

Abstract
Human immunodeficiency virus (HIV) continues to be a major contributor to morbidity and mortality worldwide, particularly in developing nations where high cost and logistical issues severely limit the use of current HIV therapeutics. This, combined HIV's high propensity to develop resistance, means that new antiviral agents against novel targets are still urgently required. We previously identified novel anti‐HIV agents directed against the nuclear import of the HIV integrase (IN) protein, which plays critical roles in the HIV lifecycle inside the cell nucleus, as well as in transporting the HIV preintegration complex (PIC) into the nucleus. Here we investigate the structure activity relationship of a series of these compounds for the first time, including a newly identified anti‐IN compound, budesonide, showing that the extent of binding to the IN core domain correlates directly with the ability of the compound to inhibit IN nuclear transport in a permeabilised cell system. Importantly, compounds that inhibited the nuclear transport of IN were found to significantly decrease HIV viral replication, even in a dividing cell system. Significantly, budesonide or its analogue flunisolide, were able to effect a significant reduction in the presence of specific nuclear forms of the HIV DNA (2‐LTR circles), suggesting that the inhibitors work though blocking IN, and potentially PIC, nuclear import. The work presented here represents a platform for further development of these specific inhibitors of HIV replication with therapeutic and prophylactic potential.
1. INTRODUCTION
Human immunodeficiency virus (HIV) remains one of the most widespread human pathogens despite the availability of highly active antiretroviral therapies. HIV has a high mutation rate, which is leading to the emergence of resistance among patients receiving these drugs resulting in a critical need to identify new targets and develop new classes of effective antiviral therapeutics (Wainberg, Mesplede, & Quashie, 2012). A key step in HIV infection is transport into the infected cell nucleus of reverse transcribed viral cDNA packaged with both viral and cellular proteins into what is known as the preintegration complex (PIC; Bukrinsky et al., 1993; Piller, Caly, & Jans, 2003). A key component of the PIC is the HIV integrase (IN) protein, which, along with other HIV‐1 proteins, plays an important role in PIC nuclear entry (Hearps & Jans, 2006); once inside the nucleus, IN mediates integration of the viral cDNA into the host cell genome, followed by subsequent synthesis of the viral mRNA from this integrated DNA. Due to this critical role of IN, IN‐targeted antiviral therapeutics that target the strand‐transfer process of integration are already utilised in the clinic, including raltegravir, elvitegravir, and dolutegravir. These compounds target IN‐DNA binding, with resistance/cross‐resistance between these various inhibitors already evident (Wainberg et al., 2012); clearly, new IN‐targeting drugs with alternative mechanisms of action are urgently needed.
We previously performed a high‐throughput screen to identify small molecule inhibitors of nuclear entry of IN, subsequently demonstrating their efficacy in limiting HIV infection (Wagstaff & Jans, 2006; Wagstaff, Rawlinson, Hearps, & Jans, 2011; Wagstaff, Sivakumaran, Heaton, Harrich, & Jans, 2012). The inhibitors were specifically directed at the interaction between IN and its cellular nuclear import receptor complex, the importin (Imp) α/β1 heterodimer (Hearps & Jans, 2006). In this study, we describe in detail a new set of novel IN nuclear transport inhibitors, which bind directly to IN as demonstrated by nuclear magnetic resonance (NMR) spectroscopy, determining that the region of inhibitor binding to IN is in the vicinity of the nuclear localisation signal (NLS) that confers interaction with Impα/β1, and showing that the extent of binding to the IN core domain correlates directly with the ability of the compound to inhibit IN nuclear transport in a permeabilised cell system. Importantly, compounds that inhibited the nuclear transport of IN were found to significantly decrease HIV viral replication, even in a dividing cell system. Significantly, two of the compounds were able to effect a significant reduction in the presence of specific nuclear forms of the HIV DNA (2‐LTR circles), suggesting that the inhibitors work though blocking IN, and potentially PIC, nuclear import. The results represent a solid platform for further development of these specific inhibitors of HIV replication with therapeutic and prophylactic potential.
2. RESULTS
2.1. Specific inhibitors of HIV‐1 IN nuclear transport bind directly to the IN core domain
We previously used high‐throughput screening to identify a number of compounds, including mifepristone, that specifically inhibited IN binding to Impα/β1 and demonstrated that they can inhibit HIV‐1 infection in a single cycle replication assay (Lundberg et al., 2013; Wagstaff et al., 2011; Wagstaff et al., 2012). Several compounds identified, such as the FDA‐approved ivermectin, were found to be broad spectrum inhibitors of Impα/β1‐dependent nuclear import and to have antiviral activity towards a range of viruses as a consequence (Lundberg et al., 2013; Wagstaff et al., 2012). To delineate the molecular interactions at play, we firstly tested the ability of mifepristone (Wagstaff et al., 2011), as well as the chemically unrelated specific IN nuclear transport inhibitor molecule budesonide, described here for the first time, to bind directly to IN using heteronuclear single quantum coherence (HSQC) NMR spectroscopy (Figure 1). The Impα‐targeting agent ivermectin was included as a negative control. IN core protein was analysed by HSQC‐NMR in the absence (Figure 1a, blue spectra) and presence (Figure 1a, red spectra) of each compound, with both budesonide and mifepristone eliciting multiple spectral perturbances (Figure 1a, example regions of interest are highlighted with coloured circles) indicative of direct interaction of the compounds with specific regions of IN. In contrast, ivermectin had no observable effect on the IN spectra by HSQC‐NMR, consistent with its mechanism of action being through binding to Impα rather than IN. Significantly, although there were some minor overlaps in the perturbations of IN induced by budesonide and mifepristone, most of the spectral perturbations of IN residues were distinct to one compound or the other. Some of these are highlighted in Figure 1, where the top panels highlight a region of interest that is perturbed by binding to budesonide but not mifepristone, and the bottom panels highlight the reverse. The results imply distinct mechanisms of action for the two compounds and suggest the existence of multiple druggable target sites on IN. Mapping the residues that showed significant (>0.02 ppm) perturbation from the Apo (unbound) IN spectra, onto the crystal structure of the IN core domain (Figure 1b, blue residues) highlighted the ability of budesonide to perturb residues mapping to and around an α‐helix located directly adjacent to the Impα/β1‐recognised NLS of IN (Figure 1b, core residues highlighted in pink), which is located directly adjacent to the binding site on IN for the critical host‐cell co‐factor LEDGF/p75 (lens epithelial derived growth factor; Berthoux, Sebastian, Muesing, & Luban, 2007; Hou et al., 2008; Latham, la, Tinetti, Chalmers, & Tachedjian, 2016; Peat, Dolezal, Newman, Mobley, & Deadman, 2014); residue 186 of the NLS is also known to contribute to IN multimerisation (Berthoux et al., 2007). Similarly, mifepristone impacted residues adjacent to the NLS on the opposite side of IN to budesonide, including those within a pocket known to be highly active for compound binding in fragment‐based drug discovery trials (the IN fragment binding pocket; see Latham et al., 2016; Peat et al., 2014; Wielens et al., 2013). These results clearly indicate direct binding of both budesonide and mifepristone to different sites on IN, both of which would appear to impact NLS‐recognition by Impα/β1.
Figure 1.
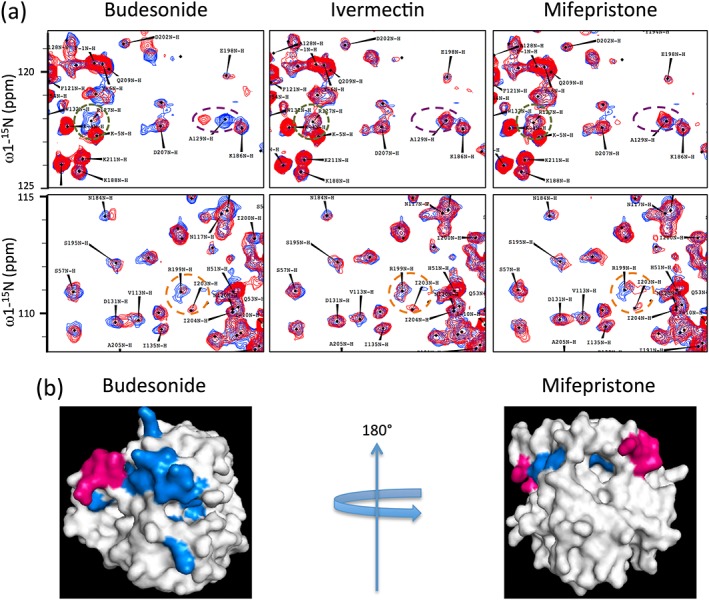
Binding of specific inhibitors of HIV‐1 IN nuclear import perturbs different regions of the IN core domain near to the NLS, as shown by heteronuclear single quantum coherence (HSQC) nuclear magnetic resonance. Nuclear transport inhibitor binding to HIV‐IN was analysed using HSQC nuclear magnetic resonance spectroscopy. (a) 15N‐1H HSQC spectrum of 15N‐labelled INCORE123 in the absence (blue) and presence (red) of the indicated compounds. Two areas are shown for each compound, with regions of interest indicated by the green, purple (upper panels) and orange (lower panels) dashed circles. Data are from a single typical experiment representative of two independent experiments. (b) Compound binding sites, defined by the residues that showed perturbations in the HSQC analysis (Blue) from (a) of the indicated IN‐specific inhibitors are superimposed onto the IN core domain crystal structure (white surface model); the four basic residues of the IN nuclear localisation signal (NLS) are highlighted in pink
2.2. IN core domain binding nuclear transport inhibitors reduce HIV IN nuclear accumulation
To confirm the ability of budesonide and mifepristone to inhibit IN nuclear import directly, we used an established in vitro nuclear import assay (Ghildyal et al., 2005; Hearps & Jans, 2006). This system allows for quantitative analysis of the kinetics of the protein transport, rather than a single qualitative end point such as those provided by similar assays (Adam, Marr, & Gerace, 1990; Adam, Sternemarr, & Gerace, 1992). Because semi‐intact cells are used, it is possible to bypass the cell membrane to test inhibitors for intracellular mechanisms directly. A routine inclusion in the assay is 70‐kDa Texas red labelled dextran (TD70), exclusion of which indicates nuclear integrity (Bernis & Forbes, 2014; Hearps & Jans, 2006). In this system, unperforated intact cells appear as large black cells (denoted by white * in Figure 2a), whereas nuclei with intact nuclear envelopes appear as smaller TD70 excluding areas (marked by white arrows, Figure 2a) and overperforated nuclei with damaged/leaky nuclear envelopes fail to exclude TD70 properly (marked by white arrowheads, Figure 2a; Bernis & Forbes, 2014; Lau et al., 2009). Based on this internal control, only isolated intact nuclei are included in the quantitation (Bernis & Forbes, 2014; Hearps & Jans, 2006; Lau et al., 2009).
Figure 2.
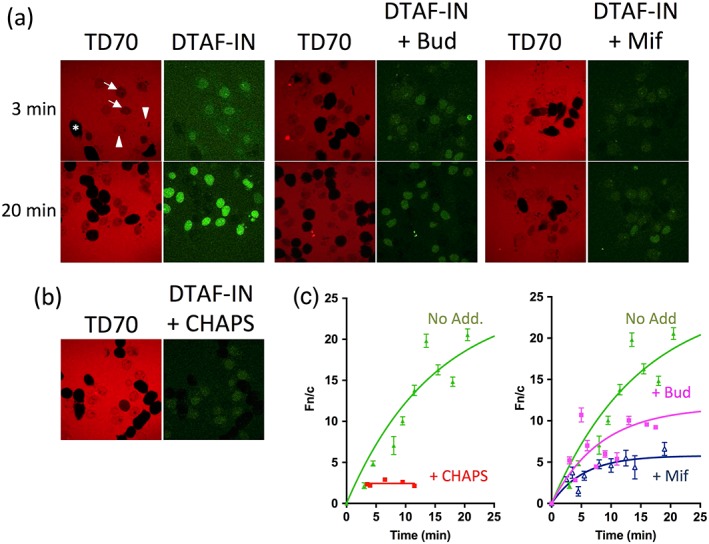
IN core domain binding inhibitors reduce HIV IN nuclear accumulation. Nuclear import of DTAF‐labelled IN (DTAF‐IN) was reconstituted in mechanically perforated hepatoma tissue culture cells in the presence of exogenous cytosol and an ATP regeneration system as described in Section 4. (a) Confocal laser scanning microscopy (CLSM) images were acquired periodically for accumulation of DTAF‐IN (green panels) into intact nuclei in the absence or presence of the indicated inhibitors. Representative images for each sample are shown at 3 min and 20 min accumulation. Nuclear integrity was confirmed by the exclusion of a Texas Red‐labelled 70‐kDa dextran (TD70, red panels), in which intact cells appear as large black cells (denoted by white *), nuclei with intact nuclear envelopes appear as smaller TD70 excluding areas (marked by white arrows) and overperforated nuclei with damaged/leaky nuclear envelopes fail to exclude the TD70 properly (marked by white arrowheads). (b) CLSM images of accumulation of DTAF‐IN in the absence of an intact nuclear membrane (indicated by the lack of exclusion of TD70), induced by the addition of 0.025% CHAPS. (c) Image analysis was performed on the CLSM images such as those in (a, b) using ImageJ (NIH) software as described in Section 4. Each point represents the mean ± scanning electron microscope (n > 10). Note that in the absence of an intact nuclear membrane, steady state is reached within minutes; therefore, the data for the CHAPS‐treated samples are only presented for the first 10 min. Bud, budesonide; Mif, mifepristone; No Add, No addition
Fluorescently labelled IN (DTAF‐IN) exhibited rapid and efficient nuclear accumulation when added to the in vitro nuclear import assay (Figure 2a, left), reaching a maximal level of accumulation >20 fold that of the cytoplasm (Figure 2c, left), consistent with what we have previously observed (Hearps & Jans, 2006). Similar experiments conducted on DTAF‐IN in the presence of budesonide or mifepristone showed a significant reduction in the level of IN nuclear accumulation from a nuclear to cytoplasmic fluorescence ratio (Fn/c) of ~33 to ~12 and 6, respectively (Figure 2a,c). These results confirm that both compounds significantly decrease the nuclear accumulation of IN, consistent with their proposed mechanism of action. IN possesses inherent DNA binding ability and, combined with the small size of the protein (32 kDa), a proportion of its nuclear accumulation can be attributed to simple diffusion and retention in the nucleus through binding to DNA or other components. This was examined as previously (Hearps & Jans, 2006) using the detergent CHAPS, which permeabilises the nuclear membrane resulting in a loss of TD70 nuclear exclusion (Efthymiadis, Shao, Hübner, & Jans, 1997), revealing as expected that DTAF‐IN accumulates in the nucleus of CHAPS‐treated nuclei to an Fn/c of ~2 (Figure 2b,c, left), indicating around two‐fold accumulation of the protein in the permeabilised nuclei compared with the cytoplasm. This value represents the minimal level of nuclear accumulation, which can be expected even if 100% of the IN active nuclear transport is inhibited. Taking this result into account, the reduction in IN nuclear accumulation by the nuclear transport inhibitory drugs, shown here for the first time, is even more significant.
2.3. Budesonide and its analogues inhibit IN binding to Imp α/β
An established AlphaScreen binding assay (Wagstaff et al., 2011; Wagstaff & Jans, 2006) was used to test the ability of budesonide to inhibit binding of Impα/β to full‐length IN protein. Increasing concentrations of budesonide were tested for their effect on the binding of purified recombinant (His)6‐tagged IN to a predimerised GST‐Impα/β heterodimer as described in Section 4. The IC50 of the inhibition was determined to be ~1 μM (Figure 3a; Table 1), consistent with the high level of inhibition observed in both the initial library screen and the in vitro nuclear transport assay (Figure 2). To confirm that budesonide is a specific inhibitor of the IN:Impα/β interaction, rather than a more general inhibitor of all Impα/β‐mediated nuclear transport, its activity towards an alternative Impα/β cargo, SV40 large tumour T‐antigen NLS (T‐ag NLS) was assessed. Results (Figure 3b) demonstrated that whereas ivermectin, a known nonspecific inhibitor of Impα/β:cargo interactions (Wagstaff et al., 2012), had significant impact on T‐ag NLS binding (IC50 = 11.1 μM), no significant difference in binding was observed for budesonside; this is consistent with the NMR results (Figure 1), whereby budesonide action can be attributed to direct binding to IN rather than through binding to Impα/β. To begin to assess which chemical moieties of budesonide are key to activity, a focussed AlphaScreen analysis was performed on commercially available compounds with >98% identity to budesonide (Figure 3; Table 1). Of 3 analogues tested, flunisolide had a similar IC50 to budesonide, whereas desisobutyryl ciclesonide (DC) was less potent (IC50 of ~9 μM) and triamcinolone acetonide (TA) had no measurable inhibitory effect. The close structural relatedness of the different compounds enables the preliminary identification of key parts of the budesonide molecule that are required for activity (see below); eg. since flunisolide and TA differ only by the placement of a single additional fluorine (highlighted in pink in the lower panels of Figure 4) on the steroidal B ring, this analysis suggests that this steroidal B ring is critical to activity.
Figure 3.
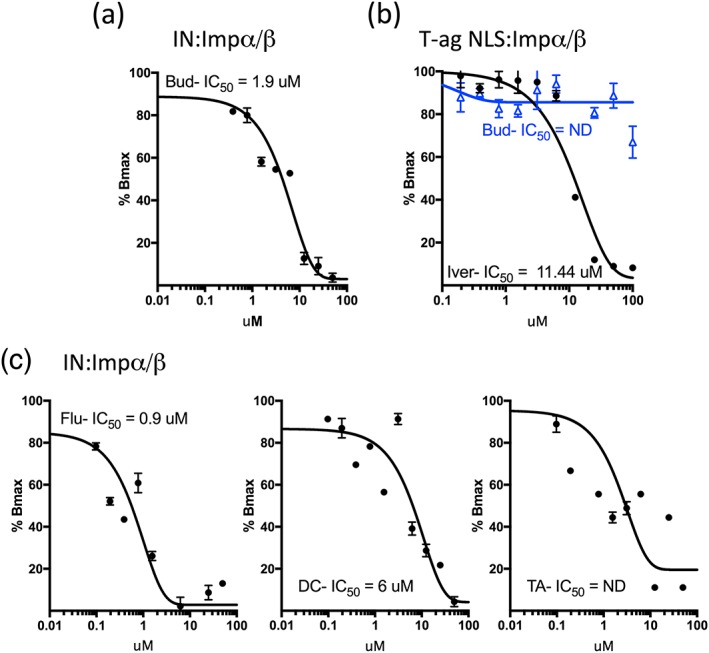
IC50 analysis for inhibition by budesonide and analogues of integrase (IN)‐Imp α/β interaction. (a) IC50 analysis was performed via AlphaScreen assay (see Section 4) using 60‐nM IN, 3.75‐nM Imp α/β and increasing concentrations of budesonide (Bud), in PBS/5% DMSO. (b) IC50 analysis was performed as per (a) using 30 nM T‐ag NLS and 7.5‐nM Imp α/β1 and increasing concentrations of Bud (blue) or Ivermectin (Iver; black). (c) IC50 analysis was performed as per (a) using 60‐nM IN, 3.75‐nM Imp α/β and increasing concentrations of flunisolide (flu, left), desisobutyryl ciclesonide (DC; middle) or triamcinolone acetonide (TA; right). Exponential binding curves were fitted using Graphpad Prism software. Each point represents the mean ± scanning electron microscope from quadruplicate assays. Graphs shown represent a single typical experiment from a series of ≥3 similar experiments
Table 1.
IC50 and anti‐HIV properties of budesonide analogues
| Compound | IC50 (n) | EC50 (n) MT‐2 cells | EC50 (n) TZMbl cells |
|---|---|---|---|
| Budesonide | 1.2 ± 0.9 μM (3) | 79.4 ± 14.9 μM (3) | 49.4 ± 5.4 μM (6) |
| Flunisolide | 1.4 ± 0.3 μM (3) | 77.5 ± 15.2 μM (3) | 105 ± 40 μM (7) |
| Desisobutyryl ciclesonide | 8.9 ± 2.5 μM (4) | NA | NA |
| Triamcinolone acetonide | ND (4) | NA | NA |
Note. Results are for the mean ± SEM (n in parentheses). ND: not able to be determined. NA: not applicable. IC50 values determined by AlphaScreen assay, EC50 values determined from infectious assays in the indicated cell systems.
Figure 4.
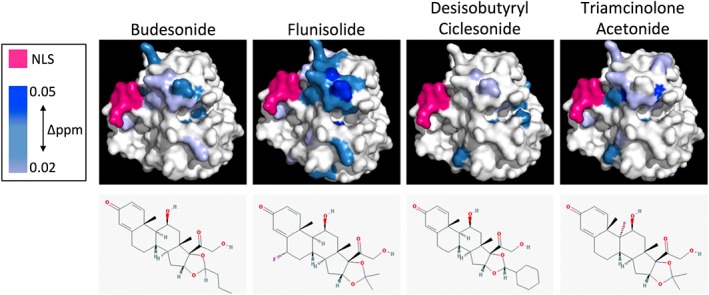
Budesonide and its analogues show varying degrees of binding to the HIV IN core domain as determine by heteronuclear single quantum coherence nuclear magnetic resonance. 15N‐1H heteronuclear single quantum coherence spectra of 15N‐labelled INCORE123 were determined in the absence and presence of the indicated compounds and superimposed onto the IN core domain crystal structure according to the extent of perturbation observed (in Δppm, scale given to the left). NLS residues are indicated in pink and the structures of each compound are indicated in the lower panels. Data are from a single typical experiment representative of two independent experiments
2.4. The level of binding of budesonide and its analogues to the HIV IN core domain varies significantly and is consistent with the degree of inhibition observed on IN binding to Impα/β
To dissect further the molecular interaction between the budesonide analogues and the IN core domain, HSQC‐NMR was once again employed (Figure 4) and the residues showing perturbations in the presence of drug mapped onto the crystal structure of the IN core domain. Perturbations were colour graduated in accordance with the strength of disturbance (measured in Δppm). As expected, all analogues showed binding to the IN core domain in the same region as budesonide. Flunisolide showed perturbation of all the same residues as budesonide, but to a much greater level and included significant perturbation of a larger number of residues, including most notably, residues in the LEDGF binding domain located adjacent to the budesonide binding site. TA showed a similar but weaker perturbation profile to budesonide and was generally more dispersed in terms of the impacted residues. DC showed a reduced perturbation profile compared with the other analogues, with very few residues showing any perturbation. These results nicely parallel those for the IC50 values (Figure 3), where flunisolide and TA showed similar or slightly reduced inhibitory activity, whereas DC failed to prevent IN binding to Impα/β, even at high concentration. As indicated above, TA and flunisolide differ only in the position of a fluorine on the steroidal B ring/chiral carbon attaching steroidal rings B and C, respectively (Figure 4, lower panels), with DC identical to budesonide except for the replacement of the propyl moiety attached to the ketal carbon of the cyclic acetyl group on the D‐ring of the steroid backbone with a cyclohexal moiety (TA and flunisolide in turn have an isopropylidene ketal containing a dimethyl substitution at this position). The clear implication is that although some chemical alterations of the budesonide scaffold are tolerated (and may in fact improve binding), their placement may reduce activity, with bulky additions, particularly at the diketal moiety attached to the steroidal D ring, likely to be less favourable.
2.5. Flunisolide inhibits nuclear accumulation of HIV IN to a greater extent than budesonide
The in vitro reconstituted nuclear transport assay was employed to test the ability of the budesonide analogues to inhibit IN nuclear transport (Figure 5). Flunisolide was found to significantly reduce the maximal nuclear accumulation of IN by c. 86% of that in its absence, in analogous fashion to budesonide (c. 83% reduction; see Figure 5c). In contrast, TA and DC inhibited maximal IN nuclear accumulation by about 50% and 20%, respectively, correlating well with the extent of their binding to the IN core domain and ability to inhibit IN‐Impα/β binding.
Figure 5.
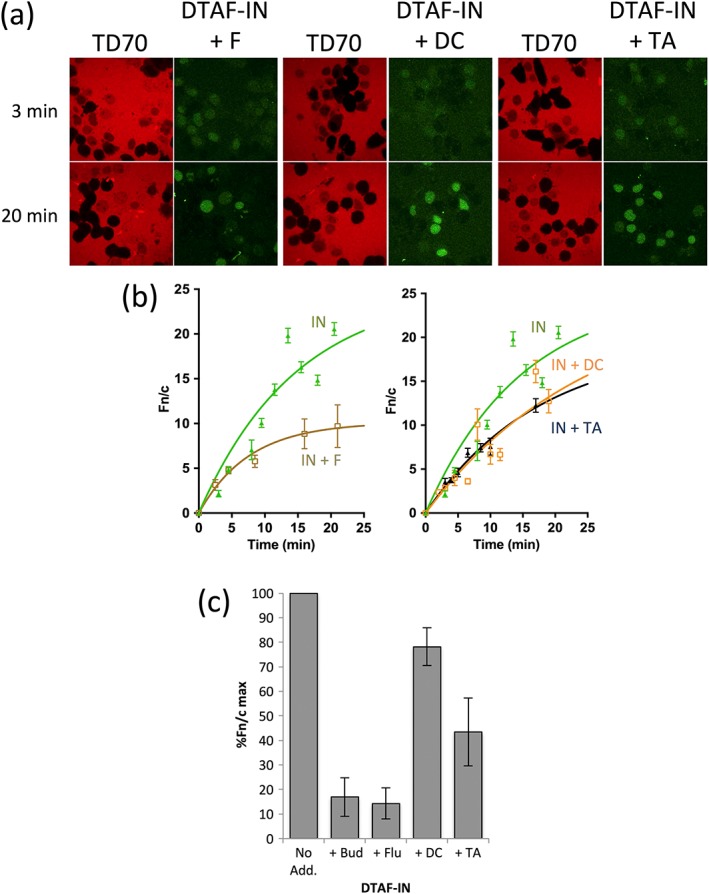
Flunisolide inhibits nuclear import of HIV IN to a greater extent than other budesonide analogues. Nuclear import of DTAF‐labelled IN (DTAF‐IN) was reconstituted in mechanically perforated hepatoma tissue culture cells in the presence of exogenous cytosol and an ATP regeneration system as described in the legend to Figure 2. (a) Confocal laser scanning microscopy images for the nuclear accumulation of DTAF‐IN in the presence of the indicated analogues. (b) Image analysis as per the legend to Figure 2c on images such as those in (a) for the accumulation of DTAF‐IN. The data presented in panels a/b were from an experiment conducted in parallel to that shown in Figure 2; together they constitute a single typical experiment from a series of three similar experiments. (c) The maximal nuclear accumulation of IN in the presence of the indicated compounds, relative to IN in the presence of DMSO alone (% Fn/c max) was determined from curves such as those in (b). The data represent the mean ± SEM (n = 3 independent experiments). No Add, No addition; B, budesonide; DC, Desisobutyryl Ciclesonide; F, flunisolide; TA, Triamcinolone Acetonide; TD70, 70 kDa Texas‐red labelled dextran
2.6. IN nuclear import inhibitors inhibit HIV‐1 infection
Infectious assays were performed to confirm that both budesonide and its most active analogue flunisolide are able to inhibit HIV‐1 infection. Budesonide was able to inhibit HIV‐1 infection robustly with an EC50 of ~50 μM (Figure 6a, Table 1) in TZM‐bl cells using a luciferase‐based assay (see Section 4) with flunisolide showing slightly lower potency (EC50 of c. 105 μM). Similar results were obtained in the more physiologically relevant MT‐2 single infectious round cell system (Figure 6b, Table 1; EC50s of ~80 μM for both compounds). Importantly, cytotoxicity results (Figure 6c/d) in both MT‐2 and TZM‐bl cells demonstrated that whereas budesonide was mildly toxic at concentrations above 200 μΜ (CC50s of 514 and 444 μM in TZM‐bl and MT‐2 cells, respectively, well above the EC50 concentration), flunisolide showed no significant toxicity at the concentrations tested (CC50 of 2,673 μM) in MT‐2 cells and only mild toxicity in TZM‐bl cells at concentrations above 1 mM). The low toxicity of flunisolide is consistent with its extensive use as a seasonal allergy spray and in patients with chronic asthma (Jackson, Polygenis, McIvor, & Worthington, 1999) without reports of side effects. Clearly, the anti‐HIV activity of the compounds cannot be attributed to cellular toxicity.
Figure 6.
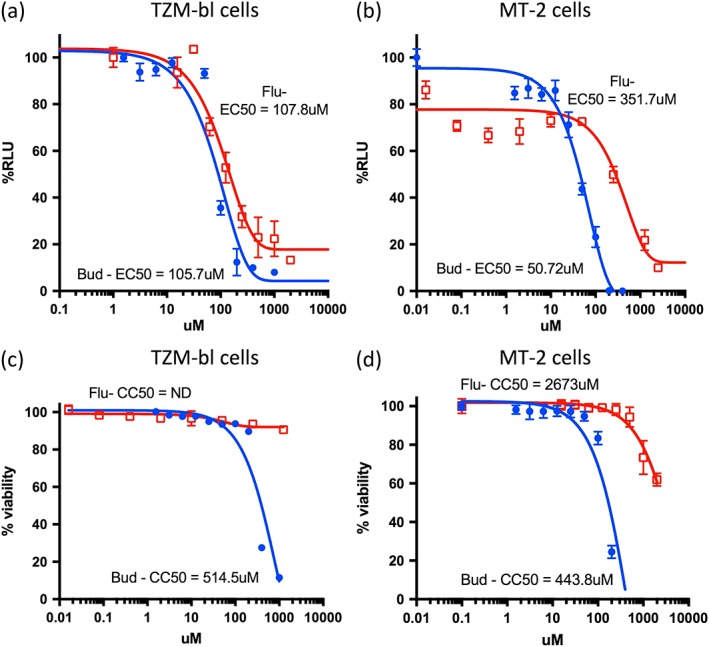
Budesonide and flunisolide inhibit HIV infection. HIV infection was performed in the presence of either budesonide (B) or flunisolide (F) in triplicate in either TZM‐bl (a) or MT‐2 (b) cells and allowed to proceed for 2 days. Relative levels of HIV virus was determined from the supernatant using a luciferase assay (as per Section 4), in TZM‐bl cells. In parallel, cytotoxicity was measured (TZM‐bl, c, and MT‐2 cells, d) in triplicate for each dose using a cytotoxicity assay as well as visual assessment using light microscopy. Dose response curves were plotted from the percent inhibition of each dosage compared with untreated controls (% Relative Luciferase Units: %RLU), where the data represent the mean ± SEM, n = 3). EC50 values were determined at noncytotoxic concentrations. Graphs represent a single typical experiment from a series of six (Tzm‐bl) or four (MT‐2) similar experiments
2.7. Budesonide and flunisolide appear to inhibit HIV‐1 PIC nuclear entry rather than the integration step of infection
Finally to confirm that the observed inhibition of HIV‐1 infection is the result of inhibition of HIV nuclear import, we preformed quantitative PCR using primers designed to detect several products of HIV‐1 reverse transcription and nuclear DNA forms including linear double stranded DNA (early HIV transcripts), near full length RT products such as those that would be incorporated into the preintegration complex (late HIV transcripts) and 2‐LTR circles (indicative of unintegrated nuclear DNA; see Figure 7a). MT‐2 cells were infected with HIV in the presence of the budesonide, flunisolide, or the anti‐HIV compounds efavirenz (an inhibitor of HIV‐1 reverse transcriptase) and raltegravir (an inhibitor of HIV‐1 integration) and samples analysed by qPCR at the indicated times for early HIV, late HIV, or 2‐LTR transcripts (Figure 7b). All results were relativised to a DMSO‐treated control, and 2‐LTR circles were expressed relative to late RT products to allow specific analysis of the nuclear import step. The full results for all compounds at all time points is included in the Supporting Information (Figure S1).
Figure 7.
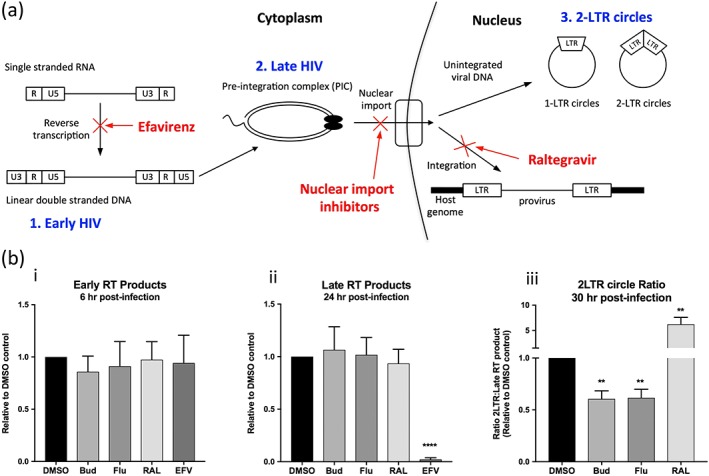
IN nuclear transport inhibitors reduce formation of nuclear forms of the HIV DNA. (a) Schematic representation of the HIV reverse transcription process highlighting the points of action of the inhibitors. Single stranded HIV RNA is first reverse transcribed into double stranded DNA (1. Early HIV transcripts), which then undergoes a complex series of rearrangements to generate the preintegration complex (PIC; 2. Late HIV transcripts). The PIC undergoes nuclear import where it is integrated in the host genome to form the provirus, from which transcription will then occur. If integration fails to occur, the unintegrated viral DNA will form a number of nuclear forms of DNA including 1‐ and 2‐LTR circles (3.2‐LTR circles). The stages of infection targeted by the commercially available HIV drugs (efavirenz and raltegravir) or the IN nuclear import inhibitors are indicated in red. (b) Quadruplicate wells of MT‐2 cells were infected with HIV in the presence of the indicated compounds and analysed by qPCR at the indicated times using specific primers for the presence of either early HIV (detected at 6 hr postinfection, Figure 7b(i)), late HIV (detected at 24‐hr postinfection, Figure 7b(ii)) or 2‐LTR transcripts (detected at 30‐hr postinfection, Figure 7b(iii)). Results were relativised to a DMSO treated control and 2‐LTR circles were further standardised to late RT products measures at the same time point. Results represent the mean ± scanning electron microscope (n = 4—budesonide/flunisolide or 3—Raltegravir/Efavirenz independent experiments), **P < 0.01, ****P < 0.0001, as compared to DMSO treated control (unpaired t‐test). RAL; raltegravir, EFV; Efavirenz, Bud; budesonide, Flu; flunisolide
As expected raltegravir had no consistent effect on the relative quantities of either the early or late HIV transcripts but produced a significant (up to six fold) increase in the ratio of 2‐LTR circles to late HIV products, consistent with its role as an IN inhibitor, thereby driving transcripts towards the formation of alternative nuclear transcripts including 1‐ and 2‐LTR circles. Conversely, efavirenz treatment significantly reduced the production of complete RT products, which was more pronounced later in infection (Figure 7b), consistent with its action as a reverse transcription inhibitor. This inhibition carried forward into a complete lack of all nuclear HIV as expected.
In contrast to both of these, budesonide and flunisolide had no significant effect on the production of either the early or late HIV transcripts but significantly reduced the ratio of 2‐LTR circles (Figure 7b), consistent with the mechanism of action of these compounds being inhibition of PIC nuclear transport.
3. DISCUSSION
This study represents the first molecular dissection of the mechanism of action of a novel class of anti‐HIV inhibitors that target IN nuclear transport (see Wagstaff et al., 2011; Wagstaff et al., 2012). We show here for the first time that budesonide, originally identified by high‐throughput screening, can inhibit IN nuclear transport through the ability to bind directly to a binding site on IN juxtaposed to the IN NLS and adjacent to the LEDGF/p75 binding site. Importantly, in this study, we were able to confirm direct inhibition of IN nuclear accumulation in an in vitro reconstituted nuclear transport assay and apparent inhibition of HIV PIC nuclear import using qPCR targeting various HIV‐1 transcripts, as well as inhibition of HIV infection, including in dividing cells (MT‐2 cells). This is an important point as the precise mechanisms behind the nuclear import of the HIV‐1 PIC remain hotly debated, with conflicting reports suggesting roles for multiple HIV‐1 proteins, including capsid (CA), IN, matrix (MA), and Vpr, as well as cellular proteins in this process (Jayappa, Ao, & Yao, 2012; Levin, Loyter, & Bukrinsky, 2011). Several reports suggest a key role for CA in particular, on the basis of CA interaction with nuclear pore components (Burdick et al., 2017; Schaller et al., 2011). Clearly, IN is not the only HIV‐1 component responsible for PIC nuclear import, but the data presented here demonstrate conclusively that efficient IN nuclear transport is required for optimal HIV‐1 replication efficiency, even in dividing cells. Whether this is due to inhibition of PIC nuclear transport or inhibition of the IN protein only remains to be determined, but it is clear that the compounds characterised here in detail represent anti‐HIV therapeutics with a unique mechanism of action.
Of several budesonide analogues examined, we identified flunisolide as a highly active analogue that can effect the same level of anti‐HIV inhibition as budesonide, but with reduced toxicity, that has been associated with budesonide use in the clinic (Iborra, Alvarez‐Sotomayor, & Nos, 2014; Saberi, Phengrasamy, & Nguyen, 2013), including potential cross‐reactions with HIV protease inhibitor drugs such as ritonavir leading to acute hepatitis or Cushing's syndrome (Saberi et al., 2013). Flunisolide, in contrast, does not appear to have these contraindications, being used extensively as a seasonal allergy spray with a high safety profile, including in HIV patients (Saberi et al., 2013), demonstrating the potential to generate a compound that is both highly active against IN nuclear transport and that maintains a high safety profile, consistent with our findings here. Importantly, the limited structure activity relationship analysis performed here suggests that maintaining a small modification at the cyclic acetyl group attached to the steroidal D‐ring appears to be important in maintaining activity and also in reducing toxicity. Fluorination would also appear to be important, but placement of the fluorine atom is critical, with fluorination at the chiral carbon between the B and C rings severely inhibiting activity, compared with fluorination of the B‐ring directly. Future medicinal chemistry strategies should focus in the first instance on further substitutions at the ketal carbon of the di‐acetyl group and also on substitution of the additional fluorine atom in flunisolide for alternative functional groups.
Given the continued emergence of HIV strains that are resistant to drugs targeted against traditional HIV targets such as HIV integration and reverse transcription, the development of new classes of therapeutics against alternative targets is critical. The identification of a druggable binding patch on IN, which is the target of budesonide and its active analogues, therefore lays the foundation for a future concerted rational drug design approach to generate a novel class of highly active nontoxic inhibitors of HIV targeting PIC nuclear transport and potentially incorporating activity against other functions of IN such as integration or LEDGF/p75 binding given the close proximity of the two binding sites. In this context, it is intriguing to note that Lysine 186 within the IN NLS has been found to play a critical role in IN multimerisation (Berthoux et al., 2007), a key factor in IN‐mediated activities such as integration (Faure et al., 2005) and nuclear import (Berthoux et al., 2007). The proximity of the budesonide drug‐binding patch to the IN NLS/multimerisation interface suggests that the IN‐binding inhibitors characterised may potentially impact IN multimerisation. In fact, a new class of IN inhibitors selectively targeting multimerisation (Sharma et al., 2014) appear to interact with residues on both sides of the multimerisation interface, including Threonine 174, which was also strongly perturbed by all of our IN inhibiting compounds. All four of the other residues identified as playing a role in multimerisation inhibition (Sharma et al., 2014) are located directly adjacent to the budesonide binding patch, implying that dual action nuclear import/multimerisation inhibitors may be able to be designed in the future. This possibility is a focus of ongoing work in this laboratory.
4. EXPERIMENTAL PROCEDURES
4.1. Small molecule Inhibitors
Budesonide (Bud), mifepristone (Mif), ivermectin, flunisolide (Flu), desisobutyryl ciclesonide (DC), and triamcinolone acetonide (TA) were purchased from Sigma.
4.2. Bacterial expression plasmids
Bacterial expression vectors encoding (His)6‐tagged IN (pINSD.His.Sol plasmid), GST‐Impα (pET30a‐Impα2), and GST‐Impβ (pET30a Impβ1) have been described (Hearps & Jans, 2006; Hubner, Xiao, & Jans, 1997; Wagstaff & Jans, 2006; Xiao, Hubner, & Jans, 1997).
4.3. Cell culture
The hepatoma tissue culture (HTC) rat hepatoma cell line was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) foetal calf serum, L‐glutamine, penicillin, and streptomycin. For in vitro transport assay experiments, HTC cells were trypsinised and seeded onto glass coverslips 2 days prior to use to achieve a confluency of 70% at the time of experimentation. The MT‐2 cell line (Harada, Koyanagi, & Yamamoto, 1985), TZM‐bl indicator cell line (Wei et al., 2002), and 293 T cells were obtained through the NIH AIDS Research and Reference Reagent Program. MT‐2 cells were cultured in RF‐10 (RPMI 1640 supplemented with 10% foetal bovine serum, 100‐U ml−1 penicillin, 100‐μg ml−1 streptomycin, and 2‐mM glutamine). TZM‐bl and 293 T cells were cultured in DMEM‐10 (DMEM supplemented with 10% FBS, 100‐U ml−1 penicillin, 100‐μg ml−1 streptomycin, and 2‐mM glutamine). All cells were maintained in a humidified 37°C incubator with 5% CO2.
4.4. Protein purification and Impα/β1 dimerisation
His6‐tagged IN protein was expressed in BL21 (DE3) bacteria as previously (Hearps & Jans, 2006). Imp proteins were purified from bacteria as GST‐fusion proteins under native conditions as described in Hubner et al. (1997), Wagstaff and Jans (2006), and Xiao et al. (1997). Where appropriate, Impα and Impβ were predimerised at 13.6 μM for 15 min at room temperature to generate the Impα/β heterodimer for binding studies.
4.5. Labelling of IN protein
IN protein (500 μg) was fluorescently labelled by incubation with 0.4 mg ml−1 of the fluorescent dye DTAF (5‐(4,6‐dichlorotriazinyl)‐aminofluorescein, Molecular Probes) dissolved in 250‐mM bicine ([N,N‐bis(2‐hydroxyethyl)‐glycine) buffer, pH 9.5, for 90 min at room temperature. Excess dye was removed via buffer exchange (PD‐10 column, Amersham) and dialysis. For use in in vitro experiments, the final salt concentration was reduced by diluting the labelled protein 1:15 in native buffer containing 150‐mM NaCl and reconcentrating the protein as above.
4.6. Biotinylation of Imp proteins
Impα was biotinylated for use in the AlphaScreen assay as previously (Wagstaff & Jans, 2006), using the sulfo‐NHS‐biotin (sulfo‐N‐hydroxysuccinimide biotin) reagent (Pierce). Briefly, 3.5 mg of Imp was incubated with 250 μl of sulfo‐NHS‐biotin (1 mg dissolved in 150 μl of water) on ice for 2 hr. Unbound biotin was removed via a PD‐10 column (GE Healthcare) and the resulting biotinylated protein was concentrated in an Amicon‐30 concentration device.
4.7. HSQC NMR
The IN construct used for NMR comprised the IN catalytic domain (residues 50–212) containing mutations Q53E,C56S,W131E,F185K,Q209E (Fitzkee, Masse, Shen, Davies, & Bax, 2010). The coding sequence was cloned into plasmid vector pET28 into the NdeI and BamHI sites and expressed in BL21(DE3) RIL Escherichia coli using 15N‐autoinduction media (Studier, 2005) and purified as described (Fitzkee et al., 2010). Samples analysed using NMR contained 240‐μM protein in 25‐mM HEPES pH 6.8, 150‐mM NaCl, 40‐mM MgCl2, and 10% 2H2O in a sample volume of 150 μl in 3‐mm NMR tubes. 1H,15N‐HSQC spectra were acquired at 25°C on a Bruker AVANCE 600‐MHz NMR spectrometer fitted with a CryoProbe, with spectral dimensions 160 (F1) × 2048 (F2) with 16 scans per increment and a recycle delay of 1.3 s. Compound addition was made from 100‐mM d6‐DMSO stocks.
4.8. In vitro nuclear transport assay
Nuclear import of fluorescently labelled IN was investigated in vitro using mechanically perforated HTC cells as previously described (Jans, Jans, Briggs, Sutton, & Trapani, 1996). Briefly, mechanical perforation was used to remove the plasma membrane of cells, but leave the nuclear membrane intact, and the perforated cells inverted on to a microscope slide over a chamber of artificial “cytoplasm” containing rabbit reticulocyte lysate, an ATP regenerating system (0.125‐mg ml−1 creatine kinase, 30‐mM creatine phosphate, and 2‐mM ATP), 70‐kDa Texas Red‐conjugated dextran (to assess nuclear integrity), 2‐μM DTAF‐labelled IN protein and IB buffer (110‐mM KCl, 5‐mM NaHCO3, 5‐mM MgCl2, 1‐mM EGTA, 0.1‐mM CaCl2, 20‐mM HEPES, and 1‐mM DTT, pH 7.4) in a final volume of 5 μl. Where required, 5‐μM individual nuclear import inhibitor compounds or 0.025% CHAPS were added to the chamber. CHAPS is used to estimate the extent to which IN protein is bound to nuclear components. Cells were imaged over time by confocal laser scanning microscopy (CLSM) using an Olympus FV1000 CLSM with a 100× oil immersion lens. Digitised images were analysed using the ImageJ software (NIH) to determine the nuclear to cytoplasmic fluorescence ratio (Fn/c), calculated using the equation Fn/c = (Fn − Fb)/(Fc − Fb), where Fn, Fb, and Fc represent the nuclear, background (from a nonperforated cell), and cytoplasmic fluorescence values, respectively. Nuclear import kinetics were plotted using Graphpad Prism software, and exponential curves were fitted for DTAF‐IN in the absence or presence of the indicated compounds.
4.9. AlphaScreen‐based binding assay
The AlphaScreen assay was performed in triplicate in 384‐well white opaque plates (PerkinElmer) essentially as previously (Wagstaff & Jans, 2006). The assay was quantified on a PerkinElmer EnVision plate reader, triplicate values were averaged, and titration curves (sigmoidal) were plotted using the Graphpad Prism graphing program. Values in the “hooking zone,” where quenching of the signal occurs because of too much of either binding partner, were excluded from the final plot as previously (Wagstaff & Jans, 2006).
4.10. Virus strain
NL4.3, a CXCR4 (X4)‐using laboratory strain of HIV‐1 (Adachi et al., 1986), was derived by transfection of 293 T cells with pDRNL using the calcium phosphate method (Yap et al., 2007). Virus supernatant was collected and stored at −80°C following propagation by infection at least three times in MT‐2 cells prior to use in anti‐HIV assays.
4.11. HIV inhibition and cytotoxicity assays in TZM‐bl cells
For the HIV inhibition assays, NL4.3 stock virus with known titre (Wapling, Moore, Sonza, Mak, & Tachedjian, 2005) was diluted in DMEM‐10 supplemented with 20‐μg ml−1 DEAE‐Dextran (GE Healthcare) in 96‐well flat bottom TC plates (Nunclon, Denmark) containing monolayers of TZM‐bl cells seeded 24 hr prior to infection at 2 × 104 cells per well. The cells were incubated with 300–500 infectious units of HIV per well for 1 hr at 37°C in 5% CO2. For the cytotoxicity assay, TZM‐bl monolayers were treated as above but the virus omitted. Serial dilutions of the inhibitors were prepared in DMSO, followed by dilution in DMEM‐10 (0.1% DMSO final concentration), and added in triplicate to both the infected (HIV Inhibition) and noninfected TZM‐bl cells (cytotoxicity) and incubated for 2 days at 37°C in 5% CO2. Equivalent concentrations of DMSO or media alone were used as controls. HIV inhibition was determined using the Steady‐Glo luciferase assay system (Promega) as described previously (Tyssen et al., 2010). Cytotoxicity was determined for each inhibitor using the CellTitre 96 AQueous One Solution cell proliferation reagent (Promega) as described previously (Pauwels et al., 1988). EC50 values (half maximal effective concentration) and CC50 values (half maximal cytotoxic concentration) were calculated for each inhibitor by nonlinear regression analysis (GraphPad Prism 7.0 software).
4.12. HIV inhibition and cytotoxicity assay in MT‐2 cells (single cycle replication)
NL4.3 stock virus was added to 10 × 106 MT‐2 cells in a 10‐ml centrifuge tube at a MOI (multiplicity of infection) of 0.1 in RF‐10 medium. Mock‐infected cells were treated similarly in parallel. Following incubation for 2 hr at 37°C in a humidified incubator with 5% CO2 atmosphere, cells were pelleted for 5 min at 600 g (Beckman Allegra X‐12R centrifuge), washed once in 10‐ml RF‐10 before resuspension in 5‐ml RF‐10, and seeded into 96‐well flat bottom TC plates at a concentration of 1 × 105 cells per well. Serial dilutions of the inhibitors (in DMEM‐10) or media alone were added (triplicate wells) and the cells incubated for 30 hr at 37°C in 5% CO2. Cell supernatant was collected from all wells and assayed for HIV infectivity by adding to TZM‐bl reporter cells and using the Steady‐Glo luciferase assay system as described above. Cytotoxicity was determined using the CellTitre 96 AQueous One Solution cell proliferation reagent (Promega) as per the TZM‐bl cell system and EC50 and CC50 values were calculated in the same fashion.
4.13. qPCR assay for analysis of HIV reverse transcription products
qPCR analysis to examine the production of HIV reverse transcription intermediates was performed using a modified method of that utilised by Mbisa and colleagues (2009). Briefly, MT2 cells were infected as above, but with an additional incubation of the virus stocks with 10‐units ml−1 DNAse1 and 40‐units ml−1 benzonase in the presence of 4‐mM MgCl2 for 1 hr at 37°C prior to infection to remove any potential plasmid DNA carry over from the transfection. Cells were infected in the presence of DMSO (0.1% final) without or with 100‐μM budesonide, 250‐μM flunisolide, 10‐μM raltegravir, or 0.1‐μM efavirenz. Concentrations chosen were nontoxic, but effective in HIV‐1 inhibition. Infected cells were harvested at various time points postinfection by centrifugation at 1,000 g for 5 min. The cell pellet was washed once in PBS and DNA extracted using the Qiagen DNA Blood kit according to the manufacturer's instructions. Residual plasmid DNA was removed by treatment with 0.5‐μl Dpn1 per 100 μl of DNA for 2 hr at 37°C. qPCR analysis was performed using primers specific for early (the R‐U5, minus strand strong stop DNA; Fwd: GCCTCAATAAAGCTTGCCTTGA, Rev: TGACTAAAAGGGTCTGAGGGATCT), late (U5‐Psi element, complete RT product; Fwd: TGTGTGCCCGTCTGTTGTGT, Rev: GAGTCCTGCGTCGAGAGATC) and 2‐LTR (U5‐U3, 2‐LTR circle formation in the nucleus; Fwd: AACTAGGGAACCCACTGCTTAAG, Rev: TCCACAGATCAAGGATATCTTGTC) HIV RT products. Serial dilutions of pDRNL XN HIV (from 107 to 1 copy per 10 μl) were used as standards for the early and late qPCR products. 2‐LTR standards were generated by amplification of the 2‐LTR region from infected MT‐2 cells using the 2‐LTR Fwd and Rev primers (above) and the product cloned into pTOPO. Amplification was performed using Brilliant III SYBR qPCR mix (Agilent) according to the manufacturer's instructions and results normalised to infected DMSO‐treated control samples within each experiment.
Supporting information
Figure S1. IN nuclear transport inhibitors reduce formation of nuclear forms of the HIV DNA‐ Full data set. Quadruplicate wells of MT‐2 cells were infected with HIV in the presence of the indicated compounds and analysed by qPCR at the indicated times using specific primers for the presence of either early HIV, late HIV or 2‐LTR transcripts. Results were relativised to the DMSO treated control within each experiment. Results represent the mean ± SEM (n = 4 – Budesonide/Flunisolide or 3 –Raltegravir/Efavirenz independent experiments), ** p < 0.01, *** p < 0.001, as compared to DMSO treated control (unpaired t‐test). RAL; Raltegravir, EFV; Efavirenz, Bud; Budesonide, Flu; Flunisolide
ACKNOWLEDGEMENTS
This work was supported by a grant from the Australian Centre for HIV and Hepatitis Virology Research. K. M. W. is a National Breast Cancer Foundation Career Development Fellow (ECF‐17‐007). D. A. J. is a National Health and Medical Research Council Senior Principal Research Fellow (APP1103050). The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pINSD.His.Sol was from Dr. Robert Craigie, pNL4‐3 was from Dr. Malcolm Martin and TZM‐bl was from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc.
Wagstaff KM, Headey S, Telwatte S, et al. Molecular dissection of an inhibitor targeting the HIV integrase dependent preintegration complex nuclear import. Cellular Microbiology. 2019;21:e12953 10.1111/cmi.12953
Contributor Information
Kylie M. Wagstaff, Email: kylie.wagstaff@monash.edu.
David A. Jans, Email: david.jans@monash.edu
REFERENCES
- Adachi, A. , Gendelman, H. E. , Koenig, S. , Folks, T. , Willey, R. , Rabson, A. , & Martin, M. A. (1986). Production of acquired immunodeficiency syndrome‐associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. Journal of Virology, 59(2), 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam, S. A. , Marr, R. S. , & Gerace, L. (1990). Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. The Journal of Cell Biology, 111(3), 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam, S. A. , Sternemarr, R. , & Gerace, L. (1992). Nuclear‐protein import using digitonin‐permeabilized cells. Methods in Enzymology, 219, 97–110. [DOI] [PubMed] [Google Scholar]
- Bernis, C. , & Forbes, D. J. (2014). Analysis of nuclear reconstitution, nuclear envelope assembly, and nuclear pore assembly using Xenopus in vitro assays. Methods in Cell Biology, 122, 165–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoux, L. , Sebastian, S. , Muesing, M. A. , & Luban, J. (2007). The role of lysine 186 in HIV‐1 integrase multimerization. Virology, 364(1), 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky, M. I. , Sharova, N. , McDonald, T. L. , Pushkarskaya, T. , Tarpley, W. G. , & Stevenson, M. (1993). Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proceedings of the National Academy of Sciences of the United States of America, 90(13), 6125–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick, R. C. , Delviks‐Frankenberry, K. A. , Chen, J. , Janaka, S. K. , Sastri, J. , Hu, W. S. , & Pathak, V. K. (2017). Dynamics and regulation of nuclear import and nuclear movements of HIV‐1 complexes. PLoS Pathogens, 13(8), e1006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiadis, A. , Shao, H. , Hübner, S. , & Jans, D. A. (1997). Kinetic characterization of the human retinoblastoma protein bipartite nuclear localization sequence (NLS) in vivo and in vitro. A comparison with the SV40 large T‐antigen NLS. The Journal of Biological Chemistry, 272(35), 22134–22139. [DOI] [PubMed] [Google Scholar]
- Faure, A. , Calmels, C. , Desjobert, C. , Castroviejo, M. , Caumont‐Sarcos, A. , Tarrago‐Litvak, L. , … Parissi, V. (2005). HIV‐1 integrase crosslinked oligomers are active in vitro. Nucleic Acids Research, 33(3), 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzkee, N. C. , Masse, J. E. , Shen, Y. , Davies, D. R. , & Bax, A. (2010). Solution conformation and dynamics of the HIV‐1 integrase core domain. The Journal of Biological Chemistry, 285(23), 18072–18084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildyal, R. , Ho, A. , Wagstaff, K. M. , Dias, M. M. , Barton, C. L. , Jans, P. , … Jans, D. A. (2005). Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin β1 independent of importin α. Biochemistry, 44(38), 12887–12895. [DOI] [PubMed] [Google Scholar]
- Harada, S. , Koyanagi, Y. , & Yamamoto, N. (1985). Infection of HTLV‐III/LAV in HTLV‐I‐carrying cells MT‐2 and MT‐4 and application in a plaque assay. Science, 229(4713), 563–566. [DOI] [PubMed] [Google Scholar]
- Hearps, A. C. , & Jans, D. A. (2006). HIV‐1 integrase is capable of targeting DNA to the nucleus via an importin α/β‐dependent mechanism. Biochemical Journal, 398(3), 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Y. , Mcguinness, D. E. , Prongay, A. J. , Feld, B. , Ingravallo, P. , Ogert, R. A. , … Howe, J. A. (2008). Screening for antiviral inhibitors of the HIV integrase‐LEDGF/p75 interaction using the AlphaScreen luminescent proximity assay. Journal of Biomolecular Screening, 13(5), 406–414. [DOI] [PubMed] [Google Scholar]
- Hubner, S. , Xiao, C. Y. , & Jans, D. A. (1997). The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T‐antigen nuclear localization sequence by importin. Journal of Biological Chemistry, 272(27), 17191–17195. [DOI] [PubMed] [Google Scholar]
- Iborra, M. , Alvarez‐Sotomayor, D. , & Nos, P. (2014). Long‐term safety and efficacy of budesonide in the treatment of ulcerative colitis. Clinical and Experimental Gastroenterology, 7, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, L. D. , Polygenis, D. , McIvor, R. , & Worthington, I. (1999). Comparative efficacy and safety of inhaled corticosteroids in asthma. The Canadian Journal of Clinical Pharmacology, 6(1), 26–37. [PubMed] [Google Scholar]
- Jans, D. A. , Jans, P. , Briggs, L. J. , Sutton, V. , & Trapani, J. A. (1996). Nuclear transport of granzyme B (fragmentin‐2). Dependence of perforin in vivo and cytosolic factors in vitro. The Journal of Biological Chemistry, 271(48), 30781–30789. [DOI] [PubMed] [Google Scholar]
- Jayappa, K. D. , Ao, Z. , & Yao, X. (2012). The HIV‐1 passage from cytoplasm to nucleus: The process involving a complex exchange between the components of HIV‐1 and cellular machinery to access nucleus and successful integration. International Journal of Biochemistry and Molecular Biology, 3(1), 70–85. [PMC free article] [PubMed] [Google Scholar]
- Latham, C. F. , la, J. , Tinetti, R. N. , Chalmers, D. K. , & Tachedjian, G. (2016). Fragment based strategies for discovery of novel HIV‐1 reverse transcriptase and integrase inhibitors. Current Topics in Medicinal Chemistry, 16(10), 1135–1153. [DOI] [PubMed] [Google Scholar]
- Lau, C. K. , Delmar, V. A. , Chan, R. C. , Phung, Q. , Bernis, C. , Fichtman, B. , … Forbes, D. J. (2009). Transportin regulates major mitotic assembly events: From spindle to nuclear pore assembly. Molecular Biology of the Cell, 20(18), 4043–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, A. , Loyter, A. , & Bukrinsky, M. (2011). Strategies to inhibit viral protein nuclear import: HIV‐1 as a target. Biochimica et Biophysica Acta, 1813(9), 1646–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg, L. , Pinkham, C. , Baer, A. , Amaya, M. , Narayanan, A. , Wagstaff, K. M. , … Kehn‐Hall, K. (2013). Nuclear import and export inhibitors alter capsid protein distribution in mammalian cells and reduce Venezuelan Equine Encephalitis Virus replication. Antiviral Research, 100(3), 662–672. [DOI] [PubMed] [Google Scholar]
- Mbisa, J. L. , Delviks‐Frankenberry, K. A. , Thomas, J. A. , Gorelick, R. J. , & Pathak, V. K. (2009). Real‐time PCR analysis of HIV‐1 replication post‐entry events. Methods in Molecular Biology, 485, 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels, R. , Balzarini, J. , Baba, M. , Snoeck, R. , Schols, D. , Herdewijn, P. , … de Clercq, E. (1988). Rapid and automated tetrazolium‐based colorimetric assay for the detection of anti‐HIV compounds. Journal of Virological Methods, 20(4), 309–321. [DOI] [PubMed] [Google Scholar]
- Peat, T. S. , Dolezal, O. , Newman, J. , Mobley, D. , & Deadman, J. J. (2014). Interrogating HIV integrase for compounds that bind‐a SAMPL challenge. Journal of Computer‐Aided Molecular Design, 28(4), 347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller, S. C. , Caly, L. , & Jans, D. A. (2003). Nuclear import of the pre‐integration complex (PIC): The Achilles heel of HIV? Current Drug Targets, 4(5), 409–429. [DOI] [PubMed] [Google Scholar]
- Saberi, P. , Phengrasamy, T. , & Nguyen, D. P. (2013). Inhaled corticosteroid use in HIV‐positive individuals taking protease inhibitors: A review of pharmacokinetics, case reports and clinical management. HIV Medicine, 14(9), 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller, T. , Ocwieja, K. E. , Rasaiyaah, J. , Price, A. J. , Brady, T. L. , Roth, S. L. , … Towers, G. J. (2011). HIV‐1 capsid‐cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathogens, 7(12), e1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A. , Slaughter, A. , Jena, N. , Feng, L. , Kessl, J. J. , Fadel, H. J. , … Kvaratskhelia, M. (2014). A new class of multimerization selective inhibitors of HIV‐1 integrase. PLoS Pathogens, 10(5), e1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F. W. (2005). Protein production by auto‐induction in high density shaking cultures. Protein Expression and Purification, 41(1), 207–234. [DOI] [PubMed] [Google Scholar]
- Tyssen, D. , Henderson, S. A. , Johnson, A. , Sterjovski, J. , Moore, K. , la, J. , … Tachedjian, G. (2010). Structure activity relationship of dendrimer microbicides with dual action antiviral activity. PLoS One, 5(8), e12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff, K. M. , & Jans, D. A. (2006). Intramolecular masking of nuclear localization signals: Analysis of importin binding using a novel AlphaScreen‐based method. Analytical Biochemistry, 348(1), 49–56. [DOI] [PubMed] [Google Scholar]
- Wagstaff, K. M. , Rawlinson, S. M. , Hearps, A. C. , & Jans, D. A. (2011). An AlphaScreen(R)‐based assay for high‐throughput screening for specific inhibitors of nuclear import. Journal of Biomolecular Screening, 16(2), 192–200. [DOI] [PubMed] [Google Scholar]
- Wagstaff, K. M. , Sivakumaran, H. , Heaton, S. M. , Harrich, D. , & Jans, D. A. (2012). Ivermectin is a specific inhibitor of importin α/β‐mediated nuclear import able to inhibit replication of HIV‐1 and dengue virus. The Biochemical Journal, 443(3), 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg, M. A. , Mesplede, T. , & Quashie, P. K. (2012). The development of novel HIV integrase inhibitors and the problem of drug resistance. Current Opinion in Virology, 2(5), 656–662. [DOI] [PubMed] [Google Scholar]
- Wapling, J. , Moore, K. L. , Sonza, S. , Mak, J. , & Tachedjian, G. (2005). Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. Journal of Virology, 79(16), 10247–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X. , Decker, J. M. , Liu, H. , Zhang, Z. , Arani, R. B. , Kilby, J. M. , … Kappes, J. C. (2002). Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T‐20) monotherapy. Antimicrobial Agents and Chemotherapy, 46(6), 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielens, J. , Headey, S. J. , Rhodes, D. I. , Mulder, R. J. , Dolezal, O. , Deadman, J. J. , … Scanlon, M. J. (2013). Parallel screening of low molecular weight fragment libraries: Do differences in methodology affect hit identification? Journal of Biomolecular Screening, 18(2), 147–159. [DOI] [PubMed] [Google Scholar]
- Xiao, C. Y. , Hubner, S. , & Jans, D. A. (1997). SV40 large tumor antigen nuclear import is regulated by the double‐stranded DNA‐dependent protein kinase site (serine 120) flanking the nuclear localization sequence. Journal of Biological Chemistry, 272(35), 22191–22198. [DOI] [PubMed] [Google Scholar]
- Yap, S. H. , Sheen, C. W. , Fahey, J. , Zanin, M. , Tyssen, D. , Lima, V. D. , … Tachedjian, G. (2007). N348I in the connection domain of HIV‐1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Medicine, 4(12), e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IN nuclear transport inhibitors reduce formation of nuclear forms of the HIV DNA‐ Full data set. Quadruplicate wells of MT‐2 cells were infected with HIV in the presence of the indicated compounds and analysed by qPCR at the indicated times using specific primers for the presence of either early HIV, late HIV or 2‐LTR transcripts. Results were relativised to the DMSO treated control within each experiment. Results represent the mean ± SEM (n = 4 – Budesonide/Flunisolide or 3 –Raltegravir/Efavirenz independent experiments), ** p < 0.01, *** p < 0.001, as compared to DMSO treated control (unpaired t‐test). RAL; Raltegravir, EFV; Efavirenz, Bud; Budesonide, Flu; Flunisolide