

Abstract
FLT3‐ITD–mutated acute myeloid leukemia (AML) has very high risk of relapse and is associated with poor outcome following allogeneic hematopoietic‐cell transplant (allo‐HCT). This two‐part, phase 1, multicenter, open‐label, sequential‐group, dose‐escalation study aimed to determine dose‐limiting toxicities (DLTs), maximum tolerated dose (MTD), and safety/tolerability of quizartinib, a selective and highly potent FLT3 inhibitor, when administered as maintenance therapy after allo‐HCT. Thirteen subjects with documented FLT3‐ITD–mutated AML in morphological remission following allo‐HCT received one of two quizartinib dihydrochloride dose levels (DL): 40 mg/d (DL1; n = 7) and 60 mg/d (DL2; n = 6), administered orally in 28‐day cycles for up to 24 cycles. Median age of participants was 43 years. All subjects received human leukocyte antigen (HLA)‐matched allo‐HCT. One subject treated at DL1 and 1 treated at DL2 had DLTs that required drug interruption (grade 3 gastric hemorrhage and grade 3 anemia, respectively). Ten subjects (77%) received quizartinib for >1 year; 5 (38%) completed 24 cycles. Four subjects (31%) discontinued quizartinib due to adverse events. One subject (8%) experienced relapse during cycle 1 and discontinued treatment. Most common grade 3/4 adverse events were neutropenia (23%), anemia (15%), leukopenia (15%), lymphopenia (15%), and thrombocytopenia (15%). This study demonstrated acceptable tolerability and early evidence of reduced relapse rate following allo‐HCT with quizartinib maintenance compared to historical cohorts. No MTD was identified, but 60 mg daily was selected as highest dose for continuous daily administration based on randomized comparison of daily 30 and 60 mg doses in relapsed/refractory AML.
1. INTRODUCTION
AML is the most common acute leukemia in adults and a heterogenous malignancy involving clonal proliferation of abnormally or poorly differentiated hematopoietic cells of myeloid lineage.1, 2 Despite recent progress in the understanding of the biology of AML, improvements in patient outcomes have been limited over the last 30 years, and 5‐year survival rate remains poor (∼25%).2, 3, 4 While approximately 60% to 80% of subjects with AML achieve complete remission (CR) in response to standard of care induction/consolidation chemotherapy, disease relapse occurs in a majority of patients and is a major cause of treatment failure. Subjects with relapsed AML have poor outcomes.1, 2, 4, 5
Mutations of the FMS‐like tyrosine kinase 3 (FLT3) receptor are among the most common molecular alterations in subjects with AML, particularly in individuals presenting with normal karyotype.6, 7, 8, 9 Internal tandem duplication (ITD) mutations of FLT3 occur in approximately 25% of subjects with newly diagnosed AML, resulting in constitutive activation of FLT3 kinase activity and the promotion of cellular proliferation and survival that is associated with high leukemic burden.10, 11, 12, 13, 14 Importantly, FLT3‐ITD is a driver mutation in AML, and its presence identifies a high‐risk population of patients with resistance to induction chemotherapy,14, 15, 16 as well as with shorter duration of remission, increased risk of relapse, and increased mortality in subjects who respond to standard of care chemotherapy.8, 11, 14, 15, 16, 17, 18 These data highlight the great need for more effective therapies in subjects with FLT3‐ITD–mutated AML.
For subjects with FLT3‐ITD–mutated AML who achieve morphologic remission following induction chemotherapy, consolidation therapy with allo‐HCT has been shown to be associated with lower rates of relapse versus chemotherapy or autologous HCT (auto‐HCT).19, 20, 21 For example, individuals with biallelic FLT3‐ITD and a FLT3‐ITD to wild‐type FLT3 allelic ratio >0.6 have a risk of relapse of >90% following consolidation with chemotherapy or auto‐HCT versus 40%‐50% following allo‐HCT.22 However, subjects with FLT3‐ITD–mutated AML still have an elevated risk of relapse and poor outcomes after allo‐HCT compared with FLT3‐ITD‐negative subjects, with reported incidence of relapse ranging between 30% and 59% vs 15%‐25%, respectively.19, 21, 23, 24, 25 These findings provide a strong rationale for the targeting of FLT3‐ITD as a potential means of improving therapeutic outcomes in this patient population.
While maintenance therapy is not currently a standard management strategy for AML, it is being investigated in this patient population and has the potential to improve outcomes by preventing and/or delaying relapse.26 For instance, recent studies suggest that maintenance therapy using FLT3‐targeted therapies may improve post‐HCT outcomes in patients with FLT3‐ITD–mutated AML, possibly by eliminating minimal residual disease (MRD) following consolidation therapy.26, 27, 28, 29, 30 Quizartinib is a novel, orally administered, selective and highly potent inhibitor of FLT3 that has demonstrated clinical activity in phase 1/2 studies of subjects with newly diagnosed and relapsed and refractory (R/R) AML harboring FLT3‐ITD mutations.31, 32, 33, 34, 35, 36, 37, 38, 39 Here, we report the results of a phase 1 study—the first to evaluate the safety and tolerability of quizartinib as maintenance therapy in subjects with FLT3‐ITD–mutated AML who underwent allo‐HCT.
2. METHODS
2.1. Study design
Study 2689‐CL‐0011 (NCT01468467) was a 2‐part, phase 1, multicenter, open‐label, sequential‐group, dose‐escalation trial of quizartinib monotherapy administered as maintenance therapy following allo‐HCT in subjects with AML. The study protocol was approved by Institutional Review Board/Independent Ethics Committee at participating centers. All subjects signed an informed consent form prior to screening. The primary objectives of the study were determination of the DLTs of quizartinib, definition of MTD, and evaluation of the safety and tolerability of quizartinib when given as maintenance therapy after allo‐HCT. Secondary objectives of the study were evaluation of the efficacy of quizartinib in subjects who receive maintenance therapy after allo‐HCT, evaluation of transplant‐related outcomes in subjects who receive quizartinib as maintenance therapy after allo‐HCT, and characterization of the pharmacokinetics (PK) of quizartinib and AC886 (the pharmacologically active metabolite of quizartinib)
2.2. Part 1: Quizartinib dose escalation
In part 1 of the study, quizartinib dose escalation was planned in up to 3 cohorts (3–6 subjects treated per cohort) with assessment of DLTs during the first 2 cycles of treatment until an MTD was identified. Quizartinib dosing started within 30 to 60 days post HCT.
Quizartinib dihydrochloride was administered orally (solution or tablet formulations) at least 1 hour before or 2 hours after a meal in 28‐day cycles for up to 24 cycles. The first dose level tested was quizartinib dihydrochloride 40 mg daily (equivalent to 35.4 mg quizartinib free base) and the second dose level tested was quizartinib dihydrochloride 60 mg daily (equivalent to 53 mg quizartinib free base). Subject doses were assigned manually using a modified 3 + 3 (rolling six) design that allowed for concurrent enrollment of 2 to 6 subjects into a cohort based on number of subjects currently enrolled and evaluable, number experiencing a DLT, and number still at risk for developing a DLT (subjects with data pending). Enrollment of subjects into dose cohorts was determined by a dose‐escalation committee consisting of the study sponsor's medical monitor and the principal investigator at each participating center, after review of data available for previously enrolled subjects who were treated at the most current dose level being evaluated. Dose‐escalation rules were further defined by whether the MTD had or had not been exceeded in the trial overall. Dose escalation was allowed if 0 out of 3 to 5 or ≤ 1 out of 6 subjects experienced a DLT. Intrapatient dose escalation/re‐escalation were not permitted. Dose de‐escalation was allowed if ≥2 out of 2 to 6 or ≤1 out of 6 subjects experienced a DLT
The following quizartinib dose modifications were permitted: (1) treatment interruptions, (2) hematologic toxicities: neutropenia (absolute neutrophil count [ANC] <500/mm3) or thrombocytopenia (platelets <10,000/mm3). If unrelated to leukemia, quizartinib was held until ANC ≥1000/mm3 and platelets ≥25,000/mm3 and then resumed at next lowest quizartinib dose level. If not resolved within 14 days, or occurred in a subject treated at 30 mg, treatment was discontinued. If unrelated to leukemia, treatment was discontinued, (3) donor chimerism: 20% decrease from baseline in donor CD3 chimerism at any time point during the study. If chimerism increased by 20%, subject could resume at next lowest quizartinib dose level. If 20% reduction was persistent or occurred in a subject treated at 30 mg, treatment was discontinued. Treatment could be interrupted for recurrent reduction of 20% only after 20% increase (recovery) of chimerism. Treatment was discontinued if there was a third recurrence, (4) other toxicities: any toxicity considered a DLT or grade 3/4 toxicity considered possibly/probably related to study drug that persisted >48 hours without resolution to grade ≤ 2. Subjects could resume treatment at next lowest quizartinib dose level if toxicity recovered to ≤grade 1 within 14 days of interruption. If not resolved within 14 days, or occurred in a subject treated at 30 mg, treatment was discontinued, (5) QTcF prolongation: asymptomatic grade 3 QTcF prolongation. Quizartinib was held until QTcF was ≤30 ms above baseline and resumed at next lowest quizartinib dose level. Dose reductions could continue with each cycle for QTcF prolongation; however, reductions below 30‐mg quizartinib were not permitted. If QTcF did not return to ≤30 ms above baseline within 14 days, treatment was discontinued.
DLTs were defined were defined as occurrence of any of the following events during the first 2 treatment cycles which were considered possibly or probably related to study drug: (1) any grade ≥3 nonhematologic toxicity persisting >48 hours without resolution to grade ≤2 (excluding alopecia, anorexia, or fatigue; grade 3 nausea, vomiting, or diarrhea that was managed to grade ≤2 with standard antiemetic or antidiarrheal medications used at prescribed dose within 7 days of onset; grade 3 mucositis that resolved to grade ≤2 within 7 days of onset; grade 3 fever with neutropenia, with or without infection; and grade 3 infection), (2) hematologic toxicities, including peripheral ANC) <500/mm3 (grade 4 unrelated to leukemia (assessed by marrow aspirate or biopsy) or that could not be attributed to a concomitant medication, or platelet count <10,000/mm3, (3) any confirmed grade ≥3 QTcF prolongation (QTcF >500 ms), (4) any other study drug–related toxicity occurring after the first dose of quizartinib and causing interruption of study drug for >14 days or discontinuation of study drug.
MTD was defined as highest dose of quizartinib associated with the occurrence of a DLT in fewer than 33% of the subjects in a cohort and was estimated to be dose level at which ≤1 out of 6 subjects experienced a DLT and below the lowest dose level at which ≥2 out of 2 to 6 subjects experience a DLT.
2.3. Part 2: MTD evaluation
Part 2 of the study was designed to evaluate the safety, efficacy, and PK of quizartinib at the MTD in an expanded patient cohort. However, the study was terminated before the start of part 2 due to end of collaboration agreement between the trial sponsors Ambit Biosciences and Astellas Pharma Inc., and total study enrollment consisted of 13 subjects in part 1.
2.4. Eligibility
Subjects age ≥18 years with morphologically documented AML as defined by the World Health Organization (WHO) criteria,9 and who received a high‐dose or a reduced‐intensity conditioning allo‐HCT during first or second morphologic remission (CR1 or CR2; defined as <5% marrow blasts) and without active central nervous system AML within 14 days prior to first dose of study drug), within 30 to 60 days prior to first dose of study drug, and with donor CD3 chimerism >50% at screening and meeting the following criteria were enrolled: HLA matched related or unrelated donor with only single allele disparity for HLAs allowed, Karnofsky performance status ≥60, ANC >1000/mm3 and platelet count >50,000/mm3 without platelet transfusion support within 2 weeks prior to first dose of study drug, adequate renal, hepatic, and coagulation parameters.
Exclusion criteria included: disease relapse during prior treatment with quizartinib; active grade ≥2 graft versus host disease (GVHD); concurrent chemotherapy, immunotherapy, or radiotherapy within 21 days prior to the first dose of quizartinib; any antineoplastic therapy considered to be investigational within 30 days or 5 half‐lives prior to the first dose of study drug; requirement for treatment with concomitant drugs that prolong QT/QTc interval or with strong CYP3A4 inhibitors or inducers (excluding immunosuppressants, antibiotics, antifungals, and antivirals used as standard of care post‐transplant; drugs used to prevent or treat infections; or other drugs considered essential for care of the subject); requirement for treatment with anticoagulant therapy; positive test for human immunodeficiency virus, hepatitis C, or hepatitis B surface antigen; major surgery within 4 weeks prior to first dose of study drug; uncontrolled or significant cardiovascular disease; active acute fungal, bacterial, or other infection that is unresponsive to therapy; medical, psychiatric, addictive, or other kind of condition that compromises subject's ability to give written informed consent and/or to comply with study procedures.
Prior treatment with a FLT3‐targeted inhibitor before SCT and >1 HCT were allowed. Baseline cytogenetic information was categorized according to the UK Medical Research Council classification.7 FLT3‐ITD mutation status was analyzed per local testing methods by each participating center prior to HCT using bone marrow aspirate samples collected with the same procedure and at the same time as collection of marrow for disease assessment. A whole blood sample was collected if aspirate was not available or sufficient. Subjects were considered FLT3‐ITD–positive if allelic ratio ≥10% and FLT3 wild type for allelic ratios <10%. Per protocol amendment, subjects were required to have a positive FLT3‐ITD test performed by the local center prior to HCT to be eligible for study enrollment. However, this requirement was removed on November 15, 2012, following enrollment of the first 7 subjects though all subsequent patients enrolled had a FLT3‐ITD mutation. Classification of subjects according to FLT3‐ITD levels (i.e., high, low) was not performed. Assessment of minimal residual disease following HCT was not required prior to initiating therapy with quizartinib.
2.5. Tolerability and safety assessments
Safety assessments were performed at baseline and throughout the study. Primary safety variables included AEs, DLTs, acute and chronic GVHD evaluations, physical examinations, vital signs, electrocardiograms (ECGs), CBCs, chemistry evaluations, coagulation (PT, PTT, INR) evaluations, and urinalyses. All AEs were graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) v. 4.03.40 Routine laboratory assessments for hematology, chemistry, coagulation, and urinalysis were collected and analyzed at local accredited laboratories within participating centers.
2.6. Efficacy assessments
Bone marrow aspirates and/or biopsies were required for determination of AML status, including morphology. Bone marrow biopsies could be omitted at the discretion of the investigator if the aspirate was considered adequate. Criteria used for disease assessment have been published.41, 42 Response was assessed at each disease assessment, with consideration of hematology labs, bone marrow evaluation, transfusion status, and cytogenetics, if appropriate. Time points for disease assessment were per institutional standard for post‐transplant monitoring and with consideration of the date of transplant.
2.7. PK assessments
Plasma concentrations of quizartinib and AC886 were determined using a validated liquid chromatography with tandem mass spectrometry (LC/MS/MS) assay methodology by BASi (West Lafayette, IN, USA). PK analyses were performed for subjects receiving at least 1 dose of quizartinib and for whom sufficient concentration data were available to facilitate derivation of at least 1 primary PK parameter. PK parameters included area under the curve (AUC), maximum concentration (C max), trough concentration (C trough), and time to peak concentration (T max).
2.8. Statistical analyses
Planned enrollment for the study was approximately 30 subjects, with up to 18 subjects in the dose‐escalation phase (part 1) and approximately 12 subjects in the MTD evaluation phase (part 2), allowing for treatment of at least 18 subjects at the MTD. All data processing, summarization, and analyses were performed using SAS® Version 9.1 or higher.
Safety analyses consisted of data summaries for AEs, DLTs, and other safety parameters and were conducted on all subjects receiving at least 1 dose of quizartinib. AEs were coded to system organ class and preferred term using MedDRA terminology. Number and percent of subjects experiencing 1 or more AE(s) were summarized by cohort. Relationship to study drug and severity of AEs were also summarized. AEs leading to permanent discontinuation of study drug, and serious AEs (SAEs) were summarized by NCI‐CTCAE grade and relationship to study drug. Laboratory parameters, including hematology, urinalysis, serum chemistry, and coagulation were summarized by cohort using descriptive statistics and by evaluating shifts in change from baseline and data listings of clinically significant abnormalities. Vital signs and ECG parameters and clinically significant changes from baseline were summarized by cohort using descriptive statistics
Efficacy data analyses were conducted on all subjects who received at least 1 dose of quizartinib, had no major protocol deviations related to efficacy, and had at least 1 nonmissing post‐baseline efficacy measurement. Duration of confirmed CR, duration of CR, DFS, and overall survival (OS) were summarized using descriptive statistics. The survival curve and the median for time‐to‐event variables were estimated using the Kaplan‐Meier method and were reported along with the corresponding 95% confidence interval. Transplant‐related outcomes were analyzed and summarized using descriptive statistics. Numbers and percentages of transplant rejection, GVHD, chimerism, and treatment‐related mortality (TRM) were summarized by cohort.
Plasma concentrations and PK parameters were summarized by cohort using descriptive statistics, including number of subjects, mean, standard deviation, minimum, median, maximum, geometric mean, and coefficient of variation (CV) of the mean and geometric mean.
3. RESULTS
3.1. Demographics
A total of 13 subjects were enrolled between June 2012 and January 2015. One subject dosed at 40 mg/d experienced relapse during cycle 1 and discontinued treatment. For this reason, 1 additional subject was recruited for a total of 7 subjects and 6 evaluable subjects dosed at 40 mg/d. Six subjects received 60 mg/d (Supporting Information Figure S1). All subjects discontinued therapy: 5 (38%) due to completion of all 24 cycles of therapy, 4 (31%) due to SAEs (1 subject with corneal epithelium defect; 1 subject with Epstein‐Barr–associated lymphoproliferative disorder; 1 subject with neutropenia; and 1 subject with pneumonia, GVHD, and peritoneal hemorrhage), 2 (15%) due to investigator discretion, and 1 (8%) due to disease progression. For 1 remaining subject (8%) the cause of discontinuation was listed as unspecified; however, the subject completed the protocol‐specified 24 cycles of treatment. Baseline characteristics were generally similar between the 2 treatment groups (Table 1). Median age was 43 (range, 23, 61) years. All 13 subjects were positive for FLT3‐ITD mutations at diagnosis by local testing, prior to receiving an HLA‐matched allogeneic transplant. Twelve subjects (92%) had intermediate or unfavorable cytogenetic risk, and risk was unknown for 1 subject (8%). Six subjects (46%) had detectable NPM1 mutation. Ten subjects (77%) received HCT transplant from an unrelated donor. Ten subjects (77%) received HCT while in CR1. One subject previously failed an allogeneic transplant from her sibling, and the current transplant was from an unrelated donor.
Table 1.
Baseline patient characteristics
| Starting dose | |||
|---|---|---|---|
|
40 mg N = 7 |
60 mg N = 6 |
Total N = 13 (%) |
|
| Age, median years (range) | 43.0 (27, 59) | 49.5 (23, 61) | 43.0 (23, 61) |
| Sex, n | |||
| Male | 3 | 4 | 7 (54) |
| Female | 4 | 2 | 6 (46) |
| Cytogenetic risk,a n | |||
| Favorable | 0 | 0 | 0 |
| Intermediate | 5 | 3 | 8 (62) |
| Unfavorable | 1 | 3 | 4 (31) |
| Unknown | 1 | 0 | 1 (8) |
| NPM1 mutation, n | |||
| Yes | 4 | 2 | 6 (46) |
| No | 3 | 4 | 7 (54) |
| Prior Transplant | 1 | 0 | 1 (8) |
| Prior FLT3 inhibitor treatment, n | |||
| Yes | 3 | 4 | 7 (54) |
| No | 4 | 2 | 6 (46) |
| Allogeneic transplant donor type, n | |||
| Related | 4 | 0 | 4 (31) |
| Unrelated | 3 | 6 | 9 (69) |
| Outcome of transplant, n | |||
| Continued CR | 6 | 6 | 12 (92) |
| Relapse | 1 | 0 | 1 (8) |
CR, complete remission; NPM1, Nucleophosmin 1.
Grimwade D, Walker H, Harrison G, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML 11 Trial. Blood. 2001;98(5):1312–1320.
3.2. Disposition and treatment exposure
At 40 mg/d, 1 of 7 subjects (14%) had a DLT, 4 (57%) had dose interruption due to AEs, and 2 (29%) had dose reduction as allowed per protocol to 30 mg/d. Median number of treatment cycles was 24.0 (min, max: 1–24). Median daily dose per subject was 39.2 mg (min, max: 28–42). At 60 mg/d, 1 of 6 subjects (17%) had a DLT, 3 (50%) had dose interruption due to AEs, and 2 (33%) had dose reduction (1 to 40 mg/d and 1 to 30 mg/d). Median number of treatment cycles was 20.5 (min, max: 10–24). Median daily dose per subject was 56.9 mg (min, max: 31–60).
3.3. Pharmacokinetics
Pharmacokinetic analyses were limited by small number of subjects and samples. Following administration of 40 mg or 60 mg quizartinib once‐daily, median time to peak quizartinib plasma concentration (t max) occurred between 2.00 h and 3.21 h on days 1 and 15, with peak plasma concentrations (C max) approximately 3‐fold to 4‐fold higher on Day 15 compared to Day 1 (Figure 1A,B), and an approximately 5‐fold increase in geometric mean AUC0–24 at both doses during multiple dosing. By contrast, plasma concentrations of AC886 were variable with t max occurring over a large range (from 15.25 h after 60 mg to 23.67 h after 40 mg), and suggesting delayed formation of the metabolite. AC886 C max was approximately 5‐fold to 8‐fold higher on Day 15 compared to Day 1 (Figure 1C,D), with an approximately 6‐fold (40 mg) to 9‐fold (60 mg) increase in geometric mean AUC0–24 during multiple dosing. Plasma quizartinib and AC886 trough concentrations reached steady state by approximately day 15 following daily administration. Relatively high variability was observed for quizartinib and AC886 trough concentrations, reflecting the small number of subjects providing samples. However, mean quizartinib and AC886 trough concentrations remained relatively consistent from cycle 2 onward at >300 ng/mL and >75 ng/mL, respectively (Figure 1E,F).
Figure 1.
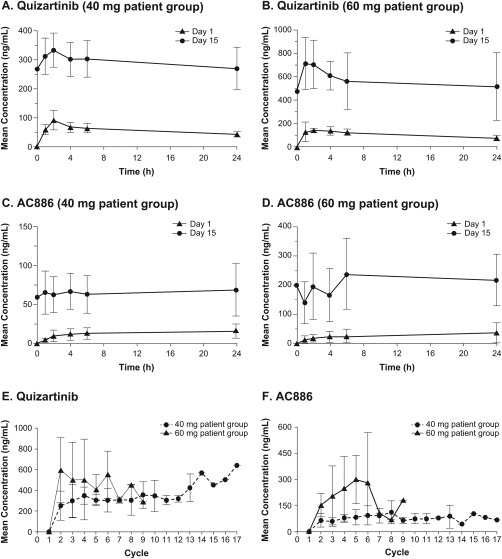
Mean Quizartinib/AC886 Peak and Trough Plasma Concentrations. Mean quizartinib plasma peak concentrations increased 3‐fold to 4‐fold after 15 days of continued daily quizartinib administration at 40 mg/d [Panel A] and 60 mg/d [Panel B]. Mean AC886 plasma peak concentrations increased 5‐fold to 8‐fold after 15 days of continued daily quizartinib administration at 40 mg/d [Panel C] and 60 mg/d [Panel D]. Mean plasma trough concentrations of quizartinib [Panel E] and AC886 [Panel F] remained consistent from cycle 2 onward following continuous administration of quizartinib after allo‐HCT
3.4. Safety
There were no documented cases of reductions in donor chimerism reported in any subjects. Use of G‐CSF was reported for 6 subjects (46%). No subjects had platelet counts below the threshold value of <20 × 109 while on study drug (Supporting Information Figure S2). Two subjects (15%) had ANC values below the threshold value of <500 × 106. One subject had a nadir ANC value of 360 × 106 on study day 29 and spent 4.5% of study duration below the ANC threshold value. A second subject had a nadir ANC value of 200 × 106 on study day 114, with undetectable ANC counts on study day 141, and spent 24.8% of study duration below the ANC threshold value. Neither subject recovered above the ANC threshold value (500 × 106) during the study.
Rates of grade 3/4 AEs were similar for both treatment groups. The most common grade 3/4 AEs were hematologic (Table 2) and included neutropenia (23%), leukopenia (15%), anemia (15%), thrombocytopenia (15%), and lymphopenia (15%). Other grade 3/4 AEs reported included corneal epithelium defect, retinal infarction, gastric hemorrhage, and pneumonia. A total of 13 GVHD events were reported in a total of 9 subjects (69%): 5 (38%) grade 1, 3 (23%) grade 2, and 1 (8%) grade 3. Ten events were classified as acute GVHD and 3 were classified as chronic GVHD. Seven subjects (54%) experienced grade 1/2 QTcF prolongation (>450 to ≤500 ms), 3 were treated at 40 mg and 4 were treated at 60 mg. Six of these 7 subjects were taking co‐medications associated with QT prolongation (1 subject moxifloxacin, 1 subject azithromycin, 3 subjects prochlorperazine, 1 subject azithromycin and prochlorperazine). There were no reports of grade 3 QTcF prolongation (>500 ms) or treatment‐emergent AE of ECG QT prolongation for any subject. One subject in each dose group had a maximum QTcF change from baseline of >60 msec. No subjects had dose reductions or interruptions due to QTcF prolongation events. There were no cases of Torsade de pointes. One subject who discontinued treatment on study day 574 experienced a grade 5 SAE of peritoneal hemorrhage on study day 610, which resulted in death. This event was considered not related to study treatment.
Table 2.
Most common grade 3 or 4 treatment‐related adverse events by dose cohort
| Starting Dose | |||
|---|---|---|---|
|
40 mg N = 7 |
60 mg N = 6 |
TOTAL N = 13 (%) |
|
| Hematologic, n | |||
| Neutropenia | 2 | 1 | 3 (23) |
| Leukopenia | 1 | 1 | 2 (15) |
| Anemia | 1 | 1 | 2 (15) |
| Thrombocytopenia | 1 | 1 | 2 (15) |
| Lymphopenia | 2 | 0 | 2 (15) |
| Nonhematologic, n | |||
| Corneal epithelium defect | 0 | 1 | 1 (8) |
| Retinal infarction | 1 | 0 | 1 (8) |
| Gastric hemorrhage | 1 | 0 | 1 (8) |
| Pneumonia | 0 | 1 | 1 (8) |
| Investigations, n | |||
| Lymphocyte count decreased | 0 | 1 | 1 (8) |
| Neutrophil count decreased | 0 | 1 | 1 (8) |
| White blood cell count decreased | 0 | 1 | 1 (8) |
| GVHD on study,a n | 0 | 1 | 1 (8) |
GVHD, graft versus host disease.
This instance of GVHD was deemed not to be related to study drug.
3.5. Efficacy
Ten subjects (77%) received treatment with quizartinib for >1 year, 6 (46%) received treatment for close to 2 years (95 to 99 weeks) (Figure 2A). Five subjects (38%) completed quizartinib treatment. Relapse was observed in only 1 subject (8%) receiving quizartinib, which occurred in the first cycle. Ten subjects were still alive at the end of the study, and 3 had died (1 disease progression, 1 peritoneal hemorrhage, 1 unknown causes). OS ranged from approximately 13 weeks to 142 weeks, with 9 subjects (69%) surviving ≥50 weeks and 4 subjects (31%) surviving >2 years (104 weeks). There was no significant difference in OS between treatment groups (Figure 2B).
Figure 2.
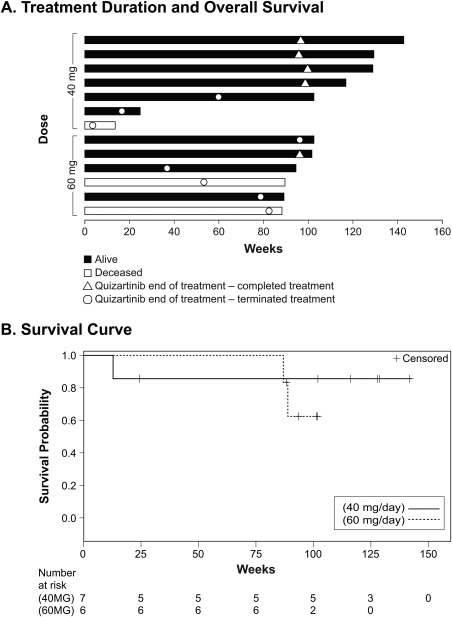
Treatment Duration and Overall Survival. A. Six subjects (46%) received treatment for almost 2 years (95–99 weeks). Ten subjects (77%) were alive at end of study. Nine subjects (69%) survived >50 weeks and 4 (31%) survived >2 years. B. There was no significant difference in OS between the 2 treatment groups
4. DISCUSSION
The prognosis for subjects with FLT3‐ITD–mutated AML is very poor, with higher risk of relapse and shorter interval prior to relapse and an estimated median survival <5 months after first relapse.43, 44 Moreover, some studies have demonstrated an increase in FLT3‐ITD mutant allelic ratio in subjects with relapsed AML.45, 46, 47 Thus, the presence of FLT3‐ITD mutation defines a high‐risk population of AML in need of more efficacious therapies. The phase 3 RATIFY study demonstrated a significant OS benefit in subjects with newly diagnosed AML harboring FLT3 mutations who received midostaurin in combination with standard first line chemotherapy.48 However, it is unclear if the improvement in outcomes observed with midostaurin is related to inhibition of FLT3 or other effects. Based on results of the RATIFY study, the US Food and Drug Administration approved midostaurin in combination with chemotherapy for treatment of adult patients with newly diagnosed AML who have FLT3 mutations. While allo‐HCT has been shown to confer improved outcomes in subjects with FLT3‐ITD–mutated AML, this patient population retains an increased frequency of relapse after transplantation, underscoring the potential selective advantage of FLT3‐ITD–mutated disease and the need for more effective therapies. A phase 1 trial evaluating the tyrosine kinase inhibitor sorafenib as maintenance therapy in subjects with FLT3‐ITD–mutated AML following HCT demonstrated 1‐year progression‐free survival (PFS) of 85% and 1‐year OS of 95%.49 However, there are currently no approved therapies for patients with AML who experience treatment failure/disease relapse following allo‐HCT.
The goal of maintenance therapy is to eradicate MRD post standard of care induction/consolidation chemotherapy. Administration of long‐term maintenance therapy has been used successfully for hematologic malignancies, including acute lymphoblastic leukemia and multiple myeloma, but it has not been integrated into standard treatment of subjects with AML.26, 50 The strong association between FLT3‐ITD mutation and poor outcomes in individuals undergoing consolidation HCT identifies a patient population with high unmet need and a potential druggable target.
Quizartinib is a novel FLT3 inhibitor that possesses greater selectivity and potency toward FLT3‐ITD versus early FLT3 inhibitors, including midostaurin, sorafenib, and sunitinib.35, 51, 52, 53, 54
This is the first study to examine the safety and activity of quizartinib when administered as maintenance therapy in subjects with FLT3‐ITD–mutated AML who underwent allo‐HCT. Post‐transplant maintenance treatment was associated with low rates of grade 3/4 AEs. Quizartinib‐associated AEs were manageable with dose adjustments and/or interruptions, and median of treatment cycles was 21. The majority of AEs reported were hematologic; however, satisfactory overall blood counts were maintained for 11 of 13 subjects throughout the duration of the trial. The 69% rate of GVHD observed following treatment with quizartinib was consistent with GVHD rates previously reported for patients with AML who underwent allo‐HCT (acute: 43%‐80%; chronic: 34%–49%).23, 55, 56, 57, 58, 59, 60 Additionally, quizartinib and was not associated with grade 3 QTcF prolongation in any subjects at doses of 40 mg/d or 60 mg/d. The rate of grade ≥2 QTcF prolongation observed in this study was consistent with the reduced rates of QTcF prolongation reported in an earlier phase 2 trial that evaluated lower doses of quizartinib (30–60 mg/d) in subjects with R/R AML.33
Steady‐state levels of quizartinib achieved in study subjects were well above the IC50 values for FLT3 inactivation and inhibition of leukemic growth in peripheral blood reported in preclinical studies,51, 52, 54 as well as within concentrations predicted to be needed for complete inhibition of leukemic growth in bone marrow.61 Although PK analyses were limited by the small number of subjects and by dose adjustments and/or interruptions in a majority of subjects, we observed a stable PK profile for both quizartinib and its active metabolite, AC886, with continuous administration—a property compatible with long‐term dosing. This contrasts with the multitarget kinase inhibitor midostaurin, for which plasma concentrations have been reported to decrease during continuous dosing, potentially due to induction of enzymes that metabolize the drug.62 Although an MTD was not reached, quizartinib dose was not escalated above 60 mg/d. While a DLT was observed at 60 mg/d, this dose was selected as MTD for continuous daily administration as it has demonstrated strong efficacy and lower rates of QT prolongation in previous quizartinib studies,33, 63 and, if necessary, dose can be reduced to help further improve tolerability.
Limitations of our study include a small patient size and lack of a control arm. The study was terminated early, and total study enrollment was limited to 13 subjects in part 1 of the study. These factors led to more limited readouts on safety, efficacy, and PK than initially planned for the study. However, encouraging safety and efficacy results were observed in this population of subjects harboring FLT3‐ITD mutations, with a majority receiving more than 1 year of treatment with quizartinib as post‐maintenance therapy following allo‐HCT. Results show acceptable tolerability of quizartinib, as well as early evidence of a reduced relapse rate following allo‐HCT (8%) compared to previously reported rates in subjects with FLT3‐ITD–mutated AML (30%‐59%),19, 21, 23, 24, 25 and support further clinical evaluation. The optimal duration of quizartinib maintenance therapy is unknown. However, given that there is elevated risk of relapse in subjects with FLT3‐ITD–mutated AML within the first 24 months following HCT,23 it would appear to be optimal to continue maintenance therapy in these patients for at least 2 years. The evaluation of quizartinib as posttransplant maintenance therapy has been included in an ongoing randomized phase 3 study (QuANTUM‐R, NCT02039726) comparing quizartinib with salvage chemotherapy in FLT3‐ITD–mutated R/R AML. Additionally, the phase 3 QuANTUM‐First study (NCT02668653) is evaluating quizartinib in combination with standard of care induction/consolidation chemotherapy and as maintenance therapy in newly diagnosed patients with FLT3‐ITD–mutated AML. These studies will provide valuable information on the benefit and optimal duration of quizartinib maintenance therapy in AML patients harboring FLT3‐ITD mutation.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This study was designed by the principal investigator (BMS) and was funded by Daiichi‐Sankyo. Data were collected by the investigators and analyzed by biostatisticians employed by the sponsor. All authors participated in study conduct, data interpretation, writing, reviewing, and/or amending the manuscript. All authors have completed a disclosure declaration. The following authors have indicated a financial, or other, interest that is relevant to the subject matter covered in this article: DT and GG are former employees of the study sponsor. BO has received research funding from AROG Pharmaceuticals, Inc. SK has served as a consultant or in an advisory role for the study sponsor. SK and GG have received honoraria, research funding, or other remuneration from the study sponsor. Preliminary results of this study were previously presented at the 2014 American Society of Hematology annual meeting; December 6–9, 2014; San Francisco, CA. Editorial assistance was provided by Roy Garcia, PhD, ETHOS Health Communications, Yardley, Pennsylvania, with financial assistance from Daiichi‐Sankyo, Edison, New Jersey, in compliance with international guidelines on Good Publication Practice.
Sandmaier BM, Khaled S, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am J Hematol. 2018;93:222–231. 10.1002/ajh.24959
REFERENCES
- 1. De Kouchkovsky I, Abdul‐Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. [DOI] [PubMed] [Google Scholar]
- 3. Alibhai SM, Leach M, Gupta V, et al. Quality of life beyond 6 months after diagnosis in older adults with acute myeloid leukemia. Crit Rev Oncol Hematol. 2009;69(2):168–174. [DOI] [PubMed] [Google Scholar]
- 4. National Cancer Institute . Surveillance Epidemiology and End Results (SEER) Program. Cancer Stat Facts: Acute Myeloid Leukemia (AML). Available online at: https://seercancergov/statfacts/html/amyl.html 2017. Accessed November 8, 2017.
- 5. Estey EH, Thall PF, Pierce S, et al. Randomized phase II study of fludarabine + cytosine arabinoside + idarubicin +/‐ all‐trans retinoic acid +/‐ granulocyte colony‐stimulating factor in poor prognosis newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Blood. 1999;93(8):2478–2484. [PubMed] [Google Scholar]
- 6. Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. [DOI] [PubMed] [Google Scholar]
- 7. Grimwade D, Mrozek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am. 2011;25(6):1135–1161. [DOI] [PubMed] [Google Scholar]
- 8. Natonal Comprehensive Cancer Network . NCCN Clinical Practice Guidelines in Oncology: Acute Myeloid Leukemia. Version 3; 2017. Available online at: https://www.nccn.org. Accessed November 7, 2017.
- 9. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues 4th ed. Lyon, France; IARC/WHO Press; 2008.
- 10. Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N‐RAS gene mutations in acute myeloid leukemia. Blood. 1999;93(9):3074–3080. [PubMed] [Google Scholar]
- 11. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–1759. [DOI] [PubMed] [Google Scholar]
- 12. Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100(1):59–66. [DOI] [PubMed] [Google Scholar]
- 13. Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3‐activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. [DOI] [PubMed] [Google Scholar]
- 14. Whitman SP, Archer KJ, Feng L, et al. Absence of the wild‐type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–7239. [PubMed] [Google Scholar]
- 15. Kayser S, Schlenk RF, Londono MC, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain‐1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114(12):2386–2392. [DOI] [PubMed] [Google Scholar]
- 16. Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematol Am Soc Hematol Educ Program. 2013;2013:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boissel N, Cayuela JM, Preudhomme C, et al. Prognostic significance of FLT3 internal tandem repeat in patients with de novo acute myeloid leukemia treated with reinforced courses of chemotherapy. Leukemia. 2002;16(9):1699–1704. [DOI] [PubMed] [Google Scholar]
- 18. Fey MF, Buske C, Group EGW. Acute myeloblastic leukaemias in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2013;24(Suppl 6):vi138–vi143. [DOI] [PubMed] [Google Scholar]
- 19. Bornhauser M, Illmer T, Schaich M, et al. Improved outcome after stem‐cell transplantation in FLT3/ITD‐positive AML. Blood. 2007;109(5):2264–2265. [DOI] [PubMed] [Google Scholar]
- 20. DeZern AE, Sung A, Kim S, et al. Role of allogeneic transplantation for FLT3/ITD acute myeloid leukemia: outcomes from 133 consecutive newly diagnosed patients from a single institution. Biol Blood Marrow Transplant. 2011;17(9):1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gale RE, Hills R, Kottaridis PD, et al. No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML10 and 12 trials. Blood. 2005;106(10):3658–3665. [DOI] [PubMed] [Google Scholar]
- 22. Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood. 2016;127(1):62–70. [DOI] [PubMed] [Google Scholar]
- 23. Brunet S, Labopin M, Esteve J, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol. 2012;30:735–741. [DOI] [PubMed] [Google Scholar]
- 24. Schmid C, Labopin M, Socie G, et al. Outcome of patients with distinct molecular genotypes and cytogenetically normal AML after allogeneic transplantation. Blood. 2015;126(17):2062–2069. [DOI] [PubMed] [Google Scholar]
- 25. Sengsayadeth SM, Jagasia M, Engelhardt BG, et al. Allo‐SCT for high‐risk AML‐CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplant. 2012;47(12):1535–1537. [DOI] [PubMed] [Google Scholar]
- 26. Baer MR. Is there a role for maintenance therapy in acute myeloid leukaemia? Best Pract Res Clin Haematol. 2009;22(4):517–521. [DOI] [PubMed] [Google Scholar]
- 27. Fiedler W, Serve H, Dohner H, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2004;105(3):986–993. [DOI] [PubMed] [Google Scholar]
- 28. Röllig C, Müller‐Tidow C, Hüttmann A, et al. Sorafenib versus placebo in addition to standard therapy in younger patients with newly diagnosed acute myeloid leukemia: results from 267 patients treated in the randomized placebo‐controlled SAL‐Soraml trial. Blood. 2014;124:6. [Google Scholar]
- 29. Schiller GJ, Tuttle P, Desai P. Allogeneic hematopoietic stem cell transplantation in FLT3‐ITD‐positive acute myelogenous leukemia: the role for FLT3 Tyrosine kinase inhibitors post‐transplantation. Biol Blood Marrow Transplant. 2016;22(6):982–990. [DOI] [PubMed] [Google Scholar]
- 30. Stone RM, Mandrekar S, Sanford BL, et al. The multi‐kinase inhibitor Midostaurin (M) prolongs survival compared with Placebo (P) in combination with daunorubicin (D)/Cytarabine (C) Induction (ind), high‐dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute Myeloid Leukemia (AML) Patients (pts) Age 18–60 with FLT3 Mutations (muts): An International Prospective Randomized (rand) P‐Controlled Double‐Blind Trial (CALGB 10603/RATIFY. Blood. 2015;126:6. 26138538 [Google Scholar]
- 31. Altman JK, Foran JM, Pratz KW, et al. Results of a phase 1 study of quizartinib (AC220, ASP2689) in combination with induction and consolidation chemotherapy in younger patients with newly diagnosed acute myeloid leukemia. Blood. 2013;122:623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burnett AK, Bowen D, Russell N, et al. AC220 (Quizartinib) can be safely combined with conventional chemotherapy in older patients with newly diagnosed acute myeloid leukaemia: experience from the AML18 pilot trial. Blood. 2013;122:622. [Google Scholar]
- 33. Cortes JE, Perl AE, Dombret H, et al. Final results of a phase 2 open‐label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥ 60 years of age with FLT3 ITD positive or megative relapsed/refractory acute myeloid leukemia. Blood. 2012;120:48. [Google Scholar]
- 34. Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS‐like tyrosine kinase 3‐internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cortes JE, Perl AE, Dombret H, et al. Response rate and bridging to hematopoietic stem cell transplantation (HSCT) with quizartinib (AC220) in patients with FLT3‐ITD positive or negative relapsed/refractory AML after secondline chemotherapy or previous bone marrow transplant. J Clin Oncol. 2013;31:7012. [Google Scholar]
- 36. Dohner H, Perf A, Rousselot P, et al. Efficacy and safety of Quizartinib in patients age ≥60 years with FLT3‐ITD‐positive relapsed/refractory acute myeloid leukemia (AML). Haematologica. 2013;98:233. [Google Scholar]
- 37. Hills RK, Gammon G, Trone D, et al. Quizartinib significantly improves overall survival in FLT3‐ITD positive AML patients relapsed after stem cell transplantation or after failure of salvage chemotherapy: a comparison with historical AML database (UK NCRI data). Blood. 2015;126:2557. [Google Scholar]
- 38. Levis M, Martinelli G, Perl AE, et al. The benefit of treatment with quizartinib and subsequent bridging to HSCT for FLT3‐ITD(+) patients with AML. J Clin Oncol. 2014;32:7093. [Google Scholar]
- 39. Russel N, Tallman MS, Goldberg S, et al. Quizartinib (AC220) in patients with FLT3‐ITD(+) relapsed of refractory acute myeloid leukemia: final results of a randomized phase 2 study. Haematologica. 2014;99:36. [Google Scholar]
- 40. National Institute Health, Institute NC . Cancer Therapy Evaluation Program (CTEP): Common Terminology Criteria for Adverse Events (CTCAE) v4.0 Stat Fact Sheets. Available online at: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed November 7, 2017.
- 41. Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21:4642–4649. [DOI] [PubMed] [Google Scholar]
- 42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33‐positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–3254. [DOI] [PubMed] [Google Scholar]
- 43. Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117(12):3294–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ravandi F, Kantarjian H, Faderl S, et al. Outcome of patients with FLT3‐mutated acute myeloid leukemia in first relapse. Leuk Res. 2010;34(6):752–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kottaridis PD, Gale RE, Langabeer SE, et al. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100(7):2393–2398. [DOI] [PubMed] [Google Scholar]
- 46. Nakano Y, Kiyoi H, Miyawaki S, et al. Molecular evolution of acute myeloid leukaemia in relapse: unstable N‐ras and FLT3 genes compared with p53 gene. Br J Haematol. 1999;104(4):659–664. [DOI] [PubMed] [Google Scholar]
- 47. Shih LY, Huang CF, Wu JH, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100(7):2387–2392. [DOI] [PubMed] [Google Scholar]
- 48. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017;377(5):454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen YB, Li S, Lane AA, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms‐like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant. 2014;20(12):2042–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Canaani J, Luger SM. Revisiting maintenance therapy in acute myeloid leukemia with novel agents. Curr Opin Hematol. 2016;23(2):175–180. [DOI] [PubMed] [Google Scholar]
- 51. Chao Q, Sprankle KG, Grotzfeld RM, et al. Identification of N‐(5‐tert‐butyl‐isoxazol‐3‐yl)‐N′‐{4‐[7‐(2‐morpholin‐4‐yl‐ethoxy)imidazo[2,1‐b][1, 3]benzothiazol‐2‐yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS‐like tyrosine kinase‐3 (FLT3) inhibitor. J Med Chem. 2009;52(23):7808–7816. [DOI] [PubMed] [Google Scholar]
- 52. Gunawardane RN, Nepomuceno RR, Rooks AM, et al. Transient exposure to quizartinib mediates sustained inhibition of FLT3 signaling while specifically inducing apoptosis in FLT3‐activated leukemia cells. Mol Cancer Ther. 2013;12(4):438–447. [DOI] [PubMed] [Google Scholar]
- 53. Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abdul Wahid SF, Ismail NA, Mohd‐Idris MR, et al. Comparison of reduced‐intensity and myeloablative conditioning regimens for allogeneic hematopoietic stem cell transplantation in patients with acute myeloid leukemia and acute lymphoblastic leukemia: a meta‐analysis. Stem. Cells Dev. 2014;23(21):2535–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Flowers ME, Inamoto Y, Carpenter PA, et al. Comparative analysis of risk factors for acute graft‐versus‐host disease and for chronic graft‐versus‐host disease according to National Institutes of Health consensus criteria. Blood. 2011;117(11):3214–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Horowitz MM, Gale RP, Sondel PM, et al. Graft‐versus‐leukemia reactions after bone marrow transplantation. Blood. 1990;75(3):555–562. [PubMed] [Google Scholar]
- 58. Inamoto Y, Flowers ME, Lee SJ, et al. Influence of immunosuppressive treatment on risk of recurrent malignancy after allogeneic hematopoietic cell transplantation. Blood. 2011;118(2):456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Storb R, Gyurkocza B, Storer BE, et al. Graft‐versus‐host disease and graft‐versus‐tumor effects after allogeneic hematopoietic cell transplantation. J Clin Oncol. 2013;31:1530–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weisdorf D, Zhang MJ, Arora M, et al. Graft‐versus‐host disease induced graft‐versus‐leukemia effect: greater impact on relapse and disease‐free survival after reduced intensity conditioning. Biol Blood Marrow Transplant. 2012;18(11):1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Levis MJ, Cortes JE, Gammon GM, et al. Laboratory and clinical investigations to identify the optimal dosing strategy for quizartinib (AC220) monotherapy in FLT3‐ITD‐positive (+) relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood. 2016;128:4042. [Google Scholar]
- 62. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small‐molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. [DOI] [PubMed] [Google Scholar]
- 63. Cortes J, Tallman MS, Schiller G, et al. Results of a phase 2 randomized, open‐label, study of lower doses of Quizartinib (AC220; ASP2689) in subjects with FLT3‐ITD positive relapsed or refractory acute myeloid leukemia (AML). Blood. 2013;122:494‐494. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information
Supporting Information