the promoters driving the transgenes, resulting in different gene expression levels in transduced cells.
Figure 1.
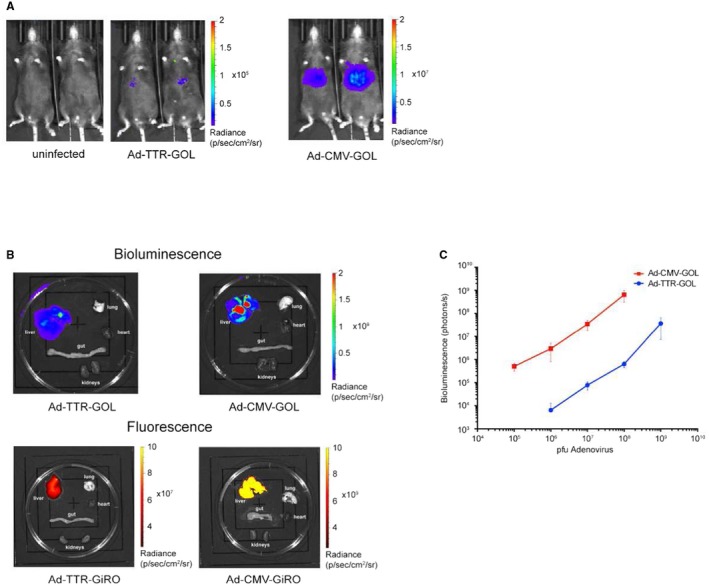
Hepatotropism of recombinant adenoviruses. (A) Bioluminescence imaging of mice infected with of Ad‐CMV‐GOL (109 PFU) or Ad‐TTR‐GOL (109 PFU) at day 2 after infection. (B) Bioluminescence and fluorescence imaging of isolated organs of infected mice. For bioluminescence measurement of luciferase, mice were infected with Ad‐CMV‐GOL (109 PFU) or Ad‐TTR‐GOL for 2 days. For fluorescence measurement of iRFP720, mice were infected with Ad‐CMV‐GiRO (109 PFU) or Ad‐TTR‐GiRO for 2 days. (C) Quantification of bioluminescence in mice infected with indicated doses of Ad‐CMV‐GOL (red) or Ad‐TTR‐GOL (blue) (n = 5 mice per virus and concentration). Data show mean ± SD. (A‐C) All experiments were performed at least three times and were reproducible.
Relevance of Hepatocyte‐Restricted Antigen Expression for Induction of Antigen‐Specific CD8 T‐Cells
Viral gene expression in secondary lymphoid organs promotes strong adaptive immunity, whereas hepatocyte‐restricted antigen presentation to naïve CD8 T‐cells may lead to their clonal deletion.7, 19 We used in vivo bioluminescence imaging to follow antiviral immunity, eliminating luciferase‐expressing hepatocytes that decrease the liver bioluminescence signal intensity. Infections with 105‐108 plaque‐forming units (PFU) of Ad‐CMV‐GOL were followed by an increase of the dose‐dependent bioluminescence signal until day 5 post injection (p.i.) (Fig. 2A). Thereafter, we detected in all mice a decline in bioluminescence down to the detection limit by day 18 p.i. (Fig. 2A). Consistent with the elimination of infected hepatocytes, we did not detect EGFP‐expressing hepatocytes any more at day 18 p.i. (Fig. 2B). Antibody‐mediated depletion of CD8 T‐cells, but not CD4+ or NK1.1+ T‐cells (Supporting Fig. S2A), prevented elimination of Ad‐CMV‐GOL‐infected hepatocytes (Supporting Fig. S2B), identifying CD8 T‐cells as key effector cells for antiviral immune surveillance.
Figure 2.
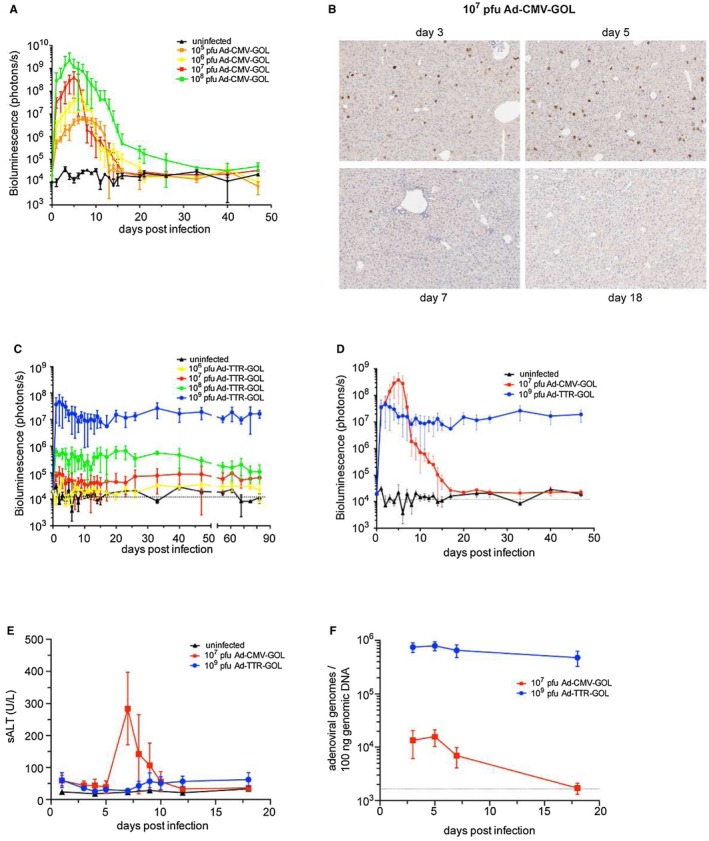
Viral persistence after infection with Ad‐TTR‐GOL. (A) Quantification of bioluminescence in mice after infection with increasing doses of Ad‐CMV‐GOL (105 to 108 PFU). (B) Liver immunohistochemistry for EGFP expression at indicated time points after infection with Ad‐CMV‐GOL (107 PFU). (C) Quantification of bioluminescence in mice after infection with increasing doses of Ad‐TTR‐GOL (106 to 109 PFU). (D) Overlay of results from bioluminescence imaging of Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (109 PFU) infected mice. (E) Time kinetics of serum ALT levels of mice after infection with Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (109 PFU). (F) Time kinetics of adenoviral genome copies by quantitative real‐time PCR in livers after infection with Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (109 PFU). (A,C‐F) Data show mean ± SD of five mice per group. All experiments have been performed at least three times and were reproducible.
We next characterized Ad‐TTR‐GOL infection. Regardless of the initial infection dose with 106 to 109 PFU/mouse Ad‐TTR‐GOL, the hepatic bioluminescence signal remained unchanged over 3 months p.i. (Fig. 2C), indicating a lack of protective CD8 T‐cell immunity after Ad‐TTR‐GOL infection. We assume that Ad‐TTR‐GOL and Ad‐CMV‐GOL both infect hepatocytes with similar efficiency, as they are identical except for the promoter driving the expression cassette. However, we lack formal proof because the low levels of virus‐encoded EGFP in Ad‐TTR‐GOL‐infected hepatocytes were below the limit of detection by immunohistochemistry or flow cytometry. Thus, regardless of the number of infected hepatocytes covering a range of three log10 infectious virus doses, CD8 T‐cell immunity either cleared Ad‐CMV‐GOL or failed to control Ad‐TTR‐GOL‐infection, suggesting that T‐cell exhaustion by high numbers of infected hepatocytes did not cause the persistent infection.
We next compared the course of Ad‐CMV‐GOL and Ad‐TTR‐GOL infection in mice and compared the infection dose of 107 PFU for Ad‐CMV‐GOL and 109 PFU for Ad‐TTR‐GOL that matched the luciferase/ovalbumin expression levels (Fig. 2D). Ad‐CMV‐GOL infection was accompanied by transient increase in serum alanine aminotransferase (sALT) levels peaking at day 7 p.i., whereas no sALT increase was observed after Ad‐TTR‐GOL infection (Fig. 2E). To further substantiate that the decrease of in vivo bioluminescence signal was the result of elimination of adenovirus‐infected hepatocytes, we quantified adenoviral genomes in liver tissue. Consistent with the 100‐fold difference in infectious dose, we found on day 3 p.i. 100‐fold lower levels of adenoviral DNA in livers of Ad‐CMV‐GOL compared with Ad‐TTR‐GOL‐infected mice (Fig. 2F). Although hepatic adenoviral DNA levels at day 18 after Ad‐CMV‐GOL infection fell below the detection limit, they remained stable after Ad‐TTR‐GOL infection, which is consistent with the failure of the immune response to clear infected hepatocytes (Fig. 2F). Taken together, these data demonstrate that Ad‐CMV‐GOL infection is cleared by CD8 T‐cells within 18 days, whereas Ad‐TTR‐GOL infection persists, suggesting failure of CD8 T‐cell immunity to eliminate Ad‐TTR‐GOL‐infected hepatocytes.
Ad‐CMV‐GOL but not Ad‐TTR‐GOL Infection Leads to Strong Induction of Antigen‐Specific CD8 T‐Cell Activation
Hepatocytes are the main target of intravenously injected adenoviruses.20 However, as infection with Ad‐CMV‐GOL was followed by immune‐mediated clearance of infected hepatocytes, we wondered whether Ad‐CMV‐GOL may have caused strong antigen‐specific CD8 T‐cell priming in lymphoid tissues. Therefore, we isolated spleens of infected mice for ex vivo bioluminescence imaging. We detected splenic luciferase expression after Ad‐CMV‐GOL, but only marginal luciferase expression after Ad‐TTR‐GOL infection (Fig. 3A), and by flow‐cytometry EGFP expression in isolated splenocytes. There were few EGFP‐expressing cells in the spleen after Ad‐CMV‐GOL infection, which were CD11c/CD11b double positive and accounted for 0.8% of the CD11c+ cells (Fig. 3B). This is consistent with either infection or uptake of soluble antigen by monocyte‐derived DCs (moDCs) in the spleen that may prime CD8 T‐cells. Because we detected adenoviral DNA for both viruses in splenic isolates and EGFP mRNA only after Ad‐CMV‐GOL infection (not shown) and did not detect EGFP expression in splenic moDCs after Ad‐TTR‐GOL‐infection, we assume that antigen‐presenting cells were infected and expressed the antigens for antigen presentation to CD8 T‐cells if driven by the CMV promoter (Fig. 3B). To obtain evidence for priming of antigen‐specific CD8 T‐cells, we cocultured CD11c+ spleen cells from Ad‐CMV‐GOL‐infected mice with naïve ovalbumin‐specific TCR‐transgenic CD8 T‐cells (OT‐1). The moDCs from noninfected mice activated naïve OT‐1 CD8 T‐cells only after the addition of ovalbumin, as measured by upregulation of the activation markers CD25 and CD44. Naïve antigen‐specific CD8 T‐cells were strongly activated by moDCs from Ad‐CMV‐GOL but not from Ad‐TTR‐GOL‐infected mice (Fig. 3C). Along this line, the moDCs from Ad‐CMV‐GOL‐infected mice induced proliferation and increased CD8 T‐cell numbers (Fig. 3D,E). There was only weak proliferation of antigen‐specific CD8 T‐cells after coculture with moDCs from Ad‐TTR‐GOL‐infected mice (Fig. 3D,E). Finally, antigen‐specific CD8 T‐cells activated by moDCs from Ad‐CMV‐GOL—but not from Ad‐TTR‐GOL‐infected mice—produced interleukin (IL)‐2 and interferon (IFN)‐γ (Fig. 3F). Taken together, these results indicate that CD8 T‐cell priming by moDCs after Ad‐CMV‐GOL infection led to proliferation and effector CD8 T‐cell differentiation, whereas Ad‐TTR‐GOL infection alone caused weak proliferation of antigen‐specific CD8 T‐cells.
Figure 3.
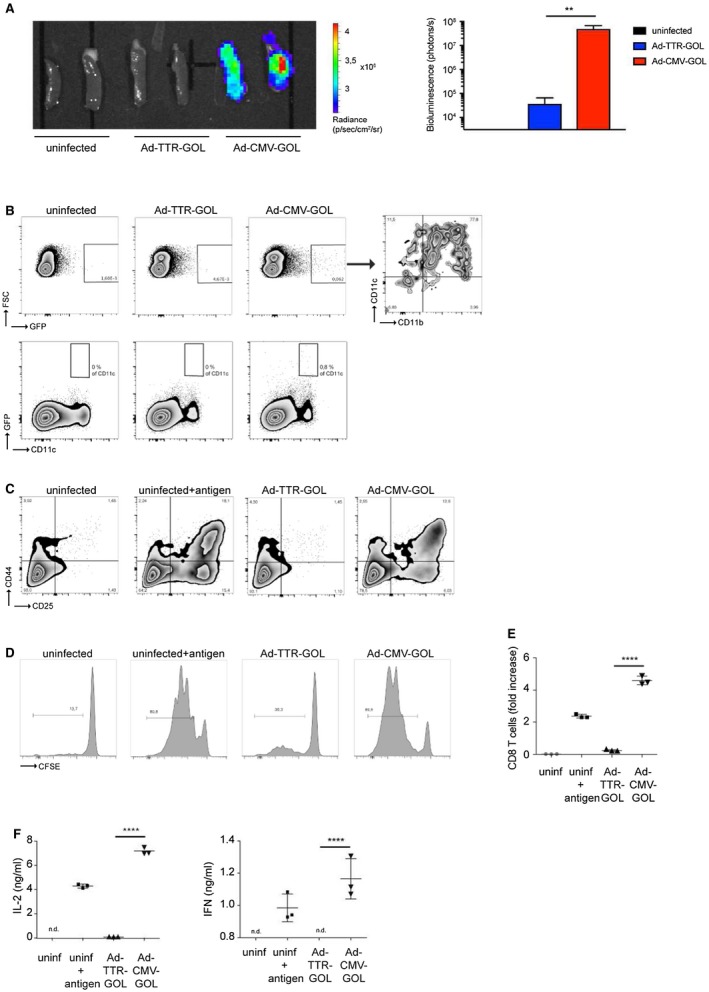
CD8 T‐cell priming after adenoviral infection. (A) Quantification of bioluminescence in spleens at day 2 after infection with Ad‐CMV‐GOL (109 PFU) or Ad‐TTR‐GOL. (B) Flow‐cytometric analysis of GFP expression of splenic cells from (A). (C) Flow‐cytometric quantification of CD25 and CD44 expression levels on ovalbumin‐specific OT‐1 T‐cells cocultured with CD11c+ splenic cells from (A). (D) Flow‐cytometric detection of CFSE dilution to quantify proliferation of ovalbumin‐specific OT‐1 T‐cells from (C). (E) Quantification of the total number of ovalbumin‐specific CD8 T‐cells from (C). (F) Detection of T‐cell–derived IL‐2 and IFN‐γ in supernatants from (C). All experiments were performed at least three times and were reproducible.
Dynamics of Expansion and Phenotype of Antigen‐Specific CD8 T‐Cells During Acute Viral Hepatitis
Because effector CD8 T‐cells eliminated virus‐infected hepatocytes, we next addressed the question whether priming and expansion of CD8 T‐cells differed between Ad‐CMV‐GOL and Ad‐TTR‐GOL infection. To quantitatively follow antigen‐specific CD8 T‐cell expansion, we initially transferred a very low number (100) of naïve CD44lowCD62Lhi OT‐1 CD8 T‐cells bearing CD45.1 as congenic marker. One day after transfer, we infected mice with Ad‐TTR‐GOL (107 or 109 PFU) or Ad‐CMV‐GOL (107 PFU) and followed the expansion of antigen‐specific CD8 T‐cells in the spleen and liver. From day 3 after Ad‐CMV‐GOL infection, we observed a rapid increase in the number of CD45.1+ CD8 T‐cells in the spleen until day 7 p.i. (Fig. 4A), coinciding with the sALT peak (Fig. 3E). Similar kinetics for CD45.1+ CD8 T‐cell expansion were observed in the liver, which was then followed by a decline of T‐cell numbers at day 18 p.i. (Fig. 4B) when infection was cleared. At the peak of the CD8 T‐cell response toward Ad‐CMV‐GOL infection, we found approximately 8 × 105 antigen‐specific CD8 T‐cells in the spleen and 2 × 106 in the liver (Fig. 4A,B). This demonstrates a more than 10,000‐fold increase of antigen‐specific CD8 T‐cell numbers after Ad‐CMV‐GOL infection. In contrast, after Ad‐TTR‐GOL infection with 109 PFU, leading to identical overall antigen expression like 107 PFU Ad‐CMV‐GOL, the number of splenic antigen‐specific CD8 T‐cells increased at a slower rate and only reached numbers of about 103 (Fig. 4A). In liver, Ad‐TTR‐GOL infection was followed by a slower and less‐pronounced 150‐fold increase of antigen‐specific CD8 T‐cells from day 3 to day 7 p.i., with a maximum of 104 cells (Fig. 4B). Like in the spleen, no decrease in the number of antigen‐specific CD8 T‐cells was observed at day 18 p.i. (Fig. 4B). After infection with 107 PFU Ad‐TTR‐GOL, leading to identical numbers of infected hepatocytes like in 107 PFU Ad‐CMV‐GOL‐infected mice, we failed to detect expansion of antigen‐specific CD8 T‐cells, neither in the liver nor in the spleen (Fig. 4A,B). Thus, our results reveal a much slower and less pronounced dynamic of antigen‐specific CD8 T‐cell responses in the spleen and liver after Ad‐TTR‐GOL compared with Ad‐CMV‐GOL infection.
Figure 4.
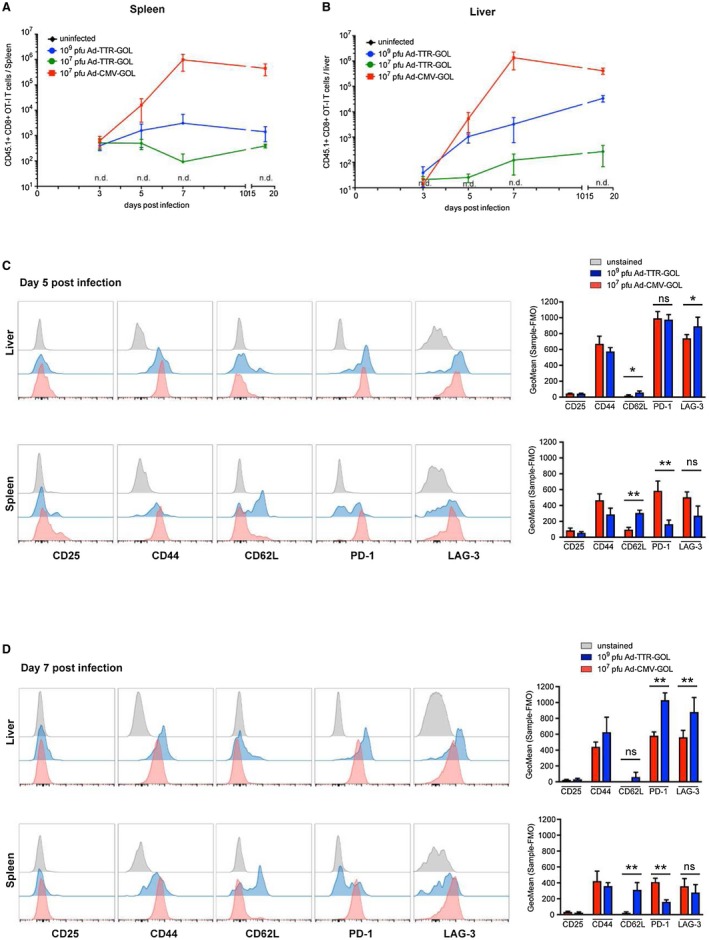
Phenotype and quantity of antigen‐specific CD8 T‐cells in liver and spleen after adenoviral infection. (A,B) Quantification of transferred CD45.1+ OT‐1 T‐cells (gated on living CD45.1+ and CD8+ cells) by flow‐cytometric analysis in spleen (A) or liver (B) at indicated time points after infection with Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (107 or 109 PFU) (n = 4). (C) Flow‐cytometric analysis and quantification of transferred CD45.1+ OT‐1 T‐cells (gated on living CD45.1+ and CD8+ cells) isolated from liver or spleen on day 5 after infection with Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (109 PFU). Each plot is a representative plot of a total of four. Quantification shows mean ± SD (n = 4). (D) Flow‐cytometric analysis on day 7 after infection as in (C). (A‐D) All experiments were performed at least three times and were reproducible.
We next characterized the phenotype of antigen‐specific CD8 T‐cells at days 5 and 7 p.i. In the absence of expansion after 107 PFU Ad‐TTR‐GOL infection, we did not obtain sufficient numbers of antigen‐specific CD8 T‐cells for phenotypic characterization. Therefore, we compared the CD8 T‐cell phenotypes arising after infection with Ad‐CMV‐GOL (107 PFU/mouse) or with Ad‐TTR‐GOL (109 PFU/mouse). After infection with either virus, all antigen‐specific CD8 T‐cells in the spleen and liver expressed high levels of CD44, compatible with being antigen‐experienced, but only some splenic but not hepatic antigen‐specific CD8 T‐cells transiently expressed the IL‐2‐receptor CD25 at day 5 p.i. Only after Ad‐TTR‐GOL infection we found some CD62Lhi antigen‐specific CD8 T‐cells in the spleen, whereas the few cells in the liver were CD62Llow (Fig. 4C,D). In contrast, after Ad‐CMV‐GOL infection, all antigen‐specific CD8 T‐cells were CD62Llow in the spleen and liver (Fig. 4C,D). Already at day 5 p.i., antigen‐specific CD8 T‐cells expressed high levels of PD‐1 and LAG‐3 in the liver of both Ad‐CMV‐GOL‐infected or Ad‐TTR‐GOL‐infected mice. Although after Ad‐TTR‐GOL infection the PD‐1 expression levels increased in hepatic antigen‐specific CD8 T‐cells from day 5 to day 7, it was slightly downregulated in the Ad‐CMV‐GOL‐infected mice (Fig. 4C,D). Interestingly, after Ad‐TTR‐GOL infection we found PD‐1low and PD‐1hi CD8 T‐cell populations in the spleen. Although the ratio between PD‐1low to PD‐1hi antigen‐specific T‐cells was approximately 1:1 at day 5, the ratio shifted to 2:1 at day 7 (Fig. 4C,D), suggesting that these T‐cells remained in the spleen and did not encounter their cognate antigen in the liver anymore. The CD62LhiCD44hi surface phenotype and localization of antigen‐specific CD8 T‐cells within lymphoid tissue after Ad‐TTR‐GOL infection suggests that these cells were initially cross‐primed by liver sinusoidal endothelial cells in the liver.14 At day 7 after Ad‐TTR‐GOL infection, we found high expression levels for LAG‐3 and PD‐1 on antigen‐specific T‐cells in the liver (Fig. 4D), correlating with a lack of T‐cell immunity to eliminate Ad‐TTR‐GOL‐infected hepatocytes. Thus, failure to control Ad‐TTR‐GOL infection correlates with slower kinetics of antigen‐specific CD8 T‐cell expansion, lower total antigen‐specific CD8 T‐cell numbers, and higher expression levels of PD‐1 and LAG‐3.
Phenotype and Functionality of Antigen‐Specific CD8 T‐Cells After Failure to Clear Acute Viral Infection from Hepatocytes
We next analyzed the phenotype of CD8 T‐cells at day 18 after viral infection (i.e., shortly after clearance of Ad‐CMV‐GOL or established persistence of Ad‐TTR‐GOL‐infection). There were no changes in CD25 or CD44 expression levels on antigen‐specific CD8 T‐cells after Ad‐CMV‐GOL compared with Ad‐TTR‐GOL infection (Fig. 5A). Although we found higher PD‐1 and LAG‐3 expression on hepatic antigen‐specific CD8 T‐cells after Ad‐TTR‐GOL infection (Fig. 5A), the PD‐1/LAG‐3 expression on splenic antigen‐specific CD8 T‐cells did not differ after Ad‐CMV‐GOL or Ad‐TTR‐GOL infection (Fig. 5A). Again, splenic antigen‐specific CD8 T‐cells after Ad‐TTR‐GOL infection were CD62LhiPD‐1low (Fig. 5A), suggesting that these cells remained localized within the spleen and did not migrate to the liver. Furthermore, hepatic PD‐1hiCD8 T‐cells also co‐expressed high levels of other inhibitory molecules like LAG‐3, Tim‐3, CD160, and CTLA‐4 (Fig. 5B). In contrast, antigen‐specific CD8 T‐cells found after Ad‐CMV‐GOL infection showed low‐level expression for all inhibitory molecules and partly expressed KLRG‐1 (Fig. 5B). This is similar to the phenotype of virus‐specific CD8 T‐cells seen during chronic HBV that also co‐express PD‐1 and CD160 and are negative for KLRG‐1 (Fig. 5C). To characterize their functionality, we restimulated antigen‐specific PD‐1+LAG‐3+Tim‐3+CD160+CD8 T‐cells. Clearly, these cells showed remarkably reduced IFN‐γ and TNF production compared with antigen‐specific CD8 T‐cells from livers of Ad‐CMV‐GOL‐infected mice (Fig. 5D). Taken together, Ad‐TTR‐GOL infection and development of persistent hepatocyte infection is characterized by co‐expression of inhibitory receptors on hepatic antigen‐specific CD8 T‐cells lacking effector function.
Figure 5.

Characterization of antigen‐specific CD8 T‐cells during persistent infection. (A) Flow‐cytometric analysis and quantification of transferred CD45.1+ OT‐1 T‐cells (gated on living CD45.1+ and CD8+ cells) isolated from liver or spleen on day 18 after infection with Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (109 PFU) (n = 4). Each plot is a representative plot of a total of four. Quantification shows mean ± SD. (B) Contour plot of flow‐cytometric analysis of PD‐1, LAG‐3, Tim‐3, CD160, CTLA‐4, and KLRG‐1 co‐expression on ovalbumin‐specific CD8 T‐cells isolated from liver on day 18 after infection as in (A). (C) Contour plot of flow‐cytometric analysis of PD‐1, CD160, and KLRG‐1 expression in intrahepatic bulk or HBV‐tetramer‐positive CD8 T‐cells from patients chronically infected with HBV. (D) Flow‐cytometric analysis following restimulation of isolated CD45.1+ CD8 T‐cells by ovalbumin‐derived SIINFEKL peptide on day 18 after infection with Ad‐CMV‐GOL (107 PFU) or Ad‐TTR‐GOL (109 PFU). (A‐D) All experiments were performed at least three times and were reproducible.
Ad‐TTR‐GOL Infected Hepatocytes Escape CD8 T‐Cell Immune Surveillance
The difference in outcome of infection (i.e., Ad‐CMV‐GOL controlled by antigen‐specific CD8 T‐cells and Ad‐TTR‐GOL establishing persistent infection in the presence of low numbers of dysfunctional antigen‐specific CD8 T‐cells) may originate from inefficient priming of antigen‐specific CD8 T‐cells after Ad‐TTR‐GOL infection. Therefore, we tested whether this could be overcome by co‐infection of both viruses. Mice received both viruses, Ad‐TTR‐GOL (108 PFU) and Ad‐CMV‐GiRO (107 PFU), a virus dose that induced protective immunity when applied alone (Fig. 3A and Fig. 6A‐C). Recombinant Ad‐CMV‐GiRO is identical to Ad‐CMV‐GOL but lacks luciferase expression, allowing the selective detection of the bioluminescence signal from Ad‐TTR‐GOL infection during co‐infection. After co‐infection, Ad‐TTR‐GOL infection persisted, whereas all Ad‐CMV‐GiRO‐infected hepatocytes were eliminated after 18 days p.i (Fig. 6A‐C). Of note, GFP is not detected after Ad‐TTR‐GOL infection due to low TTR promoter–driven expression levels (Fig. 6B,C). Numbers of hepatic antigen‐specific CD8 T‐cells did not differ between Ad‐CMV‐GiRO and Ad‐CMV‐GiRO/Ad‐TTR‐GOL co‐infected mice (Fig. 6D), suggesting efficient priming of CD8 T‐cell immunity also after co‐infection. Despite efficient elimination of Ad‐CMV‐GiRO‐infected hepatocytes in co‐infected mice, hepatic antigen‐specific CD8 T‐cells at day 18 p.i. displayed a dysfunctional phenotype co‐expressing inhibitory receptors PD‐1+Lag‐3+Tim‐3+CTLA‐4+ and being KLRG1neg (Fig. 6D). This is consistent with a selective failure to eliminate Ad‐TTR‐GOL‐infected hepatocytes and led us to investigate whether effector CD8 T‐cells were capable of eliminating Ad‐TTR‐GOL‐infected hepatocytes.
Figure 6.
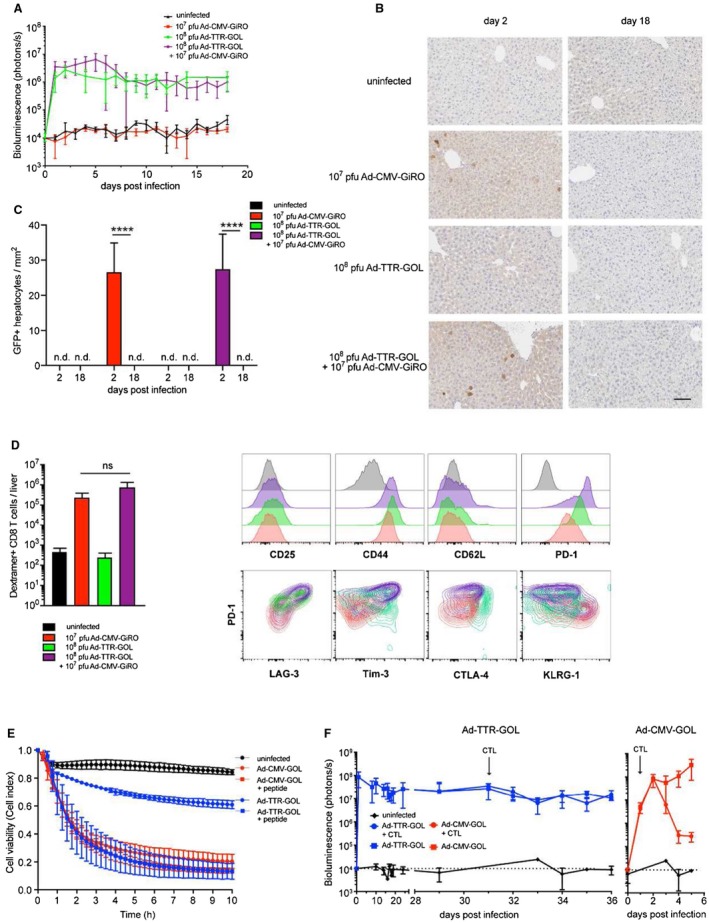
Antigen expression levels in infected hepatocytes shapes antiviral effector functions of CD8 T‐cells. (A) Quantification of bioluminescence in livers of mice infected with Ad‐CMV‐GiRO (107 PFU), Ad‐TTR‐GiRO (108 PFU), or co‐infected with Ad‐TTR‐GiRO (108 PFU) plus Ad‐CMV‐GiRO (107 PFU). (B) Liver immunohistochemistry for eGFP expression at day 2 and day 18 after infection as in (A). Scale bar = 100 µm. (C) Immunohistochemical quantification of infected eGFP+ hepatocytes at day 2 and day 18 after infection as in (A‐B). (D) Flow‐cytometric quantification and phenotypic analysis of endogenous OT‐1 T‐cells (gated on living SIINFEKL‐dextramer+ CD8+ cells) isolated from livers of mice 18 days after infection as in (A‐C). (E) Cell killing assay of in vitro cocultured primary murine hepatocytes and activated ovalbumin‐specific CD8 T‐cells (ratio 1:10). Hepatocytes were infected with moi = 5 by Ad‐TTR‐GOL or Ad‐CMV‐GOL. Infected hepatocytes were pulsed with 200 nM SIINFEKL peptide where indicated. Lyse of target cells was detected using xCELLigence technology. (F) Bioluminescence imaging of Ad‐TTR‐GOL (109 PFU) or Ad‐CMV‐GOL (107 PFU) infected mice before and after transfer of 10 million in vitro activated ovalbumin‐specific CD8 T‐cells. (A‐F) All experiments were performed at least three times and were reproducible.
We determined the efficiency of effector CD8 T‐cells to kill Ad‐CMV‐GOL‐infected or Ad‐TTR‐GOL‐infected hepatocytes. In vitro–generated antigen‐specific effector CD8 T‐cells rapidly (approximately 0.5‐1.5 hours) eliminated Ad‐CMV‐GOL‐infected hepatocytes in vitro (Fig. 6E). In contrast, they eliminated only 30% to 40% of Ad‐TTR‐GOL‐infected hepatocytes (Fig. 6E). We reasoned that low antigen expression in Ad‐TTR‐GOL‐infected hepatocytes resulted in low numbers of peptide‐loaded H2‐Kb‐molecules, and that this caused the failure of effector CD8 T‐cells to kill. To overcome this assumed limitation of subcritical H2‐Kb‐restricted antigen presentation, we increased the levels of peptide‐loaded H2‐Kb on Ad‐TTR‐GOL‐infected hepatocytes through the addition of peptide. This led to a strong increase in the killing of Ad‐TTR‐GOL‐infected hepatocytes by effector CD8 T‐cells up to the level of killing of Ad‐CMV‐GOL‐infected hepatocytes (Fig. 6E). There was no further increase in the killing of Ad‐CMV‐GOL‐infected hepatocytes after the addition of peptide (Fig. 6E). Together with the in vivo data on the killing of Ad‐CMV‐GiRO, but not Ad‐TTR‐GOL‐infected hepatocytes, these data suggest that effector CD8 T‐cells need to recognize a critical level of peptide‐loaded major histocompatibility complex (MHC) I molecules on the surface of infected hepatocytes to execute cytotoxic effector function, and that hepatocytes with low antigen expression escape from effector CD8 T‐cell killing. To further support this notion, we adoptively transferred in vitro generated antigen‐specific effector CD8 T‐cells into Ad‐TTR‐GOL‐infected or Ad‐CMV‐GOL‐infected mice. Effector CD8 T‐cells rapidly decreased hepatic bioluminescence over three log10 steps in Ad‐CMV‐GOL‐infected mice, whereas the same T‐cells failed to control Ad‐TTR‐GOL infection (Fig. 6F). Thus, the development of persistent Ad‐TTR‐GOL infection is not only caused by weak CD8 T‐cell priming but is also the result of immune escape of infected hepatocytes from CD8 T‐cell killing.
Discussion
Antiviral CD8 T‐cell immunity is key to infection control and elimination of virus‐infected hepatocytes. Yet, the mechanisms favoring protective versus dysfunctional virus‐specific CD8 T‐cell immunity in the liver have not been fully elucidated. To dissect the mechanisms underlying dysfunctional CD8 T‐cell immunity against virus‐infected hepatocytes, we have cloned identical recombinant adenoviruses co‐expressing in equimolar ratios of the reporters luciferase and EGFP together with ovalbumin either in a hepatocyte‐restricted manner using the TTR promoter (Ad‐TTR‐GOL) or using the CMV promoter (Ad‐CMV‐GOL). Therefore, we combined the known hepatotropism of adenoviruses with hepatocyte‐selective gene expression through the TTR promoter, similar to the combination of hepatotropism and hepatrophism used by HBV.23 The outcome of infection by these otherwise identical viruses was completely different: Ad‐CMV‐GOL infection was efficiently cleared, whereas infection with Ad‐TTR‐GOL persisted, independent of the numbers of infected hepatocytes covering a 4log10 range of infection dose from 105 to 109 PFU/mouse, thus going far beyond the range of antigen‐expressing hepatocytes studied so far.24 Linking ovalbumin, EGFP, and luciferase genes by 2A sequences in recombinant adenoviruses results in equimolar gene expression and allows to quantitatively and most sensitively detect infection and immune‐mediated clearance of infected hepatocytes by in vivo bioluminescence imaging.22, 25 Use of replication‐deficient adenoviruses allows us to focus on the antigens expressed, as no adenoviral antigens/epitopes are expressed/presented. This highly sensitive imaging technique enabled us to characterize the critical determinants of CD8 T‐cell immunity being relevant for elimination of virus‐infected hepatocytes.
Ad‐TTR‐GOL infection differed from Ad‐CMV‐GOL infection, as it generated 100‐fold lower antigen expression and always developed into persistent infection, whereas Ad‐CMV‐GOL infection was always cleared by antiviral CD8 T‐cell immunity. This was only related to the different promoters driving the different expression levels of genes delivered through adenoviral vectors. Previously, high numbers of antigen‐expressing hepatocytes were claimed to cause CD8 T‐cell dysfunction, although only a range between 10% and 50% of antigen‐expressing hepatocytes were investigated.24, 26, 27 In a humanized murine model, the load of HBV antigens was determined to influence antiviral immunity.28 However, in these mice the human liver graft and the human immune system were not human leukocyte antigen (HLA)‐matched, rendering it unlikely to detect HLA‐specific T‐cell responses.28 In contrast, our study revealed that numbers of infected hepatocytes are not relevant for initiation of CD8 T‐cell immunity, which most likely occurred in secondary lymphoid organs independent of the Ad‐CMV‐GOL infection dose. Our study further identified initiation of dysfunctional antiviral CD8 T‐cell immunity upon hepatocyte‐selective expression of a soluble antigen in the context of a physiologic T‐cell–receptor repertoire, which reflects the situation during infection with hepatotropic viruses.
Other experimental systems were used in the past to study mechanisms of viral immune evasion that leads to persistence of infection. This includes infection with different strains of LCMV, in which clone WE is rapidly cleared and clone‐13 persists due to interferon‐induced impairment of immunity and induction of dysfunctional antiviral T‐cells through high antigen levels.29, 30, 31 In addition, the mode of viral gene delivery to hepatocytes determines the outcome of infection: Hydrodynamic injection of HBV genomes leading to inflammation led to the elimination HBV antigen‐expressing hepatocytes, whereas the delivery of HBV genomes by adenoviral or adeno‐associated viral vectors causes persistent infection.32, 33 Therefore, our approach to compare identical viruses differing only in cell‐type selective antigen expression provides the chance to gain insight into liver‐specific regulation of antiviral immunity, independent of differences between virus‐intrinsic mechanisms of immune evasion or different modes of infection.
During the early phase of infection, we found different dynamics of antiviral CD8 T‐cell immunity. After Ad‐CMV‐GOL infection, we detected a rapid and massive increase of antigen‐specific CD8 T‐cells in liver and spleen. In contrast, Ad‐TTR‐GOL infection, which generated similar total bioluminescence and antigen‐expression levels as Ad‐CMV‐GOL infection, led to slower and less prominent T‐cell expansion, indicating less efficient priming of antigen‐specific CD8 T‐cells. Indeed, we found that Ad‐CMV‐GOL infection was accompanied by eGFP expression in monocyte‐derived DCs in the spleen and that these cells primed the CD8 T‐cells. In the spleen, a specialized macrophage population was identified that is required for induction of protective CD8 T‐cell immunity,19, 34 pointing toward the necessity of T‐cell priming in lymphoid tissues to control viral infection. Recently, the role of monocyte/macrophage and DC adenoviral transduction for strong antiviral immunity has been demonstrated,35 for which a CMV reporter‐driven gene expression is required. After infection with Ad‐TTR‐GOL leading to selective antigen expression by hepatocytes, however, only priming by hepatocytes or alternatively cross‐priming of antigens secreted from infected hepatocytes by LSECs or immature DCs can lead to CD8 T‐cell activation. However, CD8 T‐cell priming by hepatocytes causes clonal T‐cell deletion and cross‐priming by immature DCs fails to generate effector CD8 T‐cells.6, 7, 36, 37, 38 Only cross‐priming by LSECs promotes CD8 T‐cell survival and development of memory CD62LhiCD8 T‐cells that relocate to lymphoid tissues like central memory T‐cells.14 Our finding of persistent Ad‐TTR‐GOL infection that splenic antigen‐specific CD8 T‐cells were CD62Lhi indicates that cross‐priming by LSECs may have been involved. The increase in number of antigen‐specific CD8 T‐cells after Ad‐TTR‐GOL, albeit at much lower than after Ad‐CMV‐GOL infection, demonstrates that even under conditions of hepatocyte‐restricted expression of soluble antigen that is associated with the development of persistent infection, activation and expansion of antigen‐specific CD8 T‐cells occur.
Beyond weak priming of antiviral CD8 T‐cell immunity, we found a so‐far unappreciated immune escape of hepatocytes expressing low antigen levels from CD8 T‐cell killing. This deficient elimination of Ad‐TTR‐GOL‐infected hepatocytes raised the question why CD8 T‐cells were not able to kill these cells. Possibly, the number of antigen‐specific CD8 T‐cells in the liver after Ad‐TTR‐GOL infection may be too low to eliminate infected hepatocytes, or they may be dysfunctional. This notion is supported by our finding that antigen‐specific CD8 T‐cells after Ad‐TTR‐GOL infection continuously co‐expressed high levels of inhibitory receptors like PD‐1, LAG‐3, Tim‐3, CD160 and CTLA‐4, which impair CD8 T‐cell effector functions after stimulation through the T‐cell receptor.11, 39, 40, 41, 42, 43 Furthermore, they were refractory toward re‐activation, similar to hepatic HBV‐specific CD8 T‐cells found in patients with chronic HBV.12, 44, 45, 46 Of note, only hepatic, but not splenic antigen‐specific CD62Lhi CD8 T‐cells, showed this co‐expression of inhibitory receptors, suggesting that the latter did not localize to the liver and was not exposed to the tolerogenic signals in the context of Ad‐TTR‐GOL infection. Together these results indicate that dysfunction of antigen‐specific CD8 T‐cells during persistent infection was rather induced locally in the liver and not by circulating factors triggering dysfunction at the systemic level.
If CD8 T‐cells were dysfunctional because of insufficient T‐cell priming during Ad‐TTR‐GOL infection, we wondered whether co‐infection with Ad‐CMV‐GiRO, which initiated protective antiviral CD8 T‐cell immunity, could reverse T‐cell dysfunction in Ad‐TTR‐GOL‐infected mice. After co‐infection with both viruses, Ad‐CMV‐GiRO was efficiently eliminated and antigen‐specific CD8 T‐cell numbers in the liver were comparable. However, even under these conditions of protective antiviral CD8 T‐cell immunity, Ad‐TTR‐GOL‐infected hepatocytes were not eliminated. We therefore reasoned that the low antigen‐expression levels per infected hepatocytes may have caused escape from CD8 T‐cell killing. Indeed, fully activated effector CD8 T‐cells failed to kill Ad‐TTR‐GOL‐infected hepatocytes in vitro, but after the addition of specific peptide killing was as efficient as against Ad‐CMV‐GOL‐infected hepatocytes. Because expression levels of inhibitory receptors were similar on antigen‐specific CD8 T‐cells during the early phase of Ad‐CMV‐GOL and Ad‐TTR‐GOL infection, lower TCR signals in relation to high inhibitory signals might be responsible for the lack of effector function in vivo. However, even adoptive transfer of fully activated antigen‐specific effector T‐cells did not control Ad‐TTR‐GOL infection, suggesting that low‐level antigen expression allows escape from killing by effector CD8 T‐cells. The constant low‐level TCR signaling induced by Ad‐TTR‐GOL‐infected hepatocytes may drive PD‐1 upregulation on T‐cells, and increased expression of co‐inhibitory receptors are then rather the consequence, but not the cause of persistent infection. However, identification of the mechanisms underlying the upregulation of inhibitory molecules during Ad‐TTR‐GOL infection requires further analysis.
The relevance of immune escape by hepatocytes expressing only low level of viral antigens for chronic viral hepatitis remains to be defined. Hepatitis B virus, for instance, replicates from its transcriptional template the HBV cccDNA, but tools are lacking to quantify on a single‐cell level viral gene expression and how this translates into numbers of peptide‐loaded MHCI molecules on the cell surface. T‐cell receptor–like antibodies detecting MHCI‐peptide complexes have been developed, but flow cytometry with its detection limit of approximately 500 molecules per cell, is not suitable to detect low numbers of MHCI‐peptide complexes in the range of 50 to 500 per cell. Nevertheless, the numbers of MHCI‐peptide molecules appear to determine the range of effector functions of HBV‐specific CD8 T‐cells: high numbers triggering cytokine expression and degranulation, and 10‐fold lower numbers triggering only degranulation.47 Our study expands on this correlation, demonstrating that Ad‐CMV‐GOL infection triggers cytokine expression and T‐cell killing activity, whereas 100‐fold lower numbers of MHCI‐peptide complexes after Ad‐TTR‐GOL infection caused weak TCR signaling, leading to T‐cell exhaustion. It is therefore possible that infected hepatocytes expressing low‐level viral antigens may pave the way toward persistent infection by inducing T‐cell exhaustion.
In conclusion, the Ad‐TTR‐GOL‐infection model characterized by hepatocyte‐restricted antigen expression in combination with low‐level antigen expression allowed us to identify the mechanisms involved in the failure of antiviral immunity to eliminate infected hepatocytes. Inefficient CD8 T‐cell priming and escape of infected hepatocytes (with low‐level antigen presentation) from effector T‐cell killing represent two independently acting, yet synergistic, mechanisms for persistence of hepatocyte infection. Development of therapeutic vaccination strategies against chronic viral hepatitis may face similar immune escape mechanisms.
Potential conflict of interest
Nothing to report.
Supporting information
Supported by the Deutsche Forschungsgemeinschaft (SFB TRR 179, to D.W. and P.K.).
References
Author names in bold designate shared co‐first authorship.
- 1. Crispe IN. The liver as a lymphoid organ. Ann Rev Immunol 2009;27:147‐163. [DOI] [PubMed] [Google Scholar]
- 2. Thomson AW, Knolle PA. Antigen‐presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 2010;10:753‐766. [DOI] [PubMed] [Google Scholar]
- 3. Knolle PA, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 2014;146:1193‐1207. [DOI] [PubMed] [Google Scholar]
- 4. WHO . Hepatitis B. WHO fact sheet No. 204. Geneva, Switzerland: WHO; 2015. [Google Scholar]
- 5. WHO . Hepatitis C WHO fact sheet No. 164. Geneva, Switzerland: WHO; 2015. [Google Scholar]
- 6. Bertolino P, Trescol‐Biemont MC, Rabourdin‐Combe C. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur J Immunol 1998;28:221‐236. [DOI] [PubMed] [Google Scholar]
- 7. Holz LE, Benseler V, Bowen DG, Bouillet P, Strasser A, O'Reilly L, et al. Intrahepatic murine CD8 T‐cell activation associates with a distinct phenotype leading to Bim‐dependent death. Gastroenterology 2008;135:989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stross L, Gunther J, Gasteiger G, Asen T, Graf S, Aichier M, et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 2012;56:873‐883. [DOI] [PubMed] [Google Scholar]
- 9. Pallett LJ, Gill US, Schurich A, Quaglia A, Sinclair LV, Jover‐Cobos M, et al. Metabolic regulation of hepatitis B immunopathology by myeloid‐derived suppressor cells. Nat Med 2015;21:591‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peppa D, Gill US, Quaglia A, Sinclair LV, Jover‐Cobos M, Schurich A, et al. Up‐regulation of a death receptor renders antiviral T cells susceptible to NK cell‐mediated deletion. J Exp Med 2013;210:99‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD‐1, 2B4, CD160 and KLRG1 on exhausted HCV‐specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog 2010;6:e1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the Tim‐3/galectin‐9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One 2012;7:e47648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sumpter TL, Lunz JG III, Castellaneta A, Matta B, Tokita D, Turnquist HR, et al. Dendritic cell immunobiology in relation to liver transplant outcome. Front Biosci (Elite Ed) 2009;1:99‐114. [DOI] [PubMed] [Google Scholar]
- 14. Böttcher JP, Schanz O, Wohlleber D, Abdullah Z, Debey‐Pascher S, Staratschek‐Jox A, et al. Liver‐primed memory T cells generated under noninflammatory conditions provide anti‐infectious immunity. Cell Rep 2013;3:779‐795. [DOI] [PubMed] [Google Scholar]
- 15. Wohlleber D, Kashkar H, Gartner K, Frings MK, Odenthal M, Hegenbarth S, et al. TNF‐induced target cell killing by CTL activated through cross‐presentation. Cell Rep 2012;2:478‐487. [DOI] [PubMed] [Google Scholar]
- 16. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med 2016;213:1819‐1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol 2012;12:201‐213. [DOI] [PubMed] [Google Scholar]
- 18. Faure‐Dupuy S, Lucifora J, Durantel D. Interplay between the Hepatitis B virus and innate immunity: from an understanding to the development of therapeutic concepts. Viruses 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol 2011;13:51‐57. [DOI] [PubMed] [Google Scholar]
- 20. Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008;132:397‐409. [DOI] [PubMed] [Google Scholar]
- 21. Palmiter RD, Gagnon J, Walsh KA. Ovalbumin: a secreted protein without a transient hydrophobic leader sequence. Proc Natl Acad Sci U S A 1978;75:94‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus‐1 in human cell lines, zebrafish and mice. PLoS One 2011;6:e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Ann Rev Pathol 2006;1:23‐61. [DOI] [PubMed] [Google Scholar]
- 24. Cebula M, Riehn M, Hillebrand U, Kratzer RF, Kreppel F, Koutsoumpli G, et al. TLR9‐mediated conditioning of liver environment is essential for successful intrahepatic immunotherapy and effective memory recall. Mol Ther 2017;25:2289‐2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stabenow D, Frings M, Truck C, Gärtner K, Förster I, Kurts C, et al. Bioluminescence imaging allows measuring CD8 T cell function in the liver. Hepatology 2010;51:1430‐1437. [DOI] [PubMed] [Google Scholar]
- 26. Ochel A, Cebula M, Riehn M, Hillebrand U, Lipps C, Schimbeck R, et al. Effective intrahepatic CD8+ T‐cell immune responses are induced by low but not high numbers of antigen‐expressing hepatocytes. Cell Mol Immunol 2016;13:805‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tay SS, Wong YC, McDonald DM, Wood NA, Roediger B, Sierro F, et al. Antigen expression level threshold tunes the fate of CD8 T cells during primary hepatic immune responses. Proc Natl Acad Sci U S A 2014;111:E2540‐E2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dusseaux M, Masse‐Ranson G, Darche S, Ahodantin J, Li Y, Fiquet O, et al. Viral load affects the immune response to HBV in mice with humanized immune system and liver. Gastroenterology 2017;153:1647‐1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013;340:202‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013;340:207‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Utzschneider DT, Legat A, Zehn D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol 2013;14:603‐610. [DOI] [PubMed] [Google Scholar]
- 32. Huang LR, Gäbel YA, Graf S, Arzberger S, Kurts C, Heikenwalder M, et al. Transfer of HBV genomes using low doses of adenovirus vectors leads to persistent infection in immune competent mice. Gastroenterology 2012;142:1447‐1450. [DOI] [PubMed] [Google Scholar]
- 33. Dion S, Bourgine M, Michel ML. Adeno‐associated virus‐mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA‐A2 and HLA‐DR1 molecules. J Virol 2013;87:5554‐5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shaabani N, Khairnar V, Duhan V, Zhou F, Tur RF, Häussinger D, et al. Two separate mechanisms of enforced viral replication balance innate and adaptive immune activation. J Autoimmun 2016;67:82‐89. [DOI] [PubMed] [Google Scholar]
- 35. Maler MD, Nielsen PJ, Stichling N, Cohen I, Ruzsics Z, Wood C, et al. Key role of the scavenger receptor MARCO in mediating adenovirus infection and subsequent innate responses of macrophages. MBio 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurts C. Cross‐presentation: inducing CD8 T cell immunity and tolerance. J Mol Med (Berl) 2000;78:326‐332. [DOI] [PubMed] [Google Scholar]
- 37. Sumpter TL, Abe M, Thomson AW. Dendritic cells, the liver, and transplantation. Hepatology 2007;46:2021‐2031. [DOI] [PubMed] [Google Scholar]
- 38. Kurts C, Robinson BW, Knolle PA. Cross‐priming in health and disease. Nat Rev Immunol 2010;10:403‐414. [DOI] [PubMed] [Google Scholar]
- 39. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009;10:29‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMahan RH, Golden‐Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, et al. Tim‐3 expression on PD‐1+ HCV‐specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte‐directed in vitro cytotoxicity. J Clin Invest 2010;120:4546‐4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG‐3 in acute and chronic LCMV infection. Int Immunol 2010;22:13‐23. [DOI] [PubMed] [Google Scholar]
- 42. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim‐3 and PD‐1 in CD8 T‐cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 2010;107:14733‐14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Golden‐Mason L, Palmer BE, Kassam N, Townshend‐Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim‐3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol 2009;83:9122‐9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bengsch B, Martin B, Thimme R. Restoration of HBV‐specific CD8+ T cell function by PD‐1 blockade in inactive carrier patients is linked to T cell differentiation. J Hepatol 2014;61:1212‐1219. [DOI] [PubMed] [Google Scholar]
- 45. Schurich A, Khanna P, Lopes AR, Han KJ, Peppa D, Micco L, et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen‐4 on apoptosis‐Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011;53:1494‐1503. [DOI] [PubMed] [Google Scholar]
- 46. Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T‐cell responses can be restored by blocking programmed death‐1 pathway in chronic hepatitis B. Gastroenterology 2010;138:682‐693. [DOI] [PubMed] [Google Scholar]
- 47. Gehring AJ, Sun D, Kennedy PT, Nolte‐'t Hoen E, Lim SG, Wasser S, et al. The level of viral antigen presented by hepatocytes influences CD8 T‐cell function. J Virol 2007;81:2940‐2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials