

Abstract
Objective
To characterize baseline gene expression and pharmacodynamically induced changes in whole blood gene expression in 1,760 systemic lupus erythematosus (SLE) patients from 2 phase III, 52‐week, randomized, placebo‐controlled, double‐blind studies in which patients were treated with the BAFF‐blocking IgG4 monoclonal antibody tabalumab.
Methods
Patient samples were obtained from SLE patients from the ILLUMINATE‐1 and ILLUMINATE‐2 studies, and control samples were obtained from healthy donors. Blood was collected in Tempus tubes at baseline, week 16, and week 52. RNA was analyzed using Affymetrix Human Transcriptome Array 2.0 and NanoString.
Results
At baseline, expression of the interferon (IFN) response gene was elevated in patients compared with controls, with 75% of patients being positive for this IFN response gene signature. There was, however, substantial heterogeneity of IFN response gene expression and complex relationships among gene networks. The IFN response gene signature was a predictor of time to disease flare, independent of anti–double‐stranded DNA (anti‐dsDNA) antibody and C3 and C4 levels, and overall disease activity. Pharmacodynamically induced changes in gene expression following tabalumab treatment were extensive, occurring predominantly in B cell–related and immunoglobulin genes, and were consistent with other pharmacodynamic changes including anti‐dsDNA antibody, C3, and immunoglobulin levels.
Conclusion
SLE patients demonstrated increased expression of an IFN response gene signature (75% of patients had an elevated IFN response gene signature) at baseline in ILLUMINATE‐1 and ILLUMINATE‐2. Substantial heterogeneity of gene expression was detected among individual patients and in gene networks. The IFN response gene signature was an independent risk factor for future disease flares. Pharmacodynamic changes in gene expression were consistent with the mechanism of BAFF blockade by tabalumab.
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease for which there is a substantial unmet medical need, despite improvements in overall management of the disease. Although the pathogenesis of SLE remains incompletely understood, multiple roles for B cells in the pathogenesis of the disease are well established, including the production of autoantibodies directed against self antigens, such as antinuclear antibodies (ANAs) and anti–double‐stranded DNA (anti‐dsDNA) antibodies 1, 2. An important role for a B cell–activating cytokine known as B lymphocyte stimulator or BAFF, which is a member of the tumor necrosis factor superfamily, has been implicated in the pathogenesis of SLE as well as other autoimmune diseases and lymphoproliferative disorders, including lymphoma 3, 4, 5, 6, 7.
BAFF has a key role in B cell maturation and supports autoantibody production and disease development in animal models of SLE 3, 4, 5, 6, 7. The levels of BAFF serum protein and messenger RNA (mRNA) have been reported to be elevated in SLE, with associations between BAFF mRNA levels and measures of disease activity 6, 7. These and other findings led to the development and recent testing in SLE of 3 biologic treatments targeted to BAFF: belimumab, blisibimod, and tabalumab 8, 9, 10, 11, 12. The monoclonal antibody (mAb) belimumab demonstrated efficacy in phase III trials in patients with SLE, using the SLE Responder Index 4 (SRI‐4) as the primary end point 8, 9. Blisibimod, a “peptibody,” showed some signal of efficacy in a small phase II study 10. The primary end point of achieving SRI‐5 improvement with tabalumab was met in ILLUMINATE‐2 but not in ILLUMINATE‐1, both of which are large phase III clinical trials in SLE patients 11, 12.
Studies of mRNA expression in SLE, using peripheral blood mononuclear cells or whole blood samples from SLE patients, have shown increased expression of genes responsive to interferons (IFNs), especially type I IFNs, which are known collectively as the IFN response gene signature 13, 14, 15. Importantly, activation of specific IFN response gene(s) and the magnitude of their responses have varied between different studies of SLE. Aside from such studies in which the expression of various combinations of IFN response genes in SLE is described, the full characterization of peripheral blood gene expression has been limited 16, 17. There has been no previous opportunity to acquire data for comprehensive gene expression profiling in cohorts of SLE patients as large as those from ILLUMINATE‐1 and ILLUMINATE‐2.
The current study examined baseline and pharmacodynamically induced gene expression changes in whole blood RNA from 2 phase III, 52‐week, randomized, placebo‐controlled, double‐blind, parallel studies of 1,760 SLE patients treated with the BAFF‐blocking IgG4 mAb, tabalumab. At baseline in both ILLUMINATE‐1 and ILLUMINATE‐2, 75% of SLE patients had an elevated IFN response gene signature. Those in both studies who had the IFN response gene signature had significantly more immunologic and hematologic system involvement, as defined by the Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA) version of the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) organ domain score 18. They also had a higher use of corticosteroids and immunosuppressant medication. There was significant heterogeneity of expression among individual IFN response genes and complex patterns of interactions in gene networks. The IFN signature was an independent predictor of the risk of disease flares as defined by the SELENA–SLEDAI flare index. Pharmacodynamic changes in gene expression occurred in immunoglobulin and B cell–related genes, consistent with changes in anti‐dsDNA, C3, and immunoglobulin levels and congruent with the mechanism of BAFF blockade by tabalumab.
PATIENTS AND METHODS
Study design and patient population
Control samples were obtained from healthy blood donors. Patient samples were obtained from SLE patients from ILLUMINATE‐1 and ILLUMINATE‐2, 2 phase III, 52‐week, randomized, placebo‐controlled, double‐blind, parallel studies of the efficacy and safety of tabalumab in patients with moderate‐to‐severe SLE. Tabalumab is a fully human IgG4 mAb that blocks the activity of soluble and membrane‐bound BAFF. In both treatment arms (tabalumab and placebo), standard‐of‐care therapy with immunosuppressive drugs that were being administered at the time of enrollment was continued throughout the trial 11, 12. All patients met ≥4 of the updated American College of Rheumatology revised criteria for the classification of SLE 19. A severe SLE flare was defined using the SELENA‐SLEDAI flare index. The protocol was approved by the appropriate authorized independent review boards in accordance with the Declaration of Helsinki. All patients and control subjects provided written informed consent.
Blood collection and clinical laboratory analyses
Whole blood was collected in Tempus tubes (Thermo Fisher Scientific) at baseline, week 16, and week 52 11, 12. Total B cells were enumerated by flow cytometry (performed at Covance Laboratories). Complement components C3 and C4 and total serum IgA, IgM, and IgG were measured at Covance Laboratories. ANA and anti‐dsDNA antibody testing was performed at Quest Laboratories. Baseline serum BAFF levels were measured using an assay that measures total BAFF, including soluble and membrane‐bound forms, at Pacific Biomarkers.
RNA isolation, analysis, and hybridization
Total RNA was extracted from whole blood using a PerfectPure RNA Blood Kit as recommended by the manufacturer (Five Prime Therapeutics). RNA quality was assessed postextraction using RiboGreen (Life Technologies) and a Bioanalyzer RNA chip (Agilent Technologies). An Affymetrix WT Plus Reagent Kit was used for the preparation of complementary DNA (cDNA). Labeled cDNA was hybridized to the GeneChip Human Transcriptome Array 2.0 (HTA 2.0) according to the manufacturer's instructions.
Microarray analysis
The Affymetrix HTA 2.0 array with 70,753 transcript clusters was used for gene expression analysis. Annotation data were retrieved from Affymetrix NA34 build (www.Affymetrix.com). Only transcript clusters that represented a single protein‐coding gene were considered in this analysis (n = 19,818) 20, 21, 22. Complete methodologic details are provided in Supplementary Material (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). Unprocessed files containing raw data and the corresponding metadata have been deposited in the GEO database repository (GEO accession no. GSE0000) (online at http://www.ncbi.nlm.nih.gov/geo/).
NanoString analysis
Select IFN response gene expression findings using HTA 2.0 arrays were tested for correlation by mRNA expression profiling using a NanoString nCounter gene expression system (NanoString Technologies) 23.
Gene coexpression analysis
Weighted correlation network analysis (WGCNA), a standard approach to describing groups of genes whose expression is correlated (coexpressed), was performed 24, 25. Briefly, preservation of modules between the group with a high IFN signature and the group with a low IFN signature was evaluated by Fisher's exact test. For network visualization of the SLE Yellow module, only edges from the topologic overlap matrix ≥0.45 are shown in Figure 2, providing a simplified view of the most connected genes.
Figure 2.
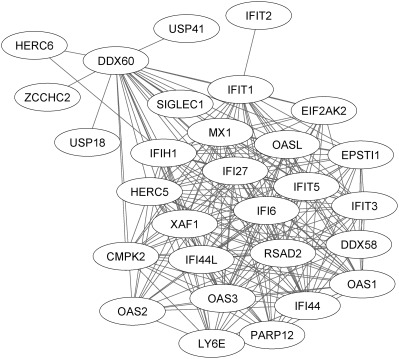
Characterization of network connectivity between highly expressed interferon (IFN) response genes. As shown by the lines connecting individual genes, a network of highly correlated genes is expressed in the systemic lupus erythematosus (SLE) cohorts, the majority of which have been identified previously as IFN response genes. There is, however, substantially greater complexity of the IFN network than has been previously described (for additional information, see Patients and Methods, and Supplementary Material, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). Briefly, the Weighted correlation network analysis module (“Yellow module”) containing this network is correlated with all 3 clinical end points: (IFN signature [ρ = 0.90, P < 2 × 10−308], Safety of Estrogens in Lupus Erythematosus National Assessment version of the SLE Disease Activity Index [ρ = 0.22, P = 5 × 10−21], anti‐DNA activity [ρ = 0.32, P = 4 × 10−43]) (see Supplementary Figure 2 and Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract) and is enriched for the MetaCore‐defined SLE disease pathway (P = 1.14 × 10−32 by hypergeometric test) and the gene ontology (GO) “response to type I interferon” gene set (GO:0034340 [P = 6.60 × 10−36]) (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract).
Statistical analysis
Complete methodologic details on the statistical analysis plan and its execution are shown in Supplementary Patients and Methods, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract 26, 27, 28, 29.
RESULTS
Characteristics of the patients studied
The samples studied were obtained from a subset of patients who participated in ILLUMINATE‐1 (876 of 1,164) and ILLUMINATE‐2 (880 of 1,124), including only those patients who had given written informed consent for genetic testing and for whom samples were available 11, 12. The ILLUMINATE‐1 patients studied were predominantly female (93% female and 7% male) with a mean age of 41 years (range 18–87 years). The majority of patients were Caucasian (67% Caucasian, 12% African descent, and 19% American Indian/Alaska Native). In ILLUMINATE‐2, the patients were also predominantly female (92% female and 8% male), with a mean age of 43 years (range 18–83 years). Overall, the majority of patients were white (72% white, 14% African descent, and 10% American Indian/Alaska Native). Patients in both trials were stratified according to anti‐dsDNA antibody status at the time of randomization. The treatment and placebo groups were well balanced in both studies, and additional demographic and baseline clinical features have been previously reported 11, 12.
Baseline gene expression in SLE patients compared with healthy controls
The most significant differences at baseline in gene expression between SLE patients and healthy controls are shown in Table 1, ranked in the order of fold difference. The top‐ranked genes were IFN response genes. Baseline expression of other genes that strongly differentiated SLE patients from control subjects included those involved with immunoglobulin production, modification or binding, ubiquitination, and poly(ADP‐ribose) polymerization. The number and magnitude of change of genes that were down‐regulated in SLE patients compared with healthy blood donors were small. Supplementary Table 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract) includes down‐regulated genes with the greatest fold change, which ranged from −1.4 to −1.9.
Table 1.
Gene expression in SLE patients versus controls at baselinea
| Gene name | Gene description | q value | Fold difference |
|---|---|---|---|
| IFI44L | Interferon‐induced protein 44‐like | 2.01 × 10−15 | 8.13 |
| IFI27 | Interferon, alpha‐inducible protein 27 | 3.91 × 10−13 | 8.01 |
| RSAD2 | Radical S‐adenosyl methionine domain containing 2 | 1.42 × 10−16 | 6.47 |
| IFI44 | Interferon‐induced protein 44 | 1.43 × 10−15 | 5.44 |
| IFIT1 | Interferon‐induced protein with tetratricopeptide repeats 1 | 2.53 × 10−13 | 4.51 |
| HERC5 | HECT and RLD domain containing E3 ubiquitin protein ligase 5 | 4.39 × 10−15 | 3.77 |
| EPSTI1 | Epithelial stromal interaction 1 | 2.52 × 10−16 | 3.55 |
| OAS3 | 2′‐5′‐oligoadenylate synthetase 3, 100 kd | 9.66 × 10−15 | 3.29 |
| IFI6 | Interferon, alpha‐inducible protein 6 | 3.85 × 10−15 | 3.2 |
| DDX60 | DEAD (Asp‐Glu‐Ala‐Asp) box polypeptide 60 | 1.47 × 10−13 | 2.91 |
| OAS1 | 2′‐5′‐oligoadenylate synthetase 1, 40/46 kd | 4.5 × 10−15 | 2.8 |
| USP18 | Ubiquitin specific peptidase 18 | 5.91 × 10−13 | 2.86 |
| IFIT3 | Interferon‐induced protein with tetratricopeptide repeats 3 | 5.92 × 10−15 | 2.78 |
| OAS2 | 2′‐5′‐oligoadenylate synthetase 2, 69/71 kd | 1.58 × 10−12 | 2.74 |
| OASL | 2′‐5′‐oligoadenylate synthetase‐like | 9 × 10−16 | 2.68 |
| PLSCR1 | Phospholipid scramblase 1 | 6.9 × 10−20 | 2.67 |
| CMPK2 | Cytidine monophosphate (UMP‐CMP) kinase 2, mitochondrial | 9.66 × 10−15 | 2.55 |
| XAF1 | XIAP associated factor 1 | 7.89 × 10−14 | 2.49 |
| MX1 | Myxovirus (influenza virus) resistance 1, interferon‐inducible protein p78 | 2.22 × 10−12 | 2.47 |
| EIF2AK2 | Eukaryotic translation initiation factor 2‐alpha kinase 2 | 5.12 × 10−16 | 2.46 |
| USP41 | Ubiquitin specific peptidase 41 | 6.27 × 10−13 | 2.39 |
| IFIH1 | Interferon induced with helicase C domain 1 | 8.13 × 10−14 | 2.28 |
| SERPING1 | Serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | 1.31 × 10−14 | 2.25 |
| IGHA1 | Immunoglobulin heavy constant alpha 1 | 6.84 × 10−5 | 2.16 |
| SAMD9L | Sterile alpha motif domain containing 9‐like | 2.63 × 10−15 | 2.16 |
| LY6E | Lymphocyte antigen 6 complex, locus E | 1.05 × 10−13 | 2.14 |
| IFIT2 | interferon‐induced protein with tetratricopeptide repeats 2 | 7.69 × 10−13 | 2.12 |
| RTP4 | Receptor (chemosensory) transporter protein 4 | 3.64 × 10−12 | 2.08 |
| TNFAIP6 | Tumor necrosis factor, alpha‐induced protein 6 | 1.37 × 10−10 | 2.08 |
| MMP8 | Matrix metallopeptidase 8 (neutrophil collagenase) | 0.001315 | 2.06 |
| IFIT5 | Interferon‐induced protein with tetratricopeptide repeats 5 | 4.32 × 10−12 | 2.05 |
| DDX58 | DEAD (Asp‐Glu‐Ala‐Asp) box polypeptide 58 | 1.05 × 10−13 | 2.04 |
| HERC6 | HECT and RLD domain containing E3 ubiquitin protein ligase family member 6 | 7.35 × 10−11 | 2.04 |
| SIGLEC1 | Sialic acid binding Ig‐like lectin 1, sialoadhesin | 1.87 × 10−12 | 2.04 |
| CARD17 | Caspase recruitment domain family, member 17 | 4.33 × 10−9 | 1.96 |
| SPATS2L | Spermatogenesis associated, serine‐rich 2‐like | 3.59 × 10−12 | 1.96 |
| FCGR1C | Fc fragment of IgG, high affinity Fc receptor (CD64), pseudogene | 1.92 × 10−11 | 1.92 |
| ZCCHC2 | Zinc finger, CCHC domain containing 2 | 2.18 × 10−11 | 1.87 |
| DDX60L | DEAD (Asp‐Glu‐Ala‐Asp) box polypeptide 60‐like | 6.8 × 10−13 | 1.86 |
| CCR1 | Chemokine (C‐C motif) receptor 1 | 3.92 × 10−11 | 1.85 |
| PARP12 | Poly (ADP‐ribose) polymerase family, member 12 | 9.63 × 10−12 | 1.84 |
| PARP9 | Poly (ADP‐ribose) polymerase family, member 9 | 3.24 × 10−15 | 1.82 |
| GBP1 | Guanylate binding protein 1, interferon‐inducible | 8.56 × 10−11 | 1.81 |
| PARP14 | Poly (ADP‐ribose) polymerase family, member 14 | 1.5 × 10−10 | 1.78 |
| IGLC7 | Immunoglobulin lambda constant 7 | 0.000513 | 1.77 |
| CEACAM1 | Carcinoembryonic antigen‐related cell adhesion molecule 1 (biliary glycoprotein) | 3.43 × 10−13 | 1.75 |
| CCL2 | Chemokine (C‐C motif) ligand 2 | 6.22 × 10−7 | 1.73 |
| IGKV1D‐27 | Immunoglobulin kappa variable 1D‐27 | 0.001748 | 1.73 |
| IGHV3‐9 | Immunoglobulin heavy variable 3‐9 | 0.00233 | 1.72 |
| CXCL10 | Chemokine (C‐X‐C motif) ligand 10 | 3.64 × 10−6 | 1.71 |
Genes are ranked from high to low based on the difference in expression between patients with systemic lupus erythematosus (SLE) and healthy control subjects. Statistical significance is shown as a q value.
Subgroups of SLE patients identified at baseline among patients expressing high versus low levels of IFN response genes
As shown in Table 1, in the SLE patient population, the most prevalent up‐regulated genes were those that are responsive to stimulation by type I IFNs, the so‐called IFN response genes. A predefined panel of 164 IFN response genes was selected for testing based on an analysis of the literature (a full list of the 164‐gene panel and final 34 genes selected is shown in Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). At least 3 broad patterns of gene expression can be visualized on the 164‐gene expression heatmaps (Figures 1A and B). Expression data were pooled from ILLUMINATE‐1 and ILLUMINATE‐2 and normalized using the mean of a set of housekeeping genes to account for a small study/array batch effect on gene expression. An IFN response gene signature was estimated from the pooled data using the first principal component derived from 34 of the highest co‐varying 164 IFN response genes. At baseline, 75% of the SLE patients in ILLUMINATE‐1 (Figure 1A) and ILLUMINATE‐2 (Figure 1B) exhibited increased expression of the IFN response gene signature compared with healthy controls (Figure 1C).
Figure 1.
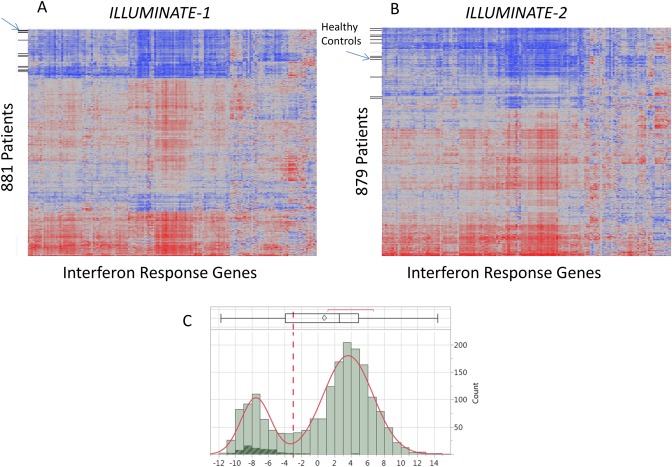
Estimation of the interferon (IFN) response gene signature in 164 individual prespecified genes. A total of 1,760 patients (881 from ILLUMINATE‐1 and 879 from ILLUMINATE‐2) and 60 control subjects were studied. A and B, The 164 prespecified genes are represented on the x‐axes; control subjects and patients are represented on the y‐axes for ILLUMINATE‐1 (A) and ILLUMINATE‐2 (B). At least 3 broad patterns of gene expression can be visualized on both 164‐gene expression heatmaps, although when reduced to a 34‐gene signature this could be clearly discerned as high and low expression subgroups. C, Distribution of IFN response gene expression among patients and controls. Of the 1,760 patients studied, 75% had elevated expression of IFN response genes compared with healthy controls (see Patients and Methods for additional details). Data are depicted at the top as a box plot, where the box represents the 25th to 75th percentiles, the line within the box represents the median, and the lines outside the box represent the 10th and 90th percentiles. The diamond represents results from healthy controls.
NanoString confirmation of findings from HTA 2.0 exon array using probes for individual genes
The custom NanoString assays generated for this study were used to examine sequences from key probe sets on exon arrays (as opposed to full genes). There was a highly statistically significant correlation between mRNA expression using HTA 2.0 exon array and individual genes probed using NanoString (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). These results and published studies support the validity of findings for the HTA 2.0 exon arrays 23.
Comparison of IFN response gene–positive and IFN response gene–negative subgroups at baseline
As shown in Table 2, analysis of organ system involvement (as defined by SELENA–SLEDAI organ domain scores), complete blood count, and concomitant medication use between the IFN response gene–positive and IFN response gene–negative groups showed a number of statistically significant differences between the 2 groups. There were significantly higher proportions of patients with organ involvement in the immunologic and hematologic systems in the IFN signature–positive groups in both ILLUMINATE‐1 and ILLUMINATE‐2 at baseline. There was a slightly lower proportion of the patients with musculoskeletal system involvement in the IFN response gene–positive group in both ILLUMINATE‐1 and ILLUMINATE‐2. Numerically small imbalances in the proportions of SELENA–SLEDAI vascular and renal organ systems were present but were not consistently observed in both studies.
Table 2.
Baseline organ system involvement, complete blood cell count, and concomitant medications in the IFN response gene–positive and IFN response gene–negative subgroupsa
| ILLUMINATE‐1 | ILLUMINATE‐2 | |||
|---|---|---|---|---|
| IFN response gene negative (n = 211) | IFN response gene positive (n = 668) | IFN response gene negative (n = 221) | IFN response gene positive (n = 660) | |
| SELENA–SLEDAI organ system involvement | ||||
| Central nervous system | 2 (0.9) | 13 (1.9) | 8 (3.6) | 11 (1.7) |
| Vascular | 9 (4.3) | 51 (7.6) | 6 (2.7) | 61 (9.3)b |
| Musculoskeletal | 194 (91.9) | 567 (84.9)b | 206 (93.2) | 574 (87.1)b |
| Renal | 11 (5.2) | 68 (10.2)b | 16 (7.2) | 52 (7.9) |
| Mucocutaneous | 200 (94.8) | 613 (91.8) | 195 (88.2) | 598 (90.7) |
| Cardiorespiratory | 17 (8.1) | 54 (8.1) | 20 (9) | 51 (7.7) |
| Immunologic | 94 (44.5) | 515 (77.1)b | 101 (45.7) | 497 (75.4)b |
| Constitutional | 1 (0.5) | 10 (1.5) | 3 (1.4) | 17 (2.6) |
| Hematologic | 7 (3.3) | 83 (12.4)b | 5 (2.3) | 56 (8.5)b |
| Hematology, mean ± SD | ||||
| Leukocytes, ×1,000/μl | 7.2 ± 2.7 | 5.5 ± 2.1b | 7.2 ± 2.7 | 5.6 ± 2.2b |
| Lymphocytes, ×1,000/μl | 1.8 ± 0.8 | 1.2 ± 0.6b | 1.8 ± 0.8 | 1.3 ± 0.6b |
| Monocytes, ×1,000/μl | 0.4 ± 0.1 | 0.3 ± 0.1b | 0.4 ± 0.2 | 0.3 ± 0.2b |
| Neutrophils, ×1,000/μl | 4.9 ± 2.4 | 3.8 ± 1.8b | 4.8 ± 2.3 | 3.9 ± 1.9b |
| Platelets, ×1,000/μl | 285.1 ± 84 | 273.4 ± 88 | 281.2 ± 81 | 262.4 ± 83b |
| Medications | ||||
| Antimalarials | 170 (80.6) | 551 (82.5) | 189 (85.5) | 554 (83.9) |
| Any immunosuppressant | 116 (55) | 424 (63.5)b | 93 (42.1) | 395 (59.8)b |
| Corticosteroids | 152 (72) | 587 (87.9)b | 154 (69.7) | 579 (87.7)b |
Concomitant medications allowed at baseline were stable doses of antimalarials (e.g., chloroquine or hydroxychloroquine), immunosuppressants (e.g., azathioprine, methotrexate, mycophenolate, cyclophosphamide, or cyclosporine), and corticosteroids. Continuous variables were compared by 2‐sample t‐test. Discrete variables were compared by Fisher's exact test. Except where indicated otherwise, values are the number (%). SELENA–SLEDAI = Safety of Estrogens in Lupus Erythematosus National Assessment–Systemic Lupus Erythematosus Disease Activity Index.
P < 0.05 versus interferon (IFN) response gene negative.
Coexpression analysis and the IFN response gene subnetworks characterized in SLE
The large size of this data set provided a unique opportunity to examine the relationships between expression of multiple genes at the individual patient level as well as the global gene coexpression relationships across the entire SLE population. A WGCNA‐generated gene subnetwork is shown in Figure 2 24, 25. The WGCNA produced a set of coexpression modules that significantly correlate with 3 key clinical end points (the type I IFN signature, SELENA–SLEDAI, and anti‐dsDNA antibodies), and these are annotated by Gene Ontology terms including immune response, IFN signaling, and cell cycle (see Supplementary Results, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). A striking result from the coexpression analysis is the high degree of conservation of module structure across the IFN signature. Figure 2 shows a network of the most correlated genes among all SLE patients. These results demonstrate the previously unreported complexity of IFN response gene expression networks among individual patients and across the SLE population as a whole. There is complex coordinate expression of gene families, with large numbers of members that are highly expressed in SLE 30, 31. Interestingly, in certain select cases, the connectivity is limited (e.g., IFIT2, USP41, HERC6, ZCCHC2, and USP18), while the connectivity for the others is extensive (e.g., MX1, OAS1, OAS2, and IFIT5) (see also Supplementary Material, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract).
Association between high IFN‐inducible gene expression at baseline and time to severe disease flare
Gene expression profiling results were compared with baseline clinical features to determine whether these could be used as prognostic biomarkers for clinical response. Results of analysis using a Cox proportional hazards model and backward elimination of IFN response gene profile expression and the other baseline measurements of race, geographic region, sex, SELENA–SLEDAI, C3, C4, and anti‐dsDNA are shown in Table 3. Baseline characteristics prognostic of time to severe flare events for ILLUMINATE‐1 included the following: baseline higher disease activity measured by SELENA–SLEDAI (relative risk [RR] 7.3, P = 0.0001), IFN response gene signature (RR 5.6, P = 0.0015), presence of anti‐dsDNA antibodies (RR 1.8, P = 0.02), and low C3 level (RR 14.3, P = 0.0005). The ILLUMINATE‐2 study showed a similarly increased RR for SELENA–SLEDAI score (RR 35.5, P < 0.0001), IFN response gene signature (RR 5.9, P = 0.0002), anti‐dsDNA antibodies (RR 2.0, P = 0.002), and geographic region (RR 1.84, P = 0.01). Taken together, these studies demonstrated that the IFN response gene signature was prognostic of future severe disease flares independent of other baseline patient covariates.
Table 3.
Cox proportional hazards regression modeling of baseline variables to predict a severe disease flare over 52 weeks in the ILLUMINATE‐1 and ILLUMINATE‐2 studiesa
| RR | χ2 | P | |
|---|---|---|---|
| ILLUMINATE‐1 | |||
| IFN response gene signature | 5.6 | 10.1 | 0.0015 |
| Baseline SLEDAI | 7.3 | 16.5 | <0.0001 |
| Baseline low C3 | 14.2 | 12.3 | 0.0005 |
| Anti‐dsDNA | 1.8 | 5.2 | 0.0232 |
| ILLUMINATE‐2 | |||
| IFN response gene signature | 5.9 | 13.9 | 0.0002 |
| Geographic region | 1.8 | 10.6 | 0.01 |
| Baseline SLEDAI | 35.5 | 30.2 | <0.0001 |
| Anti‐dsDNA | 2.0 | 9.6 | 0.002 |
Baseline covariate screening was performed using backward elimination (P to leave = 0.05) with Cox proportional hazards regression for ILLUMINATE‐1 and ILLUMINATE‐2. Baseline covariates included in the full model were age, sex, interferon (IFN) response gene signature, geographic region of the US/Canada/Europe, and serum C3, C4, and anti–double‐stranded DNA (anti‐dsDNA) antibody levels. Disease flares were defined by the Safety of Estrogens in Lupus Erythematosus National Assessment version of the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) flare index. Risk ratios (RRs) across the range of variables are shown.
Stable IFN response gene expression over 52 weeks of study
No change was apparent in elevated gene expression profiles at baseline over the 52‐week duration of the study in the placebo or tabalumab treatment group. This IFN signature was also not significantly down‐modulated in patients who showed improvement in disease activity, regardless of treatment.
Gene expression profile at baseline does not predict the response to tabalumab
Whether examined as a single predefined gene or in multiple gene combinations, no associations between gene expression at baseline and the clinical response to tabalumab treatment (i.e., no tailoring biomarker was identified) were observed (data not shown). The IFN gene signature at baseline did not predict greater improvement at week 52 as measured by the primary end point (SRI‐5) in placebo‐treated or tabalumab‐treated patients. When the analysis was restricted to those patients who received tabalumab, the responders did not have statistically significant differences in BAFF levels compared with the nonresponders (data not shown).
Pharmacodynamic gene expression changes are consistent with the predicted mechanism of action of BAFF blockade by tabalumab
A total of 410 statistically significant drug‐induced pharmacodynamic changes in gene expression were identified. The 20 most significant of these posttreatment expression changes are summarized in Table 4, listed according to the categories of immunoglobulin and B cell–related gene changes. As expected, some of these changes appeared to reflect the tabalumab‐induced impact on the B cell number. For example, the CD20 cell number as enumerated by flow cytometry was significantly correlated with CD20/CD19 mRNA expression; however, changes were observed in gene expression among many other transcripts that were not clearly associated with B cells (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). For example, mRNA transcripts that are associated with cell cycle and cell signaling were also detected (see Supplementary Table 3), and cell cycle associations were similarly observed in the WGCNA (see Supplementary Table 4).
Table 4.
Pharmacodynamically induced changes in gene expressiona
| Fold change in gene expression | |||||
|---|---|---|---|---|---|
| ILLUMINATE‐1 | ILLUMINATE‐2 | ||||
| Gene symbol | Gene name | Baseline to week 16 | Baseline to week 52 | Baseline to week 16 | Baseline to week 52 |
| BLK | B lymphoid tyrosine kinase | −1.09 | −1.25 | −1.09 | −1.30 |
| BLNK | B‐cell linker | −1.24 | −1.42 | −1.21 | −1.41 |
| CD19 | CD19 molecule | −1.14 | −1.24 | −1.16 | −1.31 |
| CD20 | CD20, membrane‐spanning 4‐domains, subfamily A | −1.35 | −1.95 | −1.36 | −1.95 |
| CD200 | CD200 molecule | −1.31 | −1.52 | −1.39 | −1.65 |
| CD22 | CD22 molecule | −1.36 | −1.85 | −1.41 | −1.99 |
| CD40 | CD40 molecule, TNF receptor superfamily member 5 | −1.10 | −1.14 | −1.12 | −1.18 |
| CD72 | CD72 molecule | −1.29 | −1.48 | −1.34 | −1.51 |
| CD79A | CD79a molecule, immunoglobulin‐associated alpha | −1.31 | −1.65 | −1.29 | −1.71 |
| CD79B | CD79b molecule, immunoglobulin‐associated beta | −1.16 | −1.26 | −1.15 | −1.28 |
| IGHD2‐15 | Immunoglobulin heavy diversity 2‐15 | −1.59 | −1.86 | −1.77 | −1.91 |
| IGHD3‐10 | Immunoglobulin heavy diversity 3‐10 | −1.99 | −2.45 | −1.76 | −2.17 |
| IGHD3‐16 | Immunoglobulin heavy diversity 3‐16 | −1.64 | −1.83 | −1.72 | −1.91 |
| IGHD3‐3 | Immunoglobulin heavy diversity 3‐3 | −2.13 | −2.79 | −2.00 | −2.43 |
| IGHM | Immunoglobulin heavy constant mu | −1.59 | −2.14 | −1.61 | −2.22 |
| IGHV1‐18 | Immunoglobulin heavy variable 1‐18 | −1.60 | −1.69 | −1.61 | −1.79 |
| IGHV3‐13 | Immunoglobulin heavy variable 3‐13 | −1.60 | −1.81 | −1.57 | −1.83 |
| IGHV3‐15 | Immunoglobulin heavy variable 3‐15 | −1.74 | −2.01 | −1.72 | −2.06 |
| IGHV3‐23 | Immunoglobulin heavy variable 3‐23 | −1.64 | −1.88 | −1.68 | −1.98 |
| IGHV3‐53 | Immunoglobulin heavy variable 3‐53 | −1.45 | −1.68 | −1.52 | −1.66 |
Statistically significant pharmacodynamically induced gene expression changes following tabalumab treatment were observed for 410 genes and were consistent with the predicted mechanism of action of BAFF blockade impacting immunoglobulin genes, B cell–related genes, and other genes with immune function. The largest changes associated with treatment were observed in immunoglobulin‐ and B cell–related genes. The B cell–related genes and immunoglobulin genes that exhibited the largest overall fold changes from baseline to week 16 and from baseline to week 52 are shown. All results shown had an overall q value of <0.00001.
DISCUSSION
ILLUMINATE‐1 and ILLUMINATE‐2 were global, phase III, 52‐week, randomized, placebo‐controlled, double‐blind, parallel studies of patients with moderate‐to‐severe SLE treated with the fully human IgG4 BAFF‐targeting mAb, tabalumab. The patients participating in ILLUMINATE‐1 and ILLUMINATE‐2 were predominantly white females with moderate disease activity and a mean disease duration of 9 years. Gene expression at baseline was similar to that observed in previous studies of SLE, demonstrating a statistically significant increase in expression of IFN response genes in 75% of the patients studied 13, 14, 15, 16, 17. The genes with the greatest fold increase as compared with healthy matched controls were the IFN response genes. Previously published studies of SLE, although smaller in size, have also shown increased expression of IFN response genes, particularly in patients with more active disease and the presence of high levels of autoantibodies 13, 14, 15, 16, 17. Observations from these 2 large, independent cohorts substantially extend those from previous studies by directly comparing IFN response gene signature–positive versus IFN response gene signature–negative groups among these 1,760 patients. Statistically significant associations were observed in both cohorts between the presence of the IFN signature and a cluster of clinical, immunologic, and laboratory manifestations of SLE at baseline (Table 2).
In addition to the IFN response genes, other genes were highly expressed in the lupus population compared with healthy controls, including those genes involved with immunoglobulin production, immunoglobulin modification, immunoglobulin binding, ubiquitination, and poly(ADP‐ribose) polymerization (Table 1). This is consistent with lymphoid cell proliferation and high levels of autoantibody production seen in SLE and aligns with current views of the overall pathogenesis of SLE 1, 2. Consistent with high expression of ubiquitination, recent genome‐wide association studies have demonstrated an association between the IFN signature and select genotypes of the ubiquitin enzyme gene, UBE2L3, suggesting that this high‐expression phenotype may be controlled by regulatory genes associated with SLE 30, 31, 32, 33.
The large size of the SLE data set presented here provides a unique opportunity to examine the relationships between the expression of multiple genes at the individual patient level and to examine global gene expression relationships across the entire SLE population (Figures 1 and 2). We observed that although there are distinctive individual gene expression patterns, there is a network of strongly correlated genes, the majority of which have been identified previously as IFN response genes (Figure 1 and Table 1). However, in select cases, the connectivity between these is limited when the WGCNA was used to construct and compare gene coexpression modules (Figure 2 and Supplementary Material, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). The expression of IFIT2, USP41, HERC6, ZCCHC2, and USP18 revealed limited connectivity, while connectivity with other IFN response genes was extensive.
Overall, a high degree of similarity of network connectivity was observed between the expression of these groups of genes in both the subgroup with high expression of the IFN response gene and the subgroup with low expression of the IFN response gene. These findings are consistent with those from a recent genetic association study demonstrating that transcriptional factors are highly represented among the susceptibility genes in SLE 33 and from a model in which aberrantly regulated gene networks, controlling expression of the innate and adaptive immune systems, are central to disease pathogenesis in SLE. The novel findings observed in the ILLUMINATE studies of complex gene networks in SLE are reminiscent of the gene networks observed in psoriatic skin lesions, in which mRNA transcripts from >1,000 individual genes have been shown to demonstrate coordinate expression in skin plaques and can be substantially modulated with highly effective therapy 34.
In the current study, we took advantage of newly available technologic advances to provide a more detailed analysis of gene expression in SLE. The recently developed Affymetrix HTA 2.0 array allows for a rapid survey of relative expression of large numbers of gene transcripts using multiple probe sets (70,753 transcript clusters). In addition, to further support HTA 2.0 array results, a newly developed method for quantifying gene expression, NanoString, was tested 23. We observed that there were highly significant correlations between mRNA expression using the HTA 2.0 exon array and regions of individual genes probed using NanoString (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract) 23. This high‐resolution expression data set may be of value to other investigators studying SLE using the data deposited into the Gene Expression Omnibus repository 35.
The relationship between gene expression at baseline and future clinical response was also a focus of the current study. Results of the analysis using the Cox proportional hazards model and backward elimination of IFN response gene profile expression and the other baseline measurements of race, geographic region, sex, SELENA‐SLEDAI, C3, C4, and anti‐dsDNA are shown in Table 3. In ILLUMINATE‐1 and ILLUMINATE‐2, the IFN response gene signature was an independent risk factor for severe disease flare (Table 3). In ILLUMINATE‐1, the IFN response gene signature, the baseline SELENA‐SLEDAI, the baseline C3 level, and anti‐dsDNA were independent risk factors for severe flare. In ILLUMINATE‐2, baseline SELENA–SLEDAI conferred the greatest risk ratio (RR 35), while the IFN response gene signature, anti‐dsDNA, and geographic region conferred RRs of 5.9, 2.0, and 1.8, respectively (Table 3). Taken together, these observations support the IFN response gene signature as being an independent predictor of clinical disease flare.
Although some previous reports have described an association between an IFN response gene signature and disease activity, subsequent studies have failed to consistently confirm these findings 36, 37, 38, 39, 40. Comparison of published studies can be challenging due to differences in the patient populations and the methods defining the IFN gene response signature. Previous studies have varied regarding the specific IFN response genes that have been interrogated, the methods used to measure gene expression, and the scoring systems used to classify patients as IFN response gene signature positive or IFN response gene signature negative.
The current study provided the opportunity to address the relationship between disease activity and gene expression in a large, controlled clinical trial setting, using standardized approaches. In this study, the elevated gene expression profile seen at baseline for IFN response genes remained stable over the 52‐week duration of the study in both the standard‐of‐care and tabalumab arms. Although the IFN response gene signature did not change with the modest effects of tabalumab or background standard‐of‐care medication, it remains possible that it can be changed with therapies specifically targeting the IFNs or the IFN receptor, via blocking intracellular signaling by the IFN receptor or by robustly impacting other mechanisms involved in disease pathogenesis. Delineating the response in the IFN response gene signature–positive and IFN response gene signature–negative populations may lead to identification of patient subpopulations in which a precision‐medicine approach can successfully be applied 35.
The findings reported here are also consistent with recent observations of a genetic association between SLE and factors in the IFN pathway. They support the concept that the IFN response gene signature is a heritable risk factor for SLE and may be less sensitive as a disease activity measure 41, 42. Consistent with this, high serum levels of IFNα have been shown to be a heritable trait in both SLE patients and their first‐degree relatives 40, 41.
Mechanistically, the data reported here are consistent with a model in which dysregulation of IFN expression in SLE, once initiated, tends to be self‐perpetuating. This persistence of IFN expression could be the result of epigenetic changes that are induced in genes/regulator elements controlling expression of the IFN response genes. Also consistent with this model are the observations described herein and reported in the literature that expression of the IFN response gene signature is associated with more severe disease including disease flares, high levels of anti‐dsDNA antibodies, and low serum complement levels (see Tables 2 and 3) 40, 41.
A major focus of the current study was to determine which genes were modulated following tabalumab treatment. A total of 410 statistically significant drug‐induced pharmacodynamic changes in gene expression were identified. The 20 most significant of these are summarized in Table 4 and are categorized as B cell–associated and immunoglobulin‐associated gene changes. As expected, some of these changes appear to reflect a tabalumab‐induced pharmacodynamic impact on the B cell number. For example, the CD20 cell number as enumerated by flow cytometry was significantly correlated with CD20/CD19 mRNA expression, consistent with the observed decrease in IgG, IgM, and IgA levels and the decreased number of anti‐dsDNA antibodies during treatment with tabalumab 11, 12. However, changes in other transcripts (Table 4) were less clearly associated with B cells (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract). For example, changes in mRNA transcripts that are associated with cell cycle and cell signaling were also detected. This is consistent with WGCNA modules associated with clinical correlates (Supplementary Table 4, http://onlinelibrary.wiley.com/doi/10.1002/art.39950/abstract), which also identified cell cycle module connectivity in addition to connectivity to humoral immune responses, antigen processing and presentation, and IFN pathways.
A limitation to the present study is that although pooled components of whole blood mRNA were interrogated using the HTA 2.0 array, and selective genes were confirmed using the alternative method of NanoString, only general inferences regarding the individual cellular sources contributing to the total pool of mRNA can be made. Currently, no published, broadly accepted, lineage‐specific mRNA transcripts have been identified for immune cells, and therefore only general inferences can be derived from any study that examined whole blood mRNA 43, 44. Identification and characterization of gene expression profiles for specific cell lineages would require correlation with alternative methods such as flow cytometry to enumerate the cell populations being studied (as was done on a limited basis in the current study) or ideally, by characterizing mRNA expression on flow cytometry–sorted cell lines or individual cells 45. Characterizing mRNA for individual cell lines and/or single cells will be of considerable interest for future studies but remains technically challenging.
No association between changes in gene expression and the clinical response to tabalumab treatment was observed. Neither BAFF mRNA expression in whole blood nor the serum BAFF protein level predicted response to treatment with tabalumab.
This large study confirms increased expression of IFN response genes in SLE patients compared with healthy controls, with 75% of the patients participating in the tabalumab phase III trials having the IFN gene signature. In both studies, patients with the IFN signature had significantly more SELENA‐SLEDAI score–defined immunologic and hematologic system involvement, and more patients in the IFN response gene–positive group used corticosteroids and immunosuppressant medications. There was substantial heterogeneity of expression among individual IFN response genes, and complex relationships were identified among IFN gene networks. The IFN signature remained stable over 52 weeks of study and was not reduced when patients showed improvement. The high baseline IFN signature was an independent predictor of deterioration as measured by time to flare but did not predict improvement as measured by the SRI‐5. Pharmacodynamic changes in gene expression associated with tabalumab treatment were extensive and occurred predominantly, although not exclusively, in B cell–related and immunoglobulin genes, consistent with changes in other pharmacodynamic markers including B cell number, anti‐dsDNA autoantibody level, and serum immunoglobulin levels.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Hoffman had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Hoffman, Merrill, Alarcón‐Riquelme, Petri, Dow, Nantz, Higgs.
Acquisition of data
Hoffman, Dow, Nantz, Nisenbaum, Schroeder, Komocsar, Perumal, Rocha, Higgs.
Analysis and interpretation of data
Hoffman, Merrill, Alarcón‐Riquelme, Petri, Dow, Perumal, Linnik, Airey, Liu, Rocha, Higgs.
ROLE OF THE STUDY SPONSOR
This study was designed by Eli Lilly and Company, with consultation with an academic advisory board. Eli Lilly and Company performed data analysis and routine laboratory testing, and provided site monitoring of the clinical trial and external vendors, as well as overall data interpretation. All authors and Eli Lilly and Company personnel wrote, reviewed, and approved the manuscript, while the authors maintained control over the final content. Publication of this article was not contingent upon approval by Eli Lilly and Company.
Supporting information
Table S1. Down regulated genes at baseline: SLE versus normal controls.
Table S2. Pre‐selected 164 genes included in the interferon responsive gene signature.
Table S3. Pharmacodynamic‐induced changes in gene expression*
Table S4. Gene co‐expression module annotation and clinical correlations*
Figure S1. Gene expression measured using NanoStringTM and compared to HTA2.0 gene expression arrays confirming that findings are strongly correlated when testing the same sample using either method (n=2647). NanoString TM results are shown as log2 adjusted units and Affymetrix units shown as log2 intensity. Results are shown here for the following genes: a) IFI27, alpha‐inducible protein 27, an interferon responsive gene (correlation r=0.984); b) RSAD2, radical S‐adenosyl methionine domain containing 2, an interferon responsive gene (correlation r=0.974; c) MS4A1, the B cell expression marker CD20 (correlation r=0.927); IGHA1, immunoglobulin heavy chain A1, a gene highly expressed by plasma cells (correlation r=0.970). The slight sigmoidal shape of the IGHA1 plot suggests that the expression measured by the HTA2.0 array is saturating.
The primers used were: IFI27, GAGCAACTGGACTCTCCGGATTGACCAAGTTCATCCTGGGCTCCATTGGGTCTGCCATTGCGGCTGTCATTGCGAGGTTCTACTAGCTCCCTGCCCCTCG; RSAD2, ATGCGCTTTCTGAACTGTAGAAAGGGACGGAAGGACCCTTCCAAGTCCATCCTGGATGTTGGTGTAGAAGAAGCTATAAAATTCAGTGGATTTGATGAAA;MS4A1, ACCCAGAAATTCAGTAAATGGGACTTTCCCGGCAGAGCCAATGAAAGGCCCTATTGCTATGCAATCTGGTCCAAAACCACTCTTCAGGAGGATGTCTTCA;IGHA1, AGGATGCCTCCGGGGACCTGTACACCACGAGCAGCCAGCTGACCCTGCCGGCCACACAGTGCCTAGCCGGCAAGTCCGTGACATGCCACGTGAAGCACTA.
Figure S2. Overlap between High and Low interferon signature subgroup modules is shown. The figure highlights similar gene co‐expression structure across the interferon signature subgroups. Module color and size information is labeled on the axes. The matrix is populated with overlap size. Minus log10 of p‐values from Fisher Exact Tests for overlap size are visualized by the white to red color scale from 0 to 40 (=40 if > 40); this scale is shown on the far right of the figure. Most modules from the High SLE subgroup significantly overlap with just one (or two) modules (converging number shown in the boxes outlined in pink to red) from the Low SLE subgroup, indicating preservation of co‐expression structure. Additional details are provided in Supplemental File SF1.
Supplemental File SF1
ACKNOWLEDGMENTS
We gratefully acknowledge the contributions of the patients as well as the investigators and their study teams who participated in this study.
ClinicalTrials.gov identifiers: NCT01205438 and NCT01196091.
Supported by Eli Lilly and Company. Dr. Alarcón‐Riquelme's work was supported in part by the Innovative Medicines Initiative Joint Undertaking (grant 115565) and European Federation of Pharmaceutical Industries Association companies in‐kind contributions.
Dr. Hoffman owns stock or stock options in Eli Lilly and Company. Dr. Merrill has received consulting fees, speaking fees, and/or honoraria from Eli Lilly and Company (less than $10,000). Dr. Alarcón‐Riquelme has received consulting fees, speaking fees, and/or honoraria from Eli Lilly and Company (less than $10,000). Dr. Petri has received consulting fees from Eli Lilly and Company (less than $10,000). Drs. Dow, Nisenbaum, Schroeder, Perumal, Linnik, Airey, Liu, Rocha, and Higgs and Ms Komocsar own stock or stock options in Eli Lilly and Company.
[The copyright line for this article was changed on 12 February 2019 after original online publication.]
REFERENCES
- 1. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet 2014;384:1878–88. [DOI] [PubMed] [Google Scholar]
- 2. D'Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet 2007;369:587–96. [DOI] [PubMed] [Google Scholar]
- 3. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 1999;189:1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cancro MP, D'Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Invest 2009;119:1066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF‐binding receptors during human B cell differentiation. J Immunol 2007;179:7276–86. [DOI] [PubMed] [Google Scholar]
- 6. Stohl W, Metyas S, Tan SM, Cheema GS, Oamar B, Roschke V, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum 2003;48:3475–86. [DOI] [PubMed] [Google Scholar]
- 7. Petri M, Stohl W, Chatham W, McCune WJ, Chevrier M, Ryel J, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum 2008;58:2453–9. [DOI] [PubMed] [Google Scholar]
- 8. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomized, placebo‐controlled, phase 3 trial. Lancet 2011;377:721–31. [DOI] [PubMed] [Google Scholar]
- 9. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. A phase III, randomized, placebo‐controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 2011;63:3918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Furie RA, Leon G, Thomas M, Petri MA, Chu AD, Hislop C, et al. A phase 2, randomized, placebo‐controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate‐to‐severe systemic lupus erythematosus, the PEARL‐SC study. Ann Rheum Dis 2015;74:1667–75. [DOI] [PubMed] [Google Scholar]
- 11. Isenberg DA, Petri M, Kalunian K, Tanaka Y, Urowitz MB, Hoffman RW, et al. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: results from ILLUMINATE‐1, a 52‐week, phase III, multicentre, randomised, double‐blind, placebo‐controlled study. Ann Rheum Dis 2016;75:323–31. [DOI] [PubMed] [Google Scholar]
- 12. Merrill JT, van Vollenhoven RF, Buyon JP, Furie RA, Stohl W, Morgan‐Cox M, et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B‐cell activating factor, in patients with systemic lupus erythematosus: results from ILLUMINATE‐2, a 52‐week, phase III, multicentre, randomised, double‐blind, placebo‐controlled study. Ann Rheum Dis 2016;75:332–40. [DOI] [PubMed] [Google Scholar]
- 13. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon‐inducible gene expression signature in peripheral blood cells of patients with severe systemic lupus. Proc Natl Acad Sci U S A 2003;100:2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003;197:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon‐α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 2005;52:1491–503. [DOI] [PubMed] [Google Scholar]
- 16. Kennedy WP, Maciuca R, Wolslegel K, Tew W, Abbas A, Chaivorapol C, et al. Association of the interferon signature metric with serologic disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci Med 2015;2:e000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carpintero MF, Martinez L, Fernandez I, Romero AC, Mejia C, Zang YJ, et al. Diagnosis and risk stratification in patients with anti‐RNP autoimmunity. Lupus 2015;24:1057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. M Petri, MY Kim, KC Kalunian, J Grossman, BH Hahn, LR Sammaritano, et al, for the OC-SELENA Trial. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005;353:2550–8. [DOI] [PubMed] [Google Scholar]
- 19. Hochberg MC, for the Diagnostic and Therapeutic Criteria Committee of the American College of Rheumatology . Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 20. Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol 2010;6:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guerra SG, Vyse TJ, Cunninghame Graham DS. The genetics of lupus: a functional perspective. Arthritis Res Ther 2012;14:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003;31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color‐coded probe pairs. Nat Biotechnol 2008;26:317–25. [DOI] [PubMed] [Google Scholar]
- 24. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol 2011;7:e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Statistical Soc 1995;57:289–300. [Google Scholar]
- 27. Liu A, Qu Y, Tang L, Ting N, Tsong Y, editors. A framework of statistical methods for identification of subgroups with differential treatment effects in randomized trials Applied statistics in biomedicine and clinical trials design: selected papers from 2013 ICSA. New York: Springer Verlag; 2015. [Google Scholar]
- 28. Leon AC, Heo M. Sample sizes required to detect interactions between two binary fixed‐effects in a mixed‐effects linear regression model. Comput Stat Data Anal 2009;53:603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eisenberg E, Levanon EY. Human housekeeping genes, revisited [published erratum appears in Trends Genet 2014;30:119–20]. Trends Genet 2013;29:569–74. [DOI] [PubMed] [Google Scholar]
- 30. Kariuki SN, Ghodke‐Puranik Y, Dorschner JM, Chrabot BS, Kelly JA, Tsao BP, et al. Genetic analysis of the pathogenic molecular sub‐phenotype interferon‐α identifies multiple novel loci involved in systemic lupus erythematosus. Genes Immun 2015;16:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chiche L, Jourde‐Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol 2014;66:1583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramos PS, Williams AH, Ziegler JT, Comeau ME, Guy RT, Lessard CJ, et al. Genetic analyses of interferon pathway–related genes reveal multiple new loci associated with systemic lupus erythematosus. Arthritis Rheum 2011;63:2049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bentham J, Morris DL, Cunninghame Graham DS, Pinder DL, Tombleson P, Behrens TW, et al. Genetic association analyses implicated aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 2015;47:1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krueger JG, Fretzin S, Suarez‐Farinas M, Haslett PA, Phipps KM, Cameron GS, et al. IL‐17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol 2012;130:145–54.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hofmann‐Apitius M, Alarcón‐Riquelme ME, Chamberlain C, McHale D. Towards the taxonomy of human disease. Nat Rev Drug Discov 2015;14:75–6. [DOI] [PubMed] [Google Scholar]
- 36. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon‐α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis 2016;75:196–202. [DOI] [PubMed] [Google Scholar]
- 37. Aranow C, Kamen DL, Dall'Era M, Massarotti EM, Mackay MC, Koumpouras F, et al. Randomized double‐blind, placebo‐controlled trial of the effect of vitamin D3 on the interferon signature in patients with systemic lupus erythematosus. Arthritis Rheumatol 2015;67:1848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Petri M, Singh S, Tesfasyone H, Dedrick R, Fry K, Lal P, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 2009;18:980–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landolt‐Marticorena C, Bonventi G, Lubovich A, Ferguson C, Unnithan T, Su J, et al. Lack of association between the interferon‐α signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann Rheum Dis 2009;68:1440–6. [DOI] [PubMed] [Google Scholar]
- 40. Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, et al. Association of increased interferon‐inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum 2006;54:2951–62. [DOI] [PubMed] [Google Scholar]
- 41. Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, et al. Network analysis of associations between serum interferon‐α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum 2011;63:1044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN‐α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 2007;8:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Streicher K, Morehouse CA, Groves CJ, Rajan B, Pilataxi F, Lehmann KP, et al. The plasma cell signature in autoimmune disease. Arthritis Rheumatol 2014;66:173–84. [DOI] [PubMed] [Google Scholar]
- 44. Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 2011;144:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 2016;165:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Down regulated genes at baseline: SLE versus normal controls.
Table S2. Pre‐selected 164 genes included in the interferon responsive gene signature.
Table S3. Pharmacodynamic‐induced changes in gene expression*
Table S4. Gene co‐expression module annotation and clinical correlations*
Figure S1. Gene expression measured using NanoStringTM and compared to HTA2.0 gene expression arrays confirming that findings are strongly correlated when testing the same sample using either method (n=2647). NanoString TM results are shown as log2 adjusted units and Affymetrix units shown as log2 intensity. Results are shown here for the following genes: a) IFI27, alpha‐inducible protein 27, an interferon responsive gene (correlation r=0.984); b) RSAD2, radical S‐adenosyl methionine domain containing 2, an interferon responsive gene (correlation r=0.974; c) MS4A1, the B cell expression marker CD20 (correlation r=0.927); IGHA1, immunoglobulin heavy chain A1, a gene highly expressed by plasma cells (correlation r=0.970). The slight sigmoidal shape of the IGHA1 plot suggests that the expression measured by the HTA2.0 array is saturating.
The primers used were: IFI27, GAGCAACTGGACTCTCCGGATTGACCAAGTTCATCCTGGGCTCCATTGGGTCTGCCATTGCGGCTGTCATTGCGAGGTTCTACTAGCTCCCTGCCCCTCG; RSAD2, ATGCGCTTTCTGAACTGTAGAAAGGGACGGAAGGACCCTTCCAAGTCCATCCTGGATGTTGGTGTAGAAGAAGCTATAAAATTCAGTGGATTTGATGAAA;MS4A1, ACCCAGAAATTCAGTAAATGGGACTTTCCCGGCAGAGCCAATGAAAGGCCCTATTGCTATGCAATCTGGTCCAAAACCACTCTTCAGGAGGATGTCTTCA;IGHA1, AGGATGCCTCCGGGGACCTGTACACCACGAGCAGCCAGCTGACCCTGCCGGCCACACAGTGCCTAGCCGGCAAGTCCGTGACATGCCACGTGAAGCACTA.
Figure S2. Overlap between High and Low interferon signature subgroup modules is shown. The figure highlights similar gene co‐expression structure across the interferon signature subgroups. Module color and size information is labeled on the axes. The matrix is populated with overlap size. Minus log10 of p‐values from Fisher Exact Tests for overlap size are visualized by the white to red color scale from 0 to 40 (=40 if > 40); this scale is shown on the far right of the figure. Most modules from the High SLE subgroup significantly overlap with just one (or two) modules (converging number shown in the boxes outlined in pink to red) from the Low SLE subgroup, indicating preservation of co‐expression structure. Additional details are provided in Supplemental File SF1.
Supplemental File SF1