

Abstract
We evaluated the effects of therapeutic and supratherapeutic doses of tramadol hydrochloride on the corrected QT (QTc) interval in healthy adults (aged 18‐55 years) in a randomized, phase I, double‐blind, placebo‐ and positive‐controlled, multiple‐dose, 4‐way crossover study. Participants were randomized to receive 1 of 4 treatments (A‐D), 1 each in 4 treatment periods (1‐4), separated by a washout period (7‐15 days). Treatment A comprised tramadol 400 mg (therapeutic dose) on days 1 through 3, tramadol 100 mg and moxifloxacin‐matched placebo on day 4, and placebo on all 4 days. Treatment B comprised tramadol 600 mg (supratherapeutic dose) on days 1 through 3, and tramadol 150 mg and moxifloxacin‐matched placebo on day 4. Treatment C comprised placebo on days 1 through 4 and moxifloxacin‐matched placebo on day 4. Treatment D comprised placebo on days 1 through 4 and moxifloxacin 400 mg on day 4. Of 68 participants enrolled, 57 (83.8%) completed the study. Both therapeutic and supratherapeutic doses of tramadol were shown to be noninferior to placebo regarding their effect on QTc prolongation. Sixty‐one of 68 (89.7%) participants reported at least 1 treatment‐emergent adverse event (mild); nausea was the most frequently reported treatment‐emergent adverse event. Summarizing, tramadol at doses up to 600 mg/day did not cause clinically relevant QTc interval prolongation in healthy adults.
Keywords: cardiac repolarization, electrocardiogram, moxifloxacin, pharmacodynamics, pharmacokinetics, QT/QTc prolongation, tramadol hydrochloride
Opioid drugs are known to be associated with adverse cardiac effects, as they bind to opioid‐specific receptors located in the central nervous system, and in many other organs including cardiovascular tissue.1, 2, 3 This may cause cardiac membrane hyperpolarization, leading to QT interval prolongation4 and possibly ventricular arrhythmia known as torsades de pointes (TdP), ultimately leading to sudden cardiac death.5
Inhibition of the rapidly activating delayed rectifier potassium channel encoded by the human ether‐a‐go‐go related‐gene (hERG) also causes QT prolongation. Therefore, in vitro hERG assays constitute a key element in the assessment of the TdP‐causing potential of drugs.6 Many opioids such as codeine, buprenorphine, and fentanyl have been shown to be capable of blocking hERG potassium channels in vitro,7 indicating that QT prolongation and TdP could be a safety concern for drugs belonging to the opioid class.8
Tramadol hydrochloride is an analgesic acting through both opioid and nonopioid mechanisms.9 The pharmaceutical form of tramadol exists as a racemic mixture: (+)‐tramadol inhibits the reuptake of serotonin and (−)‐tramadol prevents norepinephrine reuptake.10, 11, 12 Tramadol is extensively metabolized in the liver via cytochrome P450 enzymes, resulting in more than 20 phase 1 and phase 2 metabolites.10, 13, 14 The phase 1 metabolites O‐desmethyltramadol (M1) produced via O‐demethylation of tramadol by CYP2D6 and mono‐N‐desmethyltramadol (M2) produced via N‐demethylation of tramadol by CYP2B6 and CYP34A are the main metabolites.11, 13, 15 M1 and M2 undergo further metabolism to secondary metabolites before excretion.11 The opioid receptor‐mediated analgesic effects are mainly attributed to M1. Although both tramadol and its metabolite M1 act as selective μ‐receptor agonists, M1 has a 300‐fold higher affinity for the μ‐opioid receptor than tramadol itself.16 Approximately 90% of tramadol and its metabolites are excreted through the urine; the remaining 10% is excreted through feces.13 Since tramadol is metabolized to M1 primarily by CYP2D6, a highly polymorphic CYP450 enzyme,17 genetic variations in CYP2D6 may affect the pharmacokinetic (PK) and pharmacodynamic (PD) properties of tramadol.18, 19, 20, 21 Therefore, tramadol may result in an exaggerated therapeutic effect or adverse effect when a CYP450 inducer is added, while on other hand, a therapeutic failure could be seen as a result of little or no metabolism of tramadol if a CYP450 inhibitor is combined with tramadol.22
Since its approval by the US Food and Drug Administration (FDA) in 1995 for the management and treatment of pain, tramadol overdose has been associated with sporadic instances of QT prolongation in younger men as reported through Periodic Adverse Drug Experience Reports (years 2009 and 2010). A review of the postmarketing adverse event (AE) reports in the FDA's AE reporting system database also indicated a plausible association between tramadol use and QT prolongation/TdP at therapeutic doses or overdose. However, in preclinical studies, tramadol has been shown to be a weak hERG inhibitor with a half maximal inhibitory concentration of 129 μM (39 μg/mL; data on file). In view of these facts, and in line with the recommendations made by the International Council for Harmonization E14 guidance,23 a thorough PD study evaluating the effect of tramadol immediate‐release (IR) formulation on QT/QT interval corrected for heart rate (QTc) was requested by the FDA. The aim of this study was to determine if tramadol IR, at therapeutic and supratherapeutic dose levels, has a threshold pharmacologic effect on cardiac repolarization in healthy participants, as assessed by QT/QTc prolongation.
Methods
Study Population
Healthy men and women aged 18−55 years with a body mass index between 18 and 30 kg/m² (inclusive) were enrolled in this study. Participants had to have normal findings on a standard electroencephalogram (under resting conditions and during hyperventilation and normal intermittent photic stimulation) and a 12‐lead electrocardiogram (ECG).
Key exclusion criteria included personal or family history of epileptic seizures or convulsions; history of head trauma with loss of consciousness, central nervous system infection, or loss of consciousness of unknown origin; history of additional risk factors for TdP or a family history of short QT syndrome, long QT syndrome, sudden unexplained death at a young age (≤40 years), drowning, or sudden infant death syndrome in a first‐degree relative.
Study Design
This randomized, phase I, double‐blind, 4‐way crossover, placebo‐ and positive‐controlled, multiple‐dose study was conducted from December 2014 to August 2015 at a single site in the United States (Quintiles Phase 1 Services, Overland Park, Kansas). The study included a pretreatment phase (or screening phase up to 35 days, during which the participants were examined to determine if they met the inclusion criteria of the study, and informed consent was obtained from eligible participants), a double‐blind treatment phase (with 4 treatment periods [1 to 4], a washout period of 7 to 15 days following the last dose of the study drug in each period), and a posttreatment phase (end‐of‐study procedures on day 5 of period 4 or at the time of early withdrawal). The total study duration for each participant was up to a maximum of 100 days.
Eligible participants were randomized to receive 4 study treatments (A to D), 1 in each period (1 to 4) (Figure 1A). Treatment A comprised tramadol 400 mg/day (maximum therapeutic dose; each dose included 2 tramadol IR 50‐mg tablets) on days 1 to 3 (100 mg every 6 hours), and a single dose of tramadol 100 mg on day 4, for a total of 13 doses. Participants received 1 capsule of tramadol‐matched placebo on all 4 days, and a moxifloxacin‐matched placebo capsule on day 4 only. Treatment B comprised tramadol 600 mg/day (supratherapeutic dose; each dose included three tramadol IR 50 mg tablets) on days 1 to 3 (150 mg every 6 hours). A single dose of tramadol 150 mg and a moxifloxacin‐matched placebo capsule were given on day 4. Treatment C (placebo) comprised tramadol‐matched placebo capsules on days 1 to 4 (3 capsules every 6 hours) and a single moxifloxacin‐matched placebo capsule on day 4. Treatment D (moxifloxacin) comprised tramadol‐matched placebo capsules on days 1 to 4 (3 capsules every 6 hours) and a single dose of moxifloxacin 400 mg on day 4. All medications were given orally. The study drugs were overencapsulated to maintain the double‐blind.
Figure 1.
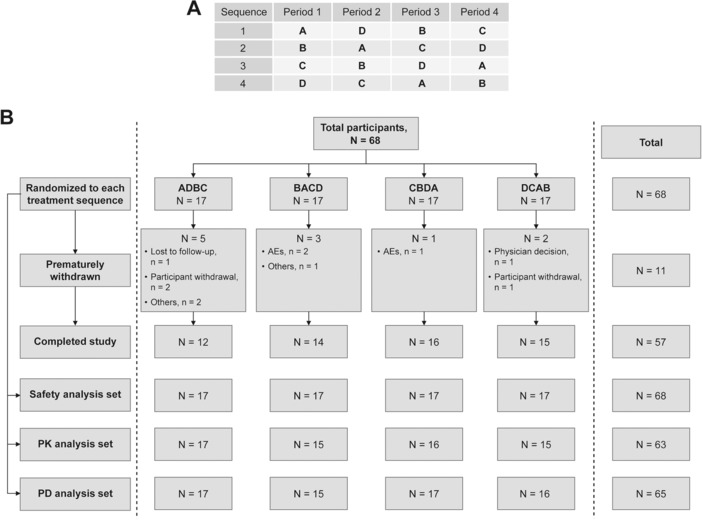
Study design and participant disposition. Tramadol 400 (treatment A): 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; tramadol 600 (treatment B): 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150‐mg dose on day 4; placebo (treatment C): placebo every 6 hours on days 1 to 3, and a single placebo dose on day 4; moxifloxacin (treatment D): placebo every 6 hours on days 1 to 3, and a single moxifloxacin 400‐mg dose on day 4. AEs, adverse events; N, number of participants; PD, pharmacodynamic, PK, pharmacokinetic.
Treatment with tramadol 600 mg/day as a supratherapeutic dose for duration of at least 48 hours was selected based on an acceptable safety data profile observed with this dose in a previous multiple ascending dose study24 and as agreed upon by the FDA. Moxifloxacin, known to prolong QT/QTc intervals, was used as a positive control to establish the assay sensitivity.
This study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with Good Clinical Practices and applicable regulatory requirements. The study protocol was reviewed and approved by the MidLands Institutional Review Board (Overland Park, Kansas). All the participants provided written informed consent to participate in the study. The study is registered at www.clinicaltrials.gov (NCT02307864).
Study Objectives
The primary objective of this study was to assess the effects of multiple doses of tramadol IR at therapeutic and supratherapeutic doses on the ECG QTc interval in healthy adults. The secondary objectives were to assess the effects of these doses of tramadol on ECG morphology and ECG intervals other than corrected QTcs in healthy adults. The PK of both enantiomers of tramadol and its metabolite M1, and their relationship with QT/QTc changes, along with the safety and tolerability of tramadol, were also evaluated.
Study Assessments
Pharmacodynamics
For PD evaluations, the heart rate (HR) and QT, RR, QRS, and PR intervals of the ECG were measured in triplicates from continuous Holter monitor at predefined time points before dosing on day 1 and after dosing on day 4. Three sets of measurements were averaged for analyses.
The measured QT data were corrected for HR using Fridericia (QTcF), Bazett (QTcB), and study‐specific power (QTcS) correction methods at each time point. For each participant, treatment, and time point of measurement, following variables, were calculated: baseline QTc (average of the 3 sets of QTc values obtained at 1 hour, 30 minutes, and 10 minutes prior to dosing on day 1 of each treatment period); ∆QTc (change from baseline QTc at each time point); ΔΔQTc (difference in ΔQTc between each treatment and placebo at each time point); and ΔQTc,Tmax (change from baseline in QTc for each dose of tramadol at the individual participant's day 4 tmax) for each analyte. The difference in ΔQTc,Tmax between each dose of tramadol (ΔΔQTc,Tmax) was also calculated for each analyte.
Pharmacokinetics
Blood samples (4 mL each) were collected prior to dose 1 and doses 6 to 13, and at predefined time points following dose 13 on day 4 (0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, and 24 hours postdose) in each treatment period. Samples were analyzed using liquid chromatography–tandem mass spectrometry to determine plasma concentrations of the (+) and (−) enantiomers of tramadol and its metabolite M1 as mentioned earlier.24 The concentration range of the calibrator samples used for the assay was 1 to 250 ng/mL, and the lower limit of quantitation was 1 ng/mL. Samples from moxifloxacin treatment were collected at 2, 2.5, 3, and 4 hours postdose.
The following PK parameters were estimated for each analyte at steady state using standard noncompartmental methods: maximum observed plasma concentration during a dosing interval (Cmax,ss), time to maximum observed plasma concentration (tmax,ss), minimum observed plasma concentration during a dosing interval (Cmin,ss), trough (predose) plasma concentration (Ctrough,ss), and area under the plasma concentration‐time curve during a dosing interval (AUCtau,ss). Phoenix WinNonlin Professional Version 6.4 (Pharsight, Certara L.P., St Louis, Missouri) was used for PK analysis.
Pharmacogenomics
For pharmacogenomic evaluations, ethylenediaminetetraacetic blood samples were collected from all participants and genomic DNA was prepared from blood samples according to standard protocol. Locus‐specific DNA fragments were amplified by polymerase chain reaction and subsequently sequenced. Genotyping was carried out through the analysis of single polymorphisms and results referred only to specific positions (g.100C > T, g.1023C > T, g.1707delT, g.1846G > A, g.2549delA, g.2615_2617delAAG, g.2850C > T, g.2988G > A, g.4180G > C). The presence of a deletion (CYP2D6*5) or duplication/multiplication (CYP2D6*MxN) of the CYP2D6 gene was tested by a special long‐range polymerase chain reaction (Eurofins Medigenomix GmbH, Ebersberg, Germany).16 Selected PK parameters (Cmax,ss and AUCtau,ss) of the (+) and (–) enantiomers of tramadol and its M1 metabolite were explored for association with the variations in the CYP2D6 gene.
Safety Evaluations
Safety evaluations were performed throughout the study and included type, incidence, and severity of AEs, treatment‐emergent adverse events (TEAEs), changes in clinical laboratory parameters (hematology, serum chemistry, and urinalysis), ECG findings, vital sign measurements, physical examinations, and pulse oximetry measurements. All ECGs were performed in triplicate, as closely as possible in succession, but no more than 2 minutes apart. An average of the ECG interval values from a triplicate (as measured by the ECG recorder's automated algorithm) was used as a single observation at every postdose time point, unless superseded by investigator's manual overread.
Statistical Analysis
Sample Size Estimation
The sample size was calculated assuming an intraparticipant standard deviation of 10 milliseconds for ΔQTcF based on the results of previous thorough QT/QTc studies using a crossover design. Using a standard deviation of 10 milliseconds for 52 participants, the probability that the upper limit of the 2‐sided 90% confidence interval (CI) for the difference in mean ΔQTcF between each dose of tramadol and placebo (∆∆QTcF) at a single time point would fall below 10 milliseconds was estimated to be 80%, when the true difference in means equaled 5 milliseconds.
The assay sensitivity was assessed by evaluating the difference in mean ΔQTcF between moxifloxacin and placebo (ΔΔQTcF) at predefined time points between 2 and 4 hours postdose (2, 2.5, 3, and 4 hours). With an intrasubject standard deviation of 10 milliseconds and a sample size of 52 participants, the probability that the lower limit of the 2‐sided 97.5%CI (adjusted for multiplicity) for ΔΔQTcF at a predefined single time point would be greater than 5 milliseconds was estimated to be 80%, when the true difference in means was ≥11.2 milliseconds.
Assuming an early withdrawal rate of approximately 20% over the 4 treatment periods, 68 participants were randomized. Additional participants were to be enrolled and randomized to ensure that 52 participants completed the study, if required.
Study Analysis Sets
All participants who had baseline and postdose PD (Holter ECG) data available for at least 1 treatment were included in the PD analysis set. All participants who had PK data available for a tramadol treatment and had not missed doses 6 to 13 required to achieve the steady‐state concentration during the treatment were included in the PK analysis set. All randomized participants who received at least 1 dose of study drug and had post‐baseline safety data available were included in the safety analysis set.
Pharmacodynamic Analysis
The ECG parameters over time were listed for each participant and treatment. Change from baseline values and placebo‐subtracted change from baseline values were summarized by the treatment and time point. Summary statistics were generated for all participants.
The change from baseline in QTc intervals at the individual participant's plasma tmax,ss (ΔQTc,Tmax) for each analyte and the difference in ΔQTc,Tmax between each dose of tramadol and placebo was also summarized using descriptive statistics.
Pharmacokinetic Analysis
The PK results were summarized, and descriptive statistics were generated for each treatment. If ≥5 participants in the tramadol arm missed 1 or more of scheduled doses 2 to 5, an additional descriptive summary of PK analyte concentrations was to be prepared for participants receiving all scheduled doses vs participants who missed 1 or more doses. If a participant had a sampling time deviation of ≥20% of its nominal time, the concentration for that time point was not included in the descriptive statistics of the PK analyte concentrations or in mean concentration‐time figures, but was included in the PK analysis, which used actual sampling times.
Pharmacokinetic–Pharmacodynamic Analysis
The statistical assessment of the plasma concentration and ΔΔQTcF relationship included a linear mixed‐effects model for calculated ΔΔQTcF values, with a fixed effect for both doses of tramadol plasma concentration, and participant intercept and concentration slope as random effects. The predicted values of ΔΔQTcF (along with 90%CIs) were estimated at the mean Cmax values of each analyte for each dose of tramadol.
Primary Analysis of Central Tendency
QTcF was the primary QT correction method, QTcS was secondary, and QTcB was used only for reference purposes. Mixed‐effect models with sequence, treatment period, time point of measurement, treatment by time point interaction as fixed effect, and participant as a random effect were fitted to ΔQTcF data. Two‐sided 90%CIs for the difference (ΔΔQTcF) between each treatment and placebo were derived at each time point. The statistical analysis aimed to evaluate whether tramadol was noninferior to placebo with respect to its effect on QTc prolongation. The null hypothesis of QTcF prolongation was to be rejected if the upper limit of 2‐sided 90%CI for the difference from baseline in least squares means between each dose of tramadol and placebo was less than 10 milliseconds at all the time points.
Assay sensitivity was assessed using the model described above by evaluating the difference in mean ∆QTcF between moxifloxacin and placebo (∆∆QTcF), and was considered to be established if the lower limits of the 2‐sided 97.5%CIs for the differences in mean ΔQTcF between moxifloxacin and placebo (ΔΔQTcF) exceeded 5 milliseconds at 1 or more time points (ie, at the predefined time points 2, 2.5, 3, and 4 hours).
Mixed‐effect models similar to ΔQTcF were fitted to the ΔQTcS, ΔQTcB, ΔQT, ΔHR, ΔRR, ΔQRS, and ΔPR data. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, North Carolina).
Results
Participant Disposition and Demographics
Of the 68 participants enrolled, 17 participants were allocated to each of the 4 treatment sequences, and 57 (83.8%) participants completed the study (Figure 1B). The majority of the participants were white (40 [58.8]), and men (48 [70.6%]). The median age (range) was 30 years (18–55 years), median weight 76.7 kg (53.3–99.1 kg) and median body mass index 26 kg/m2 (18.5–29.9 kg/m2) (Table S1).
Overall, 63 participants received tramadol 400 mg/day, 64 participants received tramadol 600 mg/day, 59 participants received placebo, and 62 participants received moxifloxacin.
The demographic and baseline characteristics were comparable between the participants in all treatment sequences (Table S1).
Pharmacodynamics
ECG Intervals Versus Time Profiles
Mean QTcF values showed a decrease from baseline for placebo at all time points. The change from baseline value (∆QTcF) was lower for placebo than for both tramadol treatments at all time points. No difference was observed in the mean QTcF value between tramadol 400 mg/day and 600 mg/day doses. Also, the mean change from baseline value was close to no‐difference reference line or lower than baseline across time points. Mean QTcF values were highest for moxifloxacin and change from baseline values showed a positive increase from baseline up to 4 hours postdosing. The results for QTcS and QTcB were similar to QTcF.
Noninferiority of Tramadol to Placebo
Following multiple dosing with tramadol, the highest upper limit of the 2‐sided 90%CI for ∆∆QTcF over the 24‐hour postdose measurement interval was 7.8 milliseconds for the 400 mg/day dose and 8.8 milliseconds for the 600 mg/day dose, which were below the protocol‐specified 10‐millisecond limit, thereby demonstrating noninferiority of tramadol to placebo (Table 1). Similar results were obtained for ΔΔQTcS where both values were below the protocol‐specified 10‐millisecond limit (8.7 milliseconds for the 400 mg/day dose and 9.4 milliseconds for the 600 mg/day dose).
Table 1.
Treatment Comparison of Time‐Matched Change From Baseline QTcF (ΔΔQTcF) for Tramadol Treatments Relative to Placebo on Day 4 (PD Analysis Set)
| Tramadol 400–Placebo | Tramadol 600–Placebo | |||
|---|---|---|---|---|
| Scheduled Time After Most Recent Dose | Mean Difference (ms) | 2‐sided 90%CI (ms) | Mean Difference (ms) | 2‐sided 90%CI (ms) |
| 0.5 h | 3.7 | (1.3, 6.0) | 4.5 | (2.2, 6.9) |
| 1 h | 2.9 | (0.5, 5.2) | 4.1 | (1.8, 6.5) |
| 1.5 h | 4.2 | (1.8, 6.5) | 4.3 | (1.9. 6.6) |
| 2 h | 4.5 | (2.1, 6.8) | 5.3 | (3.0, 7.6) |
| 2.5 h | 3.9 | (1.6, 6.2) | 5.4 | (3.0, 7.7) |
| 3 h | 4.7 | (2.4, 7.0) | 5.3 | (3.0, 7.6) |
| 3.5 h | 4.0 | (1.7, 6.3) | 5.0 | (2.7, 7.3) |
| 4 h | 3.8 | (1.5, 6.1) | 5.0 | (2.6, 7.3) |
| 6 h | 5.3 | (3.0, 7.7) | 5.1 | (2.7, 7.4) |
| 8 h | 5.5 | (3.2, 7.8) | 6.5 | (4.1, 8.8) |
| 12 h | 1.7 | (–0.6, 4.0) | 2.3 | (–0.1, 4.6) |
| 24 h | 2.5 | (0.2, 4.9) | 2.5 | (0.1, 4.8) |
CI, confidence interval; ms, milliseconds; PD, pharmacodynamics.
Mean difference: least squares mean difference; tramadol 400: 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; tramadol 600: 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150‐mg dose on day 4; placebo: placebo every 6 hours on days 1 to 3, and a single placebo dose on day 4.
Assay Sensitivity
Assay sensitivity for the noninferiority test was also established as the smallest lower limit of 2‐sided 97.5%CI (adjusted for multiplicity) for ∆∆QTcF at 4 hours after dosing with moxifloxacin on day 4 was 7.5 milliseconds, thus exceeding the protocol‐specified 5‐millisecond limit; postdosing values at other time points, that is, 2 hours, 2.5 hours, and 3 hours, were 7.6 milliseconds, 8.3 milliseconds, and 8.9 milliseconds, respectively.
QT Correction for Ventricular Heart Rate
The regression line of QTcF vs RR was almost horizontal across treatments (tramadol 400 mg/day: r 2 = 0.001 and tramadol 600 mg/day: r 2 = 0), which supports the choice of Fridericia as primary correction method for QT interval (Figure 2).
Figure 2.
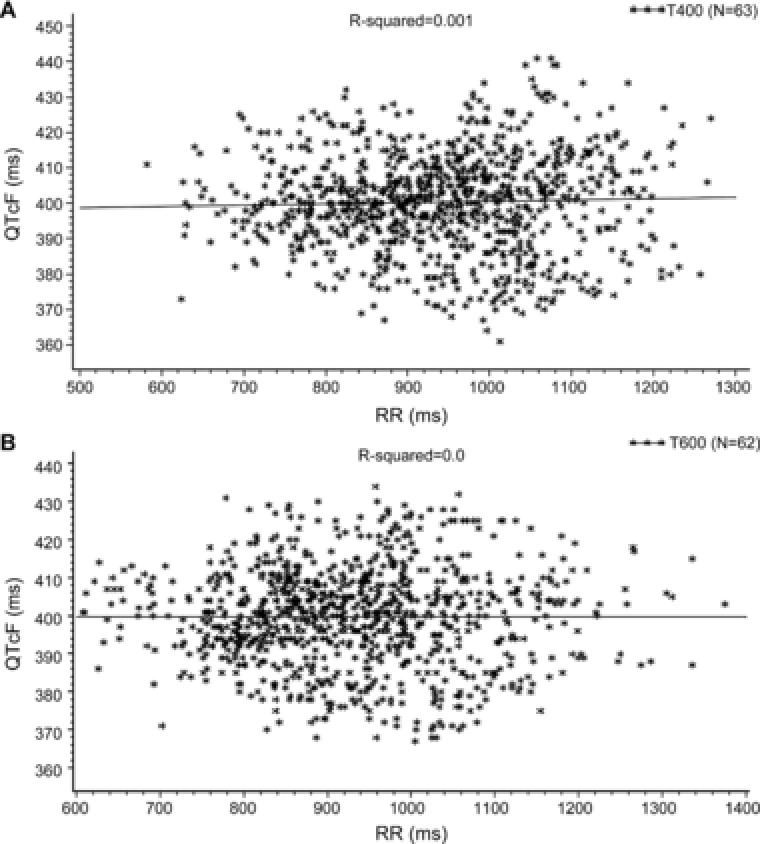
Corrected QT interval (QTcF) against RR for tramadol doses (A: tramadol 400 mg and B: tramadol 600 mg) on linear scale (PD analysis set). Solid line represents the population regression line. PD, pharmacodynamic. T400/Tramadol 400: 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; T600/Tramadol 600: 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150‐mg dose on day 4.
Outlier Analysis
No clinically noteworthy absolute QTcF or QTcS values (ie, values >450 milliseconds) were recorded during the study. However, 1 participant (tramadol 600 mg/day) experienced clinically noteworthy ΔQTcF and ΔQTcS values (>30 milliseconds and ≤60 milliseconds), while another participant (moxifloxacin) experienced a clinically noteworthy ΔQTcS value (>30 milliseconds and ≤60 milliseconds).
One participant in the tramadol 400 mg/day group, had a single clinically noteworthy HR value of 103 bpm at 12 hours postdose on day 4.
No changes of clinical interest were observed for other ECG intervals, QRS and PR.
Pharmacokinetics
The plasma concentrations of (+) and (−) enantiomers of tramadol and its metabolite M1 peaked at approximately 2.0 to 2.5 hours (tmax) following dose 13 at 72 hours, and thereafter the mean concentrations of all analytes showed a monophasic decline (Figure 3).
Figure 3.
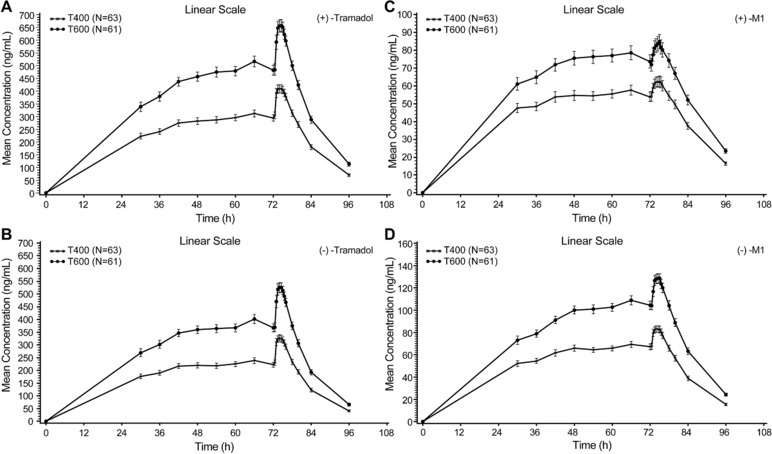
Mean plasma concentration–time profile of tramadol and its metabolite M1 (PK analysis set). Bars represent the standard error. M1, O‐desmethyl tramadol; PK, pharmacokinetic. T400/Tramadol 400: 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; T600/Tramadol 600: 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150‐mg dose on day 4.
A 50% increase in tramadol dose (from 400 mg/day to 600 mg/day) resulted in an approximately dose proportional increase in mean exposure to tramadol and its metabolite M1. The mean metabolite‐to‐parent ratios for AUCtau,ss and Cmax,ss ranged from 13.3% to 17.4% for the (+)‐M1 enantiomer, and 24.7% to 28.1% for the (−)‐M1 enantiomer (Table 2).
Table 2.
Pharmacokinetic Parameters for the (+) and (–) Enantiomers of Tramadol and M1 Metabolite on Day 4 for Tramadol Treatments (PK Analysis Set)
| Parametera | (+)−Tramadol | (–)−Tramadol | (+)−M1 | (–)−M1 |
|---|---|---|---|---|
| Tramadol 400 (N = 63) | ||||
| AUCtau,ss, ng h/mL | 2228 (663.3) | 1733 (511.5) | 355.7 (131.4) | 460.6 (133.7) |
| Cmax,ss, ng/mL | 468.5 (129.6) | 379.7 (104.5) | 66.9 (25.3) | 91.6 (24.4) |
| tmax, ss, hb | 2 (1.0, 6.0) | 2 (1.0, 6.0) | 2 (0.0, 6.0) | 2 (0.0, 4.0) |
| Ctrough, ss, ng/mL | 295 (96.3) | 222.4 (75.3) | 53.8 (20.7) | 67.2 (20.5) |
| Cmin, ss, ng/mL | 279.5 (94.6) | 207.4 (70.7) | 50.8 (19.0) | 62.1 (20.1) |
| MR (AUCtau,ss), % | NA | NA | 17.4 (7.7) | 28.1 (8.7) |
| MR (Cmax,ss), % | NA | NA | 15.5 (6.8) | 25.4 (7.6) |
| Tramadol 600 (N = 61) | ||||
|---|---|---|---|---|
| AUCtau, ss, ng h/mL | 3522 (959.7) | 2759 (771.9) | 474.7 (180.1) | 708.8 (184.8) |
| Cmax, ss, ng/mL | 726 (183.6) | 588.9 (151.1) | 88.8 (33.2) | 139.2 (34.4) |
| tmax, ss, hb | 2 (1.0, 6.0) | 2 (1.0, 6.0) | 2.5 (0.0, 6.0) | 2 (0.0, 6.0) |
| Ctrough, ss, ng/mL | 481.6 (151.9) | 366.9 (124.6) | 73.6 (29.3) | 104.4 (30.0) |
| Cmin, ss, ng/mL | 450.6 (140.1) | 337.2 (108.5) | 68.8 (26.5) | 97 (27.5) |
| MR (AUCtau,ss), % | NA | NA | 14.7 (6.8) | 26.9 (7.7) |
| MR (Cmax,ss), % | NA | NA | 13.3(6.2) | 24.7 (6.8) |
AUCtau, ss, area under the plasma concentration‐time curve during a dosing interval (tau) at steady state; Cmax, ss, maximum observed plasma concentration during a dosing interval at steady state; Cmin,ss, minimum observed plasma concentration during a dosing interval at steady state; Ctrough,ss, trough plasma concentration before dosing (predose) at steady state; M1, O‐desmethyl tramadol; MR, metabolite/parent ratio; N, number of observations included in descriptive statistics for each pharmacokinetic parameter; NA, not applicable; PK, pharmacokinetic; SD, standard deviation; tmax, ss, time to reach the maximum plasma concentration during a dosing interval at steady state.
Tramadol 400: 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; tramadol 600: 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150‐mg dose on day 4.
All values are presented as mean (SD).
tmax,ss is presented as median (range).
Missed tramadol doses 2 to 5 did not impact the steady state of enantiomers of both tramadol and M1 and the mean concentrations of the PK analytes. No PK samples had a time deviation of ≥20% at a nominal time point. Following dosing on day 4, after peaking at approximately 2.0 to 2.5 hours, mean concentrations of all analytes showed a monophasic decline.
Since the QT results for the moxifloxacin treatment arm of the study were as expected, the retained moxifloxacin samples did not require analysis.
Pharmacokinetic–Pharmacodynamic Analysis
QTc Interval Changes at tmax,ss
The mean ΔΔQTc values at tmax,ss were not noticeably higher than those observed at any other time point for any of the 4 PK analytes. While the mean increase in ΔΔQTcF at tmax,ss ranged from 3.3 to 4.9 milliseconds, it ranged from 2.8 to 4.4 milliseconds for ΔΔQTcS across tramadol treatments and PK analytes.
Concentration Versus QTc Relationship
No relevant relationship was observed between ΔΔQTcF/ΔΔQTcS and concentration of any PK analyte (Figure 4; Tables S2 and S3). Linear mixed‐effect models were fitted to the data using ΔΔQTcF as the dependent variable and the concentration of tramadol (both enantiomers and metabolites separately) as the main covariate. In these models, the slopes of the exposure‐response relationship were slightly positive but statistically insignificant for both tramadol enantiomers and their metabolites (Table S2).
Figure 4.
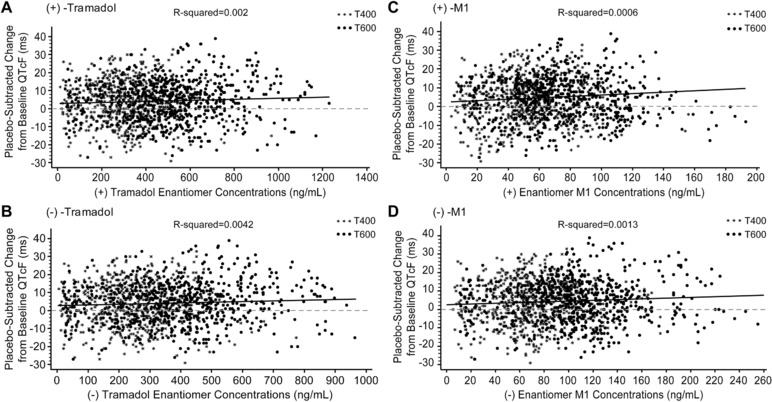
Individual ΔΔQTcF values vs tramadol and M1 enantiomer concentrations (PD analysis set). Solid line represents the regression line from statistical model; dashed line represents the no‐effect reference line. M1, O‐desmethyl tramadol; PD, pharmacodynamic. T400/Tramadol 400: 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; T600/Tramadol 600: 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150‐mg dose on day 4.
Pharmacogenomics
Participants were classified according to their CYP2D6 status. Based on composite genotype, a majority of participants (48 of 68; 70.6%) were predicted to be extensive metabolizers (EMs, the predominant phenotype); 4 (5.9%) participants were predicted as ultrarapid metabolizers (UMs); for 2 (2.9%) participants the metabolizer status could not be unambiguously determined, and they were defined as either EMs or UMs. Nine (13.2%) participants were predicted as intermediate metabolizers (IMs) and 3 (4.4%) participants as poor metabolizers (PMs). One participant's genotype (1.5%) could not be determined, and another participant withdrew from the study before genotyping.
The PK exposures were found to be related to participant composite genotype, ordered by the functional status of the alleles, and the predicted phenotype status. The exposures (median Cmax,ss) for (+)‐tramadol were lowest for EM/UM and EM/EM participants and increased with increasingly impaired CYP2D6 function (Table S4). The pattern was opposite for metabolite (+)‐M1, with exposures being highest for EM/UM participants and lowest for PM/PM participants.
One participant with a clinically noteworthy HR value and 1 with a clinically noteworthy ΔQTcF value were both EMs with phenotype EM/IM. The participants at both ends of the CYP2D6 activity range, that is, EM/UM and PM/PM, did not have noteworthy changes in QTcF with tramadol treatment.
The CYP2D6 activity had an effect on (+)‐M1 exposure in both tramadol 400 mg/day and 600 mg/day groups, with PM/PM participants having approximately 90% lower metaboliteto‐parent ratios (2.4% and 2.3%, respectively) compared to EM/UM participants (27.8% and 25.8%, respectively) (Table S4 ).
Safety
One participant (1 of 68; 1.5%) had a pretreatment AE, and 61 of 68 (89.7%) participants had at least 1 TEAE (Table 3). A majority of the TEAEs reported were mild in severity. Three participants (3 of 68; 4.4%) who withdrew prematurely due to TEAEs were all in tramadol 600 mg/day group (3 of 64; 4.7%). No serious AEs or deaths were reported.
Table 3.
Incidence of TEAEs Reported in ≥10% of Participants (Safety Analysis Set)
| Number of Participants (%) | |||||
|---|---|---|---|---|---|
| TEAEs | Tramadol 400 (N = 63) | Tramadol 600 (N = 64) | Placebo (N = 59) | Moxifloxacin (N = 63) | Total (N = 68) |
| Total number of participants with TEAEs,a n (%) | 51 (81.0) | 55 (85.9) | 26 (44.1) | 26 (41.3) | 61 (89.7) |
| Cardiac disorders | 5 (7.9) | 4 (6.3) | 6 (10.2) | 5 (7.9) | 16 (23.5) |
| Supraventricular tachycardia | 4 (6.3) | 2 (3.1) | 4 (6.8) | 3 (4.8) | 10 (14.7) |
| Ear and labyrinth disorders | 2 (3.2) | 6 (9.4) | 0 (0.0) | 0 (0.0) | 7 (10.3) |
| Vertigo | 2 (3.2) | 6 (9.4) | 0 (0.0) | 0 (0.0) | 7 (10.3) |
| Eye disorders | 2 (3.2) | 3 (4.7) | 1 (1.7) | 1 (1.6) | 7 (10.3) |
| Gastrointestinal disorders | 38 (60.3) | 47 (73.4) | 8 (13.6) | 8 (12.7) | 55 (80.9) |
| Nausea | 31 (49.2) | 43 (67.2) | 4 (6.8) | 6 (9.5) | 48 (70.6) |
| Vomiting | 15 (23.8) | 21 (32.8) | 1 (1.7) | 1 (1.6) | 27 (39.7) |
| Constipation | 3 (4.8) | 8 (12.5) | 1 (1.7) | 0 (0.0) | 9 (13.2) |
| General disorders and administration site conditions | 19 (30.2) | 22 (34.4) | 8 (13.6) | 6 (9.5) | 33 (48.5) |
| Feeling hot | 10 (15.9) | 16 (25.0) | 4 (6.8) | 0 (0.0) | 22 (32.4) |
| Fatigue | 6 (9.5) | 5 (7.8) | 1 (1.7) | 0 (0.0) | 10 (14.7) |
| Injury, poisoning and procedural complications | 1 (1.6) | 2 (3.1) | 3 (5.1) | 2 (3.2) | 8 (11.8) |
| Metabolism and nutrition disorders | 3 (4.8) | 6 (9.4) | 0 (0.0) | 1 (1.6) | 7 (10.3) |
| Nervous system disorders | 30 (47.6) | 36 (56.3) | 9 (15.3) | 13 (20.6) | 48 (70.6) |
| Dizziness | 15 (23.8) | 22 (34.4) | 2 (3.4) | 4 (6.3) | 30 (44.1) |
| Headache | 11 (17.5) | 13 (20.3) | 6 (10.2) | 9 (14.3) | 25 (36.8) |
| Somnolence | 10 (15.9) | 16 (25.0) | 2 (3.4) | 1 (1.6) | 21 (30.9) |
| Tremor | 5 (7.9) | 2 (3.1) | 0 (0.0) | 1 (1.6) | 7 (10.3) |
| Psychiatric disorders | 6 (9.5) | 12 (18.8) | 2 (3.4) | 0 (0.0) | 16 (23.5) |
| Euphoric mood | 4 (6.3) | 6 (9.4) | 1 (1.7) | 0 (0.0) | 8 (11.8) |
| Renal and urinary disorders | 5 (7.9) | 9 (14.1) | 1 (1.7) | 0 (0.0) | 12 (17.6) |
| Urinary hesitation | 3 (4.8) | 7 (10.9) | 0 (0.0) | 0 (0.0) | 9 (13.2) |
| Respiratory, thoracic, and mediastinal disorders | 5 (7.9) | 6 (9.4) | 3 (5.1) | 1 (1.6) | 12 (17.6) |
| Hiccups | 3 (4.8) | 5 (7.8) | 0 (0.0) | 0 (0.0) | 7 (10.3) |
| Skin and subcutaneous tissue disorders | 23 (36.5) | 36 (56.3) | 1 (1.7) | 0 (0.0) | 40 (58.8) |
| Pruritus | 22 (34.9) | 33 (51.6) | 0 (0.0) | 0 (0.0) | 38 (55.9) |
N, total number of participants; n, number of participants experiencing specific TEAE; TEAE, treatment‐emergent adverse event.
Tramadol 400: 100 mg tramadol every 6 hours (400 mg/day) on days 1 to 3, and a single 100‐mg dose on day 4; Tramadol 600: 150 mg tramadol every 6 hours (600 mg/day) on days 1 to 3, and a single 150 mg dose on day 4; Placebo: placebo every 6 hours on days 1 to 3, and a single placebo dose on day 4; Moxifloxacin: placebo every 6 hours on days 1 to 3, and a single moxifloxacin 400‐mg dose on day 4.
The numbers of participants in each column cannot be added because a participant may have had more than 1 AE. A participant experiencing multiple occurrences of an AE was counted, at most, once per system organ class and preferred term for each treatment and once for “overall.” AEs occurring during the washout period between treatments were attributed to the last treatment received. AEs were encoded using the Medical Dictionary for Regulatory Activities version 17.1.
A greater proportion of participants experienced TEAEs in tramadol 400 mg/day (51 of 63; 81.0%) and 600 mg/day (55 of 64; 85.9%) groups vs placebo (26 of 59; 44.1%) or moxifloxacin (26 of 63; 41.3%) groups. Nausea was the most frequently reported TEAE in 31 of 63 (49.2%) participants in the tramadol 400 mg/day group, 43 of 64 (67.2%) participants in tramadol 600 mg/day group, 4 of 59 (6.8%) participants in placebo group, and 6 of 63 (9.5%) participants in moxifloxacin group. In both the tramadol 400 mg/day and 600 mg/day groups, most of the nausea events were reported within 6 hours of first dose administration, and most of the vomiting events were reported 6 to 12 hours and 12 to 18 hours after the first dose.
Further, 57 of 68 (83.8%) participants experienced at least 1 treatment‐related AE. The number of such participants was more than double in tramadol 400 mg/day (48 of 63; 76.2%) and tramadol 600 mg/day (54 of 64; 84.4%) groups compared to placebo (15 of 59; 25.4%) or moxifloxacin (11 of 63; 17.5%) groups.
In general, clinical laboratory tests, vital signs, physical examination, and ECGs were similar between the tramadol and placebo groups and across both doses of tramadol. A decrease in oxygen saturation, considered to be related to the study drug, was reported in 3 of 63 (4.7%) participants on tramadol 600 mg/day. However, we did not measure the concentration of tramadol and its metabolites at the time of decrease in oxygen saturation in these participants and hence any association between tramadol and/or its metabolite exposure and oxygen desaturation could not be determined.
No new safety concerns were identified.
Discussion
The results of this thorough QT/QTc study show that tramadol at doses of 400 mg/day (therapeutic dose) and 600 mg/day (supratherapeutic dose) did not have an effect on QTc prolongation of regulatory concern (as established by the International Council for Harmonization E14 guidance).23 The selection of a supratherapeutic dose of tramadol as a 1.5‐fold higher dose than its therapeutic dose was based on an acceptable safety data profile observed with this dose (600 mg) in a previous multiple ascending dose study.24 Also, a dose higher than 600 mg/day could not be used because of safety concerns associated with high doses of tramadol, such as seizures and respiratory depression.25 The assay sensitivity was established using moxifloxacin 400 mg as the positive control. Similarly, the PK‐PD analyses also revealed no relationship between the plasma concentration of tramadol and changes in the QT interval. The slope of the exposure‐response relationship was slightly positive but statistically insignificant for both enantiomers of tramadol and their metabolites.
Although tramadol is an extensively used centrally acting analgesic and is considered to be safe26 compared to other opioids like methadone,27, 28, 29, 30 tramadol overdose may cause some toxicity or side effects.10 The observations from the current study indicate that tramadol does not demonstrate a proarrhythmic effect or QT prolongation at either a therapeutic or supratherapeutic dose and are consistent with the published literature for therapeutic doses.8
Due to safety concerns associated with a higher supratherapeutic dose of tramadol, it was not possible to characterize the QT effects at high concentrations of tramadol (>600 mg/day) and M1, which can be a limitation of this study. Moreover, tramadol was found to be a weak hERG inhibitor with a half maximal inhibitory concentration of 129 μM (39 μg/mL), and the results of preclinical hERG inhibition suggested that tramadol might start producing QT prolongation only at exposures 5‐ to 6‐fold of the therapeutic concentrations (data on file). However, we did not conduct a hERG study on the M1 metabolite of tramadol.
Tramadol is metabolized primarily by CYP2D6, and differential activity of CYP2D6 can result in variable metabolism of tramadol.10, 17 The levels of tramadol could be about 20% higher, while those of its metabolite M1 could be 40% lower in PM compared to EM.31 The PK exposure thus seems to be dependent upon the participants’ composite genotype for CYP2D6, ordered by the functional status (activity score) of the alleles, and the predicted phenotype status. In the present study as well, the PMs had an approximately 90% lower metabolite‐to‐parent ratio compared with UMs. The UMs could be at a higher risk of tramadol toxicity owing to higher plasma levels of M1, resulting in increased analgesic effects, and nausea compared with EMs.32 Besides analyzing the effects of genotype on tramadol exposure, the effect of overencapsulation of the study drug on tramadol exposure parameters could have also been determined. But this was not planned in our study and hence could be a limitation of this study.
Overall, both therapeutic and supratherapeutic doses of tramadol demonstrated a safety and tolerability profile consistent with the known safety profile of tramadol.11, 33 Most of the TEAEs were mild in severity as described previously,34 and no new TEAEs were observed. However, the incidences of TEAEs, treatment‐related AEs, and study discontinuations were more frequent in tramadol 600 mg/day vs tramadol 400 mg/day group. This was expected, as the supratherapeutic dose of tramadol results in higher peak plasma levels.35
In conclusion, tramadol did not cause clinically relevant prolongation of the QTc interval in this thorough QT/QTc study at either a therapeutic or supratherapeutic dose up to 600 mg/day. No new safety signals were observed based on the assessments of ECG morphology or intervals, TEAEs, or changes in laboratory parameters.
Author Contributions
J.M. was the clinical pharmacokinetic leader contributing to the data analysis and interpretation. J.N. and J.A. were the statistical lead and had primary roles in the statistical analyses and data interpretation. S. Francke was pharmacogenomics lead for this study and contributed to the design, analysis, and interpretation of genetic results in this study. B.D. and S.V. were the study responsible physicians involved in all aspects of the study and contributed to data interpretation, development, and review of the manuscript. T.M. and S. Fonseca provided strategic direction and were also involved in scientific review of the manuscript.
All authors contributed to the data interpretation, development, and review of this manuscript and confirm that they have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
All authors met International Committee of Medical Journal Editors criteria. They had access to the study data, provided direction and comments on the manuscript, made the final decision about where to publish these data, and approved submission to this journal.
Supporting information
Table S1. Demographic and Baseline Characteristics (Safety Analysis Set)
Table S2. Model Estimates of Relationship Between QTc and Plasma PK Analyte Concentrations on Day 4 (PK‐PD Analysis Sets)
Table S3. Model Predictions of Relationship Between QTc and Plasma PK Analyte Concentrations at Cmax,ss (Day 4) (PK‐PD Analysis Sets)
Table S4. Descriptive Statistics for Plasma (+) Enantiomers of Tramadol and M1 Metabolite by CYP2D6 Status (PK Analysis Set)
Acknowledgments
The authors thank Dr. Ramandeep Singh (Tata Consultancy Services) for writing assistance and Dr. Ellen Baum (Janssen Research and Development, LLC) for additional editorial assistance in the preparation of this manuscript. We would like to thank the study participants, without whom the study would have never been accomplished.
Declaration of Conflicting Interests
J.M., J.N., and J.A. are employees of JRD, LLC, Raritan, New Jersey, USA. S. Francke was an employee of JRD, LLC, Spring House, Pennsylvania, USA, at the time of study. T.M. was an employee of Quintiles Phase I Services, Overland Park, Kansas, USA, at the time of study and has now retired. B.D. was an employee of Janssen Scientific Affairs, LLC, Titusville, New Jersey, USA, at the time of study. S.V. and S. Fonseca are employees of JRD, LLC, Titusville, New Jersey, USA.
Funding
The study was sponsored by Janssen Scientific Affairs, LLC, Valeant Pharmaceuticals International Inc., and Cipher Pharmaceuticals Inc.
References
- 1. Martell BA, Arnsten JH, Krantz MJ, Gourevitch MN. Impact of methadone treatment on cardiac repolarization and conduction in opioid users. Am J Cardiol. 2005;95(7):915–918. [DOI] [PubMed] [Google Scholar]
- 2. Sobanski P, Krajnik M, Shaqura M, Bloch‐Boguslawska E, Schäfer M, Mousa SA. The presence of mu‐, delta‐, and kappa‐opioid receptors in human heart tissue. Heart Vessels. 2014;29(6):855–863. [DOI] [PubMed] [Google Scholar]
- 3. Chen A, Ashburn MA. Cardiac effects of opioid therapy. Pain Med. 2015;16(Suppl 1):S27–S31. [DOI] [PubMed] [Google Scholar]
- 4. Yap YG, Camm AJ. Drug induced Qt prolongation and torsades de pointes. Heart. 2003;89(11):1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trinkley KE, Page RL 2, Lien H, Yamanouye K, Tisdale JE. QT interval prolongation and the risk of torsades de pointes: essentials for clinicians. Curr Med Res Opin. 2013;29(12):1719–1726. [DOI] [PubMed] [Google Scholar]
- 6. Hancox JC, McPate MJ, El Harchi A, Zhang HY. The hERG potassium channel and hERG screening for drug‐induced torsades de pointes. Pharmacol Ther. 2008;119(2):118–132. [DOI] [PubMed] [Google Scholar]
- 7. Katchman AN, Mcgroary KA, Kilborn MJ, et al. Influence of opioid agonists on cardiac human ether‐a‐go‐go related gene K(+) currents. J Pharmacol Exp Ther. 2002;303(2):688–694. [DOI] [PubMed] [Google Scholar]
- 8. Fanoe S, Jensen GB, Sjøgren P, Korsgaard MP, Grunnet M. Oxycodone is associated with dose‐dependent QTc prolongation in patients and low‐affinity inhibiting of hERG activity in vitro. Br J Clin Pharmacol. 2009;67(2):172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pergolizzi JV Jr, Taylor R Jr, Raffa RB. Extended‐release formulations of tramadol in the treatment of chronic pain. Expert Opin Pharmacother. 2011;12(11):1757–1768. [DOI] [PubMed] [Google Scholar]
- 10. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395–400. [PubMed] [Google Scholar]
- 11. Vazzana M, Andreani T, Fangueiro J, et al. Tramadol hydrochloride: pharmacokinetics, pharmacodynamics, adverse side effects, co‐administration of drugs and new drug delivery systems. Biomed Pharmacother. 2015;70:234–238. [DOI] [PubMed] [Google Scholar]
- 12. Barkin RL. Extended‐release tramadol (ULTRAM ER): a pharmacotherapeutic, pharmacokinetic, and pharmacodynamic focus on effectiveness and safety in patients with chronic/persistent pain. Am J Ther. 2008;15(2):157–166. [DOI] [PubMed] [Google Scholar]
- 13. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004; 43(13):879–923. [DOI] [PubMed] [Google Scholar]
- 14. Wu WN, Mckown LA, Liao S. Metabolism of the analgesic drug ULTRAM (R) (tramadol hydrochloride) in humans: API‐MS and MS/MS characterization of metabolites. Xenobiotica. 2002;32(5):411–425. [DOI] [PubMed] [Google Scholar]
- 15. Ardakani YH, Rouini MR. Pharmacokinetics of tramadol and its three main metabolites in healthy male and female volunteers. Biopharm Drug Dispos. 2007;28(9):527–534. [DOI] [PubMed] [Google Scholar]
- 16. Gillen G, Haurand M, Kobelt DJ, Wnendt S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human μ‐opioid receptor. Naunyn Schmiedeberg Arch Pharmacol. 2000;362(2):116–121. [DOI] [PubMed] [Google Scholar]
- 17. Gan SH, Ismail R, Wan Adnan WA, Zulmi W. Impact of CYP2D6 genetic polymorphism on tramadol pharmacokinetics and pharmacodynamics. Mol Diag Ther. 2007;11(3):171–181. [DOI] [PubMed] [Google Scholar]
- 18. Stamer UM, Stuber F. Codeine and tramadol analgesic efficacy and respiratory effects are influenced by CYP2D6 genotype. Anaesthesia. 2007;62(12):1294–1295. [DOI] [PubMed] [Google Scholar]
- 19. Enggaard TP, Poulsen L, Arendt‐Nielsen L, Brøsen K, Ossig J, Sindrup SH. The analgesic effect of tramadol after intravenous injection in healthy volunteers in relation to CYP2D6. Anesth Analg. 2006;102(1):146–150. [DOI] [PubMed] [Google Scholar]
- 20. Leppert W. Tramadol as an analgesic for mild to moderate cancer pain. Pharmacol Rep. 2009;61(6):978–992. [DOI] [PubMed] [Google Scholar]
- 21. Slanar O, Dupal P, Matouskova O, et al. Tramadol efficacy in patients with postoperative pain in relation to CYP2D6 and MDR1 polymorphisms. Bratisl Lek Listy. 2012;113(3):152–155. [DOI] [PubMed] [Google Scholar]
- 22. Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3)391–396. [PubMed] [Google Scholar]
- 23. Guidance for industry. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. US Department of Health and Human Services, Food and Drug Administration. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073153.pdf. Published October 2005. Accessed August 16, 2017.
- 24. DeLemos B, Richards HM, Vandenbossche J, et al. Safety, tolerability and pharmacokinetics of therapeutic and supratherapeutic doses of tramadol hydrochloride in healthy adults: a randomized, double‐blind, placebo‐controlled, multiple ascending dose study. Clin Pharmacol Drug Dev. 2017;6(6):592–603. [DOI] [PubMed] [Google Scholar]
- 25. Tramadol—update review report. World Health Organization—Expert Committee on Drug Dependence 36th meeting. http://www.who.int/medicines/areas/quality_safety/6_1_Update.pdf. Accessed January 24, 2018.
- 26. Mehrpour O, Sharifi M, Zamani N. Tramadol poisoning. In: pharmacology, toxicology and pharmaceutical science. http://www.intechopen.com/books/toxicology-studies-cells-drugs-and-environment/tramadol-poisoning. Accessed June 28, 2017.
- 27. Duthie DJR. Remifentanil and tramadol. Br J Anaesth. 1998;81:51–57. [DOI] [PubMed] [Google Scholar]
- 28. Katz WA. Pharmacology and clinical experience with tramadol in osteoarthritis. Drugs. 1996;52(suppl 3):39–47. [DOI] [PubMed] [Google Scholar]
- 29. Cruciani RA, Sekine R, Homel P, et al. Measurement of QTc in patients receiving chronic methadone therapy. J Pain Symptom Manage. 2005;29(4):385–391. [DOI] [PubMed] [Google Scholar]
- 30. Krantz MJ, Martin J, Stimmel B, Mehta D, Haigney MC. QTc interval screening in methadone treatment. Ann Intern Med. 2009;150(6):387–395. [DOI] [PubMed] [Google Scholar]
- 31. Dean L. Tramadol Therapy and CYP2D6 Genotype . Medical genetics summaries. In: Pratt V, McLeod H, Dean L, Malheiro A, Rubinstein W, editors. National Centre for Biotechnology Information (US); 2012. https://www.ncbi.nlm.nih.gov/books/NBK315950/. Accessed June 28, 2017. [PubMed]
- 32. Johansson I, Ingelman‐Sundberg M. Genetic polymorphism and toxicology—with emphasis on cytochrome p450. Toxicol Sci. 2011;120(1):1–13. [DOI] [PubMed] [Google Scholar]
- 33. Gibson TP. Pharmacokinetics, efficacy, and safety of analgesia with a focus on tramadol HCl. Am J Med. 1996;101(1A):47S–53S. [DOI] [PubMed] [Google Scholar]
- 34. Langley PC, Patkar AD, Boswell KA, Benson CJ, Schein JR. Adverse event profile of tramadol in recent clinical studies of chronic osteoarthritis pain. Curr Med Res Opin. 2010;26(1):239–251. [DOI] [PubMed] [Google Scholar]
- 35. Kizilbash A, Ngô‐Minh CT. Review of extended‐release formulations of tramadol for the management of chronic non‐cancer pain: focus on marketed formulations. J Pain Res. 2014;7:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographic and Baseline Characteristics (Safety Analysis Set)
Table S2. Model Estimates of Relationship Between QTc and Plasma PK Analyte Concentrations on Day 4 (PK‐PD Analysis Sets)
Table S3. Model Predictions of Relationship Between QTc and Plasma PK Analyte Concentrations at Cmax,ss (Day 4) (PK‐PD Analysis Sets)
Table S4. Descriptive Statistics for Plasma (+) Enantiomers of Tramadol and M1 Metabolite by CYP2D6 Status (PK Analysis Set)