

Abstract
Clobazam (CLB) is a 1,5‐benzodiazepine that has been widely used as an anxiolytic and antiseizure drug (ASD) since it was first synthesized over 50 years ago. CLB was approved in the United States in 2011 as adjunctive therapy for seizures in patients ≥2 years old with Lennox‐Gastaut syndrome. CLB pharmacokinetics (PK) have been studied in single‐ and multiple‐dose administrations in healthy subjects. Salient features include high bioavailability, linear PK, and negligible effects from coadministration of other ASDs. CLB is highly and extensively absorbed, with little effect from food; time to maximum plasma concentration is 0.5 to 4 hours following the dose. After CLB doses of 20 to 40 mg/day, the volume of distribution is 99 to 120 L, with oral clearance ranging from 1.9 to 2.3 L/h. CLB is lipophilic and distributes throughout the body after oral administration. It is metabolized in the liver by cytochrome P450 (CYP) isoenzymes CYP3A, CYP2C19, and CYP2B6, and its main active metabolite is N‐desmethylclobazam. The half‐life of CLB after a single oral dose ranges from 36 to 42 hours; the half‐life of N‐desmethylclobazam ranges from 59 to 74 hours. The metabolites of CLB are primarily excreted renally. Population PK modeling using data from healthy subjects and patients with Lennox‐Gastaut syndrome indicates that race, sex, age, weight, and renal function do not influence CLB PK. As CLB has been extensively studied since the 1970s, this review is meant to provide a consolidated and comprehensive resource on CLB PK for both prescribers and scientists alike.
Keywords: clobazam, drug‐drug interactions, Lennox‐Gastaut syndrome, N‐desmethylclobazam, pharmacodynamics, pharmacokinetics
Lennox‐Gastaut syndrome (LGS) is a severe and chronic epileptic encephalopathy with a peak age of onset between 3 and 5 years.1 In addition to multiple intractable seizure types that can increase the risk for injury—including tonic, atypical absence, and atonic (ie, “drop attacks”)—patients with LGS suffer from cognitive impairment, loss of skills, and behavioral difficulties.1, 2 Most patients with LGS require lifelong treatment with multiple antiseizure drugs (ASDs) to manage their seizures.1, 3, 4, 5
Clobazam (CLB) was first introduced globally in the 1970s as an anxiolytic and an ASD6, 7; it was designed as an alternative to the 1,4‐benzodiazepines, a drug class that includes lorazepam, clonazepam, and diazepam, which have strong sedative effects via binding to the α1 subunit of the γ‐aminobutyric acid type A (GABAA) receptor.8, 9 As a 1,5‐benzodiazepine, CLB is highly selective for the α2 subunit of the GABAA receptor associated with antiepileptic activity and is less selective for the α1 subunit.8 CLB was approved in 2011 by the US Food and Drug Administration as adjunctive treatment for seizures associated with LGS in patients 2 years of age or older.10 Numerous studies over the past 30+ years have reported the pharmacokinetic (PK) properties of CLB (including absorption, distribution, metabolism, and excretion of single‐ and multiple‐dose formulations), its interactions with other drugs, and its pharmacodynamic effects. In addition, population pharmacokinetic (PopPK) analyses have been used to model CLB PK in a range of healthy volunteer and patient populations or in different clinical contexts. Because PK data help guide appropriate dosing of therapeutics based on patient characteristics such as age, primary disease, and/or comorbidities, as well as inform about potential drug‐drug interactions (DDIs), we have reviewed various sources of CLB PK data with the goal of consolidating such information into a comprehensive reference for prescribers and scientists. This review highlights early studies from the literature and summarizes the results from recent key PK and PopPK modeling studies of CLB.
Clobazam Formulations and Clinical Dosing
In the United States, CLB is marketed as ONFI by Lundbeck LLC (Deerfield, IL) and is formulated as either scored 10‐mg or 20‐mg tablets, or as an oral solution containing 2.5 mg/mL of CLB.10 It is marketed as Frisium in European, Canadian, and Australian markets by Sanofi‐Aventis (Australia), formulated as 10‐mg tablets. Both Frisium and ONFI CLB tablets have an identical granulation composition.11 Finally, CLB is available in European countries as a 1‐mg/mL or 2‐mg/mL oral suspension, under the name Epaclob/Silocalm.12
In the pivotal phase 3 trial assessing the efficacy of CLB for the treatment of seizures associated with LGS,13 CLB was dosed by weight: patients weighing ≤30 kg initiated treatment with once‐daily CLB at 5 mg, while patients weighing >30 kg started at either 5 mg/day or 10 mg/day. Doses were escalated on a weekly basis to a target of 0.25 to 1 mg/kg/day during the maintenance period. In the open‐label extension of the pivotal trial,14 CLB doses were adjusted to achieve seizure control, resulting in a modal dose of 0.86 mg/kg/day, which is approximately 20 mg/day for a 25‐kg patient and approximately 40 mg/day for a 50‐kg patient. Consistent with this finding, a PopPK analysis of the phase 3 data found significant anticonvulsant effect with a daily 20‐mg dose of CLB, with an optimal effect at 40 mg/day.15
Given these data and for simplicity in dosing instructions, the current recommended dosing of CLB for its indicated use in the United States is based on the total daily dose (TDD; mg/day)10: children >2 years old and ≤30 kg should initiate CLB at 5 mg/day and uptitrate weekly to a target dose of 10 to 20 mg/day, divided (doses >5 mg/day should be given as divided doses, twice daily); patients >30 kg should initiate CLB at 10 mg/day and uptitrate weekly to a target of 20 to 40 mg/day. The final dose should be based on response. Titrating CLB more rapidly than weekly is not advised as CLB and N‐desmethylclobazam (N‐CLB) are long acting and take approximately 1 week to reach steady state.10
Clobazam Background
Chemical and Physiochemical Properties
CLB is a benzodiazepine with nitrogen atoms in positions 1 and 5 and a keto group at position 4 (Figure 1)8, 16; its chemical structure is defined as 7‐chloro‐1‐methyl‐5‐phenyl‐1‐H‐1,5 benzodiazepine‐2,4[3H,5H]‐dione, yielding the molecular formula C16H13N202Cl. CLB is a white or almost white crystalline powder that is highly soluble in methylene chloride and moderately soluble in water. As a lipophilic substance, CLB distributes throughout the body.8
Figure 1.
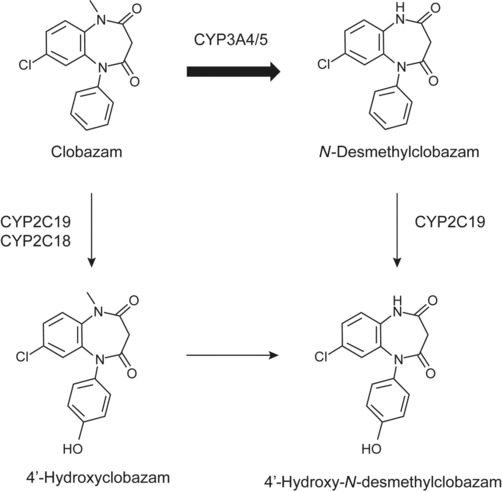
Pathways of CLB metabolism in humans. CLB, its major metabolites produced in vivo, and the primary CYP enzymes that mediate biotransformation are shown; the favored first step in the metabolism of CLB is indicated by a thicker arrow. CLB, clobazam; CYP, cytochrome P450.
Mode of Action
CLB is thought to allosterically modulate the GABAA receptor8; CLB engagement at GABAA receptors results in an influx of chloride ions, creating a hyperpolarizing, inhibitory potential.8 One predominant variant of the GABAA receptor is the α1β2γ2 configuration of subunits; modulation of this receptor activity by related drugs, the 1,4‐benzodiazepines, results in strong sedative effects by way of interaction with the α1 subunits. Because the molecular structure of CLB is distinct from these drugs, it has higher selectivity for α2‐containing GABAA receptor variants, which mediate anticonvulsant effects.8, 9, 17, 18
Clobazam Clinical Pharmacokinetics
Data Sources
A literature search was performed using the terms clobazam and pharmacokinetic in the PubMed database. Further information on CLB PK and PopPK modeling was identified from unpublished reports from the drug sponsor and from references cited in the articles obtained in the literature search. In total, over 100 English‐language articles and abstracts were reviewed; 53 published and 3 unpublished reports were used to compile the information presented here on CLB PK,11, 16, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41 use in specific patient populations,25, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 drug interactions,20, 50, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66 and PopPK analyses.47, 51, 63, 67, 68, 69
Bioanalytical Methods
Historically, various methods have been used to assess CLB PK. Early studies used gas chromatography, gas‐liquid chromatography, or fluorometric assays; these techniques were followed by the development of high‐performance liquid chromatography or electron‐capture gas or liquid chromatography. In some cases, these approaches resulted in nonspecific readouts, such that CLB and N‐CLB could not be differentiated.36 The lower limit of quantification of such techniques ranged over an order of magnitude, indicating variable sensitivity (Table S1). More sensitive assays that use add‐on mass spectrometry (MS) have been more recently implemented, including validated tandem liquid chromatography mass spectrometry (LC‐MS/MS) methods, or high‐performance liquid chromatography mass spectrometry, which provide a lower limit of quantification for both CLB and N‐CLB of approximately 1 ng/mL.11, 32, 39
Several techniques have been used to define the chemical structure of CLB and its metabolites after oral dosing. For example, following administration of radiolabeled CLB (carbon‐14 CLB), CLB and its metabolites were identified by MS, retention time versus synthetic standards, or nuclear magnetic resonance spectroscopy. More recently, CLB and N‐CLB metabolites were identified from plasma in participants receiving multiple doses of CLB via LC‐MS/MS paired with multiple reaction monitoring (data on file).
Pharmacokinetic Parameters
Multiple PK studies have evaluated the absorption, distribution, metabolism, and elimination of CLB and N‐CLB after administration of single or multiple doses of CLB in healthy subjects.20, 21, 23, 26, 27, 28, 29, 30, 31, 35, 36, 38 These studies are summarized in the following sections, with PK parameters from selected studies presented in more detail in Table 1.
Table 1.
PK Parameters of Selected Studies in Healthy Subjects After Single or Multiple Doses of CLB
| Reference (Study Type) | Treatment | Plasma Analyte | Cmax, Mean (% CV), ng/mL | Tmax, Median (range), h | AUC0‐t, Mean (% CV), ng•h/mL | t½, Mean (% CV), h | Vd/F, Mean (% CV), L | CL/F, Mean (% CV), L/h |
|---|---|---|---|---|---|---|---|---|
| OV‐101611 (bioequivalence) | CLB 4 × 5‐mg tablet, Catalent (N = 40) | CLB | 367.9 (24) | 2.0 (0.5‐4.1) | 10 693 (32) | 39.6 (36) | 99 (21) | 2.0 (27) |
| N‐CLB | 87.2 (35) | 48.0 (24‐216) | 12 731 (46) | 58.6 (30) | ND | ND | ||
| CLB 4 × 5‐mg tablet, Pharmascience (N = 39) | CLB | 364.2 (20) | 2.0 (0.5‐5.0) | 10 897 (31) | 41.5 (40) | 100 (22) | 1.9 (28) | |
| N‐CLB | 91.9 (58) | 72.0 (36‐168) | 13 638 (73) | 60.3 (29) | ND | ND | ||
| OV‐101732 (relative BA) | CLB, 20‐mg tablet (N = 18, crossover) | CLB | 392 (25) | 1.5 (0.5‐3.0) | 9384 (26) | 36.5 (34) | 109 (23) | 2.2 (28) |
| N‐CLB | 82 (34) | 48 (24‐120) | 11 451 (48) | 66.2 (34)a | ND | ND | ||
| CLB, 20‐mg solution (N = 18, crossover) | CLB | 376 (21) | 1 (0.5‐2.0) | 8871 (22) | 37.1 (30) | 119 (26) | 2.3 (24) | |
| N‐CLB | 74 (25) | 36 (36‐120) | 10 366 (34) | 62.8 (22)b | ND | ND | ||
| OV‐101839, 41 (food effect) | 4 × 5‐mg tablets crushed with applesauce (N = 16) | CLB | 373 (24) | 2 (1.0‐4.0) | 9962 (30) | 38.5 (28) | 112 (24) | 2.1 (28) |
| N‐CLB | 84 (33) | 72 (36‐72) | 12 399 (49) | 73.6 (35) | ND | ND | ||
| 4 × 5‐mg intact tablets without applesauce (N = 16) | CLB | 379 (30) | 2 (0.5‐3.0) | 9771 (31) | 37.8 (27) | 113 (28) | 2.2 (37) | |
| N‐CLB | 81 (25) | 48 (36‐168) | 11 451 (31) | 68.7 (29) | ND | ND | ||
| 1 × 20‐mg intact tablet, high‐fat meal (N = 16) | CLB | 310 (30) | 3 (1.0‐8.0) | 9987 (17) | 37.2 (26) | 108 (34) | 2.0 (21) | |
| N‐CLB | 81 (30) | 48 (36‐72) | 11 378 (27) | 69.1 (23) | ND | ND | ||
| 1 × 20‐mg intact tablet, fasted state (N = 16) | CLB | 390 (21) | 2 (1.0‐3.0) | 9579 (17) | 36.7 (30) | 112 (39) | 2.1 (22) | |
| N‐CLB | 84 (26) | 42 (36‐72) | 11 453 (24) | 71.3 (27) | ND | ND | ||
| LC‐010c (dose proportionality) | CLB tablet (N = 12, Latin square) | |||||||
| CLB 10 mg | CLB | 107 (32)d | 1.9 (1.0)d | 3518 (923)d,e | 25.5 (18)d | ND | ND | |
| CLB 20 mg | CLB | 375 (51)d | 1.9 (0.6)d | 7190 (2091)d,e | 17.4 (5)d | ND | ND | |
| CLB 40 mg | CLB | 683 (117)d | 2.6 (0.8)d | 13 980 (3608)d,e | 17.8 (5)d | ND | ND | |
| OV‐102234 (multiple dose‐1) | 40‐mg TDD CLB, duration for ≥16 days (N = 70) | CLB | 1076 (24) | 1.6 (0.6‐6.1) | 17 649 (28)f | ND | 113 (27)g | 2.1 (26) |
| N‐CLB | 2783 (71) | 4.1 (0‐12.1) | 57 693 (65)f | ND | ND | ND | ||
| 160‐mg TDD CLB, duration for ≥3 days (N = 70) | CLB | 2884 (18) | 1.9 (0.6‐6.1) | 41 389 (20)f | ND | 128 (26)g | 3.3 (20) | |
| N‐CLB | 11 020 (60) | 4.1 (0‐12.1) | 223 023 (55)f | ND | ND | ND | ||
| OV‐1038c (multiple dose‐2) | 160‐mg TDD CLB, duration for ≥3 days (N = 11) | CLB | 3091 (10) | 2.0 (1.0‐2.5) | 26 092 (13) | 38.0 (32) | 166 (23) | 3.1 (13) |
| N‐CLB | 7361 (30) | 3.0 (0‐12.0) | 84 401 (29) | 71.4 (39) | ND | ND | ||
| 120‐mg TDD CLB, duration for ≥11 daysh (N = 10) | CLB | 2629 (17) | 1.5 (0.5‐2.5) | 19 867 (19) | 37.9 (26) | 167 (23) | 3.1 (20) | |
| N‐CLB | 8090 (38) | 2.5 (1.0‐4.0) | 89 539 (39) | 81.9 (42) | ND | ND |
AUC0‐t, area under the plasma drug concentration‐time curve up to the last measurable concentration; BA, bioavailability; CLB, clobazam; CL/F, apparent clearance; Cmax, maximum plasma concentration; CV, coefficient of variation; N‐CLB, N‐desmethylclobazam; ND, not determined; SD, standard deviation; t1/2, elimination half‐life of drug; TDD, total daily dose; Tmax, time to Cmax; Vd/F, apparent volume of distribution; Vss/F, volume of distribution at steady state.
n = 15; bn = 14; cdata on file; dvariance is ±SD; eAUC is 0‐144 h after dose; fAUC is 0‐24 h after dose; gVss/F; hexcluding a poor metabolizer.
Absorption and Bioavailability
CLB is extensively absorbed at a fast to moderate rate, with time to maximum concentration ranging from 0.5 to 4 hours after single or multiple doses of orally administered CLB tablets; in fasting participants who received a single dose of CLB in oral suspension, time to maximum concentration occurred between 0.5 and 2 hours.11, 31, 32, 34, 37, 39 Overall, there is no significant difference in systemic exposure between tablet and solution‐based formulations; a bioequivalence study showed similar relative bioavailability (near 100%) between the 2 formulations and no significant effects of tablet dissolution on drug absorption (Figure 2).32 The completeness of the CLB absorption is high, with the metabolites of CLB and N‐CLB primarily excreted renally10, 33, 37 (see also Elimination).
Figure 2.
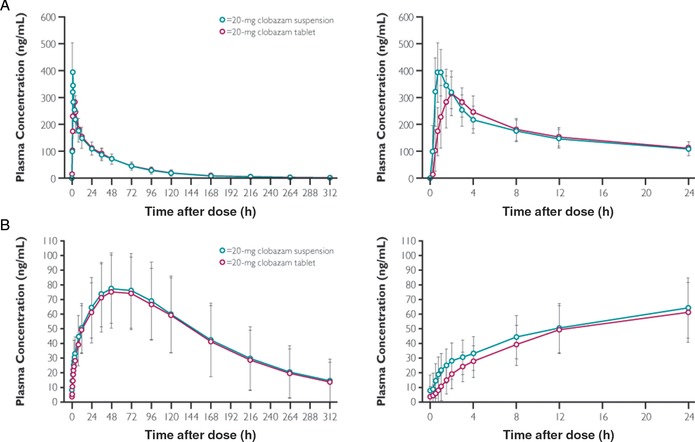
Mean (± SD) clobazam (A) and N‐desmethylclobazam (B) plasma concentrations following a single oral dose of 20‐mg clobazam suspension (test) or tablet (reference).32 In this randomized, open‐label, 2‐way crossover study, participants received a single dose of 20‐mg CLB oral suspension or tablet on study day 1 and day 15, with an intervening 14‐day washout period. PK parameters were measured from blood samples drawn predose (0 hours) and regularly through 312 hours after dose, quantified via LC‐MS/MS. The panels on the left show the entire time course; the panels on the right show finer detail for the plasma concentrations versus time in the first 24 hours. CLB, clobazam; LC, liquid chromatography; MS, mass spectrometry.
When CLB was administered either as intact tablets or crushed in applesauce, the rate and extent of CLB absorption (maximum plasma concentration [Cmax] and area under the plasma drug concentration‐time curve [AUC]) were comparable. Furthermore, when CLB tablets were administered to participants after a high‐fat meal compared with fasting, there was a minor reduction in maximum concentrations but no effect on the extent of CLB absorption.39, 41 Overall, these data indicate that there is little to no food effect on CLB absorption, consistent with previous reports.22, 24
The PK of CLB is dose proportional after both single and multiple doses (Table 1). The dose proportionality of CLB was initially demonstrated via a radiolabel study in healthy subjects; these data were in agreement with a more recent report that quantified CLB concentrations over time using LC‐MS/MS (data on file).
Distribution
For single and multiple oral doses up to 40 mg/day CLB, the mean apparent volume of distribution (Vd/F) and the mean apparent volume of distribution at steady‐state for CLB ranged from 99 to 120 L, indicating that CLB is distributed to peripheral tissues; CLB disposition is also biphasic.11, 32, 34, 39 CLB and N‐CLB bind moderately to plasma proteins in vitro (mean values of bound analyte to plasma proteins were 78.5% ± 6.2% and 72.5% ± 2.5%, respectively), independent of concentration over the range of concentrations evaluated, and the ex vivo plasma protein binding of CLB and N‐CLB is approximately 90% for both compounds.33 Because the protein binding of CLB and N‐CLB is relatively low regardless of the methods used, the differences between the results of the in vitro and ex vivo studies are not expected to be clinically important.
Clobazam, N‐CLB, and In Vitro Analysis With Transporters
In vitro results indicated that CLB and N‐CLB, the main active metabolite of CLB,70, 71 are substrates, but not inhibitors, of the P‐glycoprotein 1 efflux pump.33 Verapamil, a P‐glycoprotein 1 inhibitor, inhibits CLB and N‐CLB transport by >50%; therefore, it is not likely that CLB or N‐CLB are substrates for alternative transporters like the organic cation transporter, the organic anion transporter, or others.72 Due to the fact that CLB is highly absorbed—even in the presence of food (see also Absorption)—and penetrates the blood‐brain barrier, the in vivo effect of P‐glycoprotein 1 on the exposure of CLB is likely insignificant.
Metabolism
CLB is metabolized to over 20 metabolites in humans, including its main metabolite, N‐CLB, primarily via N‐demethylation.16 N‐CLB is approximately one‐fifth as potent as CLB, depending on the pharmacologic model (eg, in vitro or in vivo) used to determine potency.10, 18 The hepatic cytochrome P450 (CYP) enzyme primarily responsible for the conversion of CLB to N‐CLB is CYP3A; N‐CLB is further biotransformed primarily via CYP2C19‐mediated hydroxylation to 4′‐hydroxy‐N‐CLB (Figure 1).16, 25 In addition to N‐demethylation mediated by CYP3A, CLB can be alternatively metabolized to 4‐hydroxyclobazam, a minor metabolic pathway mediated by CYP2C19 and CYP2C18.25 Both CLB and N‐CLB are extensively metabolized such that <1% or <10%, respectively, is typically recovered in urine following oral CLB administration.33, 37 Genetic polymorphism in CYP genes and coadministered drugs can affect the activity of these enzymes and impact CLB metabolism (see also Specific Populations, CYP2C19 Poor Metabolizers).
Elimination
The terminal elimination half‐life (t½) of oral CLB after single or multiple doses ranges from 36 to 42 hours; the t½ of N‐CLB after oral CLB ranges from 59 to 82 hours.11, 32, 34, 39, 40 Previous studies have reported the t½ of CLB and N‐CLB to be slightly shorter (t½ of CLB, approximately 18‐24 hours; t½ of N‐CLB, approximately 36‐57 hours),20, 31 which can likely be attributed to different plasma‐sampling time schedules, performance of the analytical method, and higher lower limit of quantification in historical studies. For example, the current values were measured using longer sample collection times and higher‐sensitivity assays with more specific readouts than earlier reports, both of which could potentially impact the t½. In more recent studies, steady‐state concentrations of CLB and N‐CLB were achieved in 9 to 14 days with multiple oral doses of 120‐mg TDD CLB,33, 34 which is in agreement with timing presented in other reports in which CLB steady state was achieved in approximately 1 week19, 31 and N‐CLB steady state occurred >10 days after CLB dose.19 At steady state, N‐CLB exposure is approximately 3‐ to 5‐fold greater than that of CLB.33, 51
Following a single oral dose of carbon‐14 CLB (39 mg), elimination of carbon‐14 CLB and related metabolites was slow, and 93% of the radioactivity was recovered by 21 days after the dose. In total, 82% of CLB‐related metabolites were eliminated via urinary excretion, with only a small percentage eliminated in the feces (11%). Across single‐dose studies or after CLB 20 mg twice daily, the oral clearance of CLB ranged from 1.9 to 2.3 L/h.11, 32, 34, 39
Specific Populations
In the United States, CLB is indicated for the adjunctive treatment of seizures in patients ≥2 years of age with LGS. Specific populations within this target patient group are discussed in the following sections.
Pediatric and Geriatric Use
Early data on CLB PK in pediatric patients are limited and somewhat variable, due to small numbers of patients studied, lack of clear stratification from older patients, and the fact that patients were taking various combinations of concurrent medications, which may have affected plasma levels of CLB.21, 49, 50 In addition, patients were not routinely genotyped for liver enzyme function. In one study that included adult and pediatric patients as young as 9 months of age, CLB plasma levels were highest in patients aged 20 to 30 years, and N‐CLB concentrations increased with age up to 20 years.20 PopPK modeling is a powerful way to bypass such limitations and is discussed in another section (see also Population PK Analyses, Pediatric Dosing).
Two studies characterized the PK of CLB in adults as a function of age (young, 18–37 years; elderly, 60–72 years) and sex.43, 44 These studies found that CLB was rapidly absorbed (mean time to peak concentration, 1.5 hours) in all groups, and neither sex nor age was found to alter CLB absorption or plasma protein binding; however, an age effect was evident for oral clearance in elderly men compared with younger men. In men who were 60 to 69 years old, the total CLB clearance following a 20‐mg oral CLB dose was approximately 60% of the total clearance seen in men aged 20 to 37 years (0.36 mg/min•kg vs 0.63 mg/min•kg; P ≤ .01).43 Current recommendations advise initiating CLB therapy at 5 mg/day in geriatric patients, with slow dose escalation up to half the maximum dose; if this dose is tolerated for >21 days, elderly patients can uptitrate to a full dose.10
CYP2C19 Poor Metabolizers
The major metabolite of CLB, N‐CLB, is converted to a secondary by‐product by the polymorphic enzyme CYP2C19.25 For patients who carried at least 1 mutated copy of CYP2C19, prolonged N‐CLB elimination and higher‐than‐expected N‐CLB/CLB plasma concentration ratios (dependent on the number of mutant CYP2C19 alleles present) were observed.42, 45, 46, 48 In line with this, after a ≥40‐mg TDD of CLB, CYP2C19 poor metabolizers (PMs) and CYP2C19 extensive/intermediate metabolizers (EMs/IMs) experienced comparable systemic exposures of CLB, but CYP2C19 PMs had approximately 3‐ to 5‐fold higher plasma exposures and mean peak concentrations of N‐CLB compared with EMs/IMs.34, 63 Patients who are known CYP2C19 PMs should initiate CLB at 5 mg/day, with slow uptitration.10
Patients With Renal Impairment
Administration of a single dose (20 mg) and multiple doses (20 mg/day for 7 days) of CLB to participants with mild or moderate renal impairment did not result in clinically meaningful changes in CLB or N‐CLB exposures or renal clearance.37 Because there was no evidence of a relationship between decreasing renal function and any of the CLB or N‐CLB PK parameters following single or multiple dosing, no dose adjustment is required for patients with mild or moderate renal impairment.10
Patients With Hepatic Impairment
The PK of CLB was determined after a single oral dose of 20 mg CLB in participants with liver disease (hepatitis and cirrhosis) and in healthy controls.47 The Cmax of CLB was decreased by approximately 31% in participants with chronic liver disease; however, the mean oral clearance of CLB was approximately the same between those with liver disease and healthy subjects. Furthermore, there were no significant differences in the maximum concentrations of N‐CLB between healthy subjects and patients with liver disease; even after simulation of single doses to steady state, plasma concentrations were the same for all subjects.47 The elimination half‐life was 2 times longer in participants with liver disease compared with healthy controls.47 A PopPK analysis investigating CLB use in patients with hepatic impairment was also developed51 and is described in a later section (see also Population PK Analyses, Drug‐Drug Interaction Potential).
Drug‐Drug Interactions
Because patients with LGS may experience multiple intractable seizure types, effective treatment may require a combination of drugs to manage the disease symptoms73; however, the CYP‐dependent metabolism of CLB may be subject to interactions from other medications.74 If improperly managed, polytherapy can lead to unfavorable adverse events (AEs),75 and an understanding of the effects of concomitant medications is an important foundation for ensuring patient safety.
In Vitro Studies
In an in vitro study of human liver microsomes, CLB did not inhibit CYP enzymes 1A2, 2C8, 2C9, 2C19, 2D6, and 3A4, or the glucuronidation enzymes UGT1A1, UGT1A6, and UGT2B4. N‐CLB also did not inhibit CYP1A2, 2C8, 2C19, 2D6, 3A4, or UGT1A1, but N‐CLB showed weak inhibition of CYP2C9, UGT1A6, and UGT2B4. Furthermore, CLB and N‐CLB did not significantly increase CYP1A2 or CYP2C19 activities. Increases in CYP3A4 activity were observed at high CLB concentrations (25 μM or CLB concentration = 7519 ng/mL). CLB and N‐CLB also increased UGT1A1 mRNA but at concentrations much higher than therapeutic levels. The potential for CLB or N‐CLB to induce CYP2B6 and CYP2C8 has not been evaluated.
Clinical Studies
Effect of CYP3A/2C19 Inhibitors on CLB and N‐CLB
The effect of multiple doses of ketoconazole (a CYP3A strong inhibitor) or omeprazole (a moderate CYP2C19 inhibitor) on the single‐dose PK profile of CLB in healthy adults was evaluated (Table 2).63 CLB coadministered with a 5‐day course of once‐daily ketoconazole (400 mg) increased CLB AUC by 54%; the effect on CLB Cmax was insignificant. The change in N‐CLB AUC0‐t or Cmax when coadministered with ketoconazole was not significant. CLB coadministered with 40 mg omeprazole for 5 days led to an increase in CLB AUC of 30%; CLB Cmax was negligibly impacted. Plasma exposures of N‐CLB with omeprazole coadministration were increased 36% to 37%. While coadministration of CLB with ketoconazole or omeprazole resulted in no clinically meaningful changes in CLB PK, current dosing instructions recommend vigilance and possible dosage adjustment when CLB is coadministered with strong or moderate CYP2C19 inhibitors.10
Table 2.
PK Parameters Observed During Drug‐Drug Interaction Studies in Healthy Subjects
| Reference (Study Type) | Treatment | Plasma Analyte | Cmax, Mean (% CV), ng/mL | Tmax, Median (range), h | AUC0‐inf, Mean (% CV), ng•h/mL | t½, Mean (% CV), h | Vd/F, Mean (% CV), L | CL/F, Mean (% CV), L/hr |
|---|---|---|---|---|---|---|---|---|
| OV‐102163 (effect on CLB with CYP3A or CYP2C19 inhibitor) | 10 mg CLB + 400 mg KTZ | CLB | 190 (21) | 2.0 (0.5‐2.5) | 6432 (19) | 41.7 (32) | 94 (27) | 1.6 (20) |
| N‐CLB | 38 (43) | 84 (48‐120) | 7851 (61) | 74.0 (44) | ND | ND | ||
| 10 mg CLB | CLB | 224 (18) | 1.0 (0.5‐2.6) | 4360 (29) | 37.5 (32) | 128 (30) | 2.5 (32) | |
| N‐CLB | 38 (37) | 60 (36‐84) | 6987 (70) | 67.5 (36) | ND | ND | ||
| 10 mg CLB + 40 mg OPZ | CLB | 211 (21) | 1.3 (0.5‐4.0) | 5398 (21) | 36.7 (32) | 98 (24) | 1.9 (18) | |
| N‐CLB | 47 (34) | 60 (48‐84) | 9176 (85) | 66.7 (83) | ND | ND | ||
| 10 mg CLB | CLB | 218 (15) | 1.0 (0.5‐4) | 4239 (31) | 31.8 (37) | 107 (14) | 2.5 (23) | |
| N‐CLB | 39 (24) | 42 (24‐84) | 5549 (50) | 54.4 (38) | ND | ND | ||
| OV‐102363 (effect of CLB on CYP probe substrates) | Drug Cocktail | Caffeine | 5486 (24) | 0.5 (0.3‐1.5) | 38 528 (33) | 5.9 (26) | 46 (18) | 5.7 (32) |
| Drug Cocktail + CLB | 4676 (24) | 0.8 (0.3‐2.0) | 40 622 (29) | 6.3 (20) | 46 (17) | 5.4 (31) | ||
| Drug Cocktail | Tolbutamide | 50 806 (12) | 3.5 (1.5‐6.0) | 841 599 (35) | 10.0 (36) | 8.7 (16) | 0.7 (28) | |
| Drug Cocktail + CLB | 47 511 (13) | 3.5 (2.0‐6.0) | 746 043 (35) | 9.3 (37) | 9.0 (14) | 0.7 (27) | ||
| Drug Cocktail | Dextromethorphan | 6 (116) | 2.0 (1.0‐6.0) | 208 (235) | 14.4 (122) | 19 707 (76) | 2142 (107) | |
| Drug Cocktail + CLB | 7 (91) | 3.0 (2.0‐8.0) | 214 (205) | 14.0 (113) | 10 374 (75) | 983 (93) | ||
| Drug Cocktail | Midazolam | 20 (53) | 0.5 (0.3‐1.0) | 43 (44) | 3.9 (37) | 581 (43) | 107 (36) | |
| Drug Cocktail + CLB | 15 (43) | 0.5 (0.3‐1.0) | 31 (37) | 3.6 (29) | 687 (33) | 141 (29) |
AUC0‐inf, area under the plasma drug concentration‐time curve extrapolated to infinity; CLB, clobazam; CL/F, apparent clearance; Cmax, maximum plasma concentration; CV, coefficient of variation; CYP, cytochrome P450 enzyme; KTZ, ketoconazole; N‐CLB, N‐desmethylclobazam; ND, not determined; OPZ, omeprazole; t1/2, elimination half‐life of drug; Tmax, time to Cmax; Vd/F, apparent volume of distribution.
Effect of Other Antiseizure Drugs on CLB PK
Studies dating back to the 1980s have reported that adjunctive carbamazepine (CBZ), an inducer of CYP2C9, CYP2C19, and CYP3A4,76, 77 may alter the metabolism and/or clearance of CLB and elevate N‐CLB concentrations, resulting in 3‐ to 4‐fold higher N‐CLB:CLB ratios.57, 60 Several reports indicated that CLB coadministration with CBZ, phenytoin, or phenobarbital, and particularly CBZ and phenytoin, induced CLB metabolism and increased N‐CLB levels.20, 50, 60, 64 Finally, 2 studies examined the interaction between cimetidine (an inhibitor of several CYPs including CYP2C1976) and CLB.56, 58 Despite major differences in the doses of cimetidine administered between studies, both found that CLB exposures increased in the presence of this drug; extended duration and higher cimetidine doses significantly increased the N‐CLB maximum concentration and half‐life by 1.3‐ and 1.6‐fold, respectively.58
Effect of Alcohol on CLB PK
In healthy male subjects, coadministration of a single 20‐mg dose of CLB and alcohol (administered orally in quantities individually calculated to yield serum concentrations of approximately 1000 μg/mL) resulted in higher CLB peak serum levels (389 ng/mL) versus CLB alone (244 ng/mL), as well as an elevated AUC for CLB and alcohol (98 μg•min/mL) versus CLB alone (64 μg•min/mL). While these differences were not statistically significant, the authors caution that the effects of alcohol may be enhanced with CLB.62
Effect of CLB and N‐CLB on Other Drugs
In order to determine the effect of steady‐state concentrations of CLB on the PK profiles of multiple CYP substrates, healthy subjects were administered a drug cocktail containing caffeine (a CYP1A2 substrate), tolbutamide (a CYP2C9 substrate), dextromethorphan (a CYP2D6 substrate), and midazolam (a CYP3A substrate) (Table 2).63 Compared with the drug cocktail alone, coadministration of CLB with a single oral dose of cocktail resulted in a slight increase in the AUC of caffeine (up to 6%) and a 15% decrease in caffeine Cmax. Tolbutamide AUC and Cmax values were decreased by 11% and 7%, respectively, with CLB coadministration versus cocktail alone. Dextromethorphan AUC and Cmax values increased by 90% and 59%, respectively, consistent with some inhibition of CYP2D6. Finally, coadministration of CLB and midazolam resulted in a 27% decreased exposure to midazolam versus the cocktail alone, which indicated a weak induction of CYP3A. None of these effects (inhibition or induction) were clinically meaningful; however, dosage adjustments may be necessary when CLB is administered with drugs metabolized by CYP2D6.10
Variable effects of CLB on the metabolism of other anticonvulsants have been reported.50, 54, 60, 64 For example, some reports describe an increase in CBZ concentrations with CLB coadministration,61, 65 while other studies have found either a decrease or no change in CBZ levels with CLB coadministration.53, 55, 59 Phenytoin toxicity was described in 3 patients when CLB was added66; it is possible that the addition of phenytoin, also a CYP2C19 substrate, to CLB could saturate liver metabolizing enzymes and lead to an increase in phenytoin exposure.52 Finally, 2 reports found that CLB produced an increase in valproic acid concentrations, but this change was not considered clinically meaningful.50, 54
It should be noted that DDI information from historic clinical studies should be interpreted cautiously as the sample size for many of the studies was small and formal statistical analyses were not performed; additionally, many of these studies involved patients taking multiple concomitant ASDs. However, the findings from the DDI studies were not unexpected based on the results of in vitro studies and the characterization of the CYP enzymes responsible for CLB metabolism.
Population PK Analyses
PopPK modeling and subsequent simulations close critical gaps in the PK profile of CLB, especially from sparsely sampled patient populations. PopPK also can inform study design and optimize dosing strategies, important for specific patient populations. Details of 4 recently performed PopPK analyses are summarized in Table 3 and presented in the following sections.
Table 3.
CLB PK Parameters From Population PK Analyses
| Selected CLB PopPK Estimates | ||||||||
|---|---|---|---|---|---|---|---|---|
| PopPK Analysis | Studies Pooled for PK Data | Participants | Age (Range), yrs | Weight (Range), kg | t1/2 (hr) | V2/F (RSE, %), L | CL/F (RSE, %), L/hr | Ka (RSE, %), hr−1 |
| Integrative CLB and N‐CLB PK; PK by participant genotype; covariate analysis (effect of age, renal function, sex, race)63 | Data from healthy subjects and patients with LGS13, 83 | 439 participants; CYP2C19 genotyping available for 12 PMs, 11 IMs, and 299 EMs | 2‐74 | 11‐133 | 36 | 54.7 (5.2) | 2.5 (2.5) | 1.1 (8.7) |
| Drug‐drug interactions68 | Data from healthy subjects and patients with LGS | 171 patients | 2‐74 | 11‐113 | NA | 51.4 (17.5) | 2.8 (4.9) | 1.2 (46.8) |
| Predicting dosing in pediatric patients69 | Data from healthy subjects and patients with LGS; observational study 301 in patients with LGS; simulated study 401 | 193 patients | 0.5‐45 | NA | NA | 59.8 (15.5) | 5.5 (20.4) | 15.0(‐) |
| Determine effect of hepatic impairment on CLB PK51 | Data from healthy subjects or those with liver impairment from 5 studies | 59 participants | NA | NA | NA | 45.2 (8.0) | 2.6 (8.1) | 0.7 (12.6) |
CLB, clobazam; CL/F, apparent clearance; CYP2C19, cytochrome P450 enzyme 2C19; EM, extensive metabolizer; IM, intermediate metabolizer; Ka, first‐order rate constant of absorption; LGS, Lennox‐Gastaut syndrome; NA, not available; N‐CLB, N‐desmethylclobazam; PK, pharmacokinetics; PM, poor metabolizer; PopPK, population pharmacokinetics; RSE, relative standard error; t1/2, half‐life; V2/F, median apparent central volume of distribution.
Healthy Subjects and Patients With LGS
By pooling data from 7 studies (5 in healthy subjects and 2 in patients with LGS, including 130 children aged 2‐11 years and 45 adolescents aged 12‐17 years), a predictive analysis was developed to assess the PopPK of CLB and N‐CLB at a population level.63 PK data from 254 male and 185 female participants were pooled. CYP2C19 genotyping information was available for 322 of the 439 participants: 12 PMs, 11 IMs, and 299 EMs. The final integrative analysis for CLB was a 2‐compartment model with first‐order absorption; N‐CLB was described by a 1‐compartment model. Graphic analysis and nonlinear mixed‐effects modeling demonstrated that the final analyses adequately characterized the data. A covariate analysis indicated that the PK of CLB and N‐CLB was not influenced by age, renal function, sex, or race.63
The PopPK‐predicted CLB PK was linear, dose independent, and time invariant.63 Volume of distribution at steady state for CLB was estimated at 102 L, and t½ was 36 hours for CLB and 79 hours for N‐CLB (additional parameters summarized in Table 3). By this approach, CYP2C19 PMs had approximately 5‐fold higher plasma exposures (AUC) of N‐CLB compared with EMs/IMs, and they were slower to eliminate N‐CLB versus EMs/IMs (data on file). As such, dosage adjustments are recommended when CLB is coadministered with CYP2C19 inhibitors.10
Drug‐Drug Interaction Potential
In a second PopPK analysis, data from 2 studies (the phase 3 trial of CLB in patients with LGS and a relative bioavailability study in healthy subjects) were pooled (Table 3) to characterize the effects of ASDs that are CYP3A inducers (phenobarbital, phenytoin, CBZ), CYP2C19 inducers (valproic acid, phenobarbital, phenytoin, CBZ), or CYP2C19 inhibitors (felbamate, oxcarbazepine) on CLB and N‐CLB PK.68 CYP3A inducers did not affect CLB levels but increased N‐CLB levels by 9.4%. CYP2C19 inducers had a negligible effect on N‐CLB PK, with a 10.5% increase in apparent elimination, while CYP2C19 inhibitors did not affect N‐CLB elimination. The results of this analysis suggested that CLB and N‐CLB PK are not affected by the presence of stable dosages of CYP3A inducers, CYP2C19 inducers, or CYP2C19 inhibitors. In a subanalysis, data from patients who participated in the phase 3 trial, received concomitant valproic acid or lamotrigine, and had evaluable PK data at baseline and week 513 indicated that valproic acid and lamotrigine levels were not affected by CLB coadministration.63
Pediatric Dosing
In order to predict CLB dosing for younger pediatric patients whose ability to metabolize CLB and other drugs differs from that of adults,78, 79 a third PopPK analysis was developed that incorporated weight and ontogeny.69 Data from 4 studies, including a bioequivalence study in healthy subjects, a phase 3 efficacy trial in patients with LGS,13 an observational study in patients with LGS, and a simulated study, were pooled for this analysis. The analysis predicted PK parameters that were similar to those previously reported51 (Table 3). In addition, the predicted range of CLB/N‐CLB concentrations after 1.5 or 2.0 mg/kg CLB dosing were 2 to 3 times those reported in a previous study in which seizure improvement was demonstrated80; therefore, these dosages can be expected to yield CLB/N‐CLB concentrations that are effective in reducing seizures in pediatric patients <3 and ≥3 years of age. This PopPK analysis provides a foundation for CLB dosing regimens in pediatric patients with rare and difficult‐to‐treat epilepsies such as Dravet syndrome, for which there are no approved treatments and limited clinical trial data on potential therapies.81, 82 In addition, the analysis provides a basis for future studies evaluating CLB efficacy in this patient population.67
Effect of Hepatic Impairment on CLB PK
PK data from patients with LGS, healthy subjects, and participants with and without hepatic impairment, as well as those from a publication describing the effects of hepatic impairment on CLB PK,47 were merged to create a PopPK model to characterize the effect of hepatic impairment on CLB and N‐CLB PK (Table 3).51 The analysis predicted that hepatic impairment did not affect the clearance of CLB but may affect the PK of N‐CLB (Figure 3).51 Because the formation of N‐CLB is elimination rate limited and the apparent clearance of CLB is unaffected by hepatic impairment, patients with LGS and hepatic impairment may not require CLB dosage adjustment. However, it is advised that such patients initiate CLB at 5 mg/day and slowly uptitrate.10
Figure 3.

Hepatic function status and apparent clearance rate of CLB (A, B) and N‐CLB (C, D). Note: K40 is equivalent to the clearance rate of N‐CLB because the apparent volume of N‐CLB was fixed at 1 L. CL, clearance; CLB, clobazam; N‐CLB, N‐desmethylclobazam. Reproduced from Tolbert D, Bekersky I, Chu HM, Ette EI. An integrative population pharmacokinetics approach to the characterization of the effect of hepatic impairment on clobazam pharmacokinetics. J Clin Pharmacol. 2016;56(2):213‐222, with permission conveyed through Copyright Clearance Center, Inc.
Other Studies
Clinical Trials, Efficacy in Seizure Reduction, and Tolerability
The efficacy of CLB as adjunctive therapy for seizures in LGS was established by randomized, double‐blind, placebo‐controlled phase 283 and phase 313 trials. In these trials, patients received a target CLB dose ranging from 0.25 to 1 mg/kg/day, resulting in a significantly reduced rate of drop seizures with all levels of CLB dosage versus placebo. In the phase 3 trial, AEs reported with a ≥10% difference between placebo and any CLB group were somnolence, pyrexia, lethargy, constipation, and drooling; only the incidence of somnolence and drooling rose with higher CLB dose.13
Clobazam PK and Tolerability With Supratherapeutic Dosing
In a study examining the safety and tolerability of titrated, multiple ascending oral doses of CLB in healthy adults, CLB and N‐CLB reached steady‐state levels after 9 days with supratherapeutic dosing of 120‐mg CLB TDD maintained for 11 days (data on file). At steady state, N‐CLB exposures were approximately 5‐fold greater than that of CLB (Table 1). N‐CLB did not reach steady state after CLB TDD 160‐mg dosing for 3 days. The high CLB/N‐CLB exposures were generally well tolerated; most AEs were mild or moderate in severity (90.9%), with 16.7% of subjects (4 of 24) reporting somnolence. The single CYP2C19 PM in this study had 2‐ to 4‐fold elevated concentrations of N‐CLB relative to EMs but relatively equivalent concentrations of CLB. After 11 days on 120 mg/day CLB, this participant developed a moderate AE of delirium, which resolved fully.
In another study of healthy subjects who received either therapeutic (40 mg/day) or supratherapeutic (160 mg/day) doses of CLB, no effect on cardiac repolarization or QT interval prolongation was observed.34 Finally, recent work investigating withdrawal‐related AEs from CLB found that abrupt withdrawal at high doses (≤160 mg/day) generally resulted in mild AEs, including headache, insomnia, or tremor; withdrawal over 3 weeks is recommended, as almost no withdrawal‐related AEs occurred if the CLB dose was tapered over time.84
Conclusions
CLB, a 1,5‐benzodiazepine, is indicated in the United States as adjunctive treatment for seizures in patients ≥2 years old with LGS. Distinct from other benzodiazepines, its modulation of GABAA neurotransmission is thought to involve anticonvulsant rather than sedative receptor binding sites. It is extensively absorbed after oral administration and is metabolized in the liver primarily by CYP enzymes to produce its main active metabolite, N‐CLB, and other metabolites. CLB PK is linear and dose independent, with a mean half‐life following a single oral dose of CLB of 36.7 hours for CLB and 71.3 hours for N‐CLB. PopPK analyses predicted CLB dosing for use in pediatric population <3 years of age and characterized the effect of hepatic impairment on CLB and N‐CLB PK. In vitro, in vivo, and PopPK drug interaction studies indicate that CLB and N‐CLB PK appear to be unaffected by the presence of stable doses of most other ASDs at steady state, suggesting a low potential for DDIs. This is important for patients with LGS, as this difficult‐to‐treat epilepsy frequently requires combinations of ASDs to manage the characteristic multiple seizure types. CLB dosage may need to be adjusted in geriatric patients, known CYP2C19 PMs, and in patients with mild or moderate hepatic impairment. Overall, CLB is generally well tolerated, has no cardiac conduction concerns, and is an efficacious option for seizure reduction in patients ≥2 years old with LGS.
Supporting information
Supporting Information
Acknowledgments
Both authors contributed to drafting and revising the manuscript. Medical writing and editing support was provided by Gina Daniel, PhD, of Prescott Medical Communications Group (Chicago, IL).
Declaration of Conflicting Interests
Dr. Tolbert is an employee of Lundbeck LLC, Deerfield, IL, USA. Dr. Larsen is an employee of H. Lundbeck A/S, Valby, Denmark.
Funding
Funding for the trials, data analysis, and manuscript preparation was provided by Lundbeck LLC (Deerfield, IL).
References
- 1. Arzimanoglou A, French J, Blume WT, et al. Lennox‐Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. 2009;8(1):82–93. [DOI] [PubMed] [Google Scholar]
- 2. Camfield PR. Definition and natural history of Lennox‐Gastaut syndrome. Epilepsia. 2011;52(suppl 5):3–9. [DOI] [PubMed] [Google Scholar]
- 3. Arzimanoglou A, Resnick T. All children who experience epileptic falls do not necessarily have Lennox‐Gastaut syndrome… but many do. Epileptic Disord. 2011;13(suppl 1):S3–S13. [DOI] [PubMed] [Google Scholar]
- 4. Berg AT, Shinnar S, Testa FM, Levy SR, Smith SN, Beckerman B. Mortality in childhood‐onset epilepsy. Arch Pediatr Adolesc Med. 2004;158(12):1147–1152. [DOI] [PubMed] [Google Scholar]
- 5. Montouris GD. Rational approach to treatment options for Lennox‐Gastaut syndrome. Epilepsia. 2011;52(suppl 5):10–20. [DOI] [PubMed] [Google Scholar]
- 6. Brodie MJ, Chung S, Wade A, et al. Clobazam and clonazepam use in epilepsy: results from a UK database incident user cohort study. Epilepsy Res. 2016;123:68–74. [DOI] [PubMed] [Google Scholar]
- 7. Ng YT, Collins SD. Clobazam. Neurotherapeutics. 2007;4(1):138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sankar R. GABA(A) receptor physiology and its relationship to the mechanism of action of the 1,5‐benzodiazepine clobazam. CNS Drugs. 2012;26(3):229–244. [DOI] [PubMed] [Google Scholar]
- 9. McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3(6):587–592. [DOI] [PubMed] [Google Scholar]
- 10. ONFI® (clobazam) . Full prescribing information. Deerfield, IL, Lundbeck; 2016.
- 11. Walzer MA, Bekersky I, Reid S, Gordon J, Juan A, Tolbert D. Bioequivalence of clobazam 5‐mg test and reference tablet formulations. 2010. Annual Meeting and Exposition of the American Association of Pharmaceutical Scientists; New Orleans, LA; Published Meeting Abstracts (Poster W5369). http://abstracts.aaps.org/Published/Browse.aspx?colID=12. Accessed December 5, 2017. [Google Scholar]
- 12. Epaclob/Silocalm™ (clobazam oral suspension) . Full prescribing information. Essex, UK: Martindale Pharmaceuticals Limited; 2016. [Google Scholar]
- 13. Ng YT, Conry JA, Drummond R, Stolle J, Weinberg MA. Randomized, phase III study results of clobazam in Lennox‐Gastaut syndrome. Neurology. 2011;77(15):1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Conry JA, Ng YT, Kernitsky L, et al. Stable dosages of clobazam for Lennox‐Gastaut syndrome are associated with sustained drop‐seizure and total‐seizure improvements over 3 years. Epilepsia. 2014;55(4):558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tolbert D, Klein P, Gidal B, Chu HM, Ette E. The PK/PD basis for clobazam use in refractory seizures: is there a need for clobazam dosage reduction in the presence of other antiepileptic drugs? Paper presented at 71st Annual Meeting of the American Epilepsy Society, Scientific Exhibit; Washington, DC; December 1‐5, 2017.
- 16. Volz M, Christ O, Kellner HM, et al. Kinetics and metabolism of clobazam in animals and man. Br J Clin Pharmacol. 1979;7(suppl 1):41s–50s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wheless JW, Phelps SJ. Clobazam: a newly approved but well‐established drug for the treatment of intractable epilepsy syndromes. J Child Neurol. 2013;28(2):219–229. [DOI] [PubMed] [Google Scholar]
- 18. Jensen HS, Nichol K, Lee D, Ebert B. Clobazam and its active metabolite N‐desmethylclobazam display significantly greater affinities for alpha(2)‐ versus alpha(1)‐GABA(A)‐receptor complexes. PLOS ONE. 2014;9(2):e88456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bun H, Coassolo P, Gouezo F, Serradimigni A, Cano JP. Time‐dependence of clobazam and N‐demethylclobazam kinetics in healthy volunteers. Int J Clin Pharmacol Ther Toxicol. 1986;24(6):287–293. [PubMed] [Google Scholar]
- 20. Bun H, Monjanel‐Mouterde S, Noel F, Durand A, Cano JP. Effects of age and antiepileptic drugs on plasma levels and kinetics of clobazam and N‐desmethylclobazam. Pharmacol Toxicol. 1990;67(2):136–140. [DOI] [PubMed] [Google Scholar]
- 21. Bun H, Coassolo PH, Gouezo F, Cano JP, Dravet C, Roger J. Plasma levels and pharmacokinetics of clobazam and N‐desmethylclobazam in epileptic patients. Paper presented at Royal Society of Medicine International Congress and Symposium, York, UK; 1985.
- 22. Cenraud B, Guyot M, Levy RH, et al. No effect of food intake on clobazam absorption. Br J Clin Pharmacol. 1983;16(6):728–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davies IB, Pidgen AW, McEwen J, Robinson JD, Walker SE, Stonier PD. Rectal absorption of clobazam: pharmacokinetic aspects and possible use in epilepsy. Paper presented at Royal Society of Medicine International Congress and Symposium, York, UK; 1985.
- 24. Divoll M, Greenblatt DJ, Ciraulo DA, Puri SK, Ho I, Shader RI. Clobazam kinetics: intrasubject variability and effect of food on adsorption. J Clin Pharmacol. 1982;22(1):69–73. [DOI] [PubMed] [Google Scholar]
- 25. Giraud C, Tran A, Rey E, Vincent J, Treluyer JM, Pons G. In vitro characterization of clobazam metabolism by recombinant cytochrome P450 enzymes: importance of CYP2C19. Drug Metab Dispos. 2004;32(11):1279–1286. [PubMed] [Google Scholar]
- 26. Goggin T, Callahan N. Blood levels of clobazam and its metabolites and therapeutic effect. Paper presented at Royal Society of Medicine International Congress and Symposium, York, UK; 1985.
- 27. Guberman A, Couture M, Blaschuk K, Sherwin A. Add‐on trial of clobazam in intractable adult epilepsy with plasma level correlations. Can J Neurol Sci. 1990;17(3):311–316. [DOI] [PubMed] [Google Scholar]
- 28. Jawad S, Richens A, Oxley J. Single dose pharmacokinetic study of clobazam in normal volunteers and epileptic patients. Br J Clin Pharmacol. 1984;18(6):873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laux G, Koeppen D. Serum and cerebrospinal fluid concentration of clobazam and N‐desmethylclobazam. Int J Clin Pharmacol Ther Toxicol. 1984;22(7):355–359. [PubMed] [Google Scholar]
- 30. Ochs HR, Greenblatt DJ, Luttkenhorst M, Verburg‐Ochs B. Single and multiple dose kinetics of clobazam, and clinical effects during multiple dosage. Eur J Clin Pharmacol. 1984;26(4):499–503. [DOI] [PubMed] [Google Scholar]
- 31. Rupp W, Badian M, Christ O, et al. Pharmacokinetics of single and multiple doses of clobazam in humans. Br J Clin Pharmacol. 1979;7(suppl 1):51s–57s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sales E, Juan A, Bekersky I, Tolbert D. Relative bioavailability of clobazam oral suspension after a single 20‐mg dose in healthy volunteers. Poster 1.235. Paper presented at Annual Meeting of the American Epilepsy Society, San Diego, CA; November 30‐December 4, 2012.
- 33. Tolbert D, Bekersky I. Pharmacokinetics of N‐desmethylclobazam, the active and primary metabolite of clobazam. Poster P01.042. Paper presented at 66th Annual Meeting of the American Academy of Neurology, Philadelphia, PA; April 26‐May 3, 2014.
- 34. Tolbert D, Gordon J, Harris S, Walzer M, Bekersky I, Reid S. A thorough QT/QTc study of clobazam in healthy volunteers. Clin Ther. 2017;39(10):2073–2086. [DOI] [PubMed] [Google Scholar]
- 35. Vallner JJ, Kotzan JA, Stewart JT, Honigberg IL, Needham TE, Brown WJ. Plasma levels of clobazam after 10‐, 20‐, and 40‐mg tablet doses in healthy subjects. J Clin Pharmacol. 1980;20(7):444–451. [DOI] [PubMed] [Google Scholar]
- 36. Vallner JJ, Needham TE, Jun HW, et al. Plasma levels of clobazam after three oral dosage forms in healthy subjects. J Clin Pharmacol. 1978;18(7):319–324. [DOI] [PubMed] [Google Scholar]
- 37. Walzer M, Bekersky, I , Beconi M, Sales E, Preston RA, Gordon J, Tolbert D. Effect of renal impairment on pharmacokinetics of clobazam. Paper presented at American College of Clinical Pharmacology Annual Meeting, Baltimore, MD; September 12‐14, 2010.
- 38. Pullar T, Haigh JR, Peaker S, Feely MP. Pharmacokinetics of N‐desmethylclobazam in healthy volunteers and patients with epilepsy. Br J Clin Pharmacol. 1987;24(6):793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tolbert D, Reid S, Walzer M, et al. Bioavailability of clobazam when administered with applesauce [abstract 2.230]. Epilepsia. 2009;50(suppl 11):261. [Google Scholar]
- 40. Tolbert D, Reid S, Walzer M, et al. Oral pharmacokinetics of clobazam in normal subjects. Published Meeting Abstracts (Poster M1090). 2009 Annual Meeting of the American Association of Pharmaceutical Scientists. http://abstracts.aaps.org/Published/Browse.aspx?colID=12. Accessed February 19, 2018.
- 41. Reid S, Tolbert D, Walzer M, et al. Evaluation of the effect of food on the absorption of clobazam [abstract 138]. Thirty‐eighth annual meeting of the American College of Clinical Pharmacology. J Clin Pharmacol. 2009;49:1123. [Google Scholar]
- 42. Contin M, Sangiorgi S, Riva R, Parmeggiani A, Albani F, Baruzzi A. Evidence of polymorphic CYP2C19 involvement in the human metabolism of N‐desmethylclobazam. Ther Drug Monit. 2002;24(6):737–741. [DOI] [PubMed] [Google Scholar]
- 43. Greenblatt DJ, Divoll M, Puri SK, Ho I, Zinny MA, Shader RI. Clobazam kinetics in the elderly. Br J Clin Pharmacol. 1981;12(5):631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greenblatt DJ, Divoll M, Puri SK, Ho I, Zinny MA, Shader RI. Reduced single‐dose clearance of clobazam in elderly men predicts increased multiple‐dose accumulation. Clin Pharmacokinet. 1983;8(1):83–94. [DOI] [PubMed] [Google Scholar]
- 45. Hashi S, Yano I, Shibata M, et al. Effect of CYP2C19 polymorphisms on the clinical outcome of low‐dose clobazam therapy in Japanese patients with epilepsy. Eur J Clin Pharmacol. 2015;71(1):51–58. [DOI] [PubMed] [Google Scholar]
- 46. Kosaki K, Tamura K, Sato R, Samejima H, Tanigawara Y, Takahashi T. A major influence of CYP2C19 genotype on the steady‐state concentration of N‐desmethylclobazam. Brain Dev. 2004;26(8):530–534. [DOI] [PubMed] [Google Scholar]
- 47. Monjanel‐Mouterde S, Antoni M, Bun H, et al. Pharmacokinetics of a single oral dose of clobazam in patients with liver disease. Pharmacol Toxicol. 1994;74(6):345–350. [DOI] [PubMed] [Google Scholar]
- 48. Parmeggiani A, Posar A, Sangiorgi S, Giovanardi‐Rossi P. Unusual side‐effects due to clobazam: a case report with genetic study of CYP2C19. Brain Dev. 2004;26(1):63–66. [DOI] [PubMed] [Google Scholar]
- 49. Tedeschi G, Riva R, Baruzzi A. Clobazam plasma concentrations: pharmacokinetic study in healthy volunteers and data in epileptic patients. Br J Clin Pharmacol. 1981;11(6):619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Theis JG, Koren G, Daneman R, et al. Interactions of clobazam with conventional antiepileptics in children. J Child Neurol. 1997;12(3):208–213. [DOI] [PubMed] [Google Scholar]
- 51. Tolbert D, Bekersky I, Chu HM, Ette EI. An integrative population pharmacokinetics approach to the characterization of the effect of hepatic impairment on clobazam pharmacokinetics. J Clin Pharmacol. 2016;56(2):213–222. [DOI] [PubMed] [Google Scholar]
- 52. Dilantin® (phenytoin sodium) . Full prescribing information. New York, NY: Parke‐Davis, Division of Pfizer Inc; 2017. [Google Scholar]
- 53. Aucamp AK. Clobazam as adjunctive therapy in uncontrolled epileptic patients. Current Ther Res. 1985;37(6):1098–1103. [Google Scholar]
- 54. Cocks A, Critchley EMR, Hayward HW, Thomas D. The effect of clobazam on the blood levels of sodium valproate. Paper presented at Royal Society of Medicine International Congress and Symposium, York, UK; 1985..
- 55. Dellaportas CI, Wilson A, Rose FC. Clobazam as adjunctive treatment in chronic epilepsy. Advances in Epileptology: XVth Epilepsy International Symposium New York, NY: Raven Press; 1984:363–367. [Google Scholar]
- 56. Grigoleit HG, Hajdu P, Hundt HK, et al. Pharmacokinetic aspects of the interaction between clobazam and cimetidine. Eur J Clin Pharmacol. 1983;25(1):139–142. [DOI] [PubMed] [Google Scholar]
- 57. Levy RH, Lane EA, Guyot M, Brachet‐Liermain A, Cenraud B, Loiseau P. Analysis of parent drug‐metabolite relationship in the presence of an inducer: application to the carbamazepine‐clobazam interaction in normal man. Drug Metab Dispos. 1983;11(4):286–292. [PubMed] [Google Scholar]
- 58. Pullar T, Edwards D, Haigh JR, Peaker S, Feely MP. The effect of cimetidine on the single dose pharmacokinetics of oral clobazam and N‐desmethylclobazam. Br J Clin Pharmacol. 1987;23(3):317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schmidt D, Rohde M, Wolf P, Roeder‐Wanner U. Tolerance to the antiepileptic effect of clobazam In: Tolerance to Beneficial and Adverse Effects of Antiepileptic Drugs, edited by Frey HH. New York, NY: Raven Press; 1986:109–117. [Google Scholar]
- 60. Sennoune S, Mesdjian E, Bonneton J, Genton P, Dravet C, Roger J. Interactions between clobazam and standard antiepileptic drugs in patients with epilepsy. Ther Drug Monit. 1992;14(4):269–274. [DOI] [PubMed] [Google Scholar]
- 61. Shimizu H, Kawasaki J, Yuasa S, Tarao Y, Kumagai S, Kanemoto K. Use of clobazam for the treatment of refractory complex partial seizures. Seizure. 2003;12(5):282–286. [DOI] [PubMed] [Google Scholar]
- 62. Taeuber K, Badian M, Brettel HF, et al. Kinetic and dynamic interaction of clobazam and alcohol. Br J Clin Pharmacol. 1979;7(suppl 1):91s–97s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Walzer M, Bekersky I, Blum RA, Tolbert D. Pharmacokinetic drug interactions between clobazam and drugs metabolized by cytochrome P450 isoenzymes. Pharmacotherapy. 2012;32(4):340–353. [DOI] [PubMed] [Google Scholar]
- 64. Wang J, Hug D, Gautschi K, Wieser HG. Clobazam for treatment of epilepsy. Epilepsia. 1993;6:180–184. [Google Scholar]
- 65. Wolf P. Clobazam in drug‐resistant patients with complex focal seizures—report of an open study. Paper presented at Royal Society of Medicine International Congress and Symposium, York, UK: 1985.
- 66. Zifkin B, Sherwin A, Andermann F. Phenytoin toxicity due to interaction with clobazam. Neurology. 1991;41(2(Pt 1):313–314. [DOI] [PubMed] [Google Scholar]
- 67. Lee D, Omasta R, Canavan C, et al. Clobazam for Dravet syndrome: designing a clinical study using pharmacokinetic/pharmacodynamic modeling. Neurology. 2015;84(14 supplement):P1.260. [Google Scholar]
- 68. Tolbert D, Bekersky I, Chu HM, Ette EI. Drug‐metabolism mechanism: knowledge‐based population pharmacokinetic approach for characterizing clobazam drug‐drug interactions. J Clin Pharmacol. 2016;56(3):365–374. [DOI] [PubMed] [Google Scholar]
- 69. Tolbert D, Chu HM, Ette E. Pharmacometrics of clobazam in pediatrics: population PK modeling to predict effective clobazam doses for Dravet syndrome. Neurology. 2018;90(15):P4.265. [Google Scholar]
- 70. Hammer H, Ebert B, Jensen HS, Jensen AA. Functional characterization of the 1,5‐benzodiazepine clobazam and its major active metabolite N‐desmethylclobazam at human GABA(A) receptors expressed in Xenopus laevis oocytes. PLoS One. 2015;10(3):e0120239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Meldrum BS, Croucher MJ. Anticonvulsant action of clobazam and desmethylclobazam in reflex epilepsy in rodents and baboons. Drug Dev Res. 1982;2(suppl 1):33–38. [Google Scholar]
- 72. Giacomini KM, Huang SM, Tweedie DJ, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. van Rijckevorsel K. Treatment of Lennox‐Gastaut syndrome: overview and recent findings. Neuropsychiatr Dis Treat. 2008;4(6):1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lin JH, Lu AY. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet. 1998;35(5):361–390. [DOI] [PubMed] [Google Scholar]
- 75. Panayiotopoulos CP. A Clinical Guide to Epileptic Syndromes and Their Treatment: Based on the ILAE Classifications and Practice Parameter Guidelines. 2nd ed London: Springer; Revised 2010. [Google Scholar]
- 76. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. http://medicine.iupui.edu/clinpharm/ddis/clinical-table. Accessed February 19, 2018.
- 77. Brodie MJ, Mintzer S, Pack AM, Gidal BE, Vecht CJ, Schmidt D. Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia. 2013;54(1):11–27. [DOI] [PubMed] [Google Scholar]
- 78. Sadler NC, Nandhikonda P, Webb‐Robertson BJ, et al. Hepatic cytochrome P450 activity, abundance, and expression throughout human development. Drug Metab Dispos. 2016;44(7):984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lu H, Rosenbaum S. Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther. 2014;19(4):262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chiron C, Marchand MC, Tran A, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo‐controlled syndrome‐dedicated trial. STICLO study group. Lancet. 2000;356(9242):1638–1642. [DOI] [PubMed] [Google Scholar]
- 81. Auvin S, Cilio MR, Vezzani A. Current understanding and neurobiology of epileptic encephalopathies. Neurobiol Dis. 2016;92(Pt A):72–89. [DOI] [PubMed] [Google Scholar]
- 82. Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(suppl 2):3–9. [DOI] [PubMed] [Google Scholar]
- 83. Conry JA, Ng YT, Paolicchi JM, et al. Clobazam in the treatment of Lennox‐Gastaut syndrome. Epilepsia. 2009;50(5):1158–1166. [DOI] [PubMed] [Google Scholar]
- 84. Tolbert D, Harris SI, Bekersky I, Lee D, Isojarvi J. Withdrawal‐related adverse events from clinical trials of clobazam in Lennox‐Gastaut syndrome. Epilepsy Behav. 2014;37:11–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information