

Abstract
Congenital hyperinsulinism is a rare disease, but is the most frequent cause of persistent and severe hypoglycaemia in early childhood. Hypoglycaemia caused by excessive and dysregulated insulin secretion (hyperinsulinism) from disordered pancreatic β cells can often lead to irreversible brain damage with lifelong neurodisability. Although congenital hyperinsulinism has a genetic cause in a significant proportion (40%) of children, often being the result of mutations in the genes encoding the KATP channel (ABCC8 and KCNJ11), not all children have severe and persistent forms of the disease. In approximately half of those without a genetic mutation, hyperinsulinism may resolve, although timescales are unpredictable. From a histopathology perspective, congenital hyperinsulinism is broadly grouped into diffuse and focal forms, with surgical lesionectomy being the preferred choice of treatment in the latter. In contrast, in diffuse congenital hyperinsulinism, medical treatment is the best option if conservative management is safe and effective. In such cases, children receiving treatment with drugs, such as diazoxide and octreotide, should be monitored for side effects and for signs of reduction in disease severity. If hypoglycaemia is not safely managed by medical therapy, subtotal pancreatectomy may be required; however, persistent hypoglycaemia may continue after surgery and diabetes is an inevitable consequence in later life. It is important to recognize the negative cognitive impact of early‐life hypoglycaemia which affects half of all children with congenital hyperinsulinism. Treatment options should be individualized to the child/young person with congenital hyperinsulinism, with full discussion regarding efficacy, side effects, outcomes and later life impact.
What's new?
Congenital hyperinsulinism is a heterogeneous disorder of hypoglycaemia with high morbidity and few therapeutic options.
Investigation and treatment principles have been described in several reviews but there has been little emphasis on medium‐ to long‐term outcomes.
In the present review, we describe outcomes of congenital hyperinsulinism to provide a long‐range view of the clinical impact of current therapeutic strategies.
What's new?
Congenital hyperinsulinism is a heterogeneous disorder of hypoglycaemia with high morbidity and few therapeutic options.
Investigation and treatment principles have been described in several reviews but there has been little emphasis on medium‐ to long‐term outcomes.
In the present review, we describe outcomes of congenital hyperinsulinism to provide a long‐range view of the clinical impact of current therapeutic strategies.
Introduction
Congenital hyperinsulinism (CHI) is a rare disorder of hypoglycaemia attributable to inappropriate and dysregulated insulin secretion from the pancreas 1, 2, 3. Insulin is normally secreted in a highly regulated process by β cells within the islets of Langerhans after an increase in plasma glucose levels. In humans, glucose is transported inside β cells by facilitated diffusion, where it is metabolized to glucose‐6‐phosphate by glucokinase. Following further metabolism of glucose‐6‐phosphate, an increase in the intracellular adenosine triphosphate (ATP): adenosine diphosphate (ADP) ratio causes ATP‐sensitive K+ (KATP) channels in the β‐cell membrane to close, preventing the outward flux of K+ ions. Subsequent membrane depolarization activates voltage‐dependent Ca2+ channels in the cell membrane, and Ca2+ enters the cell, triggering Ca2+‐dependent exocytosis of insulin. In CHI, the feedback regulation of insulin by plasma glucose is disordered, causing uninhibited insulin secretion 4 and unpredictable and severe hypoglycaemia 2.
Hyperinsulinism causes not only hypoglycaemia, but also suppresses lipolysis and reduces ketones, which act as alternative fuels to maintain brain neuronal function; therefore, hypoglycaemia caused by CHI is detrimental to the brain, with adverse neurodevelopment in a third to half of children with CHI 5, 6, 7, 8, 9.
In clinical practice, a diagnosis of CHI is likely if hypoglycaemia is recurrent and glucose infusion rate increases (normal glucose infusion rate 4–6 mg/kg/min) 1, 2, 3. Diagnosis is established by the finding of detectable insulin at hypoglycaemia; however, as insulin has a short half‐life (6 min), laboratory measurements may be inaccurate and may miss the tail of a rapidly receding secretory burst. Ancillary biochemistry is often helpful to identify insulin fingerprints, i.e. low fatty acid and 3‐hydroxybutyrate levels; a response to glucagon (1.0 mg intramuscular/intravenous which increases plasma glucose levels by 1.7 mmol/l within 40 min) indicates a strong likelihood of CHI.
The incidence of CHI is recorded to be 1:50 000 in Western populations. In consanguineous populations with greater incidence of recessively inherited genetic mutations, the incidence is as high as 1:2500 10. These incidence rates do not take into account resolving/transient CHI, however, and are skewed towards the severe end of the spectrum where children may be unresponsive to conservative medical treatment. Although CHI is a rare disease, it remains one of the most common causes of recurrent and persistent hypoglycaemia in infancy.
Conundrum of hypoglycaemia
While it is well recognized that CHI is responsible for severe hypoglycaemia, the plasma glucose threshold at which treatment is required is uncertain. The definition of hypoglycaemia has been debated, with no clear numerical definition having been agreed to establish a threshold that correlates with evidence‐based outcomes. Nonetheless, there is sufficient evidence that plasma glucose <3.0 mmol/l in children with most forms of hypoglycaemia are harmful for neuronal survival 11. Considering the brain impact in CHI, it is preferable to adjust the threshold to a value nearer the normal range. Most centres aim to keep the lower limit of plasma glucose between 3.3 and 3.8 mmol/l (60–70 mg/dl) during treatment. A higher threshold mitigates against the risk of brain damage but requires greater therapeutic resources. The rationale behind the higher threshold is shown in Fig. 1.
Figure 1.
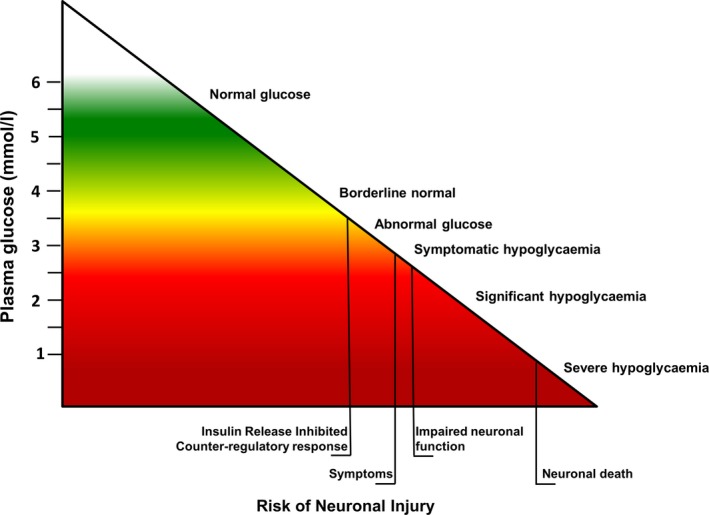
Relationship between plasma glucose and risk of neuronal injury in normal children: as glucose levels decrease, the risk of hypogycaemia‐induced neuronal damage increases. While there is no numerical definition of hypoglycaemia in children, the inverse correlation between glucose and risk supports a pragmatic clinical approach to keep glucose levels near the normal range to prevent long‐term brain damage.
Spectrum of congenital hyperinsulinism: subtypes and classification
Congenital hyperinsulinism is termed a ‘congenital’ condition, but is more a heterogeneous group of conditions underpinned by hypoglycaemia and hyperinsulinism. Presentation with hypoglycaemia is usually in the neonatal period, typically within the first week of life. Late‐onset CHI may also occur, beyond the neonatal period, in 10% of cases 12. It is not known why in genetic forms of CHI, for example, in focal CHI, presentation may be delayed.
Transient congenital hyperinsulinism
The natural history of disease varies between individuals, with different patterns of severity even in those with identified genetic forms of CHI. Hyperinsulinism causing hypoglycaemia can occur in newborn babies born small for gestational age. Although the term CHI is often applied to their condition, hypoglycaemia, no matter how severe, resolves in most cases within several weeks 12, 13. Perinatal asphyxia may also cause hyperinsulinism, which usually resolves in days and, rarely, in weeks. The mechanism of hyperinsulinism and subsequent resolution is not known. Some cases of transient CHI have been ascribed to dominantly acting mutations in ABCC8, encoding the regulatory SUR1 subunits of the β‐cell KATP channels, and responsible for diabetes later in life 14, 15.
Genetic forms of congenital hyperinsulinism
Genetic forms of CHI occur in 40% of children 12, 16, 17. The majority of genetic defects occur in the KATP channel gene ABCC8, as described above, and in KCNJ11, encoding the Kir6.2 pore‐forming subunits of the channel. These inactivating mutations in KATP channel genes cause the loss of effective KATP channel regulation by nucleotides and lipids, and dysfunctional KATP channels in the β‐cell membrane. By reducing the ability of β cells to efflux K+ through KATP channels, KATP channel gene mutations result in inappropriate membrane depolarization and unregulated Ca2+ entry, which triggers insulin release without coupling to plasma glucose levels.
Genetic mutations in GLUD1, GCK, HADH and PGM1, or inappropriate expression of HK1 or SLC16A1, all affect nutrient metabolism within β cells to cause an inappropriately raised ATP:ADP ratio, thereby closing KATP channels 2. Activating gene mutations in CACNA1D encoding the voltage‐gated Ca2+ channels have been reported; inappropriate channel activation has been postulated to cause unregulated Ca2+ entry into the β cell to trigger insulin secretion 18. Another mutation in the promoter region of PMM2 encoding phosphomannomutase 2, a key enzyme in protein glycosylation, also affects β‐cell function by unknown mechanisms 19.
Other genetic causes include mutations in, or altered expression of, transcription factors HNF4A, HNF1A and FOXA2 20, 21. The pathogenic mechanisms of transcription factor mutations are more varied, affecting development of β cells and their regulatory signalling pathways that control insulin secretion. The pathogenesis of other mutations, such as those reported in UCP2, is still debated and needs further clarification 22.
Genetic forms of CHI do not have consistent disease trajectories; while homozygous and compound heterozygous mutations are likely to suggest permanent forms of CHI, recent experience suggests reduction in the severity of hyperinsulinism over time, even in those with severe forms of disease 23. In those with CHI attributable to HNF4A mutations, hypoglycaemia switches to hyperglycaemia and maturity‐onset diabetes at a variable age 24. In those with activating GCK mutations causing CHI, some require no treatment, while others are responsive to diazoxide and yet others are severe enough to require pancreatectomy 25.
At present, for many children with CHI, no genetic mutations are identified 26. Within this group, resolution of disease is likely for many, but the time to resolution is unpredictable 12. A resolving trajectory may be evident in longitudinal follow‐up when normoglycaemia is maintained while treatment dose is gradually reduced and weaned off 12. The mechanisms and adaptations responsible for disease resolution remain unknown.
The technology enabling genetic investigations has improved rapidly. Previous reliance on KATP channel gene exon and exon–intron boundary sequencing 12 has paved the way for deep sequencing techniques and, more recently, next‐generation sequencing of the whole genome. The latter, although more time‐consuming than Sanger sequencing, is more comprehensive and covers a range of genes occurring with both high and low frequency in CHI as predetermined panels. Panel gene sequencing has several advantages over Sanger sequencing as it is better able to identify deletions causing CHI and has the potential for gene discovery through re‐evaluation of genetic output.
Histological descriptions of congenital hyperinsulinism: focal vs diffuse
Histologically, CHI is classified as either focal or diffuse (Fig. 2). Focal CHI occurs when a solitary part within the pancreas is populated predominantly by endocrine‐rich tissue, in contrast to scattered endocrine components in islets interspersed with exocrine tissue in the normal pancreas. Focal lesions occur as a result of paternally inherited mutations in ABCC8/KCNJ1, along with loss of heterozygosity in maternal alleles within the pancreas. This results in clonal expansion through loss of cell cycle repressors such as CDKN1C encoding p57kip2 and hyperfunction in mutated endocrine‐rich tissue, causing unpredictable and severe hypoglycaemia.
Figure 2.
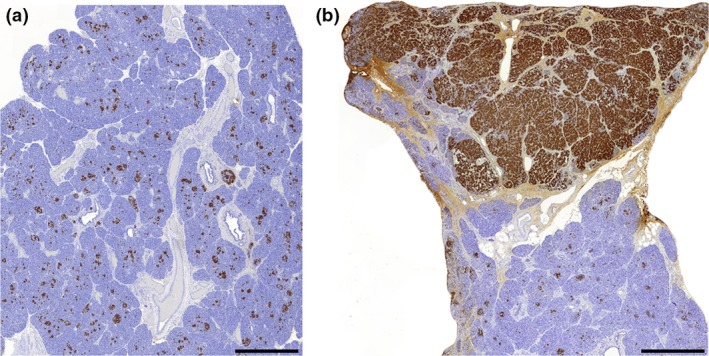
Typical histopathological appearance of diffuse and focal congenital hyperinsulinism (CHI) pancreata representing histological diversity of CHI. Panels A and B show insulin positive cells (INS+) in tissue from children with diffuse and focal disease, respectively. In diffuse disease all islet cells are similarly affected, but in focal CHI the condition is associated with expansion of INS+ cells within a defined focal lesion. Scale bar, 1 mm; magnification × 1.5.
18‐fluoro‐dopa positron‐emission tomography (PET)‐computed tomography [or PET‐magnetic resonance imaging (MRI)] is the current best investigation method with which to identify and localize focal lesions 27, although there can be lack of agreement on the results. The scan differentiates between focal and non‐focal aetiology but is unable to separate normal pancreas from that of diffuse CHI. The radiochemical 18‐fluoro‐dopa is concentrated in the abundance of neurons associated with islet cell overexpansion in the focal lesion. The scan works on the principle that dopa is converted to dopamine by dopa‐decarboxylase in the neurons. In focal lesions, there is uptake and retention of the dye in pancreatic neurons, which gives a high standard uptake value. It is not known why the neurons retain the radiochemical in focal CHI but not in diffuse CHI or in the normal pancreas.
In contrast to focal CHI, diffuse CHI is characterized by involvement of the entire endocrine pancreas. Diffuse CHI is typically caused by homozygous and compound heterozygous mutations in ABCC8/KCNJ11 acting recessively or in a dominant manner. Nuclear enlargement (greater than fourfold) of some islet cells is always found in the diffuse pancreas 28. By contrast, islet cell nucleomegaly is rarely seen in control islets. Whilst an increased incidence of nucleomegaly is pathognomonic for diffuse disease, these cells are non‐proliferative, suggesting a novel role in the pathobiology of this condition 28.
The distinction between focal CHI, diffuse CHI and normal tissue is important for frozen‐section histopathology at the time of focal lesion pancreatectomy. Tissue characteristics of focal CHI help to identify the lesion, with either a capsule or margin of normal tissue to ensure the lesion is removed in entirety. The diagnosis of focal CHI raises the prospect of a ‘cure’ if lesionectomy is successful; however, a focus may be small and hidden within the tissue of the pancreas. While palpation at the time of open laparotomy may allow a distinct firm feel if the lesion is encapsulated, the use of laparoscopic methods do not allow haptic feedback. In such cases, the search for focal lesions may be complex, with success relying on surgical skill and expertise.
Sometimes tissue histology is not characteristic of either focal or diffuse CHI and is therefore termed atypical CHI. Here, mosaic patterns of hyperfunctioning islets are present with islands of apparently normal‐appearing islets. Although atypical CHI has been described in several papers, there is no agreement on its clinical, biochemical and histological characteristics. Importantly, atypical CHI has not been associated with any known genetic cause 29, 30.
Congenital hyperinsulinism associated with other conditions
One of the problems with the classification of CHI is the heterogeneity of conditions encompassed by the diagnosis. Adding to the complexity of CHI is the association of hyperinsulinaemic hypoglycaemia with diseases such as congenital disorders of glycosylation and conditions such as Turner syndrome, Kabuki syndrome and Beckwith–Wiedemann syndrome. In each of these syndromes, chromosomal changes and differential methylation patterns suggest but do not define pathophysiology of hyperinsulinism.
Treatment
The immediate treatment goal in CHI is to stabilize plasma glucose levels and achieve normoglycaemia. This is usually achieved by additional dextrose administration. Oral dextrose is rarely able to prevent severe hypoglycaemia; in many cases high concentration dextrose is required through a central venous catheter. Children with CHI readily respond to glucagon treatment; continuous infusions of glucagon, administered intravenously, may reduce the dependence on large volumes of fluid, particularly if central venous catheter access is difficult. Early use of glucagon is therefore advocated to ensure rapid achievement of normoglycaemia without inducing fluid overload and complications such as pulmonary oedema, heart failure and electrolyte imbalance.
Glucagon
Currently glucagon formulations for intramuscular injections are diluted in saline or dextrose to provide a continuous intravenous infusion. Such infusions are used in the short term in doses ranging between 2 and 20 mcg/kg/h to achieve normoglycaemia, pending treatment with definitive agents such as diazoxide. Another typical dose is 1 mg/day as a continuous infusion; this set dose is adopted by many centres and has the potential to reduce dosing errors. Glucagon has been used in continuous subcutaneous infusions in the long‐term treatment of CHI 31; however, catheter occlusion commonly occurs as a result of fibrillation of native glucagon in slow‐moving solutions rendering treatment unsafe and ineffective. Newer glucagon formulations that are soluble and stable in saline may provide long‐term treatment options 32. While glucagon is generally effective in CHI, rare side effects need to be monitored, including the possibility of necrolytic migratory erythema (Fig. 3).
Figure 3.
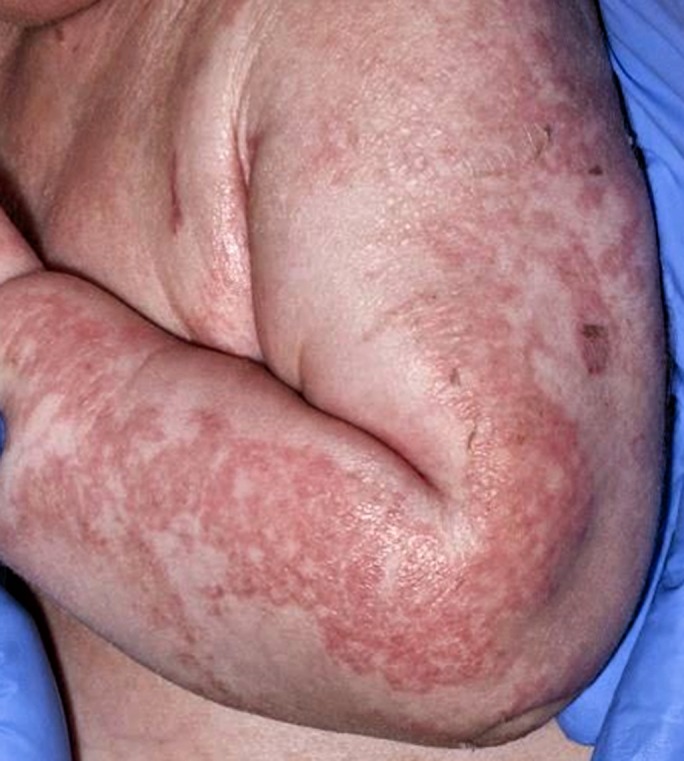
Necrolytic migratory erythema is a rare side effect of glucagon therapy. Typically the rash spreads with a red irregular border and starts to disappear within 1–2 days of stopping treatment. CHI, congenital hyperinsulinism; CT, computed tomography; SSRA, somatostatin receptor analogue; PET, positron‐emission tomography.
Diazoxide
Diazoxide is an agonist/opener of KATP channels. In islet β cells it causes repolarization of the membrane potential and reduces the risk of calcium‐mediated insulin exocytosis. Diazoxide is usually effective in children in whom KATP channels are intact, but is ineffective if KATP channels are malformed as a result of mutations in ABCC8/KCNJ11; however, exceptionally, children with recognized mutations in ABCC8 respond to diazoxide, suggesting that cell adaptations and redundancy in the standard KATP channel model determine the pathophysiology of CHI 33.
Diazoxide has a number of side effects which need careful monitoring. In the acute phase of treatment, diazoxide causes fluid accumulation which is usually offset by concomitant treatment with chlorothiazide. It is wise to restrict fluid intake in the neonatal period to <150 ml/kg/day to reduce the risk of pulmonary overload. Diazoxide is uncommonly associated with neutropaenia and thrombocytopaenia (https://bnfc.nice.org.uk/drug/diazoxide.html#sideEffects), which can complicate the risk of infection already heightened by the presence of the central venous catheter and by prolonged hospital stay. More recently diazoxide has been found to be associated with life‐threatening pulmonary hypertension 34. Diazoxide has also been associated with pericardial effusion requiring surgical drainage 35. In chronic treatment, diazoxide is complicated by excess body hair, often in doses in excess of 5 mg/kg/day (Fig. 4). Body hair can be troublesome for older children who may choose to switch to other agents to avoid this complication. Diazoxide also causes coarsening of facial features, with some families noticing facial similarities in children with prolonged treatment; therefore, while diazoxide is a convenient oral medication for CHI, its use is beset with a variety of complications. Furthermore, ~25% of children with CHI are partially or completely unresponsive to diazoxide 12. In such children, second‐line treatment with other agents, such as octreotide, may be required.
Figure 4.
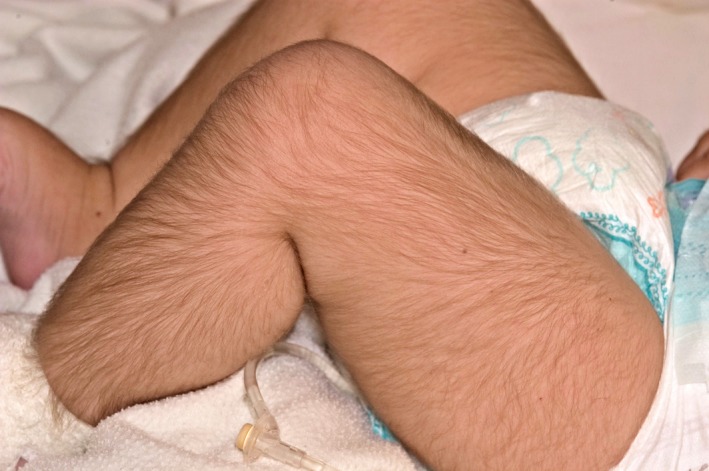
An infant on diazoxide at 10 mg/kg/day developing excess body hair, a common occurrence with diazoxide dose >5 mg/kg/day.
Somatostatin receptor analogues
Second‐line treatment involves somatostatin receptor analogues (SSRAs), typically octreotide, which act to inhibit insulin release. The drug is administered as subcutaneous or intravenous injections 4–6 times/day and can also be administered as continuous infusion by either route. Long‐acting forms of SSRAs (e.g. somatuline autogel) are also available. In some cohorts, off‐licence use of long‐acting SSRAs has been found to be beneficial for patient compliance 36.
One problem with SSRA use is tachyphylaxis, an unpredictable loss of drug efficacy after initiation of treatment; initial doses of 5 mcg/kg/day need adjustment upwards to doses at the limit of the normal range, i.e. 30–35 mcg/kg/day. In most children, treatment beyond these doses is of no therapeutic value; however, in exceptional cases, octreotide dose may be as high as 50–60 mcg/kg/day 41. The mechanisms responsible for tachyphylaxis have not been determined, but are likely to involve desensitization of receptor‐mediated responses.
In all cases on octreotide therapy, vigilance must be maintained for deterioration in hepatic function 37. In many children high alanine transaminase levels are common with both short‐ and long‐acting SSRAs 36. Octreotide slows biliary excretion, thereby causing biliary sludging, which, in time, may accumulate to form gallstones. In children, the presence of gallstones needs careful observation, with a plan for cholecystectomy if symptoms suggest cholecystitis. Octreotide occasionally causes growth failure by reducing growth hormone secretion from pituitary somatotrophs. Octreotide should be used cautiously in early life, especially in preterm babies, because of the adverse impact on splanchnic circulation and risk of necrotizing enterocolitis 38.
Long‐acting SSRAs are favoured by families on account of the monthly injection regimen without recourse to daily injections as needed for octreotide therapy 36. The experience with long‐acting SSRAs is rising worldwide; however, long‐term benefits and side effects have not been evaluated thoroughly. One problem with the use of a long‐acting SSRA is the inability to reduce dose, unlike with oral preparations such as diazoxide. Given that severity of CHI may naturally reduce over time, the use of fixed‐dose long‐acting SSRAs may be considered disproportionate. Nonetheless, in the absence of plausible and safe therapeutic alternatives, SSRAs will remain a mainstay in the treatment of CHI. A pragmatic approach to SSRA treatment would be to undertake annual assessments to test drug requirements with dose reduction as tolerated.
Experimental therapy
As hypoglycaemia in CHI is often severe and treatment choices relatively limited, a number of centres have used medication of doubtful efficacy. Nifedipine, a calcium channel antagonist, offers a theoretical basis for reducing calcium‐mediated exocytosis of insulin in β cells; however, nifedipine is rarely effective and is not commonly used as therapy for CHI 39.
The mTOR inhibitor sirolimus has been recently used, with some success, in the treatment of CHI when normoglycaemia is not achieved using standard conservative medical therapy 40. The initial success of therapy has not been replicated widely, with proposed mechanisms of action not standing up to scrutiny 41. More importantly, sirolimus, being an immunosuppressant, has been implicated in serious infections, causing more harm than benefit; therefore, at present, sirolimus is not a standard choice for treatment of diazoxide‐ and SSRA‐unresponsive CHI, although its use is expected to continue as an alternative for families who would prefer not to undertake irreversible pancreatic surgery with the inevitable risk of diabetes in later life 42.
Polyunsaturated fatty acids
Polyunsaturated fatty acids are food supplements and not classified as drugs. A small trial showed marginal benefit in children on diazoxide treatment 43. The risk of hypoglycaemia and hyperglycaemia reduced after treatment, suggesting that polyunsaturated fatty acids had the effect of reducing glycaemic flux. Polyunsaturated fatty acids cannot be advocated as standalone treatment for CHI, but they have minimal side effects and may help reduce drug dosage and therefore reduce complications and side effects.
Pancreatic surgery
In children with a focal lesion within the pancreas, lesionectomy may be curative; however, such lesions may occur in areas of the pancreas which are in proximity to important anatomical structures, such as the bile duct. Lesions in the head of the pancreas close to the C‐curve of the duodenum are less amenable to lesionectomy. In such cases, laparoscopic procedures may not be the first choice. As lesionectomy may require extensive resection to the intestine to ensure bile duct patency, one choice may be continuation of conservative medical management. Although focal lesions occur through genetically determined clonal expansion of hyperfunctioning tissue, there is emerging evidence that focal lesions may also reduce in severity and resolve over a 2–6‐year period 23.
In children with diffuse CHI, the treatment of choice is medical. With gradual reduction in severity, it is expected that hyperinsulinism will be more amenable to treatment; however, in some children, safe medical therapy is not possible and subtotal pancreatectomy may be the only choice of treatment to achieve normoglycaemia (Fig. 5). In the short term, subtotal pancreatectomy may cause exocrine insufficiency, requiring pancreatic enzyme supplements. Not all children maintain normoglycaemia in the medium term, with the incidence of hypoglycaemia being reported to be as high as 60% in some cohorts 44, 45. In the long term, children are at risk of developing hyperglycaemia and eventually diabetes, requiring insulin injection therapy. Subtotal pancreatectomy for CHI is therefore an imperfect treatment option.
Figure 5.
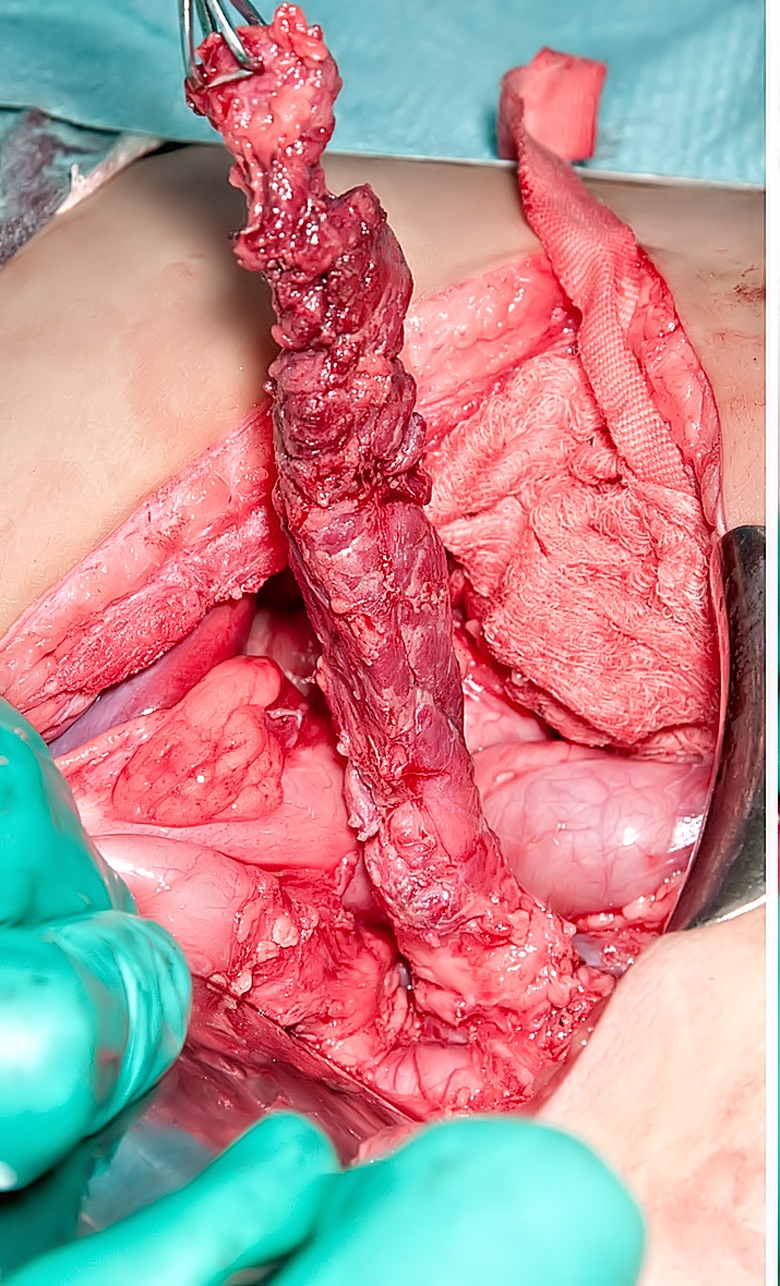
Surgical view of subtotal pancreatectomy performed using an open laparotomy approach. The tail of the pancreas has been mobilized and held vertically upwards. It is important to safeguard important structures such as the splenic artery, portal vein and bile duct during the process of dissection.
Treatment algorithms for the management of congenital hyperinsulinism
A management plan is summarized in Fig. 6. Iterative variations of this decision model will depend on local factors, such as method and turnaround of genetic analysis, medical insurance requirements and comorbidities. Exceptions to the diazoxide response‐guided gene mutation analysis include syndromic associations, possibility of HNF4A, consanguinity and family history of genetic CHI. After initial glycaemic stabilization, the assessment of genetic aetiology and diazoxide responsiveness are key steps that determine future clinical management strategies. Good response to an initial diazoxide dose <10 mg/kg/day generally indicates satisfactory KATP channel activity and therefore indicates the possibility of dose reduction over time. In those in whom there is continued requirement for diazoxide (even at a low dose) after 6 months of treatment, gene analysis should be considered to identify non‐KATP genetic forms of CHI such as GLUD1.
Figure 6.
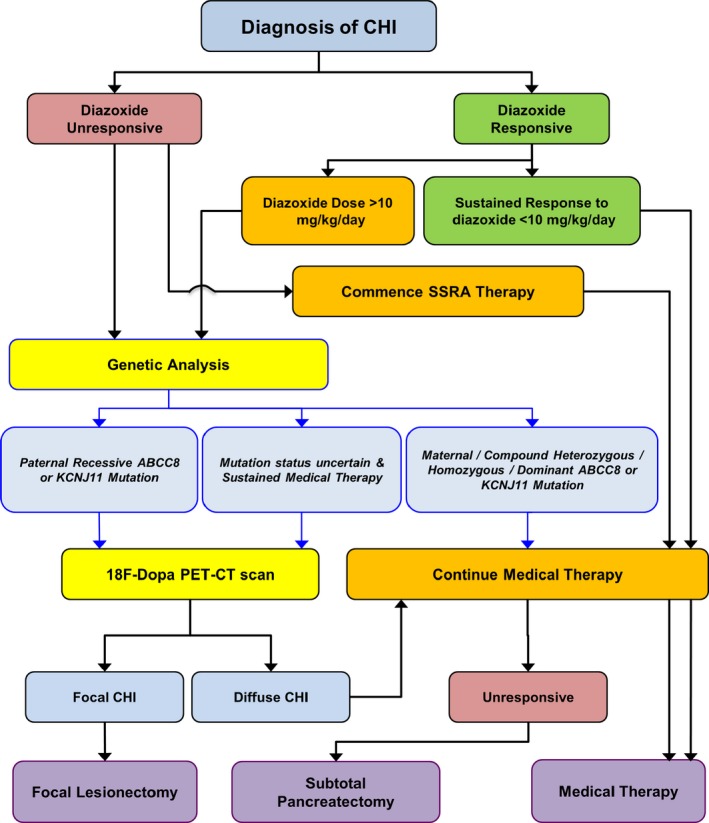
Treatment algorithm of congenital hyperinsulinism (CHI) with key components of diazoxide responsiveness and gene mutation analysis guiding clinical decisions. CT, computed tomography; SSRA, somatostatin receptor analogue; PET, positron‐emission tomography.
In contrast to other conditions, where genetic aetiology does not lead to immediate management changes, in CHI the finding of a paternal heterozygous mutation acting in a recessive manner is a clue that hypoglycaemia may be attributable to a focal lesion in the pancreas. While the additional genetic event of silencing of maternal alleles cannot be ascertained prior to surgery, the finding of a recessively acting paternal mutation should prompt 18‐fluoro‐dopa PET scanning 46. However, in some instances where the mutation is novel, it is not possible to predict recessive or dominant inheritance, the latter being associated with diffuse CHI. Another caveat to the genetic strategy delineating focal and non‐focal CHI is the reliance on blood lymphocyte DNA extraction; in the event of a mosaic mutation or a mutation occurring in low frequency, lymphocyte genomic DNA testing may fail to identify a mutation. 18‐fluorodopa PET‐CT (or PET‐MRI) scanning could therefore be considered in those in whom mutations are either uncertain or cannot be identified but continue to require diazoxide/somatostatin receptor analogue treatment without dose reduction after 1 year of diagnosis. In centres without facilities for 18‐fluorodopa PET‐CT scanning, medical therapy should be continued; however, in those in whom focal CHI remains a possibility, the aim should be to refer to a centre with scan facilities before considering pancreatectomy.
Once focal CHI has been confirmed by scanning, the logical choice is to proceed to lesionectomy, if the lesion is amenable to surgery. Exceptionally, there may be a case to consider conservative medical treatment if the patient has normoglycaemia with existing treatment 23. This choice is also applicable to those in whom a lesion is not localized in spite of scan information. However, the balance of risks favours a surgical approach over a medical one; the prospect of ‘cure’ has to be balanced against the risk of unpredictable hypoglycaemia and the long duration of medical therapy incurring the risk of life‐threatening side effects.
If investigations suggest non‐focal aetiology and hypoglycaemia is not amenable to standard medical therapy, subtotal pancreatectomy may be necessary to reduce the burden of morbidity from recurrent hypoglycaemia, prolonged hospital stay and drug side effects. Diabetes secondary to surgery is usually delayed for a few years 44 and could be reasonably well managed on modern insulin pump therapy. Some centres may advocate sparing the pancreas in return for extensive medical therapy with high‐dose treatment with SSRAs, mTOR inhibitors, enriched nutrition and continuous feeding. At present it is not known if long‐term cognitive and functional outcomes of subtotal pancreatectomy are any different from those of complicated medical therapy.
Future therapeutic options
Current treatment of CHI has not progressed from diazoxide and SSRAs used over the past two decades. Both medications are complicated by inefficacy and side effects, some of a life‐threatening nature. Experimental therapy has not shifted the burden of drug‐unresponsive CHI safely beyond subtotal pancreatectomy to justify routine clinical use. There is an urgent need to design new therapy to reduce insulin secretion and/or insulin action while maintaining normoglycaemia safely.
A number of therapeutic strategies are currently in trial planning stages. Soluble glucagon is now available for the treatment of diabetes and ready for trial in CHI. Another current drug is a specific insulin receptor allosteric antibody that reduces insulin action 47. Alternative drug targets are also in the process of exploration. The glucagon‐like peptide‐1 inverse agonist exendin 9‐39 has been shown to improve glycaemic profiles in adolescents and adults with CHI 48, but the programme of drug development has not progressed to trials in younger children. Alternative agonists of SSRAs may also be of value. Unlike octreotide, which exhibits greater selectivity for the SSR subtype 2, pasireotide is more selective for SSRA subtype 5 than 2. Pasireotide is licensed for adults with Cushing syndrome, where drug‐related hyperglycaemia is noted as a side effect 49. Pasireotide could be used for CHI, although concerns over hepatic impairment could be a limiting factor.
The development of chaperones that assist trafficking of KATP channel components from endoplasmic reticulum to cell membrane is potentially an exciting area of drug development. KATP channels are analogous in structure to the cystic fibrosis transmembrane conductance regulator, CFTR. Gene defects in CFTR associate with cystic fibrosis through altered protein‐trafficking, similarly to CHI. The corrector–potentiator combination of lumacaftor‐ivacaftor is beneficial in cystic fibrosis 50; similar strategies could be used to improve trafficking of KATP channels to improve channel function, although only limited effects of enhanced protein trafficking can be shown in isolated human CHI islets 51.
Monitoring treatment efficacy
Reduction in hypoglycaemia and maintaining normoglycaemia is a goal of treatment in CHI. Monitoring glucose levels is therefore a key investigation. It is impractical to measure plasma glucose levels several times a day using standardized laboratory methods, so alternative compact systems requiring small quantities of blood by finger prick are preferred. In the acute phase, hospital‐based point‐of‐care testing devices are useful for frequent 1–2‐hourly glucose measurements. In the community, home monitoring of plasma glucose 3–4 times/day is more convenient with handheld glucose meters, but most meters are designed for diabetes/hyperglycaemia where accuracy is important at the higher end of the plasma glucose range. The reliability in the hypoglycaemic spectrum is questionable.
Continuous glucose monitoring systems provide an alternative strategy for monitoring subcutaneous glucose levels 52. The reliability of continuous glucose monitoring has not been established in CHI but may ascertain trends at night. At present, continuous glucose monitoring continues to be an experimental tool with research applications, for example, comparing glycaemic patterns in trials 43. Sensor disconnection and incorrect glucose measurements may impede continuous glucose monitoring performance, but it is expected that precision will improve in the future.
In addition to the assessment of glucose profiles, monitoring for treatment side effects is also important. After children with CHI are discharged from hospital, specialist nurse‐led telephone/digital communication with their families may identify acute problems such as fluid overload and breathing difficulties. In regular clinic review, blood counts, electrolytes, liver function and ultrasound scans may be required to identify side effects from diazoxide and/or octreotide. Monitoring plans should be individualized and discussed with children and families for early recognition of treatment‐related complications.
Outcomes of congenital hyperinsulinism
Conservatively treated congenital hyperinsulinism: natural history
Congenital hyperinsulinism is a heterogeneous condition in which disease trajectories vary between different forms of the disease. In those children who are medically responsive, the severity of the disease decreases over time and remission may be achieved. Children with hyperinsulinism who are small for gestational age at birth may have transient disease. In children with normal or large birth weight, CHI resolves in about half of those with no genetic mutations 12. Some children with transient CHI can develop ketotic hypoglycaemia 53 at a later stage, which should be differentiated from hypoglycaemia caused by persistent hyperinsulinism.
Genetic forms of CHI are less likely to resolve than forms without identified CHI‐causing mutations; however, reduction in severity and resolution is also observed in conservatively treated children with KATP channel gene mutations 12, 23, 54, 55, not only in those with dominantly acting ABCC8/KCNJ11 mutations, but also in those with homozygous, compound heterozygous and paternally inherited ABCC8/KCNJ11 mutations causing focal CHI 23, 54.
Children with dominantly acting ABCC8 mutations may resolve but have the risk of developing diabetes in later life 14, 15. At present, it is not clear whether the switch from hypoglycaemia to normoglycaemia to hyperglycaemia is mutation‐specific. It is reasonable to advise the family on the signs and symptoms of diabetes which may develop at a later age. The mechanisms of remission are unknown; increased β‐cell apoptosis and slow, progressive loss of β‐cell mass has been suggested as a possible explanation 56. Similar mechanisms may be at play in the switch to hyperglycaemia in CHI resulting from HNF4A mutations, although the causal link to β‐cell fate is probably indirect.
The uncertainty regarding the natural history of CHI is confusing to families and adds complexity that needs to be factored into clinical decision‐making at different time points in the disease trajectory. A composite perspective with re‐evaluation of severity and treatment has to be considered in keeping with the evolving nature of the disease.
Surgical outcomes
Focal lesionectomy is the preferred treatment choice for focal CHI, with complete resection being curative in the majority of cases. In contrast, the surgical outcomes of subtotal pancreatectomy for children with diffuse CHI are less than satisfactory. In the postoperative period, some children experience a combination of hypoglycaemia and hyperglycaemia, particularly during episodes of stress, which complicates glycaemic management. Eventually, β‐cell reserve is exhausted and diabetes requiring insulin therapy becomes inevitable in adolescence. Reported rates of development of diabetes vary from 13% soon after surgery to 77% after 7 years and 96% by age 11 years 45. Another consequence of subtotal pancreatectomy is a loss of pancreatic exocrine function. Faecal elastase 1 as a measure of exocrine function is low in 72% of patients after surgery but symptomatic in 49% 45. It is important to replace pancreatic enzymes by supplements to ensure satisfactory lipid absorption and nutritional status.
Neurodevelopment
Persistent and unpredictable episodes of hypoketotic hypoglycaemia are detrimental to the vulnerable neonatal brain and are associated with adverse neurodevelopment. Although treatment for CHI in general has improved over the last decade, with rapid gene sequencing, next‐generation sequencing and advances in imaging to identify focal CHI, the risk of adverse neurodevelopment remains high; the frequency of incidence varies between 26% and 48% in different cohorts [Table 1]. While focal CHI is potentially curable, the frequency of adverse neurodevelopment is similar in children with diffuse CHI 8, 9, 57. Furthermore, adverse neurodevelopment occurs in children with transient CHI with a similar frequency to those with permanent forms of CHI 7, suggesting that the impact of hypoglycaemia is not related to the type of CHI but to a delay in diagnosis at presentation. It is important to raise awareness to treat hypoglycaemia promptly to prevent neurodevelopmental sequelae.
Table 1.
Neurodevelopmental outcomes in the different populations
| Reference Country, years | Participants, n | Surgery, % | Abnormal neurodevelopment |
|---|---|---|---|
|
Menni et al. 2001 5
France, 1982–1998 |
90 | 70 |
26% (8% severe, 18% intermediate) 18% epilepsy |
|
Meissner et al. 2003 6 Germany (and other countries) Participants born 1975–2002 |
114 | 55 |
44% (18% severe, 26% mild) 25% epilepsy |
| Jack et al. 2003 61 Australia, 1972–1998 | 55 | 54 |
45% (25% mild, 20% severe) 29% seizures 31% developmental delay +speech delay 18% cerebral palsy |
| Steinkrauss et al. 2005 62 USA, 1980–2000 | 68 | 51 | 31% (16% severe, 15% moderate) |
|
Mazor‐Aronovitch et al. 2007 54
Israel, 1982–1997 |
21 | 0 |
38% fine motor problems 33% gross motor problems 29% learning problems 19% hypotonia 4% speech problems |
|
Avatapalle et al. 2013 7
UK |
67 | 18 |
39% (69% severe) 24% epilepsy 37% speech problems 36% motor delay 15% vision impairment 12% lower limb weakness |
| Lord et al. 2015 8 USA, 1960–2008 | 121 | 100 |
48%
21% psychiatric/behavioural problems 18% speech problems 16% learning disabilities 13% seizures 11% physical impairment 10% ADHD, 2% autism |
| Helleskov et al. 2017 57 Scandinavia, Russia, Eastern Europe, 2013–2016 | 75 | 33 |
47%
23% epilepsy 15% microcephaly 13% cerebral palsy 5% visual impairment |
| Ludwig et al. 2018 9 Germany, 2008–2013 | 60 | 37 |
46.7%
39% motor problems 27% speech problems 16% cognitive 9% socio‐emotional |
Adverse neurodevelopment was observed in 26–48% of cases. The rate of neurodevelopment impairment is not reduced over time, despite the advances in imaging and genetic techniques.
Bold values indicate total percentage of patients with abnoral neurodevelopment. ADHD, attention‐deficit hyperactivity disorder.
Figure 7 shows MRI scan changes which reveal the impact of severe and unrecognized early‐life hypoglycaemia attributable to CHI. The effects of hypoglycaemia on the brain may be more subtle, for instance causing mild motor delay not detected by MRI but by formal cognitive assessment. An alternative to formal testing could be the application of parent report questionnaires testing neurobehavioural status 58. Early identification of neurodevelopmental issues may provide the opportunity to support education, health and social care contributions.
Figure 7.
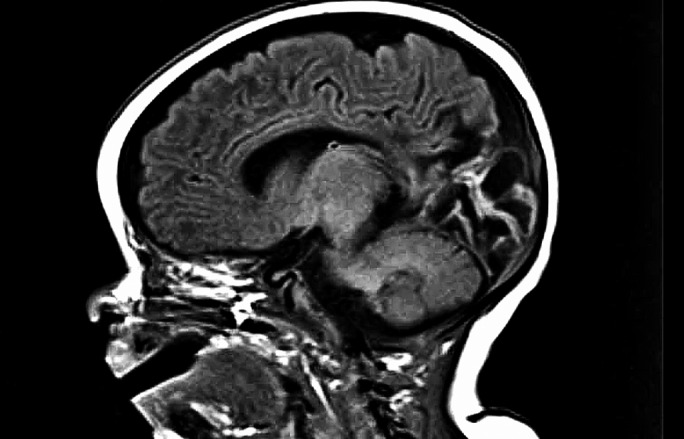
Brain MRI showing consequences of severe perinatal hypoglycaemia, with extensive bilateral areas of cystic encephalomalacia in the occipito‐parietal area and posterior temporal lobes.
Feeding
Feeding difficulties, such as sucking and swallowing difficulties, persistent vomiting, gastro‐oesophageal reflux and food refusal, represent another burden for families with CHI. Feeding difficulties occur in a significant proportion of children (33–45%) 59 and are associated with more severe forms of CHI. The cause is not clear, but likely to be multifactorial. Often children require invasive intensive treatment regimens with high concentrations of dextrose for prolonged periods, which are not conducive to normal feeding development. Medications may induce nausea and feeding intolerance, with parental anxiety adding to an already stressful scenario. A multidisciplinary team, comprising dietitians, speech and language therapists and clinical psychologists working with parents and families, is essential to manage feeding difficulties, although resolution make take several weeks and months.
Diabetes
Diabetes is a common complication of subtotal pancreatectomy, as discussed above. Long‐acting and short‐acting insulin regimens are used to correct hyperglycaemia, and individual glycaemic trends are likely to determine treatment choice. At present, there is insufficient evidence to guide choice, with both regimens causing different patterns of hypoglycaemia.
Diabetes has been well recognized in dominant genetic forms of CHI, such as with HNF4A and some ABCC8 variants 14, 15, 24. For the majority of ABCC8 dominant pathogenic variants, there is no evidence of predictability; however, in the absence of evidence of natural history for these variants, it may be wise to counsel families to be vigilant for symptoms of diabetes in later life.
Transition
Congenital hyperinsulinism is essentially a paediatric disease whose severity is greatest in early life. Nonetheless, a number of children continue to have troublesome hypoglycaemia in late adolescence. In such cases, transition to an adolescent and young adult service is envisaged. The development of a CHI‐specific transition service is recommended, with a gradual transition from children's to adult services that draws upon experiences and examples of other transition pathways 60. For those in whom transition is anticipated in the early puberty years, it is important to engage early with the young person to encourage greater understanding of his/her condition. In many young people with CHI or diabetes, understanding of disease is compromised because of adverse cognitive outcomes; the impact of side effects and adjustment to neurodevelopmental concerns are particularly pertinent at this point; therefore, it is important that transition pathway development is robust enough to address the individual and complex needs of young people and their families.
Conclusions
Congenital hyperinsulinism is a heterogeneous and complex rare disease of hypoglycaemia, often resulting in brain damage in a significant proportion of children. In spite of rapid advances, such as PET‐CT scanning, CHI remains a high‐morbidity disease with lifelong consequences that require sustained medical input. While novel therapy is in the pipeline and presumably unavailable for a few years, it is important to optimize current medical practice to safeguard brain function and quality of life for children graduating to adult life.
Funding sources
None.
Competing interests
None declared.
Diabet. Med. 36: 9–21 (2019)30246418
References
- 1. Banerjee I, Avatapalle B, Padidela R, Stevens A, Cosgrove K, Clayton P, Dunne M. Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin Endocrinol (Oxf) 2013; 78: 803–813. [DOI] [PubMed] [Google Scholar]
- 2. Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab 2016; 101: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shah P, Rahman SA, Demirbilek H, Güemes M, Hussain K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol. 2017; 5: 729–742. [DOI] [PubMed] [Google Scholar]
- 4. Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley‐Green A, Lindley KJ. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev 2004; 84: 239–275. [DOI] [PubMed] [Google Scholar]
- 5. Menni F, de Lonlay P, Sevin C, Touati G, Peigne C, Barbier V et al Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycaemia. Pediatrics 2001; 107: 476–479. [DOI] [PubMed] [Google Scholar]
- 6. Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long‐term follow‐up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol 2003; 149: 43–51. [DOI] [PubMed] [Google Scholar]
- 7. Avatapalle HB, Banerjee I, Shah S, Pryce M, Nicholson J, Rigby L et al Abnormal neurodevelopmental outcomes are common in children with transient congenital hyperinsulinism. Front Endocrinol (Lausanne) 2013; 4: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De Leon DD. High risk of diabetes and neurobehavioral deficits in individuals with surgically treated hyperinsulinism. J Clin Endocrinol Metab 2015; 100: 4133–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ludwig A, Enke S, Heindorf J, Empting S, Meissner T, Mohnike K. Formal Neurocognitive Testing in 60 Patients with Congenital Hyperinsulinism. Horm Res Paediatr 2018; 89: 1–6. [DOI] [PubMed] [Google Scholar]
- 10. Dunne MJ, Kane C, Shepherd RM, Sanchez JA, James RF, Johnson PR et al Familial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptor. N Engl J Med 1997; 336: 703–706. [DOI] [PubMed] [Google Scholar]
- 11. Thornton PS, Stanley CA, De León DD, Harris D, Haymond MW, Hussain K et al Pediatric Endocrine Society. Recommendations from the pediatric endocrine society for evaluation and management of persistent hypoglycemia in neonates, infants, and children. J Pediatr 2015; 167: 238–245. [DOI] [PubMed] [Google Scholar]
- 12. Banerjee I, Skae M, Flanagan SE, Rigby L, Patel L, Didi M et al The contribution of rapid KATP channel gene mutation analysis to the clinical management of children with congenital hyperinsulinism. Eur J Endocrinol 2011; 164: 733–740. [DOI] [PubMed] [Google Scholar]
- 13. Stanley CA, Rozance PJ, Thornton PS, De León DD, Harris D, Haymond MW et al Re‐evaluating transitional neonatal hypoglycemia: mechanism and implications for management. J Pediatr 2015; 166: 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huopio H, Reimann F, Ashcroft FM, Laakso M. A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 2003; 361 (9354): 301–307. [DOI] [PubMed] [Google Scholar]
- 15. Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC et al Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia 2011; 54: 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N et al Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 2013; 98: E355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol 2013; 168: 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H et al A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes 2017; 18: 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cabezas OR, Flanagan SE, Stanescu H, García‐Martínez E, Caswell R et al Polycystic Kidney Disease with Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol 2017; 28: 2529–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M et al Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm‐derived organ abnormalities. Hum Mol Genet 2017; 26: 4315–4326. [DOI] [PubMed] [Google Scholar]
- 21. Vajravelu ME, Chai J, Krock B, Baker S, Langdon D, Alter C, De León DD. Congenital Hyperinsulinism and Hypopituitarism Attributable to a Mutation in FOXA2. J Clin Endocrinol Metab 2018; 103: 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferrara CT, Boodhansingh KE, Paradies E, Giuseppe F, Steinkrauss LJ, Topor LS et al Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J Clin Endocrinol Metab 2017; 2017: 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salomon‐Estebanez M, Flanagan SE, Ellard S, Rigby L, Bowden L, Mohamed Z et al Conservatively treated congenital Hyperinsulinism (CHI) due to K‐ATP channel gene mutations: reducing severity over time. Orphanet J Rare Dis 2016; 11: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McGlacken‐Byrne SM, Hawkes CP, Flanagan SE, Ellard S, McDonnell CM, Murphy NP. The evolving course of HNF4A hyperinsulinaemic hypoglycaemia ‐ a case series. Diabet Med 2014; 31: e1–5. [DOI] [PubMed] [Google Scholar]
- 25. Wabitsch M, Lahr G, Marchant C, Lindner M, von Puttkamer J, Fenneberg A et al Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet Med 2007; 24: 1393–1399. [DOI] [PubMed] [Google Scholar]
- 26. Flanagan SE, Kapoor RR, Hussain K. Genetics of congenital hyperinsulinemic hypoglycemia. Semin Pediatr Surg 2011; 20: 13–17. [DOI] [PubMed] [Google Scholar]
- 27. Christiansen CD, Petersen H, Nielsen AL, Detlefsen S, Brusgaard K, Rasmussen L et al 18F‐DOPA PET/CT and 68Ga‐DOTANOC PET/CT scans as diagnostic tools in focal congenital hyperinsulinism: a blinded evaluation. Eur J Nucl Med Mol Imaging 2018; 45: 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han B, Newbould M, Batra G, Cheesman E, Craigie RJ, Mohamed Z et al Enhanced islet cell nucleomegaly defines diffuse congenital hyperinsulinism in infancy but not other forms of the disease. Am J Clin Pathol 2016; 145: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han B, Mohamed Z, Estebanez MS, Craigie RJ, Newbould M, Cheesman E et al Atypical forms of congenital hyperinsulinism in infancy are associated with mosaic patterns of immature islet cells. J Clin Endocrinol Metab 2017; 102: 3261–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sempoux C, Capito C, Bellanné‐Chantelot C, Verkarre V, de Lonlay P, Aigrain Y et al Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab 2011; 96: 3785–3793. [DOI] [PubMed] [Google Scholar]
- 31. Mohnike K, Blankenstein O, Pfuetzner A, Pötzsch S, Schober E, Steiner S et al Long‐term non‐surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Horm Res 2008; 70: 59–64. [DOI] [PubMed] [Google Scholar]
- 32. Newswanger B, Ammons S, Phadnis N, Ward WK, Castle J, Campbell RW et al Development of a highly stable, nonaqueous glucagon formulation for delivery via infusion pump systems. J Diabetes Sci Technol 2015; 9: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henquin JC, Nenquin M, Sempoux C, Guiot Y, Bellanné‐Chantelot C, Otonkoski T et al In vitro insulin secretion by pancreatic tissue from infants with diazoxide‐resistant congenital hyperinsulinism deviates from model predictions. J Clin Invest 2011; 121: 3932–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Timlin MR, Black AB, Delaney HM, Matos RI, Percival CS. Development of Pulmonary Hypertension During Treatment with Diazoxide: A Case Series and Literature Review. Pediatr Cardiol 2017; 38: 1247–1250. [DOI] [PubMed] [Google Scholar]
- 35. Avatapalle B, Banerjee I, Malaiya N, Padidela R. Echocardiography monitoring for diazoxide induced pericardial effusion. BMJ Case Rep 2012;2012: pii: bcr0320126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van der Steen I, van Albada ME, Mohnike K, Christesen HT, Empting S, Salomon‐Estebanez M et al A Multicenter Experience with Long‐Acting Somatostatin Analogues in Patients with CongenitalHyperinsulinism. Horm Res Paediatr 2018; 89: 82–89. [DOI] [PubMed] [Google Scholar]
- 37. Avatapalle B, Padidela R, Randell T, Banerjee I. Drug‐induced hepatitis following use of octreotide for long‐term treatment of congenital hyperinsulinism. BMJ Case Rep 2012; 30: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McMahon AW, Wharton GT, Thornton P, De Leon DD. Octreotide use and safety in infants with hyperinsulinism. Pharmacoepidemiol Drug Saf 2017; 26: 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Güemes M, Shah P, Silvera S, Morgan K, Gilbert C, Hinchey L, Hussain K. Assessment of Nifedipine Therapy in Hyperinsulinemic Hypoglycemia due to Mutations in the ABCC8 Gene. J Clin Endocrinol Metab 2017; 102: 822–830. [DOI] [PubMed] [Google Scholar]
- 40. Senniappan S, Alexandrescu S, Tatevian N, Shah P, Arya V, Flanagan S et al Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N Engl J Med 2014; 370: 1131–1137. [DOI] [PubMed] [Google Scholar]
- 41. Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A et al mTOR Inhibitors for the Treatment of Severe Congenital Hyperinsulinism: Perspectives on Limited Therapeutic Success. J Clin Endocrinol Metab 2016; 101: 4719–4729. [DOI] [PubMed] [Google Scholar]
- 42. Banerjee I, De Leon D, Dunne MJ. Exreme caution on the use of sirolimus for the congenital hyperinsulinism in infancy patient. Orphanet J Rare Dis 2017; 12: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skae M, Avatapalle HB, Banerjee I, Rigby L, Vail A, Foster P et al Reduced glycemic variability in diazoxide‐responsive children with congenital hyperinsulinism using supplemental omega‐3‐polyunsaturated fatty acids; a pilot trial with MaxEPA(R.) Front Endocrinol (Lausanne) 2014;5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beltrand J, Caquard M, Arnoux JB, Laborde K, Velho G, Verkarre V et al Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care 2012; 35: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arya VB, Senniappan S, Demirbilek H, Alam S, Flanagan SE, Ellard S et al Pancreatic endocrine and exocrine function in children following near‐total pancreatectomy for diffuse congenital hyperinsulinism. PLoS One 2014; 9: e98054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Otonkoski T, Näntö‐Salonen K, Seppänen M, Veijola R, Huopio H, Hussain K et al Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]‐DOPA positron emission tomography. Diabetes 2006; 55: 13–18. [PubMed] [Google Scholar]
- 47. Patel P, Charles L, Corbin J, Goldfine ID, Johnson K, Rubin P et al A unique allosteric insulin receptor monoclonal antibody that prevents hypoglycemia in the SUR‐1‐/‐ mouse model of KATP hyperinsulinism. MAbs 2018; 10: 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Calabria AC, Li C, Gallagher PR, Stanley CA, De León DD. GLP‐1 receptor antagonist exendin‐(9‐39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP‐ sensitive K+ channel. Diabetes 2012; 61: 2585–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Silverstein JM. Hyperglycemia induced by pasireotide in patients with Cushing's disease or acromegaly. Pituitary 2016; 19: 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M et al Lumacaftor‐ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N Engl J Med 2015; 373: 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Powell PD, Bellanne‐Chantelot C, Flanagan SE, Ellard S, Rooman R, Hussain K et al In vitro recovery of ATP‐sensitive potassium channels in beta‐cells from patients with congenital hyperinsulinism of infancy. Diabetes 2011; 60: 1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Henriksen MM, Andersen HU, Thorsteinsson B, Pedersen‐Bjergaard U. Hypoglycemic Exposure and Risk of Asymptomatic Hypoglycemia in Type 1 Diabetes Assessed by Continuous Glucose Monitoring. J Clin Endocrinol Metab 2018; 103: 2329–2335. [DOI] [PubMed] [Google Scholar]
- 53. Patil P, Giri D, Didi M, Senniappan S. Ketotic Hypoglycemia in Children with Previous Transient Congenital Hyperinsulinism. Indian Pediatr 2018; 55: 167–168. [PubMed] [Google Scholar]
- 54. Mazor‐Aronovitch K, Gillis D, Lobel D, Hirsch HJ, Pinhas‐Hamiel O, Modan‐Moses D et al Long‐term neurodevelopmental outcome in conservatively treated congenital hyperinsulinism. Eur J Endocrinol 2007; 157: 491–497. [DOI] [PubMed] [Google Scholar]
- 55. Martínez R, Fernández‐Ramos C, Vela A, Martínez R, Fernández‐Ramos C, Vela A et al Clinical and genetic characterization of congenital hyperinsulinism in Spain. Eur J Endocrinol 2016; 174: 717–726. [DOI] [PubMed] [Google Scholar]
- 56. Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta‐cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 2000; 49: 1325–1333. [DOI] [PubMed] [Google Scholar]
- 57. Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM et al Both Low Blood Glucose and Insufficient Treatment Confer Risk of Neurodevelopmental Impairment in Congenital Hyperinsulinism: A Multinational Cohort Study. Front Endocrinol (Lausanne) 2017; 8: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salomon‐Estebanez M, Mohamed Z, Michaelidou M, Collins H, Rigby L, Skae M et al Vineland adaptive behavior scales to identify neurodevelopmental problems in children with congenital hyperinsulinism (CHI). Orphanet J Rare Dis 2017; 12: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Banerjee I, Forsythe L, Skae M, Avatapalle HB, Rigby L, Bowden LE et al Feeding Problems Are Persistent in Children with Severe Congenital Hyperinsulinism. Front Endocrinol (Lausanne) 2016; 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Poisson J, Pitfield C, Gilbert C, Morgan K, Cunjamalay A, Misra S et al Development and implementation of congenital hyperinsulinism transition clinic pathway. Arch Dis Child 2017; 102(Suppl 3): A1–A31. [Google Scholar]
- 61. Jack MM, Greer RM, Thomsett MJ, Walker RM, Bell JR, Choong C et al The outcome in Australian children with hyperinsulinism of infancy: early extensive surgery in severe cases lowers risk of diabetes. Clin Endocrinol 2003; 58: 355–364. [DOI] [PubMed] [Google Scholar]
- 62. Steinkrauss L, Lipman TH, Hendell CD, Gerdes M, Thornton PS, Stanley CA. Effects of hypoglycemia on developmental outcome in children with congenital hyperinsulinism. J Pediatr Nurs 2005; 20: 109–118. [DOI] [PubMed] [Google Scholar]