

Abstract
The objective of this study was to evaluate the relative bioavailability of olanzapine in 3 olanzapine‐containing tablet formulations. ALKS 3831 is a fixed‐dose combination of olanzapine (OLZ, an atypical antipsychotic) and samidorphan (SAM, a μ‐opioid receptor antagonist with low intrinsic activity at δ‐ and κ‐opioid receptors), intended to provide the efficacy of OLZ while mitigating its known weight and metabolic effects. Relative bioavailability of OLZ in ALKS 3831, a bilayer tablet containing OLZ and SAM, a matching bilayer tablet containing OLZ only (OLZ), and Zyprexa (brand olanzapine [B‐OLZ]) was assessed in an open‐label study. Forty‐eight healthy volunteers were randomly assigned to receive single oral doses of ALKS 3831 (10 mg OLZ/10 mg SAM), OLZ (10 mg OLZ), and B‐OLZ (10 mg B‐OLZ) on day 1 of each treatment period. Blood samples for pharmacokinetic evaluation were collected before and after each dose. Ratios of OLZ AUC0‐∞, AUC0‐t, and Cmax were compared between treatments and tested for bioequivalence, determined by 90%CIs of the geometric mean ratios (GMRs). GMRs of OLZ AUC0‐∞, AUC0‐t, and Cmax were close to 1, and the 90%CIs of the GMRs were contained within the bioequivalence limit of 80%–125% for comparison of ALKS 3831 with B‐OLZ, ALKS 3831 with OLZ, and OLZ with B‐OLZ, demonstrating bioequivalence of OLZ in ALKS 3831, OLZ, and B‐OLZ.
Keywords: pharmacokinetics, bioavailability, bioequivalence, olanzapine, samidorphan
Schizophrenia is a complex and chronic disorder that requires long‐term treatment with antipsychotic medication, with the main goals of treatment being a reduction in symptoms, increased functionality and quality of life, and prevention of relapse.1 Olanzapine (OLZ) is regarded as one of the most efficacious antipsychotics, with well‐recognized efficacy and advantages such as decreased incidence of extrapyramidal symptoms. However, OLZ also has safety and tolerability limitations including pervasive weight gain and metabolic abnormalities that are associated with significant health risks that can affect adherence and retention of patients on OLZ therapy.2 Samidorphan (SAM) is a novel entity that intends to address the significant risk of weight gain and metabolic consequences associated with OLZ treatment. In vivo, SAM has been demonstrated to function as a μ‐opioid antagonist.3 In vitro, SAM binds with high affinity to human μ‐, κ‐, and δ‐opioid receptors and acts as an antagonist at μ‐opioid receptors, with low intrinsic activity at κ‐ and δ‐opioid receptors.4 In a phase 2 study in subjects with schizophrenia, coadministration of OLZ with SAM had antipsychotic efficacy that was similar to OLZ monotherapy, whereas significantly mitigating OLZ‐induced weight gain.5 Based on the observed ratio for weight mitigation and the safety profile observed in the phase 2 study,5 a fixed dose of SAM (10 mg) in combination with multiple dose strengths of OLZ (5, 10, 15, and 20 mg) was selected for further investigation in phase 3 studies.
In clinical studies in healthy adults, SAM was well tolerated after single oral doses of up to 55.7 mg and multiple daily doses of up to 20 mg/day and exhibited a pharmacokinetic (PK) profile suitable for a once‐daily dosing regimen.6 In addition to being developed in combination with OLZ for the treatment of schizophrenia, SAM is also under evaluation in a fixed‐dose combination with buprenorphine for the adjunctive treatment of major depressive disorder.7, 8
ALKS 3831 is a fixed‐dose combination, immediate‐release bilayer tablet with 1 layer containing OLZ and the other layer containing SAM. An OLZ‐alone bilayer tablet with an appearance identical to ALKS 3831 but containing OLZ in 1 layer and placebo in the other layer was developed for use as a matching comparator medication in double‐blind phase 3 efficacy studies. To support the use of ALKS 3831 bilayer tablets and OLZ‐alone bilayer tablets in phase 3 studies, the present pharmacokinetic study was carried out to evaluate the relative oral bioavailability of OLZ in ALKS 3831, OLZ, and Zyprexa (from here on referred to as brand olanzapine [B‐OLZ]).
Methods
Study Design and Treatment Schedule
This was a phase 1 single‐center, open‐label, randomized study with 3 treatment periods and 6 treatment sequences. The study was conducted at a single phase 1 clinic (Vince & Associates Clinical Research, an Altasciences company, Overland Park, Kansas) in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines agreed on by the International Conference on Harmonization (1997). The study protocol and informed consent forms were reviewed and approved by the clinical site's institutional review board (Midlands Independent Review Board) before any subjects were enrolled in the study. The study was designed and powered to evaluate the bioequivalence of OLZ in the 3 OLZ‐containing tablet formulations in accordance with the US Food and Drug Administration (FDA) Draft Guidance on Bioavailability and Bioequivalence Studies Submitted in new drug applications (NDAs) or investigational new drug applications (INDs)9 and Guidance on Statistical Approaches to Establishing Bioequivalence.10
Healthy, nonsmoking subjects with no clinically relevant conditions were eligible for inclusion. Key inclusion criteria were male and female subjects aged 18–40 years with a body mass index (BMI) of 18.0–30.0 kg/m2 and in good physical health, as determined by medical history, physical examination, and laboratory assessments at screening. Subjects were excluded from the study if they had a history of any clinically significant medical or psychiatric condition, an absolute neutrophil count ≤ 1.5 × 103/μL, a history of diabetes or glycated hemoglobin > 6.0%, risk of narrow‐angle glaucoma, abnormal electrocardiogram results, a history of dependence on any substance other than caffeine, positive serologic test for hepatitis B, hepatitis C, or human immunodeficiency virus, prior use of any antipsychotic medication, use of any product containing nicotine within 90 days before screening or intention to use any product containing nicotine during the course of the study, history of any movement disorder, neuroleptic malignant syndrome, or suicidality.
Subjects were screened during a 21‐day period before the first treatment period. Eligible subjects were randomly assigned in a 1:1:1:1:1:1 ratio to 1 of the 6 treatment sequences (n = 8 subjects per sequence group), in which they would receive single doses of ALKS 3831 (10 mg OLZ/10 mg SAM), OLZ (10 mg OLZ), or B‐OLZ (10 mg OLZ) on the first day (day 1) of each treatment period, with a 14‐day washout period between doses (Figure 1).
Figure 1.
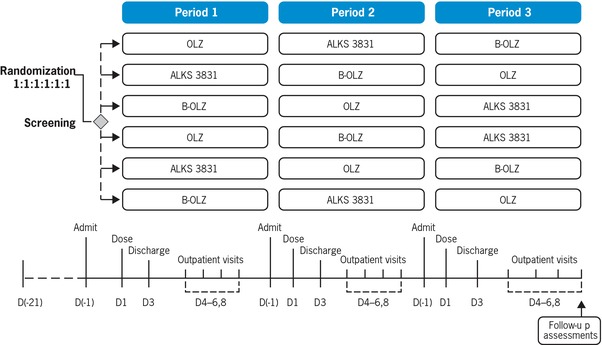
Study design schematic. There was a 14‐day washout period between doses. ALKS 3831, 10 mg OLZ/10 mg samidorphan; B‐OLZ, 10 mg OLZ; OLZ, 10 mg OLZ.
The study had 3 treatment periods for each subject (periods 1, 2, and 3) and each treatment period included a 4‐day inpatient stay and a 5‐day outpatient follow‐up (9 days total).
Subjects were admitted to the inpatient unit the night before and fasted for at least 10 hours before receiving a single dose of study drug on the morning of day 1 of each treatment period. Subjects remained as inpatients until approximately 48 hours after dosing for all inpatient PK and safety assessments to be completed and were asked to return for 4 outpatient follow‐up visits on days 4–6 and 8 for further PK and safety assessments.
Sample Collection and Analysis
Blood samples for PK assessments were collected within 15 minutes before dosing (predose) and 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 12, 16, 24, 32, 48, 72, 96, 120, and 168 hours postdose. Plasma concentrations of OLZ and SAM were analyzed using a validated liquid chromatography system with tandem mass spectrometry method in accordance with Good Laboratory Practice guidelines by an independent bioanalytic laboratory (Covance/Tandem, Salt Lake City, Utah). Briefly, OLZ and SAM were extracted from a 50.0 μL aliquot of human plasma (K2‐ethylenediaminetetraacetic acid) using a protein precipitation extraction procedure and then injected into a liquid chromatography system equipped with a Luna PFP(2) 5 μm 2 × 50 mm column (Phenomenex, Torrance, California). Mobile phase A was 0.1% formic acid in water. Mobile phase B was a mixture of methanol:acetonitrile (50:50, v/v). Mobile phase C was a mixture of water:methanol:acetonitrile (10:45:45, v/v/v). The chromatographic separation was performed using a gradient at room temperature at a flow rate of 0.500 mL/min with column backflush 1.00 mL/min. The detection was made with a tandem mass spectrometry detector. An API 5000 (MDS Sciex, Concord, Ontario, Canada) was operated in the selected reaction monitoring mode under optimized conditions; the transitions monitored were m/z 313.3→256.2 for OLZ, m/z 321.2→261.2 for olanzapine‐d8 (internal standard for OLZ), m/z 371.1→336.1 for SAM, and m/z 345.2→327.2 for naltrexone‐d3 (internal standard for SAM). OLZ and SAM concentrations were calculated with 1/x2 linear regression of peak area response ratio (analyte/internal standard) versus nominal concentrations. The assay had a linear range of 0.250 to 100 ng/mL, with a lower limit of quantitation (LLOQ) of 0.250 ng/mL. Precision and accuracy of the method were evaluated by analyzing quality control (QC) samples at the LLOQ (0.250 ng/mL), low QC (0.750 ng/mL), low‐medium QC (3.5 ng/mL), medium QC (40.0 ng/mL), and high QC (80.0 ng/mL). Precision was expressed as the percentage of coefficient of variation (% CV) of each QC concentration. Accuracy was assessed as the percentage difference from the theoretical analyte concentration (% bias). For OLZ, intra‐assay accuracy (% bias) ranged from −5.3% to 7.2%, intra‐assay precision (% CV) ranged from 0.7% to 5.6%, interassay accuracy (% bias) ranged from −2.3% to 4.0%, and interassay precision (% CV) ranged from 2.1% to 2.9%. For SAM, intra‐assay accuracy (% bias) ranged from −8.0% to 5.2%, intra‐assay precision (% CV) ranged from 0.9% to 7.4%, interassay accuracy (% bias) ranged from −4.5% to −0.1%, and interassay precision (% CV) ranged from 0% to 7.4%.
Pharmacokinetic and Statistical Analysis
Pharmacokinetic and statistical analyses were conducted in accordance with the FDA's Draft Guidance on Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs9 and Guidance on Statistical Approaches to Establishing Bioequivalence.10
PK parameters were calculated by a standard noncompartmental method using Phoenix WinNonLin, version 6.4 (Pharsight, a Certara Company, Princeton, New Jersey). The following PK parameters were determined: Cmax (maximal plasma concentration), Tmax (time to Cmax), AUC0‐t (area under the plasma concentration‐time curve from zero to the time of the last quantifiable concentration), t½ (terminal half‐life), tlag (lag time), AUC0‐∞ (area under the plasma concentration‐time curve from zero to infinity), and CL/F (apparent plasma clearance of drug after extravascular administration).
Systemic exposure parameters of OLZ (AUC0‐∞, AUC0‐t, and Cmax) were natural log‐transformed and analyzed using a mixed‐effects model with sequence, period, sex, and treatment as fixed effects and subject within sequence as a random effect. Geometric mean ratios (GMRs) and 90% confidence intervals (90%CIs) were calculated for the systemic exposure parameters. Bioequivalence of OLZ in the 3 OLZ‐containing tablet formulations would be established if 90%CIs for the ratio of population geometric means of OLZ AUC0‐∞ (AUC0‐t when appropriate) and Cmax were contained in the equivalence limits of 80%–125%. All statistical calculations were performed with SAS, version 9.4 (SAS Institute Inc., Cary, North Carolina).
Safety Assessments
Safety measures were carried out on screening days −21 to −2 and during the treatment periods and included monitoring of adverse events (AEs) and Columbia Suicide Rating Scale results (days −1 to 8 of each treatment period), weight (day −1 of each treatment period), vital signs and 12‐lead electrocardiogram parameters (days 1, 2, and 8 of each treatment period), and clinical laboratory parameters (days 1 and 8 of each treatment period).
Results
Subject Disposition and Baseline Characteristics
In total, 48 subjects were enrolled and randomized. Mean age was 26.7 years, and mean BMI was 24.2 kg/m2. Most subjects were male (58.3%) and black or African American (52.1%); see Table 1.
Table 1.
Demographic and Baseline Characteristics
| Treatment Sequence | |||||||
|---|---|---|---|---|---|---|---|
| Variable | Sequence 1 (n = 8) | Sequence 2 (n = 8) | Sequence 3 (n = 8) | Sequence 4 (n = 8) | Sequence 5 (n = 8) | Sequence 6 (n = 8) | All Subjects (n = 48) |
| Age (years), mean (SD) | |||||||
| 28.0 (3.9) | 23.6 (3.4) | 26.6 (5.1) | 27.5 (5.7) | 27.6 (6.1) | 26.8 (4.7) | 26.7 (4.9) | |
| Sex, n (%) | |||||||
| Male | 6 (75.0) | 4 (50.0) | 5 (62.5) | 4 (50.0) | 4 (50.0) | 5 (62.5) | 28 (58.3) |
| Race, n (%) | |||||||
| White | 6 (75.0) | 2 (25.0) | 3 (37.5) | 3 (37.5) | 4 (50.0) | 4 (50.0) | 22 (45.8) |
| Black or African American | 2 (25.0) | 5 (62.5) | 5 (62.3) | 5 (62.5) | 4 (50.0) | 4 (50.0) | 25 (52.1) |
| American Indian or Alaska Native | 0 | 1 (12.5) | 0 | 0 | 0 | 0 | 1 (12.5) |
| Weighta (kg), mean (SD) | |||||||
| 82.7 (12.8) | 67.1 (11.2) | 66.9 (12.8) | 71.3 (7.2) | 65.9 (11.8) | 70.7 (12.4) | 70.7 (12.3) | |
| BMI (kg/m2), mean (SD) | |||||||
| 26.7 (2.6) | 22.8 (4.2) | 23.2 (2.9) | 25.3 (2.4) | 23.1 (3.3) | 24.0 (3.2) | 24.2 (3.3) | |
BMI, body mass index; B‐OLZ, brand olanzapine; OLZ, olanzapine; SD, standard deviation.
Treatment sequence 1 = ABC; 2 = BCA; 3 = CAB; 4 = ACB; 5 = BAC; 6 = CBA; where treatments A, B, C are OLZ, ALKS 3831, and B‐OLZ, respectively.
Weight measured at baseline, which is defined as the day −1 assessment in period 1 (or the last nonmissing value before the first dose if the day −1 assessment in period 1 was missing).
All 48 subjects received at least 1 dose of study drug (ALKS 3831, OLZ, or B‐OLZ), and 45 received all 3 drugs. Five subjects discontinued before the last scheduled visit: 2 subjects discontinued during treatment periods 1 and 2 because of AEs (1 subject had an increased blood creatine phosphokinase level [this subject received B‐OLZ in period 1], and 1 subject had an increased blood creatine phosphokinase level and abnormal liver function test result [this subject received ALKS 3831 in period 1 and B‐OLZ in period 2]), 2 withdrew from the study during treatment period 3 (1 subject because of a family emergency [this subject received ALKS 3831 in period 1, B‐OLZ in period 2, and OLZ in period 3] and 1 subject did not have transportation [this subject received OLZ in period 1, B‐OLZ in period 2, and ALKS 3831 in period 3]), and 1 subject was lost to follow‐up during period 1 (this subject received B‐OLZ in period 1).
Pharmacokinetics of Olanzapine
The mean concentration–time profiles of OLZ following a single oral administration of ALKS 3831, OLZ, or B‐OLZ were superimposable (Figure 2). Key PK parameters for OLZ are summarized in Table 2.
Figure 2.
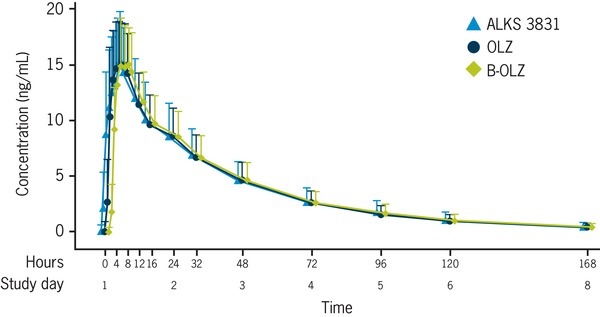
Plasma concentrations of OLZ after a single dose of ALKS 3831, OLZ, or B‐OLZ. Values displayed are mean + standard deviation. ALKS 3831, 10 mg OLZ/10 mg SAM; B‐OLZ, 10 mg OLZ; OLZ, 10 mg OLZ.
Table 2.
PK Parameters for OLZ After Single‐Dose Administration of ALKS 3831 (10 mg OLZ/10 mg SAM), OLZ (10 mg OLZ), or B‐OLZ (10 mg OLZ)
| Parameter Statistics | ALKS 3831 (n = 46) | OLZ (n = 45) | B‐OLZ (n = 48) |
|---|---|---|---|
| Cmax, ng/mL | |||
| Mean (SD) | 16.6 (4.5) | 16.7 (4.2) | 16.6 (3.8) |
| tmax, h | |||
| Median (min–max) | 7.0 (2.0–16.0) | 5.0 (2.0–12.0) | 5.0 (2.0–12.0) |
| tlag, h | |||
| Median (min–max) | 0.5 (0.0–0.6) | 0.5 (0.0–1.0) | 0.5 (0.0–1.0) |
| t½, h | |||
| Mean (SD) | 34.7 (9.2) | 34.7 (11.0) | 34.5 (8.7) |
| AUC0‐t, ng·h/mL | |||
| Mean (SD) | 610.6 (215.7) | 599.1 (187.8) | 594.3 (190.8) |
| AUC0‐∞, ng·h/mL | |||
| Mean (SD) | 652.0 (226.5) | 629.2 (205.0) | 632.6 (197.2) |
| CL/F, L/h | |||
| Mean (SD) | 17.2 (6.0) | 18.2 (8.3) | 17.4 (5.8) |
AUC0‐∞, area under the plasma concentration‐time curve from zero to infinity; AUC0‐t, area under the plasma concentration‐time curve from zero to the time of the last quantifiable concentration; B‐OLZ, brand olanzapine; CL/F, apparent plasma clearance of drug after extravascular administration; Cmax, maximal plasma concentration; max, maximum; min, minimum; OLZ, olanzapine; PK, pharmacokinetics; SD, standard deviation; t½, terminal half‐life; tlag, lag time; tmax, time to Cmax.
The rate and extent of OLZ absorption were similar following oral administration of ALKS 3831, OLZ, or B‐OLZ, with a median tlag of 0.5 hours and median tmax of 5–7 hours postdose across the 3 OLZ‐containing tablet formulations. Mean plasma Cmax values of OLZ were 16.6, 16.7, and 16.6 ng/mL, and the mean AUC0‐∞ values were 652.0, 629.2, and 632.6 ng·h/mL following a single dose of ALKS 3831, OLZ, and B‐OLZ, respectively. The t½ was also similar across the 3 tablet formulations, with a mean value of 34–35 hours. Box plots of OLZ Cmax, AUC0‐t, and AUC0‐∞ illustrate comparable systemic exposure to OLZ with ALKS 3831, OLZ, and B‐OLZ (Figure 3).
Figure 3.
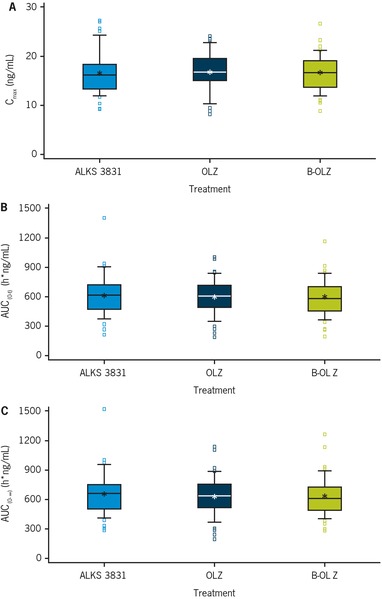
Comparison of OLZ (A) Cmax, (B) AUC0‐t, and (C) AUC0‐∞ after a single dose of ALKS 3831, OLZ, or B‐OLZ. The bottoms and tops of the boxes represent the 25th and 75th percentiles, respectively; the whiskers represent the 10th and 90th percentiles; the lines within the boxes represent the medians; the stars represent the means. The squares represent the values smaller than the 10th percentile or values greater than the 90th percentile. AUC0‐∞, area under the plasma concentration‐time curve from zero to infinity; AUC0‐t, area under the plasma concentration‐time curve from zero to the time of the last quantifiable concentration; B‐OLZ, brand olanzapine; Cmax, maximal plasma concentration; OLZ, olanzapine.
Relative Bioavailability and Bioequivalence
Statistical comparisons of relative bioavailability of OLZ between the 3 OLZ‐containing tablet formulations are summarized in Tables 3 and 4. Across all 3 exposure parameters (Cmax, AUC0‐∞, and AUC0‐t), GMRs were close to 1, and the 90%CIs of the GMRs were within the bioequivalence limit of 80%–125% for comparison of ALKS 3831 or OLZ versus B‐OLZ (Table 3) and ALKS 3831 versus OLZ (Table 4), indicating equivalent oral bioavailability or bioequivalence of OLZ following oral administration of ALKS 3831, OLZ, and B‐OLZ.
Table 3.
Bioequivalence Evaluation of OLZ for Comparison of ALKS 3831 Versus B‐OLZ and OLZ Versus B‐OLZ
| ALKS 3831 | OLZ | B‐OLZ | |
|---|---|---|---|
| Parameter Statistics | (n = 46) | (n = 45) | (n = 48) |
| AUC0‐∞, ng·h/mL | |||
| Geometric mean | 627.9 | 607.0 | 610.4 |
| Geometric mean ratio | 1.0 | 0.994 | — |
| 90%CI | (0.995–1.063) | (0.962–1.028) | — |
| AUC0‐t, ng·h/mL | |||
| Geometric mean | 594.4 | 578.9 | 578.9 |
| Geometric mean ratio | 1.0 | 1.0 | — |
| 90%CI | (0.993–1.062) | (0.967–1.034) | — |
| Cmax, ng/mL | |||
| Geometric mean | 16.3 | 16.3 | 16.4 |
| Geometric mean ratio | 0.994 | 0.995 | — |
| 90%CI | (0.954–1.036) | (0.955–1.037) | — |
AUC0‐∞, area under the plasma concentration‐time curve from zero to infinity; AUC0‐t, area under the plasma concentration‐time curve from zero to the time of the last quantifiable concentration; B‐OLZ, brand olanzapine; Cmax, maximal plasma concentration; OLZ, olanzapine; PK, pharmacokinetics.
Table 4.
Bioequivalence Evaluation of OLZ for Comparison of ALKS 3831 Versus OLZ
| ALKS 3831 | OLZ | |
|---|---|---|
| Parameter Statistics | (n = 46) | (n = 45) |
| AUC0‐∞, ng·h/mL | ||
| Geometric mean | 627.9 | 607.0 |
| Geometric mean ratio | 1.035 | — |
| 90%CI | (1.001–1.070) | — |
| AUC0‐t, ng·h/mL | ||
| Geometric mean | 594.4 | 578.9 |
| Geometric mean ratio | 1.027 | — |
| 90%CI | (0.993–1.062) | — |
| Cmax, ng/mL | ||
| Geometric mean | 16.3 | 16.3 |
| Geometric mean ratio | 0.999 | — |
| 90%CI | (0.959–1.041) | — |
AUC0‐∞, area under the plasma concentration‐time curve from zero to infinity; AUC0‐t, area under the plasma concentration‐time curve from zero to the time of the last quantifiable concentration; CI, confidence interval; Cmax, maximal plasma concentration; OLZ, olanzapine; PK, pharmacokinetics.
Safety
AEs were reported for 28 of 46 subjects (60.9%) who received ALKS 3831, 23 of 45 subjects (51.1%) who received OLZ, and 27 of 48 subjects (56.3%) who received B‐OLZ (Table 5). All AEs were mild or moderate in severity. One subject had serious AEs of increased blood creatine phosphokinase level and abnormal liver function test results during treatment period 2 (the subject received ALKS 3831 in period 1 and B‐OLZ in period 2); both were serious AEs, as the subject was hospitalized for these events, and they were classified as mild in severity by the investigator and led to discontinuation. Another subject had an AE of increased blood creatine phosphokinase level during treatment period 1 (subject received B‐OLZ in period 1), which was also classified as mild in severity and led to discontinuation.
Table 5.
Adverse Events
| Treatment | |||
|---|---|---|---|
| ALKS 3831 (n = 46), n (%) | OLZ (n = 45), n (%) | B‐OLZ (n = 48), n (%) | |
| Any AEs | 28 (60.9) | 23 (51.1) | 27 (56.3) |
| AEs by severity | |||
| Mild | 23 (50.0) | 20 (44.4) | 23 (47.9) |
| Moderate | 5 (10.9) | 3 (6.7) | 4 (8.3) |
| Severe | 0 | 0 | 0 |
| Drug‐related AEsa | 26 (56.5) | 20 (44.4) | 26 (54.2) |
| Serious AEs | 0 | 0 | 1 (2.1) |
| AEs leading to study discontinuation | 0 | 0 | 2 (4.2) |
| AEs reported by ≥5% of subjects, n (%) | |||
| Dizziness | 14 (30.4) | 11 (24.4) | 12 (25.0) |
| Somnolence | 9 (19.6) | 4 (8.9) | 9 (18.8) |
| Nausea | 7 (15.2) | 2 (4.4) | 3 (6.3) |
| Tachycardia | 6 (13.0) | 5 (11.1) | 6 (12.5) |
| Sedation | 5 (10.9) | 1 (2.2) | 2 (4.2) |
| Headache | 2 (4.3) | 2 (4.4) | 3 (6.3) |
| Blood CPK increased | 0 | 1 (2.2) | 3 (6.3) |
| Syncope | 0 | 0 | 3 (6.3) |
AE, adverse event; B‐OLZ, brand olanzapine; CPK, creatine phosphokinase; OLZ, olanzapine.
Related = definitely related, probably related, or possibly related.
The most common AE associated with all treatments was dizziness, which was reported in 30.4% of subjects treated with ALKS 3831, 24.4% with OLZ, and 25% with B‐OLZ (Table 5). Nausea and sedation were reported more frequently in the ALKS 3831 group (15.2% and 10.9%, respectively) compared with the OLZ group (4.4% and 2.2%, respectively) and the B‐OLZ group (6.3% and 4.2%, respectively).
Discussion
The Cmax and tmax as well as total systemic exposure (as measured by AUC0‐∞ and AUC0‐t) following administration of ALKS 3831, OLZ, and B‐OLZ were similar, indicating similar rate and extent of OLZ absorption for the 3 OLZ‐containing tablet formulations. The 90%CIs of the GMRs of OLZ AUC0‐∞, AUC0‐t, and Cmax were contained within the 80%–125% bioequivalence limits for comparison of ALKS 3831 versus B‐OLZ and OLZ versus B‐OLZ, indicating bioequivalence of OLZ in ALKS 3831 to B‐OLZ and bioequivalence of OLZ to B‐OLZ, therefore supporting the use of ALKS 3831 and OLZ in phase 3 clinical studies. In addition, the bioequivalence of OLZ in ALKS 3831 to OLZ further indicated that the SAM component in ALKS 3831 had no effect on the PK of OLZ. The PK of OLZ observed in this study is consistent with published data in healthy volunteers.11 Therefore, combining OLZ with SAM in a single bilayer tablet does not affect the PK and bioavailability of OLZ.
The PK of SAM observed when administered as a component of ALKS 3831 in the present study (Supplementary Table 1) was consistent with that observed in the previous studies, when SAM was administered as a single agent,6 indicating that combining OLZ with SAM in a single bilayer tablet does not affect the PK and bioavailability of SAM.
OLZ is mainly eliminated via hepatic metabolism, with 7% of the dose excreted in the urine as unchanged drug.12 Direct glucuronidation via UDP‐glucuronosyltransferase (UGT)1A4 and cytochrome P450 (CYP)‐mediated oxidation mainly by CYP1A2, is the primary metabolic pathway for OLZ.13 SAM is primarily eliminated through hepatic metabolism and renal excretion. CYP3A4‐mediated oxidation is the primary metabolic pathway for SAM.6 Based on the divergent metabolic pathways of OLZ and SAM, PK drug‐drug interactions between OLZ and SAM were not anticipated. The lack of PK drug‐drug interactions between OLZ and SAM observed in the present study, when the 2 drugs were administered in combination as a bilayer tablet, is consistent with previously published data indicating that coadministration of OLZ and SAM did not impact the PK of either drug.14
In this study, a 10 mg OLZ dose was chosen as opposed to the highest commercial maintenance dose of 20 mg because of tolerability concerns with a higher dose in healthy volunteers. The dose of ALKS 3831 used in this study was within the intended therapeutic dose range. Because the PK of OLZ is linear over the dose range of 2.5 to 20 mg11 and the SAM component in ALKS 3831 had no effect on the PK of OLZ, the bioequivalence of ALKS 3831 to B‐OLZ established in the present study can be extrapolated to other ALKS 3831 dose strengths according to FDA Guidance on Olanzapine.15
ALKS 3831 was generally well tolerated under the conditions of this study, and no safety concerns unique to ALKS 3831 compared with OLZ monotherapy were identified. The profile of AEs was generally consistent with that observed in a completed trial of ALKS 3831 in healthy volunteers.14
Conclusions
Combining OLZ with SAM in a single bilayer tablet (ALKS 3831) does not affect the PK and bioavailability of OLZ. The OLZ component in ALKS 3831 was demonstrated to be bioequivalent to B‐OLZ. ALKS 3831 was generally well tolerated, and no safety concerns unique to ALKS 3831 were noted.
Supporting information
Table S1. Summary of Pharmacokinetic Parameters for Samidorphan
Acknowledgments
The authors thank the ALK3831‐A101 study team, study participants, investigators, and study coordinators. Medical writing and editorial support for the preparation of this manuscript, under the guidance of the authors, was provided by Tabasum Mughal (ApotheCom, UK).
Declaration of Conflicting Interests
Lei Sun, David McDonnell, and Lisa von Moltke are all employees and shareholders of Alkermes, Inc. Jianjun Liu is a former employee of Alkermes, Inc.
Funding
This study was sponsored by Alkermes, Inc., Waltham, Massachusetts. Funding for editorial support was provided by Alkermes, Inc., Waltham, Massachusetts.
Role of the Study Sponsor
The study sponsor was involved in the design, collection, and analysis of the data. Interpretation of the results was by the authors, and the decision to submit the manuscript for publication in Clinical Pharmacology in Drug Development was made by the authors.
Data Sharing
Queries concerning the data reported in this study may be directed to the corresponding author, Lei Sun.
References
- 1. Javitt DC. Balancing therapeutic safety and efficacy to improve clinical and economic outcomes in schizophrenia: exploring the treatment landscape. Am J Manag Care. 2014;20(8 suppl):S166–173. [PubMed] [Google Scholar]
- 2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. [DOI] [PubMed] [Google Scholar]
- 3. Shram MJ, Silverman B, Ehrich E, Sellers EM, Turncliff R. Use of remifentanil in a novel clinical paradigm to characterize onset and duration of opioid blockade by samidorphan, a potent μ‐receptor antagonist. J Clin Psychopharmacol. 2015;35(3):242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wentland MP, Lou R, Lu Q, et al. Syntheses of novel high affinity ligands for opioid receptors. Bioorg Med Chem Lett. 2009;19(8):2289–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silverman B TM, Jiang Y, Pathak S, et al. ALKS 3831: A novel drug candidate for the treatment of schizophrenia. 2015. http://ascpmeeting.org/wp-content/uploads/2015/06/AbstractBook_Posters.pdf. Accessed May 2018.
- 6. Turncliff R, DiPetrillo L, Silverman B, Ehrich E. Single‐ and multiple‐dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers. Clin Ther. 2015;37(2):338–348. [DOI] [PubMed] [Google Scholar]
- 7. Fava M, Memisoglu A, Thase ME, et al. Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double‐blind placebo‐controlled trial. Am J Psychiatry. 2016;173(5):499–508. [DOI] [PubMed] [Google Scholar]
- 8. Ragguett RM, Rong C, Rosenblat JD, Ho RC, McIntyre RS. Pharmacodynamic and pharmacokinetic evaluation of buprenorphine + samidorphan for the treatment of major depressive disorder. Expert Opin Drug Metab Toxicol. 2018;14(4):475–482. [DOI] [PubMed] [Google Scholar]
- 9. FDA . Guidance for Industry ‐ Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs ‐ General Considerations 2014. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm389370.pdf. Accessed December 2017.
- 10. FDA . Guidance for Industry Statistical Approaches to Establishing Bioequivalence. 2001. https://www.fda.gov/downloads/drugs/guidances/ucm070244.pdf. Accessed December 2017.
- 11. Callaghan JT, Bergstrom RF , Ptak LR, Beasley CM. Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet. 1999;37:177–193. [DOI] [PubMed] [Google Scholar]
- 12. Kassahun K, Mattiuz E, Nyhart E Jr, et al. Disposition and biotransformation of the antipsychotic agent olanzapine in humans. Drug Metab Dispos. 1997;25:81–93. [PubMed] [Google Scholar]
- 13. Ring BJ, Catlow J, Lindsay TJ, et al. Identification of the human cytochromes P450 responsible for the in vitro formation of the major oxidative metabolites of the antipsychotic agent olanzapine. J Pharmacol Exp Ther. 1996;276(2):658–666. [PubMed] [Google Scholar]
- 14. Silverman BL, Martin W, Memisoglu A, et al. A randomized, double‐blind, placebo‐controlled proof of concept study to evaluate samidorphan in the prevention of olanzapine‐induced weight gain in healthy volunteers. Schizophrenia Res. 2018;195:245–251. [DOI] [PubMed] [Google Scholar]
- 15. FDA . FDA guidance on olanzapine. 2008. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm089213.pdf. Accessed October 17, 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of Pharmacokinetic Parameters for Samidorphan