

Abstract
Synthetic cannabinoids are one of the most significant groups within the category new psychoactive substances (NPS) and in recent years new compounds have continuously been introduced to the market of recreational drugs. A sensitive and quantitative screening method in urine with metabolites of frequently seized compounds in Norway (AB‐FUBINACA, AB‐PINACA, AB‐CHMINACA, AM‐2201, AKB48, 5F‐AKB48, BB‐22, JWH‐018, JWH‐073, JWH‐081, JWH‐122, JWH‐203, JWH‐250, PB‐22, 5F‐PB‐22, RCS‐4, THJ‐2201, and UR‐144) using ultra‐high pressure liquid chromatography–quadrupole time of flight–mass spectrometry (UHPLC–QTOF–MS) has been developed. The samples were treated with ß‐glucuronidase prior to extraction and solid‐phase extraction was used. Liquid handling was automated using a robot. Chromatographic separation was achieved using a C18‐column and a gradient of water and acetonitrile, both with 0.1% formic acid. Each sample was initially screened for identification and quantification followed by a second injection for confirmation. The concentrations by which the compounds could be confirmed varied between 0.1 and 12 ng/mL. Overall the validation showed that the method fulfilled the set criteria and requirements for matrix effect, extraction recovery, linearity, precision, accuracy, specificity, and stability. One thousand urine samples from subjects in drug withdrawal programs were analyzed using the presented method. The metabolite AB‐FUBINACA M3, hydroxylated metabolite of 5F‐AKB48, hydroxylated metabolite of AKB48, AKB48 N‐pentanoic acid, 5F‐PB‐22 3‐carboxyindole, BB‐22 3‐carboxyindole, JWH‐018 N‐(5‐hydroxypentyl), JWH‐018 N‐pentanoic acid, and JWH‐073 N‐butanoic acid were quantified and confirmed in 2.3% of the samples. The method was proven to be sensitive, selective and robust for routine use for the investigated metabolites.
Keywords: high resolution mass spectrometry, synthetic cannabinoids, urine screening
1. INTRODUCTION
Synthetic cannabinoids (SCs) are a group of cannabinoid receptor agonists produced as alternatives to Δ‐9‐tetrahydrocannabinol (THC), the main psychoactive compound in cannabis. The first SCs were synthesized to investigate the endogenous cannabinoid system and to explore potential new pharmaceuticals.1 In 2008, an increasingly popular recreational drug containing the SC JWH‐018 [1‐naphthyl(1‐pentyl‐1H‐indol‐3‐yl)methanone] was identified.2 Since then, legislation has evolved to criminalize the trafficking and use of this class of compounds in many countries. At the same time, though, these legislative activities have acted as a motive to produce new compounds not covered by the current legislations. In the last decade, this “race” has resulted in an increasing number of new SCs entering the market for recreational drugs. As one of the most important classes of new drugs, the ability to find and determine SCs in biological samples is important on an individual level (abuse, toxicity, law enforcement) as well as a social level (drug market trends, extent of trafficking).
Urinary screening methods of SCs based on immuno assay or chromatography with mass spectrometry (MS) detection, in particular liquid chromatography (LC) with quadrupole tandem‐MS (MS/MS) detection, have dominated in the toxicological laboratories.3 Used for analyses of a definite number of compounds, these techniques are a good choice due to their robustness, sensitivity, and selectivity. However, these methods can only identify the compounds they are designed for, and updates are not easily performed. A number of quantitative screening methods in urine by LC–MS/MS have previously been published.4, 5, 6, 7, 8 High resolution mass spectrometry (HRMS) with quadrupole time of flight (QTOF) instrumentation that acquires full spectrum data is not limited by scan/dwell times, and introducing new masses/formulas to the method will not affect the detection of the previously included ones. In addition, retrospective analysis of previously acquired data can be performed. Few articles have previously been published exploring quantitative screening of SCs using HRMS, although the technique has more frequently been used solely for qualitative targeted and non‐targeted methods.9, 10, 11 In a non‐targeted method, ideally all MS spectra plus additional MS/MS spectra are acquired for a tentative identification, and can be obtained from findings of interest after sample acquisition. The method presented in this article can be described as a dynamic quantitative and targeted screening method since MS data from the first injection are used for quantification purposes while MS/MS data for confirmation are acquired in a second injection only for confirmation of a definite panel of analytes. By this approach the targets included in the method can be adjusted in accordance to the current drugs of interest. Potential disadvantages using HRMS instrumentation are the higher cost compared to LC–MS/MS and the large size of data files generated. In addition, an efficient processing of the data requires powerful computers.
In comparison with blood, advantages of detecting metabolites of drugs of abuse in urine include the expanded detection window and the non‐invasive sampling. Quantification of metabolites can be valuable when a recent intake needs to be distinguished from residual drug excretion from a former intake. This principle is well known after intake of cannabis, and various algorithms have been developed for this purpose.12, 13, 14 For synthetic cannabinoids some data exist on the urinary pharmacokinetics and excretion rate of the metabolites of JWH‐018 and JWH‐073,6, 15 whereas for other compounds, very little is known. Thus, for synthetic cannabinoids more data are needed before a recent intake can be unequivocally distinguished from residual drug excretion. Nevertheless, gathering data from quantitative analyses of the various metabolites in serial urinary samples is a prerequisite for developing the algorithms needed. Moreover, the access of quantitative methods is crucial in order to carry out pharmacokinetic studies (ie, to estimate half‐lives, peak concentrations and detection times in urine). However, the low concentrations of unconjugated metabolites in urine often require cleavage of the glucuronidated metabolites by hydrolysis before analysis. In previously published identification and quantification assays, preparation techniques varying from simple dilution,6 salting‐out liquid–liquid extraction (LLE)10 and traditional LLE4 to more complex procedures including supported liquid extraction9 and solid‐phase extraction (SPE)5 have been used. To simplify sample preparation, automatization of this procedure has become more common.5, 6, 10
All SCs undergo metabolism to a certain extent.16 Consequently, a screening method for SCs in urine must cover the most abundant and unique metabolites if an accurate determination of the drug taken is necessary. Some SCs that are biotransformed to metabolites which are unique and unambiguously can point out the specific drug ingested. However, compounds with close structural similarities often result in several identical metabolites, but in many cases also unique secondary metabolites are produced. One such example is AM‐2201 and JWH‐018, both having the major metabolites JWH‐018 N‐pentanoic acid and JWH‐018 N‐(5‐hydroxypentyl). Nevertheless, the specific markers AM‐2201 N‐(4‐hydroxypentyl) and AM‐2201 N‐(6‐hydroxyindole) of AM‐2201 and JWH‐018 N‐(4‐hydroxypentyl) of JWH‐018 are also formed and can be used to distinguish between intake of these two.17, 18 A careful selection of metabolites is therefore required. New SCs that are biotransformed to metabolites identical to a drug that already is covered by a method are frequently introduced. Consequently, the exact intake cannot be confirmed without updating the method with new available unique markers. The introduction of AMB‐FUBINACA which gives the same metabolite as AB‐FUBINACA is an example of the latter.19
Reference standards are necessary for performing quantification. It is both a time‐consuming and a resource‐demanding process from the time a new drug is introduced on the market to the point when selected metabolites have been synthesized and can be included in a new or updated method. Potential metabolites can be identified by exposing human liver microsomes20, 21 or human hepatocytes22 to the drug in question, and analyze the residues with MS, together with urinary samples from people with known consumption of the same drug.
The aim of the present study was to develop a high throughput quantitative screening method for SCs in urine, using LC–QTOF–MS and automated sample preparation. To evaluate the feasibility of the method in clinical practice, we also aimed to describe our experience and results from analyzing a total of 1000 consecutive routine urinary samples sent to our laboratory where screening for SCs had been requested.
2. MATERIALS AND METHODS
The analytes included in this method consisted of commercially available and assumed relevant metabolites of the SCs most frequently used in Norway at the time the method was developed. The seizure statistics from the Norwegian National Criminal Investigation Service (KRIPOS) were used to choose relevant SCs. A complete list of the metabolites included, formulas, monoisotopic masses, CAS numbers, IUPAC names, and structures is given in the Supporting Information (Table S1).
2.1. Chemicals and reagents
Metabolite reference standards of JWH‐018 N‐pentanoic acid, JWH‐073 N‐butanoic acid, JWH‐122 N‐pentanoic acid, JWH‐203 N‐pentanoic acid, JWH‐210 N‐pentanoic acid, JWH‐081 N‐pentanoic acid, JWH‐250 N‐pentanoic acid, AM‐2201 N‐(5‐hydroxyindole), AB‐PINACA COOH, AB‐FUBINACA M3 and the isotope labeled d4‐JWH‐250 N‐pentanoic acid and d4‐JWH‐018 N‐pentanoic acid were purchased as solutions from Chiron (Trondheim, Norway). 5F‐PB‐22 3‐carboxyindole, 5F‐AKB48 N‐(4‐hydroxypentyl), AB‐CHMINACA 3‐carboxyindazole, AB‐CHMINACA M1A, AB‐CHMINACA M2, AB‐PINACA N‐pentanoic acid, AKB48 N‐(4‐hydroxypentyl), AKB48 N‐(5‐hydroxypentyl), AKB48 N‐pentanoic acid, BB‐22 3‐carboxyindole, AM‐2201 N‐(4‐hydroxypentyl), JWH‐018 N‐(5‐hydroxypentyl), JWH‐210 N‐(5‐hydroxyindole), JWH‐210 N‐(5‐hydroxypentyl), PB‐22 3‐carboxyindole, PB‐22 N‐(4‐hydroxypentyl), PB‐22 N‐pentanoic acid, RCS‐4 N‐(4‐hydroxypentyl)phenol, THJ‐2201 N‐pentanoic acid, UR‐144 N‐(4‐hydroxypentyl), UR‐144 N‐5‐hydroxypentyl, UR‐144 N‐pentanoic acid, and d5‐UR‐144 N‐(5‐hydroxypentyl) were from Cayman Chemicals (Ann Arbor, MI, USA). LiChrosolve® hypergrade LC–MS quality of acetonitrile and methanol in addition to LiChrosolve® water were from Merck (Darmstadt, Germany). ARISTAR® formic acid was from VWR Chemicals (Oslo, Norway). Ammonium acetate of LC–MS grade was from Sigma Aldrich (St Louis, MO, USA) and β‐glucuronidase stock solution (Helix promatia) was purchased from Roche Diagnostics (Mannheim, Germany).
2.2. Preparation of solutions
Stock solutions of the reference compounds were prepared and further diluted and combined into five different working solutions. One set was prepared for calibrators and one set for quality controls (QCs). Calibrators and QCs were prepared by fortifying blank urine with the working solutions and stored at 4°C. An overview of the calibration levels, QCs, and distribution of metabolites in working solutions are given in the Supporting Information (Table S2). A solution of internal standards was prepared by diluting stock solutions in 20% methanol (v/v) in water to a concentration of 100 ng/mL d4‐JWH‐250 N‐pentanoic acid and d4‐JWH‐018 N‐pentanoic acid and 50 ng/mL d5‐UR‐144 N‐(5‐hydroxypentyl). The buffer for sample pretreatment of 30.8 g/L ammonium acetate was prepared by dissolving the salt in water. A solution of β‐glucuronidase containing 25 000 units/mL was prepared from a stock solution. Needle wash was made from methanol/acetonitrile/isopropanol/water/formic acid (25:25:25:23:2, v/v).
2.3. Authentic samples
The method was applied on a total of 1000 consecutive routine urinary samples sent to our laboratory for which screening for SCs had been requested. These samples originated from subjects in whom an intake of SCs was suspected, mainly patients enrolled in medication‐assisted treatment programs for drug dependence and patients undergoing other forms of treatment for drug dependence. The samples were received from all over Norway and were collected through 2014 and in the first half of January 2015. At arrival at the laboratory, these samples were principally analyzed with a routine targeted LC–MS/MS method covering JWH‐018 N‐pentanoic acid, JWH‐073 N‐butanoic acid, JWH‐122 N‐pentanoic acid, JWH‐203 N‐pentanoic acid, JWH‐210 N‐pentanoic acid, JWH‐081 N‐pentanoic acid, JWH‐250 N‐pentanoic acid, and AM‐2201 N‐(5‐hydroxyindole). This method has previously been described in a publication but then with focus only on JWH‐018 N‐pentanoic acid and JWH‐073 N‐butanoic acid.6 The collection and storage of the samples selected for subsequent analysis with the present method was approved from the Regional Committee of Medical and Health Research Ethics in Mid Norway (approval No. 2014/2281). As these samples had to be anonymized prior to analysis in accordance to the approval given by the Ethics Committee we were precluded from comparing the results of these two methods.
In a subsample containing specimens from five patients who had tested positive for JWH‐018 N‐pentanoic acid and/or JWH‐073 N‐butanoic acid by the targeted LC–MS/MS method described,6 a separate approval from the Regional Committee of Medical and Health Research Ethics in Mid Norway (approval No. 2014/737) and individual consent from each patient made it possible to compare the results from that method with the present. From these patients, originating from the same drug rehabilitation clinic and having their samples collected over a short period of time after suspected drug use,6 a total of 27 samples were available.
2.4. Method optimization
The method optimization aimed at developing a general method that could detect the relatively diverse group of metabolites and also include new, similar metabolites as they become available. Different sample preparations techniques, LC conditions, and MS settings were explored and the optimization process revealed several methodical issues and challenges. An extraction based on supported liquid extraction, SLE+ from Biotage (Uppsala, Sweden) and SPE HLB PRiME from Waters were compared. The SPE resulted in better sample clean‐up and compound recovery. The HLB solid phase consisted of a water‐wettable combined hydrophilic and lipophilic polymer. This sorbent did not require conditioning and equilibrating steps, which resulted in a fast throughput and provided to some degree a more convenient protocol and was therefore chosen.
An evaporation and reconstitution step was required and two evaporation temperatures (30°C or 50°C) and reconstitution solvents (20/80 and 50/50 (v/v) mobile phase A/B) were tested to minimize the loss of compounds in these steps. Highest recovery was found with evaporation at 30°C and reconstitution in 20/80 (v/v) mobile phase A/B. Initially the eluates were collected in a well plate of plastic but this material introduced contaminants interfering with the analysis. This was most noticeable using ethyl acetate as eluent in the SLE+ process. Contaminants were avoided when plastics were replaced by a well plate consisting of glass vials.
As most SCs undergo phase II metabolism with conjugation, for example to glucuronic acid16 a hydrolysis step was required before analysis. Hydrolysis efficiency and reproducibility was tested using different conditions: 10, 25, or 30 μL of Helix promatia extract (25,000 units/mL) was added to samples fortified with 500 ng/mL of JWH‐018 N‐pentanoic acid glucuronide and UR‐144 N‐(5‐hydroxypentyl) glucuronide and incubated for one or two hours at 60°C. The efficiency of hydrolysis was determined by measuring the glucuronide and hydrolysis product in treated and untreated samples. Using 25 or 30 μL extract gave the same effective hydrolysis when incubated for 1 hour, and 25 μL was therefore chosen to minimize the contribution of enzyme to the matrix.
The chromatographic conditions achieving the best separation of isomers with identical fragmentation patterns, such as AKB48 N‐(5‐hydroxypentyl) and AKB48 N‐(4‐hydroxypentyl), as well as separating as many of the analytes as possible from endogenous compounds, was found by testing three different columns, C18, phenyl‐hexyl and biphenyl, in combination with different mobile phase set‐ups and gradients. A C18 column and a linear gradient were chosen.
In general, urine as a matrix results in high background and potential interferences affecting the continuous measurement of two lock masses maintaining the high degree of mass accuracy achieved by the LC–QTOF–MS system. Interference was observed close to m/z 121.0509 which is monitored together with m/z 922.0098 as lock masses to control mass accuracy. This resulted in a high mass error in certain spectra. Instead of using high resolution mode which compromises the dynamic range an alternative lock mass, m/z 118.0863 from trimethylglycine ([M + H]+) were chosen.
2.5. Sample preparation
All pipetting operations were performed using a Tecan Freedom Evo pipetting robot (Tecan, Männedorf, Switzerland). Urine sample, calibrator, or QC in aliquots of 600 μL was pipetted into a 2‐mL 96‐well plate. Volumes of 20 μL internal standard solution, 600 μL ammonium acetate and 25 μL β‐glucuronidase were added and the plate was incubated for 1 h at 60°C. After cooling to ambient temperature, 1000 μL of the sample was transferred to a Waters Oasis® HLB PRiME 30 mg HLB 96‐well plate (Wexford, Ireland) SPE. A positive pressure processor (Waters, Milford, MA, USA) was used to gently push the sample and the following reagents through the packing material. The SPE material was washed with 1000 μL water and 1000 μL of 10% methanol (v/v) in water in sequence following elution twice with 500 μL 10% methanol (v/v) in acetonitrile. The eluate was collected in a rack of 96 glass vials in a tray with well plate foot print (J.G. Finneran Associates Inc., Vineland, NJ, USA) and dried completely under air at 30°C prior to reconstitution with 400 μL 80/20 mobile phase A/B (v/v).
2.6. Instrumentation
Instrumental analysis was performed using a 6550 QTOF‐MS (Agilent, Santa Clara, CA, USA) with electrospray ionization (ESI) and iFunnel interface coupled with a 1290 Infinity UHPLC system from Agilent. Mobile phase A and B consisted of 0.1% formic acid in water and acetonitrile, respectively, and separation was achieved using a Zorbax Eclipse Plus C18 Rapid Resolution HD column (2.1x100 mm, 1.8 μm) from Agilent maintained at 60°C. A linear gradient with a flow of 0.30 mL/min starting at 10% mobile phase B increasing to 50% in 2 minutes, continuing to 60% in the next 6 minutes and further increasing to 95% in 1 minute was employed. This condition was maintained for 3 minutes and before the next injection the initial condition was held for 2 minutes, giving a total cycle time of 14 minutes.
Positive ionization was used with the fragmentor voltage at 375 V, capillary voltage at 3500 V, gas temperature at 150°C, gas flow at 15 L/min, nebulizer pressure at 20 psig and sheath gas temperature at 380°C. The following settings were applied for the iFunnel interface: Exit direct current of 40 V and radio frequency high pressure and low pressure at 150 V and 100 V, respectively.
All samples were first analyzed by injecting 5 μL and using the MS‐only mode acquiring full‐scan data in low mass range (1700 m/z) at a scan rate of 2 Hz and the detector in 2 GHz extended dynamic range giving a resolution (m/Δm at FWHM) of approx. 20,000 at m/z 322.0481. Presumably positive samples based on the two first identification criteria described in Section 2.7, were then injected once again with an injection volume of 10 μL using a targeted MS/MS mode with a list of precursors for acquiring MS/MS spectra. A collision energy of 10, 20, or 40 eV was applied to each precursor based on previous experiments to get a collision induced dissociation (CID) spectrum containing fragments and traces of the precursor. In this mode the instrument cycles between acquiring MS scans and MS/MS scans both in a rate of 6 Hz and with the detector in 4 GHz high resolution state (resolution of approx. 30,000 at m/z 322.0481). The computer controlling the instrument was equipped with the MassHunter Acquisition software (Acq) B.05.01 (Agilent, Santa Clara, CA, USA).
2.7. Library spectra
CID spectra were added to the in‐house library according to Broecker et al.23 This procedure involved diluting individual 1 mg/mL stock solutions of SCs in methanol to 100 ng/mL and then 1 μL was injected on a guard column with 0.1% formic acid in water and 0.1% formic acid in acetonitrile (50:50) as mobile phase. Three CID spectra of the protonated compound using collision energies of 10, 20, and 40 eV were acquired.
The acquired CID spectra were transferred to the library file using MassHunter Qualitative software (Qual) B.07.01 and MassHunter PCDL Manager B.07.01 (Agilent). In this process the fragment masses in every spectrum were corrected to their theoretical masses. Fragments with intensities lower than 1% of the most abundant mass in each spectrum were deleted.
2.8. Quantification and confirmation of compounds
Quantification and confirmation of the compounds was done by two injections where the first was using MS‐only and the second was using targeted MS/MS. Three identification criteria (ID criteria I, II, and III) with increasing degree of confidence was used. All data files of samples, calibrators and QCs from the first injection were first processed using the MassHunter Quantitative software (Quant) B.07.01. The compounds were identified based on accurate monoisotopic mass and retention time (RT) (ID criterion I). The instrument settings in the first injection gave the widest dynamic range and 20 spectra per peak which are sufficient for quantification. Calibration curves based on peak area ratios of analyte to internal standard at each concentration level were formed using linear least square regression employing 1/x or 1/x2 as weighting factor. Results of the processed data presented by the software were manually reviewed and a sample was presumed positive if above the limit of quantification (LOQ) as defined in Section 2.9.1 and additionally gave a mass match score ≥ 80 in Qual software, using profile data and “Find by Formula” (ID criterion II). This score was based on accurate mass and isotopic pattern from a database of the analytes, and only the compounds with a mass error of ±15 parts per million (ppm) and a deviation of ±0.15 minutes from the RT given in the database were considered. The mass match score was calculated using the following equation:
| (1) |
The accuracy was weighted (w) 100, abundance was weighted 80 and isotope spacing was weighted 50.
A threshold mass match score of 80 out of 100 was chosen based on experience through method development and gave only a few presumable positive findings that were not confirmed.
In case of presumable positive findings, the MS/MS spectra acquired in a second injection were compared with a spectral library holding reference CID spectra for all the compounds in the target list obtained at 10, 20, and 40 eV. This identification was done by processing the data using the Qual software tool “Identify Compounds” and the option “Search Library.” The numbers of matching and non‐matching fragments and the mass accuracy of the fragments were the criteria in the identification of the compound. A score ≥ 80 out of 100 was regarded as a definite identification (ID criterion III). An example of a positive library comparison is given in Figure S1. The minimum concentration in spiked negative samples which fulfilled this most stringent criterion was defined as the limit of confirmation (LOC). This approach may result in a quantitative finding in the first assumption but the sample ending up negative after the second injection if the LOC was higher.
2.9. Method validation
LOQ, linearity, selectivity, RT stability, carry‐over, matrix effects, recovery, precision, accuracy, and stability are parameters recommended to evaluate during method validation for forensic applications.24 All these parameters were included in the validation and the number of calibration levels, parallels and analytical runs as well as acceptance limits are described in the following paragraphs.
2.9.1. Limit of quantification and limit of confirmation
LOQ was first evaluated for each analyte by spiking blank urine to different concentration levels (0.01–5 ng/mL). The lowest concentration level giving reproducible results when analyzed at 10 days with precision (CV) < 20% and accuracy within 80%–120% of the theoretical value was defined as LOQ.
LOC was defined as the lowest concentration identified by the library search identification criteria (ID criterion III). A serial dilution of spiked urine was first analyzed to estimate this limit. Blank urine from different individuals was then spiked at three or four concentration levels equal to and around the estimated LOC (in the range of 0.01–5 ng/mL). The concentration level where the compound was identified in all urines using criterion III was set to the LOC.
2.9.2. Linearity
The linear range of every compound was explored by using the analyzed calibrators from the first four days of validation (all days within a week) at six calibration levels (except AB‐PINACA pentanoic acid, AB‐CHMINACA M1A, and RCS‐4 N‐(4‐hydroxypentyl)phenol where five levels were used) in a linear least square regression employing 1/x or 1/x2 weighting and reported as the correlation coefficient R2. The concentration range was defined from LOQ to highest calibration concentration. R2 ≥ 0.990 was regarded as accepted.
2.9.3. Selectivity
The selectivity of the method was evaluated by spiking 10 different blank urines (creatinine concentrations 34–249 mg/dL) with a mix of 28 drugs of abuse or their corresponding metabolites commonly observed in the samples sent to the laboratory for screening for drugs of abuse. The drugs were amphetamine, methamphetamine, 3,4‐methylenedioxymethamphetamine (MDMA), ephedrine, 3,4‐methylenedioxyamphetamine (MDA), para‐methoxy‐N‐methylamphetamine (PMMA), para‐methoxyamphetamine (PMA), codeine, oxycodone, morphine, methadone, tramadol, O‐desmethyl‐tramadol, ethylmorphine, 6‐monoacetylmorphine, buprenorphine, fentanyl, methadone, desmethyl‐diazepam, hydroxy‐alprazolam, 7‐amino‐nitrazepam, 7‐amino‐clonazepam, 7‐amino‐flunitrazepam, benzoylecgonine, ritalinic acid, ketamine, zolpidem, and 11‐nor‐9‐carboxy‐Δ9 THC (THC‐COOH).
2.9.4. Retention time stability
The stability of RT and relative RT (ratio of analyte RT to internal standard RT) was monitored through an analytical sequence of minimum 14 hours at three random validation days. The deviation of RT and relative RT in QC samples through the sequence to the average RT of the calibrators in the beginning of the run was calculated. RT deviation ≤1% throughout an analytical sequence up to 14 hours was accepted.
2.9.5. Carry‐over in the LC system
The carry‐over from a high concentration sample to the next was determined by injecting blank urine after a sample containing a concentration equal to its highest calibration level or at least 125 ng/mL. A carry‐over <20% of LOQ was accepted.
2.9.6. Matrix effects
To estimate the matrix effect (ME) reconstitution reagent (A) (80/20 mobile phase A/B (v/v)) and 10 extracted blank urines (B) was fortified with all compounds and analyzed to acquire the analyte signal. ME (%) was calculated as [area of B/area of A] x 100%. A value below 100% is indicative of ion suppression and a value above 100% is indicative of ion enhancement. ME values in the interval 75%–125% were regarded as acceptable for quantification of compounds lacking a dedicated isotopically marked internal standard.
2.9.7. Recovery
The extraction efficiency was estimated by comparing the signal in six blank urines fortified with all compounds after extraction (B) to the signal in the same samples fortified to the identical concentration level before extraction (C). Internal standards were added in the same amount to all samples after extraction. Recovery was calculated as [area of compound relative to internal standard in C/area of compound relative to internal standard in B] x 100%. Recoveries ≥75% were regarded as acceptable for quantification.
2.9.8. Precision and accuracy
The intra‐day precision was determined by analyzing 10 parallels of two concentration levels in the same sequence. The inter‐day precision was calculated by analyzing one sample at two different concentration levels at 10 different days over a period of five weeks. The acceptance criterion of intra‐ and inter‐sequence precision at both concentration levels was a CV ≤ 15%. The average value of the inter‐day data was used to calculate the accuracy expressed as the deviation from theoretical/nominal value. The acceptance criterion of accuracy was values in the interval 85%–115%.
2.9.9. Stability
The stability of the compounds was tested at different temperature conditions in spiked QC samples stored in glass tubes at one concentration level. Spiked QC samples were stored in darkness at 4°C to simulate the standard storage conditions from receiving a sample to its analysis. QC samples were analyzed after seven and 14 days. In addition QC samples were stored for three and five days at 25°C in darkness to simulate typical conditions during transport from the sampling location to the laboratory. Stored samples at 4°C and 25°C were analyzed together with freshly thawed samples and relative changes in concentration were reported. In addition the stability of extracted samples in the autosampler at 10°C was re‐tested at three and seven days. An interval of three days covers the maximum time that can be experienced between first and second injection as there can be a delay between the first injection via processing and the second injection. The seven‐day period was included to explore the time frame for a typical postponement due to e.g. instrument failure.
3. RESULTS
A quantitative UHPLC–QTOF–MS screening method of 35 SC metabolites with a run time of 14 minutes was achieved. A second injection with the same run time was required for confirmation by acquiring MS/MS‐spectra for library search.
3.1. Method validation
The validation parameters were within the set criteria and requirements for the majority of analytes. However, high matrix effects and insufficient recoveries question the ability to accurately quantify 14 of the investigated analytes and therefore the method must consider being semi‐quantitative for these compounds (Table 1).
Table 1.
Retention time (RT), limit of confirmation (LOC), limit of quantification (LOQ), highest limit of quantification (HLOQ), linearity (R2), and precision (intra‐ and inter‐sequence) for 35 metabolites of synthetic cannabinoids in urine. The analytes are sorted after retention time. ID refers to the numbers in Figure 1. n = number of parallels. SEMI = method is semi‐quantitative. QC = quality control. CV = coefficient of variation
| Metabolite | ID | RT | Can Originate From Intake Of: | LOC | LOQ | HLOQ | R2 | QC Low | QC High | Intra‐sequence CV (%) (n = 10) | Inter‐sequence CV (%) (n = 10) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| min. | ng/mL | ng/mL | ng/mL | ng/mL | ng/mL | lowc | highd | lowc | highd | ||||
| AB‐PINACA pentanoic acid | 1 | 3.1 | AB‐PINACA or 5F‐AB‐PINACA | – a | 10 | 320 | 0.9927 | 20 | 200 | 6.4 | 4.0 | 9.5 | 7.3 |
| AB‐CHMINACA M1ASEMI | 2 | 3.2 | AB‐CHMINACA | 10 | 10 | 320 | 0.9908 | 20 | 200 | 3.2 | 2.4 | 6.9 | 5.7 |
| RCS‐4 N‐(4‐hydroxypentyl)phenolSEMI | 3 | 3.5 | RCS‐4 | 10 | 5.0 | 160 | 0.9859 | 10 | 200 | 5.5 | 2.3 | 7.7 | 6.1 |
| AB‐FUBINACA M2SEMI | 4 | 3.6 | AB‐FUBINACA | 12 | 2.0 | 240 | 0.9909 | 20 | 200 | 5.1 | 4.8 | 6.6 | 7.1 |
| 5F PB‐22 3‐carboxyindole | 5 | 4.1 | 5F‐PB‐22 or 5F‐MDMB‐PICA | 5 | 1.0 | 120 | 0.9950 | 8.4 | 67.0 | 3.4 | 4.2 | 8.6 | 6.0 |
| RCS‐4 N‐pentanoic acid | 6 | 4.3 | RCS‐4 | 1.0 | 0.25 | 60 | 0.9940 | 0.5 | 50.0 | 16 | 4.0 | 10 | 6.1 |
| PB‐22 N‐pentanoic acidSEMI | 7 | 4.4 | PB‐22 or 5F‐PB‐22 | 2.5 | 0.25 | 50 | 0.9644 | 0.5 | 25.0 | 7.1 | 4.0 | 7.6 | 2.4 |
| JWH‐250 N‐pentanoic acid | 8 | 4.6 | JWH‐250 | 0.25 | 0.125 | 60 | 0.9936 | 0.5 | 50.0 | 3.2 | 2.1 | 11 | 10 |
| PB‐22 N‐(4‐hydroxypentyl)SEMI | 9 | 4.7 | PB‐22 | 0.5 | 0.25 | 50 | 0.9970 | 1.0 | 50.0 | 3.1 | 4.1 | 5.3 | 4.0 |
| JWH‐073 N‐butanoic acid | 10 | 5.1 | JWH‐073 or JWH‐018 | 0.5 | 0.125 | 60 | 0.9959 | 0.5 | 50.0 | 2.2 | 2.1 | 2.7 | 2.5 |
| JWH‐203 N‐pentanoic acid | 11 | 5.1 | JWH‐203 | 0.5 | 0.25 | 60 | 0.9963 | 0.5 | 50.0 | 2.2 | 2.8 | 6.6 | 2.7 |
| PB‐22 3‐carboxyindole | 12 | 5.3 | PB‐22 or CBL‐018 | 12 | 1.0 | 120 | 0.9973 | 2.0 | 100 | 3.3 | 2.1 | 9.3 | 2.3 |
| AB‐FUBINACA M3SEMI | 13 | 5.4 | AB‐FUBINACA, AMB‐FUBINACA or EMB‐FUBINACA | 0.5 | 0.5 | 120 | 0.9836 | 4.3 | 45.0 | 2.5 | 2.5 | 5.0 | 2.1 |
| AB‐CHMINACA 3‐carboxyindazoleSEMI | 14 | 5.4 | AB‐CHMINACA or AMB‐CHMINACA | 2.5 | 0.25 | 50 | 0.9957 | 0.5 | 22.5 | 8.6 | 2.4 | 7.3 | 4.5 |
| JWH‐018 N‐pentanoic acid | 15 | 5.4 | JWH‐018 or AM‐2201 | 0.5 | 0.125 | 60 | 0.9970 | 0.5 | 50.0 | 2.6 | 2.2 | 7.1 | 4.1 |
| AM‐2201 N‐(4‐hydroxypentyl)SEMI | 16 | 5.7 | AM‐2201 | 0.1 | 0.2 | 50 | 0.9962 | 0.5 | 25.0 | 2.5 | 2.1 | 5.6 | 4.6 |
| JWH‐018 N‐(5‐hydroxypentyl)SEMI | 17 | 5.7 | JWH‐018 or AM‐2201 | 0.25 | 0.25 | 50 | 0.9827 | 0.5 | 25.0 | 6.1 | 3.3 | 8.3 | 4.8 |
| JWH‐081 N‐pentanoic acid | 18 | 5.8 | JWH‐081 | 0.5 | 0.25 | 60 | 0.9951 | 0.5 | 50.0 | 3.1 | 3.1 | 4.8 | 12 |
| AM‐2201 N‐(5‐hydroxyindole)SEMI | 19 | 6.0 | AM‐2201 | 0.25 | 0.25 | 60 | 0.9855 | 0.5 | 50.0 | 4.4 | 3.8 | 8.5 | 9.1 |
| JWH‐122 N‐pentanoic acid | 20 | 6.1 | JWH‐122 or MAM‐2201 | 0.5 | 0.25 | 60 | 0.9957 | 0.5 | 50.0 | 3.7 | 3.1 | 10 | 8.6 |
| THJ‐2201 N‐pentanoic acidSEMI | 21 | 6.2 | THJ‐2201 or THJ‐018 | 0.5 | 0.25 | 50 | 0.9867 | 0.5 | 22.5 | 6.7 | 3.2 | 17 | 4.3 |
| BB‐22 3‐carboxyindole | 22 | 6.4 | BB‐22, MDMB‐CHMICA or ADB‐CHMICA | 17.5 | 2.0 | 240 | 0.9941 | 20 | 200 | 2.6 | 2.3 | 5.2 | 6.4 |
| JWH‐122 N‐(5‐hydroxypentyl)SEMI | 23 | 6.6 | JWH‐122 or MAM‐2201 | 0.5 | 0.25 | 50 | 0.9938 | 0.5 | 25.0 | 4.3 | 2.6 | 15 | 7.5 |
| AB‐PINACA COOHSEMI | 24 | 6.7 | AB‐PINACA or AMB | 1.0 | 1.0 | 120 | 0.9914 | 2.0 | 100 | 2.3 | 1.9 | 3.4 | 2.7 |
| UR‐144 N‐pentanoic acid | 25 | 6.8 | UR‐144 or XLR11 | 0.2 | 0.25 | 50 | 0.9926 | 0.5 | 25.0 | 3.4 | 1.8 | 9.7 | 2.7 |
| JWH‐210 N‐pentanoic acidSEMI | 26 | 7.2 | JWH‐210 | 0.25 | 0.25 | 30 | 0.9894 | 0.5 | 25.0 | 2.2 | 4.5 | 19 | 14 |
| UR‐144 N‐(5‐hydroxypentyl) | 27 | 7.3 | UR‐144 or XLR11 | 0.1 | 0.1 | 50 | 0.9941 | 0.5 | 25.0 | 2.8 | 2.0 | 4.7 | 2.0 |
| UR‐144 N‐(4‐hydroxypentyl) | 28 | 7.5 | UR‐144 | 0.1 | 0.1 | 50 | 0.9945 | 0.5 | 25.0 | 2.6 | 2.2 | 4.7 | 1.3 |
| AKB48 N‐pentanoic acid | 29 | 7.7 | AKB48 or 5F‐AKB48 | 0.1 | 0.1 | 50 | 0.9980 | 0.5 | 25.0 | 2.9 | 2.4 | 5.8 | 3.9 |
| JWH‐210 N‐(5‐hydroxypentyl)SEMI | 30 | 7.8 | JWH‐210 | 1.0 | 0.25 | 50 | 0.9814 | 0.5 | 25.0 | 6.3 | 3.8 | 12 | 13 |
| AB‐CHMINACA M2 | 31 | 8.1 | AB‐CHMINACA or AMB‐CHMINACA | 1.0 | 1.0 | 50 | 0.9938 | 2.0 | 100 | 2.6 | 1.6 | 6.3 | 4.7 |
| 5F‐AKB48 N‐(4‐hydroxypentyl) | 32 | 8.2 | 5F‐AKB48 | 0.04 | 0.1 | 120 | 0.9957 | 0.5 | 25.0 | 2.7 | 2.2 | 4.4 | 1.4 |
| AKB48 N‐(4‐hydroxypentyl) | 33 | 8.5 | AKB48 | 0.1 | 0.1 | 25 | 0.9924 | 0.5 | 25.0 | 2.3 | 2.0 | 6.3 | 3.1 |
| AKB48 N‐(5‐hydroxypentyl) | 34 | 8.7 | AKB48 or 5F‐AKB48 | 0.1 | 0.1 | 25 | 0.9937 | 0.5 | 25.0 | 3.4 | 2.1 | 5.0 | 2.8 |
| JWH‐210 N‐(5‐hydroxyindole)SEMI | 35 | 10.0 | JWH‐210 | 2.0 | 1.2 | 72 | 0.9376 | 2.0 | – b | 12 | – b | 17 | – b |
Not determined.
QC High ended up outside of the linear range and data of intra‐ and inter‐sequence precision are therefore left out.
Refers to the concentration shown in the QC Low column.
Refers to the concentration shown in the QC High column.
3.1.1. Chromatographic separation
Ideally the LC set‐up should manage to separate all compounds with identical masses and similar MS/MS spectra. The chromatogram of calibrator 2 containing all metabolites included in the method is displayed in Figure 1. As can be observed, several compounds elute in clusters, but these co‐eluting compounds are not isomers of each other and were separated based on their masses. The choice of chromatographic column, mobile phases and gradient made it possible to separate the isomeric pairs of the hydroxylated metabolites of AKB48, AM‐2201, JWH‐210, and UR‐144. The isomers PB‐22 N‐(4‐hydroxypentyl) and PB‐22 N‐(5‐hydroxypentyl), though, could not be baseline separated. The isomers PB‐22 N‐(4‐hydroxypentyl) and PB‐22 N‐(5‐hydroxypentyl), though, could not be baseline separated. PB‐22 N‐(4‐hydroxypentyl) which eluted first and is a more specific marker of PB‐22 intake was kept, whereas PB‐22 N‐(5‐hydroxypentyl) was excluded from the calibrators. Thus, the calibration was done based on peak height. As baseline separation was not achieved this must be regarded as semi‐quantification.
Figure 1.
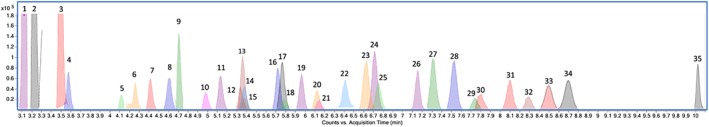
Chromatogram of calibrator 2 containing the 35 metabolites of the synthetic cannabinoids in urine. The numbers corresponds to the ID numbers shown in Table 1 [Colour figure can be viewed at wileyonlinelibrary.com]
3.1.2. Limit of quantification and limit of confirmation
The lowest concentrations detected using the different ID criteria are given in the Supporting Information (Table S3). The LOQs and LOCs of the metabolites are summarized in Table 1. AB‐PINACA pentanoic acid could not be confirmed by the library search at any of the levels explored. BB‐22 3‐carboxyindole could not be confirmed at the level of 17.5 ng/mL due to poor fragmentation and interferences in the MS/MS spectra. However, the metabolite AB‐PINACA‐COOH which showed an LOC of 2 ng/mL could be used as an alternative indicator for an intake of AB‐PINACA, although this is also a metabolite of AMB.25
3.1.3. Linearity
The LOQ and the highest calibration level for each analyte (highest limit of quantification, HLOQ) define the concentration range of the method. Correlation coefficients, LOQs and HLOQs for all compounds included in the method are given in Table 1. The correlation coefficients were above 0.990 except for RCS‐4 N‐(4‐hydroxypentyl)phenol, AB‐FUBINACA M3, AM‐2201 N‐(5‐hydroxyindole), JWH‐018 N‐(5‐hydroxypentyl), THJ‐2201 N‐pentanoic acid, JWH‐210 N‐(5‐hydroxyindole), JWH‐210 N‐(5‐hydroxypentyl) and JWH‐210 N‐pentanoic acid. JWH‐210 N‐5‐hydroxyindole showed reduced linearity and calibration level six was excluded resulting in a less broad concentration range (1.2–72 ng/mL; ie, about 50‐fold) compared to what was expected from the method optimization.
3.1.4. Selectivity and retention time stability
Urine fortified with a mixture of 28 drugs of abuse did not give any false positive results, and the analysis identified no peaks within the retention time windows fulfilling the identification criteria of any of the metabolite compounds.
The acceptance criteria were met for both RT and relative RT for all analytes with the exception of RCS‐4 N‐pentanoic acid and PB‐22 N‐(4‐hydroxypentyl), which in some sequences displayed a deviation up to 2%.
3.1.5. Carry‐over in LC system
No carry‐over above 20% of LOQ after injecting a sample containing 125 ng/mL or the highest calibration level of AB‐PINACA pentanoic acid (320 ng/mL), AB‐CHMINACA M1A (320 ng/mL), RCS‐4 N‐(4‐hydroxypentyl)phenol (160 ng/mL), and AB‐FUBINACA M2 (240 ng/mL). This was achieved using a needle wash of eight seconds between sample draw and injection.
3.1.6. Precision and accuracy
Precision expressed as relative standard deviation (%) and accuracy data expressed as bias (%) are given in Tables 1 and 2, respectively. The acceptance criterion of intra‐sequence precision (≤ 15%) at both concentration levels was achieved for all analytes. The acceptance criterion of inter‐sequence precision (≤ 15%) was achieved for all analytes except JWH‐210 N‐(5‐hydroxyindole) (17%), JWH‐210 N‐pentanoic acid (19%) and THJ‐2201 N‐pentanoic acid (17%) at low concentration. The accepted accuracy of 85%–115% was achieved for all compounds except AB‐FUBINACA M2 (84%), BB‐22‐3‐carboxyindole (79%), JWH‐210 N‐pentanoic acid (131%), and JWH‐210 N‐(5‐hydroxyindole) (119%) at low concentrations; AB‐PINACA pentanoic acid (119%), AB‐CHMINACA M1A (117%) and AM‐2201 N‐(5‐hydroxyindole) (121%) at high concentrations; and AB‐FUBINACA M3 at both low and high concentrations (119% and 135%, respectively). The QC high of JWH‐210 N‐(5‐hydroxyindole) of 100 ng/mL was outside of the linear range and data of precision and accuracy of this level were therefore left out.
Table 2.
Accuracy, matrix effects and recovery for the 35 metabolites of synthetic cannabinoids in urine. n = number of parallels. For concentrations of QC Low and QC High, see Table 1
| Accuracy (n = 10) | Matrix Effects (n = 10) | Recovery (n = 6) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite | QC Low | QC High | QC Low | QC High | QC Low | QC High | ||||
| % | % | % | CV (%) | % | CV (%) | % | CV (%) | % | CV (%) | |
| AB‐PINACA pentanoic acid | 102 | 119 | –a | –a | 123 | 119 | 98 | 13 | 105 | 33 |
| AB‐CHMINACA M1A | 95 | 117 | –a | –a | 57 | 59 | 106 | 4 | 105 | 12 |
| RCS‐4 N‐(4‐hydroxypentyl)phenol | 103 | 112 | –a | –a | 74 | 40 | 103 | 2 | 108 | 10 |
| AB‐FUBINACA M2 | 84 | 111 | –a | –a | 228 | 63 | 105 | 8 | 87 | 15 |
| 5F PB‐22 3‐carboxyindole | 92 | 100 | 101 | 24 | 88 | 14 | 106 | 6 | 103 | 22 |
| RCS‐4 N‐pentanoic acid | 95 | 115 | 88 | 33 | 108 | 27 | 106 | 7 | 99 | 11 |
| PB‐22 N‐pentanoic acid | 96 | 103 | 64 | 18 | 63 | 15 | 106 | 4 | 103 | 11 |
| JWH‐250 N‐pentanoic acid | 97 | 108 | 75 | 12 | 78 | 10 | 108 | 7 | 104 | 7 |
| PB‐22 N‐(4‐hydroxypentyl) | 96 | 108 | 62 | 15 | 72 | 12 | 101 | 5 | 98 | 8 |
| JWH‐073 N‐butanoic acid | 95 | 108 | 90 | 7 | 97 | 5 | 102 | 6 | 98 | 11 |
| JWH‐203 N‐pentanoic acid | 97 | 104 | 100 | 6 | 115 | 7 | 104 | 8 | 102 | 10 |
| PB‐22 3‐carboxyindole | 93 | 106 | 101 | 24 | 103 | 6 | 95 | 10 | 98 | 11 |
| AB‐FUBINACA M3 | 119 | 135 | 115 | 19 | 156 | 16 | 108 | 7 | 102 | 8 |
| AB‐CHMINACA 3‐carboxyindazole | 92 | 112 | 106 | 24 | 133 | 11 | 105 | 5 | 106 | 5 |
| JWH‐018 N‐pentanoic acid | 107 | 100 | 94 | 33 | 117 | 7 | 98 | 9 | 95 | 11 |
| AM‐2201 N‐(4‐hydroxypentyl) | 98 | 98.9 | 146 | 15 | 175 | 12 | 99 | 6 | 97 | 9 |
| JWH‐018 N‐(5‐hydroxypentyl) | 107 | 108 | 83 | 19 | 84 | 16 | 84 | 9 | 84 | 11 |
| JWH‐081 N‐pentanoic acid | 106 | 102 | 114 | 18 | 123 | 9 | 91 | 12 | 97 | 9 |
| AM‐2201 N‐(5‐hydroxyindole) | 105 | 121 | 149 | 17 | 262 | 13 | 74 | 9 | 86 | 7 |
| JWH‐122 N‐pentanoic acid | 102 | 104 | 95 | 19 | 112 | 19 | 83 | 13 | 84 | 14 |
| THJ‐2201 N‐pentanoic acid | 101 | 113 | 195 | 22 | 220 | 28 | 96 | 9 | 96 | 9 |
| BB‐22 3‐carboxyindole | 79 | 109 | –a | –a | 114 | 10 | 84 | 9 | 93 | 8 |
| JWH‐122 N‐(5‐hydroxypentyl) | 102 | 110 | 177 | 28 | 176 | 31 | 70 | 8 | 79 | 6 |
| AB‐PINACA COOH | 91 | 113 | 144 | 27 | 143 | 23 | 100 | 9 | 100 | 7 |
| UR‐144 N‐pentanoic acid | 93 | 100 | 121 | 19 | 115 | 12 | 103 | 6 | 101 | 8 |
| JWH‐210 N‐pentanoic acid | 131 | 102 | 91 | 8 | 99 | 3 | 69 | 18 | 76 | 13 |
| UR‐144 N‐(5‐hydroxypentyl) | 98 | 102 | 118 | 10 | 118 | 7 | 84 | 5 | 88 | 7 |
| UR‐144 N‐(4‐hydroxypentyl) | 96 | 101 | 114 | 8 | 117 | 8 | 90 | 8 | 90 | 8 |
| AKB48 N‐pentanoic acid | 105 | 107 | 100 | 6 | 110 | 3 | 92 | 9 | 93 | 11 |
| JWH‐210 N‐(5‐hydroxypentyl) | 103 | 116 | 109 | 6 | 116 | 5 | 51 | 18 | 63 | 9 |
| AB‐CHMINACA M2 | 97 | 101 | 95 | 21 | 104 | 4 | 94 | 14 | 94 | 10 |
| 5F‐AKB48 N‐(4‐hydroxypentyl) | 95 | 104 | 112 | 6 | 118 | 5 | 88 | 9 | 88 | 8 |
| AKB48 N‐(4‐hydroxypentyl) | 93 | 111 | 102 | 4 | 109 | 4 | 76 | 7 | 79 | 8 |
| AKB48 N‐(5‐hydroxypentyl) | 95 | 110 | 111 | 5 | 115 | 8 | 80 | 9 | 82 | 9 |
| JWH‐210 N‐(5‐hydroxyindole) | 119 | – b | 89 | 10 | 93 | 5 | 11 | 56 | 17 | 25 |
Matrix effect was not estimated at low concentration.
QC High ended up outside of the linear range and data of accuracy are therefore left out.
3.1.7. Matrix effects and recovery
MEs from 57% to 262% were observed (Table 2). In general, the compounds eluting early and midway through the gradient were most influenced by the matrix. There was a relatively good agreement between MEs observed at low and high concentrations. The compounds showing the highest degree of ion suppression were AB‐CHMINACA M1A (57%), PB‐22 N pentanoic acid, PB‐22 N‐(4‐hydroxypentyl) (63%) and RCS‐4 N‐(4‐hydroxypentyl)phenol (74%). The compounds showing the highest degree of ion enhancement were AM‐2201 N‐(5‐hydroxyindole), AB‐FUBINACA‐M2 and THJ‐2201 N‐pentanoic acid (220% ‐ 262%). JWH‐122 N‐(5‐hydroxypentyl, AB‐PINACA COOH, AM‐2201 N‐(4‐hydroxypentyl), AB‐FUBINACA‐M3, and AB‐CHMINACA 3‐carboxyindazole had somewhat less ion enhancement (133%–175%). The remaining 23 compounds were within the acceptance criterion. The level chosen for estimation of the ME at low concentrations for AB‐PINACA pentanoic acid, AB‐CHMINACA M1A, RCS‐4 N‐(4‐hydroxypentyl)phenol, AB‐FUBINACA‐M2, and BB‐22 3‐carboxyindole gave a signal too weak to calculate an ME value.
Recovery was above the accepted limit of 75% for all compounds except JWH‐210 N‐(5‐hydroxyindole) (10%) and JWH‐210 N‐(5‐hydroxypentyl) (51%) at both concentration levels (Table 2).
3.1.8. Stability
Concentrations were considered stable when the calculated values of the stored samples were within 20% from the initial concentration measured in the sample. The QC samples stored at 4°C and 25°C were stable (data not shown), with the exception of JWH‐210 N‐(5‐hydroxyindole) for which a decline of 25% was observed after three days of storage at 25°C. Processed samples stored at 10°C showed a decline of more than 20% after three days for JWH‐018 N‐pentanoic acid, d4‐JWH‐018 N‐pentanoic acid, JWH‐081 N‐pentanoic acid, AM‐2201 N‐(5‐hydroxyindole), JWH‐122 N‐pentanoic acid, BB‐22 3‐carboxyindole, JWH‐122 N‐(5‐hydroxypentyl), JWH‐210 N‐(5‐hydroxyindole), JWH‐210 N‐(5‐hydroxypentyl), and JWH‐210 N‐pentanoic acid (data not shown).
3.2. Results of authentic samples
One or more metabolites were quantified and confirmed in 21 of the total of 1000 samples and in two additional samples metabolites were quantified and identified with ID criterion II, giving a frequency of positive findings of 2.3%. A total of seven different metabolites were confirmed and two identified with ID criterion II. Additionally two metabolites were subsequently identified based on new reference substances. A summary of the findings, with suggestions of which drug(s) that have been ingested in each case, is given in Table 3. JWH‐018 N‐pentanoic acid, JWH‐018 N‐(5‐hydroxypentyl), and JWH‐073 N‐pentanoic acid were the most frequently confirmed metabolites. JWH‐018 N‐pentanoic acid was confirmed in 13 samples and quantified in a range from 0.5 to 10 ng/mL. JWH‐018 N‐(5‐hydroxypentyl) was confirmed in seven samples and quantified from 0.25 to 8.7 ng/mL. JWH‐073 N‐pentanoic acid was confirmed in seven samples and quantified in a range from 0.5 to 12 ng/mL. AKB‐48 N‐pentanoic acid was confirmed in six samples and quantified in a range from 0.28 to 14 ng/mL. AB‐FUBINACA M3 was confirmed in six samples and quantified in a range from 1.4 to 2300 ng/mL. 5F‐PB‐22 3‐carboxyindole was identified, but not confirmed, in three samples at a concentration range from 2.5 to 8.9 ng/mL. BB‐22 3‐carboxyindole was identified, but not confirmed, in one sample at a concentration of 12 ng/mL. In one sample metabolites from three different drugs were confirmed. Metabolites that may originate from more than one drug was confirmed in 17 of 23 samples.
Table 3.
List of samples with one or more metabolites above limit of confirmation (LOC)
| Sample Number | Metabolite I | Conc. (ng/mL) | Metabolite II | Conc. (ng/mL) | Metabolite III | Conc. (ng/mL) | Metabolite IV | Conc. (ng/mL) | Consistent with Intake of |
|---|---|---|---|---|---|---|---|---|---|
| 1 | JWH‐018 N‐pentanoic acid | < LOCa | JWH‐018 N‐(5‐hydroxypentyl) | 0.48 | JWH‐073 N‐butanoic acid | < LOCa | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 2 | 5F‐PB‐22 3‐carboxyindole | 4.8 | 5F‐PB‐22 or 5F‐MDMB‐PICA | ||||||
| 3 | AB‐FUBINACA M3 | 2300 | AKB‐48 N‐pentanoic acid | 14 | AKB‐48‐hydroxy met.c | 29d | 5F‐AKB‐48‐hydroxy met.b | –e | AB‐FUBINACA (or AMB‐FUBINACA or EMB‐FUBINACA) and 5F‐AKB‐48 |
| 4 |
AB‐FUBINACA M3 BB‐22 3‐carboxyindole |
1400 12 |
AKB‐48 N‐pentanoic acid | 6 | AKB‐48‐hydroxy met.c | 13d | 5F‐AKB‐48‐hydroxy met.b | –e | AB‐FUBINACA (or AMB‐FUBI9NACA or EMB‐FUBINACA), 5F‐AKB‐48 and BB‐22 (or MDMB‐CHMICA or ADB‐CHMICA) |
| 5 | AB‐FUBINACA M3 | 5.2 | AKB‐48 N‐pentanoic acid | 1 | AKB‐48 N‐(5‐hydroxypentyl) | 0.48 | 5F‐AKB‐48‐hydroxy met.b | –e | AB‐FUBINACA (or AMB‐FUBINACA or EMB‐FUBINACA) and 5F‐AKB‐48 |
| 6 | AKB‐48 N‐pentanoic acid | 1.3 | AKB‐48 N‐(5‐hydroxypentyl) | 0.88 | 5F‐AKB‐48‐hydroxy met.b | –e | 5F‐AKB‐48 | ||
| 7 | AB‐FUBINACA M3 | 340 | AB‐FUBINACA or AMB‐FUBINACA or EMB‐FUBINACA | ||||||
| 8 | AB‐FUBINACA M3 | 800 | AKB‐48 N‐pentanoic acid | 0.68 | AKB‐48‐hydroxy met.c | 18.6d | 5F‐AKB‐48‐hydroxy met.b | –e | AB‐FUBINACA (or AMB‐FUBINACA or EMB‐FUBINACA) and 5F‐AKB‐48 |
| 9 | 5F‐PB‐22 3‐carboxyindole | 8.9 | 5F‐PB‐22 or 5F‐MDMB‐PICA | ||||||
| 10 | AB‐FUBINACA M3 | 1.35 | AKB‐48 N‐pentanoic acid | 0.28 | AB‐FUBINACA (or AMB‐FUBINACA or EMB‐FUBINACA) together with 5F‐AKB‐48 or AKB‐48 | ||||
| 11 | 5F‐PB‐22 3‐carboxyindole | 4.9 | 5F‐PB‐22 or 5F‐MDMB‐PICA | ||||||
| 12 | JWH‐018 N‐pentanoic acid | 0.78 | JWH‐018 N‐(5‐hydroxypentyl) | < LOC | JWH‐073 N‐butanoic acid | 0.82 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 13 | JWH‐018 N‐pentanoic acid | < LOCa | JWH‐018 N‐(5‐hydroxypentyl) | 8.7 | JWH‐073 N‐butanoic acid | < LOCa | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 14 | JWH‐018 N‐pentanoic acid | 1.6 | JWH‐018 N‐(5‐hydroxypentyl) | 1.6 | JWH‐073 N‐butanoic acid | 2.2 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 15 | JWH‐018 N‐pentanoic acid | 3.5 | JWH‐018 N‐(5‐hydroxypentyl) | 0.87 | JWH‐073 N‐butanoic acid | 2.7 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 16 | JWH‐018 N‐pentanoic acid | 10 | JWH‐018 N‐(5‐hydroxypentyl) | 2.3 | JWH‐073 N‐butanoic acid | 12 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 17 | JWH‐018 N‐pentanoic acid | < LOCa | JWH‐018 N‐(5‐hydroxypentyl) | 2.0 | JWH‐073 N‐butanoic acid | < LOCa | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 18 | JWH‐018 N‐pentanoic acid | < LOC | JWH‐018 N‐(5‐hydroxypentyl) | 0.32 | JWH‐073 N‐butanoic acid | 0.57 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 19 | JWH‐018 N‐pentanoic acid | 0.50 | JWH‐018 N‐(5‐hydroxypentyl) | 0.58 | JWH‐073 N‐butanoic acid | 0.67 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 20 | JWH‐018 N‐pentanoic acid | < LOCa | JWH‐018 N‐(5‐hydroxypentyl) | 0.28 | JWH‐073 N‐butanoic acid | < LOCa | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 21 | JWH‐018 N‐pentanoic acid | < LOCa | JWH‐018 N‐(5‐hydroxypentyl) | 0.46 | JWH‐073 N‐butanoic acid | < LOCa | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 22 | JWH‐018 N‐pentanoic acid | < LOCa | JWH‐018 N‐(5‐hydroxypentyl) | 0.42 | JWH‐073 N‐butanoic acid | 0.52 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 in a mix with JWH‐073 or alone |
| 23 | JWH‐018 N‐(5‐hydroxypentyl) | 0.39 | JWH‐018 N‐(4‐hydroxypentyl)b | –e | JWH‐018 |
Analyte detected but in a concentration below the LOC.
Based on subsequent identification with additional reference substances.
Hydroxylated on the adamantyl ring.
Based on AKB‐48 N‐(5‐hydroxypentyl) calibration.
Not quantified.
4. DISCUSSION
4.1. Method validation
A screening method capable for quantification and confirmation of a variety of SC metabolites at concentrations relevant for clinical and toxicological investigations has been developed. Quantitative screening results are essential when a recent intake needs to be distinguished from residual drug excretion caused by a former intake and repeated samples are available from the same individual.14 Moreover, the access of quantitative methods is crucial in order to carry out pharmacokinetic studies (ie, to estimate half‐lives, peak concentrations, and detection times in urine). The validation of this method demonstrates a satisfactory recovery and selectivity, linearity, precision and accuracy within accepted limits for a majority of the investigated metabolites. No carry‐over following injection of high concentration samples was observed with the selected needle wash settings.
However, some limitations need to be acknowledged. Especially early eluting polar compounds suffer from more pronounced MEs, higher LOQs and LOCs, and less precise quantification. Due to poor quality of MS/MS spectra acquired for a few analytes, relatively high concentrations were needed to achieve acceptable library‐search scores, with correspondingly high LOCs. Co‐eluting isomeric species suppressing or contaminating the MS/MS spectra by introducing additional fragment masses or poor ionization and fragmentation of the precursor can cause these problems. Generally, the LOC is expected to be higher than the LOQ. For AM‐2201 N‐(4‐hydroxypentyl), 5F‐AKB48 N‐ (4‐hydroxypentyl), and UR‐144 N‐(5‐hydroxypentyl), however, the opposite was observed. This was due to MS/MS spectra acquired at concentrations lower than LOQ meeting the threshold scores of ID criterion III. Nevertheless, this had no practical impact as levels below LOQ were not confirmed with a second injection and library search.
There are limited data available on the expected concentrations of the different metabolites in urine after recreational use, but a relatively broad range of concentration levels, from under one and up to hundreds of ng/mL, has been reported.5, 7, 26 The majority of the analytes have an LOC at 1 ng/mL or below which will be sufficient to confirm them at their presumable levels in urine. The window of detection will obviously be narrower if the LOC is higher. LOC of AB‐PINACA pentanoic acid, RCS‐4 N‐(4‐hydroxypentyl)phenol, RCS‐4‐N‐pentanoic acid, AB‐FUBINACA M2, PB‐22 3‐carboxyindole, and BB‐22 3‐carboxyindole was up to 50 times higher compared to LOQs presented using LC–MS/MS based methods.4, 6, 7, 26, 27 The majority of these elute early (RTs < 4 minutes) and are more prone to ME as they co‐elute with matrix components. Higher LOC values than LOQ values were expected as the LOC is based on a more stringent identification criterion. The LOQ is in most methods based on the signal‐to‐noise ratio of the quantifier transition together with accuracy of the concentration measurement. In the presented method, the instrument is both acquiring MS and MS/MS which compromise the sensitivity. Other compounds like AKB48 N‐(4‐hydroxypentyl), AKB48 N‐(5‐hydroxypentyl), AKB48 N‐pentanoic acid, AM‐2201 N‐(4‐hydroxypentyl), JWH‐018 N‐(5‐hydroxypentyl), JWH‐203 N‐pentanoic acid, JWH‐018 N‐pentanoic acid, JWH‐210 N‐pentanoic acid, JWH‐250 N‐pentanoic acid, UR‐144 N‐5‐hydroxypentyl, UR‐144 N‐pentanoic acid, and UR‐144 N‐(4‐hydroxypentyl) had an LOC at the same level or even below the LOQ achieved in methods with a comparable panel of analytes based on LC–MS/MS.4, 5, 7, 28, 29, 30
With the exception of AB‐FUBINACA M3, the HLOQs in this method are sufficiently high to encompass the relevant levels in the positive patient samples as well as previous published levels of SCs in urine, without further dilution. In some studies it has been shown that the ingestion of JWH‐018, JWH‐122, JWH‐210, AM‐2201, UR‐144, and AB‐PINACA can result in high metabolite concentrations (approximately 200 to above 2000 ng/mL),5, 7, 26, 31 which are above the upper calibration limits of the method, but such high levels were not observed in the authentic samples in this study. Of the 23 positive samples analyzed, only four samples had levels above the linear range and therefore had to be diluted to achieve a precise quantification. These samples were diluted 1:20 with blank urine and then re‐analyzed. The method showed good selectivity indicating that other commonly abused compounds should have no influence on the quantification and confirmation of SCs. RTs were proven to be very stable within a worklist of up to 14 hours and can be used as an important ID criterion. The deviation of up to 2% seen for RCS‐4 N‐pentanoic acid and PB‐22 N‐(4‐hydroxypentyl) is within the RT window used in ID criteria and will not compromise the detection and quantification.
The majority of compounds showed MEs and recoveries within the acceptance criteria. A general sample preparation, which was chosen here, can be used for extraction of analytes with a broad spectrum of physico‐chemical properties, but a high ME and thereby unfavorable influence on the analytical quality was observed for some compounds. Choosing a sample preparation method that removes matrix more effectively may most likely decrease the MEs but also potentially reduce the recoveries of many of the analytes. The measured MEs outside the accepted range indicate that both ion suppression and ion enhancement occur. Quantifications with corresponding internal standards for all analytes would potentially compensate for the MEs. However, in a screening method this is not easily achieved and a compromise on the analytical quality for certain analytes must be accepted. Moreover, a tendency toward lower recovery for the compounds eluting late indicates that these compounds also are adsorbed strongly on the SPE sorbent. This must be taken in to account when introducing new compounds to the screening method. As a consequence of high MEs, low recoveries and the absence of dedicated isotopically labeled internal standards, the method must be regarded as semi‐quantitative for the following analytes: AB‐CHMINACA M1A, AB‐CHMINACA 3‐carboxyindazole, AB‐FUBINACA‐M2, AB‐FUBINACA‐M3, AB‐PINACA COOH, AM‐2201 N‐(4‐hydroxypentyl), AM‐2201 N‐(5‐hydroxyindole), JWH‐122 N‐(5‐hydroxypentyl), JWH‐210 N‐(5‐hydroxyindole), JWH‐210 N‐(5‐hydroxypentyl), JWH‐210 N‐pentanoic acid, PB‐22 N‐pentanoic acid, PB‐22 N‐(4‐hydroxypentyl), RCS‐4 N‐(4‐hydroxypentyl)phenol, and THJ‐2201 N‐pentanoic acid.
Our stability results of processed samples stored at 72 hours and 4°C are not in agreement with those previously reported by Scheidweiler et al, who did not reveal any degradation of the metabolites under investigation after 24 hours in room temperature.9 Previous studies of the stability and storage of naturally occurring cannabinoids in urine have proven loss of these types of compounds under different conditions.32, 33, 34, 35 In our method, the use of glass materials and the temperature of 10°C can possibly result in a reduction of analyte due to degradation or adherence to the glass surface. Injections should therefore be done directly after processing the urine samples. If samples are injected three or more days after being processed, the response of JWH‐018 N‐pentanoic acid, JWH‐081 N‐pentanoic acid, AM‐2201 N‐(5‐hydroxyindole), JWH‐122 N‐pentanoic acid, BB‐22 3‐carboxyindole, JWH‐122 N‐(5‐hydroxypentyl), JWH‐210 N‐(5‐hydroxyindole), JWH‐210 N‐(5‐hydroxypentyl), and JWH‐210 N‐pentanoic acid will be lower than freshly prepared samples. This degradation can compromise the quantitative quality of the method.
4.2. Authentic samples
In the 1000 authentic samples analyzed, a total of 10 different metabolites were confirmed or identified with ID criterion II. The majority of the chosen metabolites in the method can be produced by more than one drug (Table 1) which means that a definite identification of the ingested substance(s) is difficult. However, such a list of substances will probably never cover all possibilities as new derivatives with minor chemical modifications will continue to be synthesized. JWH‐018 N‐ pentanoic acid, JWH‐018 N‐(5‐hydroxypentyl) and JWH‐073 N‐pentanoic acid can be a result of consumption of both JWH‐018 and AM‐2201. JWH‐018 N‐(4‐hydroxypentyl) is formed after JWH‐018 consumption but small amounts of JWH‐018 can be produced when smoking AM‐2201 which may result in trace levels of JWH‐018 N‐(4‐hydroxypentyl).17, 18
Retrospectively, a reference standard of JWH‐018 N‐(4‐hydroxypentyl) was analyzed with the method and acceptable chromatographical separation from the 5‐OH isomer was achieved. When samples positive for JWH‐018 N‐(5‐hydroxypentyl) were re‐investigated also JWH‐018 N‐(4‐hydroxypentyl) was confirmed by RT and MS/MS spectrum. JWH‐018 N‐(4‐hydroxypentyl) was not quantified but the peak areas were similar to those of JWH‐018 N‐(5‐hydroxypentyl) in the same sample. The peak areas in the positive samples show that the two metabolites were formed in similar amounts, indicating that JWH‐018 and not AM‐2201 was the drug of origin. The concentrations of JWH‐018 N‐pentanoic acid and JWH‐073 N‐pentanoic acid in these samples analyzed by LC–MS/MS have previously been published by our group.6 In that study, elimination half‐lives of these compounds were determined and detection times established based on the LOQs of that method.6 The relatively high LOCs of JWH‐073 N‐pentanoic acid and JWH‐018 N‐pentanoic acid in the present study as compared to the LOQ of the LC–MS/MS method, which was 0.1 ng/mL, will result in detection times of days instead of weeks.
The pentanoic acid metabolite of AKB48 was detected in six samples. The specific metabolite of 5F‐AKB48 hydroxylated at the pentyl chain (5F‐AKB48‐N‐(4‐hydroxypentyl)) was not detected in any of the samples suggesting that our findings originated from AKB48 and not the 5‐fluoro analogue. However, the seizure statistics from KRIPOS indicate that the use of 5F‐AKB48 was more frequent than AKB48 at the time of sample collection. Previous studies have showed that both AKB48 and 5F‐AKB48 are metabolized to AKB‐48 N‐pentanoic acid and AKB48‐N‐(5‐hydroxypentyl).21, 36 Our initial findings could therefore not unambiguously determine which compounds were taken by these individuals.
A retrospective search for the general formula of hydroxylated 5F‐AKB48 (C23H30FN3O2) revealed a peak three minutes earlier than 5F‐AKB48‐N‐(4‐hydroxypentyl) in five out of the six positive samples. By acquiring CID spectra of this compound the fragmentation pattern could be compared with the literature21, 36 and reveal the structure (Figure 2). The detection of the fragments m/z 151.1117 and 133.1012 corresponding to a hydroxylated adamantyl cation [C10H15O]+ and water loss, and not the m/z 135.1168 which dominate the spectra when fragmenting the metabolite hydroxylated at the pentyl chain, strongly suggested that the metabolite was hydroxylated at the adamantyl group. Sample #10 had the lowest concentration of AKB48 N‐pentanoic acid indicating that the absence of a detected hydroxylated metabolite was sensitivity related. Three synthesized metabolites of 5F‐AKB48 hydroxylated at the adamantyl group (hydroxy‐group in position 3 and both axial and equatorial orientation in position 4) kindly donated by the Department of Forensic Genetics and Forensic Toxicology, National Board of Forensic Medicine (Linköping, Sweden) were analyzed. Chromatographic separation was achieved and RT and fragmentation pattern of the equatorial positioned structure was congruent with the peak detected in the samples. The position of the hydroxyl group on the adamantyl group influenced the fragmentation pattern significantly. The hydroxyl group at position 3 resulted in the proton to seek the carboxamide giving the dominant m/z 250.1085 and 233.1350. In position 4 the hydroxyl group is closer to the cleavage which can explain the formation of the dominating adamantyl cation (m/z 151.1117 and 133.1012). Chromatographic separation and fragmentation of the three synthesized metabolites are given in Figure 3 and NMR spectra are presented in the Supporting Information (Figure S2).
Figure 2.
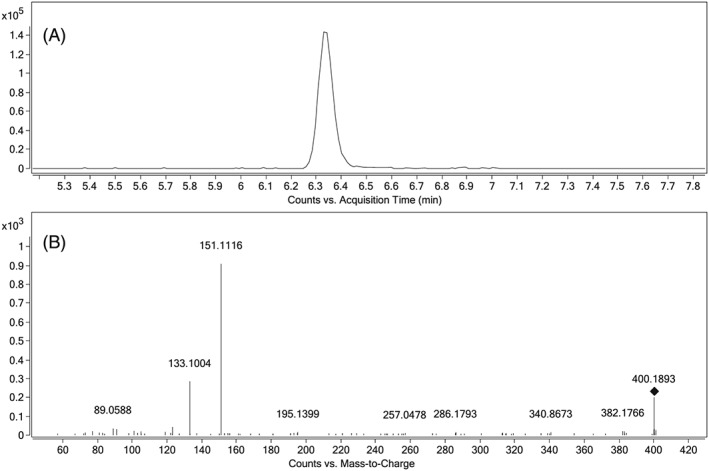
A, extracted ion chromatogram of [C23H30FN3O2 + H]+. B, a CID‐spectrum of the precursor at collision energy of 20 eV
Figure 3.
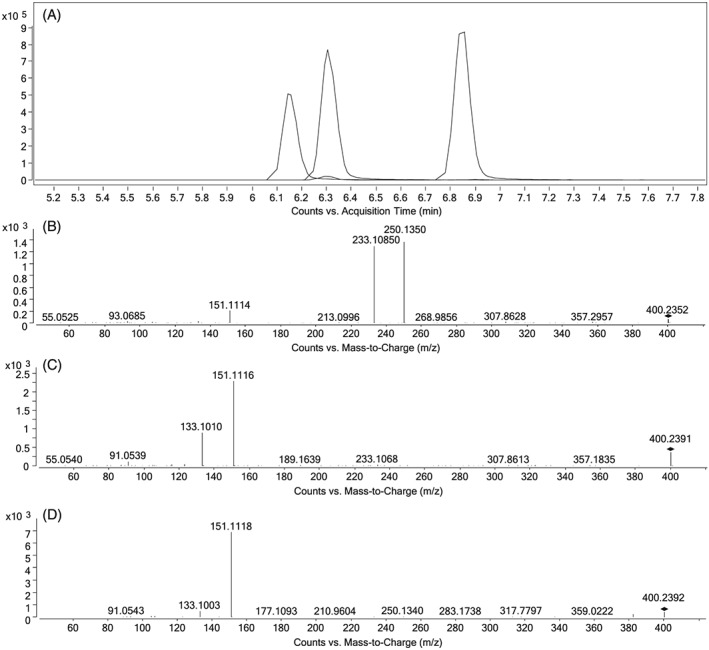
A, extracted ion chromatogram of the protonated synthesized metabolites of 5F‐AKB48 hydroxylated at different positions at the adamantyl group. B, CID‐spectrum of the first eluting compound with hydroxyl‐group in position 3. C, CID‐spectrum of second eluting compound with hydroxy‐group with equatorial orientation in position 4. D, CID‐spectrum of third eluting compound with hydroxy‐group with axial orientation in position 4. All CIDs with a collision energy of 20 eV
The detected AKB48‐OH metabolite in samples # 3, 4 and 8 eluted slightly earlier than AKB48‐N‐(5‐hydroxypentyl), but baseline separation was not achieved. The CID spectra of the precursor (C23H31N3O2, mono‐hydroxylated metabolite of AKB48) at this RT showed a fragmentation pattern typical of the AKB48 metabolite hydroxylated at the adamantyl group while the CID spectra produced at the RT of AKB48‐N‐(5‐hydroxypentyl) confirmed the presence of this metabolite as well (Figure 4). Concentration estimation of the metabolite in these samples was based on the calibration curve of AKB48‐N‐(5‐hydroxypentyl). The hydroxylated metabolite in samples #5 and #6 was confirmed to be AKB48‐N‐(5‐hydroxypentyl), indicating individual differences in the metabolic pathways. The original choice of AKB48 and 5F‐AKB48 metabolites was not sufficient for deciding the specific consumption of these drugs. The method allowed a retrospective investigation of metabolites outside of the original panel, which gave us the possibility to confirm the drug of origin to be 5F‐AKB48. The absence of AKB48‐N‐(4‐hydroxypentyl) in any of the samples supports the theory that AKB48 was not the drug of origin in any of the cases. Sample #10 was the only sample of these where distinguishing between intake of AKB48 or 5F‐AKB48 was not possible.
Figure 4.
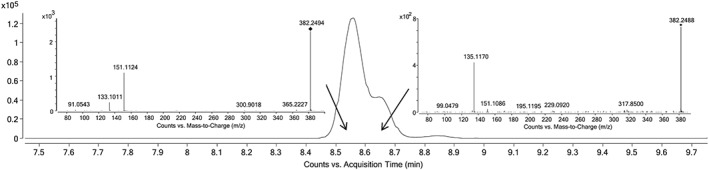
Extracted ion chromatogram of hydroxylated AKB48 [C23H31N3O2 + H]+ and a CID‐spectrum acquired of the precursor from the beginning of the peak and a CID‐spectrum from the shoulder of the peak. Both CID‐spectrum with a collision energy of 10 eV
The AB‐FUBINACA M3 metabolite was semi‐quantified in six samples with a concentration range of 1.35 to 2300 ng/mL. The samples with a concentration above the linear range were diluted 1:20 with blank urine and re‐analyzed. A carry‐over at this high concentration was not tested during validation, but no carry‐over was observed in the samples injected after the samples containing AB‐FUBINACA M3. AB‐FUBINACA M3 is formed by oxidation of the primary amide producing a carboxylic acid, while M2 is formed by oxidation at the oxobutane moiety. M3 has, in contrast to M2, previously been demonstrated to be one of top three markers of AB‐FUBINACA.20, 37 Having AB‐FUBINACA M2 as an analyte in the panel and not detecting it is an additional proof to the studies cited above of M2 being a unsuitable marker. AB‐FUBINACA itself was not included in the method, but a retrospective search for the formula of this compound returned a positive finding in samples #3, 4, and 8, which were also the samples with the highest concentrations of AB‐FUBINACA M3. The more non‐polar mother substance was not detected in samples #5, 7, and 10 demonstrating both the extensive metabolism of this compound and the increased detection time when choosing the more polar metabolites as markers. This method is to the best of the authors' knowledge the first published comprehensive screening method containing AB‐FUBINACA M3. The results show that including this marker is essential to be able to detect AB‐FUBINACA. It must be emphasized, though, that the methyl ester analogue AMB‐FUBINACA (also known as MMB‐FUBINACA)19 and the ethyl ester analogue EMB‐FUBINACA also can result in AB‐FUBINACA M3.
In five of the six samples containing AB‐FUBINACA M3 at least one metabolite of 5F‐AKB48 was also detected. This can be a result of concomitant intake of either AB‐FUBINACA, AMB‐FUBINACA or EMB‐FUBINACA and 5F‐AKB48 from two different products, but it can also be caused by intake of a product containing both drugs either sold as a mix or the one being a contamination of the other. Information from KRIPOS shows that in only one out of 11 AB‐FUBINACA seizures 5F‐AKB48 was detected in the same product. In two out of 11 seizures of AB‐FUBINACA a seizure of 5F‐AKB48 was made in the same case. As our samples were anonymized before analysis we could not determine if some of them were from the same individual(s) or from the same geographical area. A corresponding situation was seen with JWH‐073, which was always detected when any of the metabolites of JWH‐018 were present. A demethylation of JWH‐018 to JWH‐073 and further oxidation to JWH‐073‐N pentanoic acid has previously been hypothesized and cannot be ruled out.8
5F‐PB‐22 3‐carboxyindole could not be confirmed with spectral library in the two samples where a concentration below the LOC (< 5 ng/mL) was observed. The second injection, however, provided MS spectra that strongly indicated the presence of the compound at a concentration > 2.5 ng/mL even though the concentration was too low to be confirmed with ID criteria III. Neither 5F‐PB‐22 3‐carboxyindole nor BB‐22 3‐carboxyindole are specific markers of 5F‐PB or BB‐22 intake, respectively. 5F‐PB‐22 3‐carboxyindole can origin from 5F‐MDMB‐PICA38 and a biotransformation of MDMB‐CHMICA to BB‐22 3‐carboxyindole can take place.39 Other specific markers were not available as certified reference materials. In the case of BB‐22, the absence of specific metabolites for MDMB‐CHMICA and AMB‐CHMICA in biological samples must be documented to prove intake of this substance.40
In statistics provided by KRIPOS of seizures in Norway in 2014, 5F‐AKB48 was at the top with 43 seizures followed by 5F‐PB‐22, BB‐22, AB‐FUBINACA and AM‐2201 with 15, 15, 11, and 10 seizures respectively. JWH‐210, PB‐22, UR‐144, AKB48, JWH‐018, JWH‐073, AB‐CHMINACA, JWH‐122, and JWH‐081 were reported in five or fewer seizures. With the present method, metabolites of 5F‐AKB48 were found in six samples. In addition we found metabolites of five other SCs or their closely related analogs.
The introduction of new SCs to the global market puts the laboratories in a challenging position. Covering all existing and new SCs in the analytical repertoire is a labor‐intensive task, but knowledge of the current situation in a nation and the neighboring countries is a valuable tool to design relevant methods. The statistics of seized drugs of abuse in Norway in recent years show that a couple of new drugs have appeared on the marked. At the same time those dominating in 2014 are still occurring, but at a much lower frequency. This requires a frequent revision of the analytes covered by the method and potentially an addition of new compounds if standards for relevant metabolites become available. With a generic sample preparation and the analytical methodology presented here the addition of new analytes is relatively straight forward with a limited number of validation experiments depending on whether the analyte is added for qualitative or quantitative purposes. Qualitative validation should include experiments to determine LOC, selectivity, retention time, carry‐over, and stability of the new compound. For quantitative purposes additional experiments to determine LOQ, ME, recovery, precision, accuracy and linearity should be conducted.
5. CONCLUSIONS
A UHPLC–QTOF–MS method was developed and validated for quantification and confirmation of 35 metabolites of SCs. The method was based on two injections where the first facilitated the identification and quantification based on full spectra MS data and the second acquired MS/MS data for confirmation. The method showed acceptable performance for its purpose. The sensitivity expressed as LOC was sufficient to confirm the analytes at their presumable levels in urine with a few exceptions which primarily were caused by matrix effects, low recoveries or interference of MS/MS spectra used for confirmation. As a consequence of matrix effects, low recoveries and linearities below the acceptance criteria, in combination with absence of dedicated isotopically labeled internal standards, the method must be regarded as semi‐quantitative for the following analytes: AB‐CHMINACA M1A, AB‐CHMINACA 3‐carboxyindazole, AB‐FUBINACA‐M2, AB‐FUBINACA‐M3, AB‐PINACA COOH, AM‐2201 N‐(4‐hydroxypentyl), AM‐2201 N‐(5‐hydroxyindole), JWH‐018 N‐pentanoic acid, JWH‐122 N‐(5‐hydroxypentyl), JWH‐210 N‐(5‐hydroxyindole), JWH‐210 N‐(5‐hydroxypentyl), JWH‐210 N‐pentanoic acid, PB‐22 N‐pentanoic acid, PB‐22 N‐(4‐hydroxypentyl), RCS‐4 N‐(4‐hydroxypentyl)phenol, and THJ‐2201 N‐pentanoic acid. Presence of AB‐PINACA pentanoic acid could not be confirmed by MS/MS‐spectra.
Relatively generic method settings were chosen to cover a broad range of analytes. This is an advantage if the panel is to be expanded and updated as new SCs are introduced to the marked, but can also result in compromised analytical performance as were demonstrated by those analytes not meeting the defined validation criteria. The validated method was applied to 1000 authentic samples from subjects undergoing drug treatment programs. Interpretation of the analytical results revealed the need for the method to contain specific urine markers if the exact compounds have to be decided. This is a challenge as the availability of commercially synthesized metabolites is limited and the constant release of structurally similar compounds which are biotransformed to metabolites identical to analytes already present in the method. As shown by the presented method, retrospectively processing previously analyzed samples based on new information can detect additional important metabolites that later can be confirmed and included in the method. The presented method is an approach to the analytical challenges that the evolving drug market brings. The targets in the method have to be adjusted according to the drugs used and the current legislation.
Supporting information
Figure S1. Acquired CID of AB‐FUBINACA M3 in A, positive sample and C, the library CID of AB‐FUBINACA M3 B is a comparison of the two
Figure S2‐A1 to S2‐C3. 1H, 13C, and 19F‐NMR spectra were recorded on a Varian Mercury 300 MHz instrument at 25°C in CDCl3, MeOH‐d4 or acetone‐d6.[1]
Table S1. The name, formula, accurate mass, CAS‐number, IUPAC name, and structure of 35 metabolites of synthetic cannabinoids and three deuterium‐labeled internal standards in the method
Table S2. Concentration levels of the 35 metabolites in the six calibration levels and two quality control levels distributed in five groups of working solutions
Table S3. The 35 metabolites of synthetic cannabinoids in urine and the lowest concentration detected using the different ID criteria where (+) or (‐) means detected/not detected at the correspondent concentration. Criterion I means identified by mass accuracy and RT using MassHunter Quantitative, criterion II means identified using Find by formula algorithm with a minimum score of 80 and criterion III means confirmed by MS/MS library spectra both using MassHunter Qualitative
Gundersen POM, Spigset O, Josefsson M. Screening, quantification, and confirmation of synthetic cannabinoid metabolites in urine by UHPLC–QTOF–MS. Drug Test Anal. 2019;11:51–67. 10.1002/dta.2464
REFERENCES
- 1. Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes (Lond). 2006;30(S1):S13‐S18. 10.1038/sj.ijo.0803272 [DOI] [PubMed] [Google Scholar]
- 2. Auwärter V, Dresen S, Weinmann W, Müller M, Pütz M. Ferreirós N. ‘Spice’ and other herbal blends: harmless incense or cannabinoid designer drugs? J Mass Spectrom. 2009;44(5):832‐837. 10.1002/jms.1558 [DOI] [PubMed] [Google Scholar]
- 3. Meyer MR, Maurer HH. Review: LC coupled to low‐ and high‐resolution mass spectrometry for new psychoactive substance screening in biological matrices ‐ Where do we stand today? Anal Chim Acta. 2016;927:13‐20. 10.1016/j.aca.2016.04.046 [DOI] [PubMed] [Google Scholar]
- 4. Borg D, Tverdovsky A, Stripp R. A fast and comprehensive analysis of 32 synthetic cannabinoids using Agilent triple quadrupole LC–MS‐MS. J Anal Toxicol. 2017;41(1):6–16. 10.1093/jat/bkw104. [DOI] [PubMed] [Google Scholar]
- 5. Jang M, Shin I, Kim J, Yang W. Simultaneous quantification of 37 synthetic cannabinoid metabolites in human urine by liquid chromatography‐tandem mass spectrometry. Forensic Toxicol. 2015;33(2):221‐234. 10.1007/s11419-015-0265-x [DOI] [Google Scholar]
- 6. Hegstad S, Westin AA, Spigset O. Detection times of carboxylic acid metabolites of the synthetic cannabinoids JWH‐018 and JWH‐073 in human urine. J Anal Toxicol. 2015;39(4):280‐286. 10.1093/jat/bkv013 [DOI] [PubMed] [Google Scholar]
- 7. Scheidweiler KB, Huestis MA. Simultaneous quantification of 20 synthetic cannabinoids and 21 metabolites, and semi‐quantification of 12 alkyl hydroxy metabolites in human urine by liquid chromatography‐tandem mass spectrometry. J Chromatogr A. 2014;1327:105‐117. 10.1016/j.chroma.2013.12.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moran CL, Le V‐H, Chimalakonda KC, et al. Quantitative measurement of JWH‐018 and JWH‐073 metabolites excreted in human urine. Anal Chem. 2011;83(11):4228‐4236. 10.1021/ac2005636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scheidweiler KB, Jarvis MJY, Huestis MA. Nontargeted SWATH acquisition for identifying 47 synthetic cannabinoid metabolites in human urine by liquid chromatography‐high‐resolution tandem mass spectrometry. Anal Bioanal Chem. 2015;407(3):883‐897. 10.1007/s00216-014-8118-8 [DOI] [PubMed] [Google Scholar]
- 10. Kronstrand R, Brinkhagen L, Birath‐Karlsson C, Roman M, Josefsson M. LC‐QTOF‐MS as a superior strategy to immunoassay for the comprehensive analysis of synthetic cannabinoids in urine. Anal Bioanal Chem. 2014;406(15):3599‐3609. 10.1007/s00216-013-7574-x [DOI] [PubMed] [Google Scholar]
- 11. Sundström M, Pelander A, Angerer V, Hutter M, Kneisel S, Ojanperä I. A high‐sensitivity ultra‐high performance liquid chromatography/high‐resolution time‐of‐flight mass spectrometry (UHPLC‐HR‐TOFMS) method for screening synthetic cannabinoids and other drugs of abuse in urine. Anal Bioanal Chem. 2013;405(26):8463‐8474. 10.1007/s00216-013-7272-8 [DOI] [PubMed] [Google Scholar]
- 12. Huestis MA, Cone EJ. Differentiating new marijuana use from residual drug excretion in occasional marijuana users. J Anal Toxicol. 1998;22(6):445‐454. [DOI] [PubMed] [Google Scholar]
- 13. Smith ML, Barnes AJ, Huestis MA. Identifying new cannabis use with urine creatinine‐normalized THCCOOH concentrations and time intervals between specimen collections. J Anal Toxicol. 2009;33(4):185‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westin AA, Huestis MA, Aarstad K, Spigset O. Urinary excretion of 11‐Nor‐9‐Carboxy‐Δ9‐tetrahydrocannabinol in a pregnant woman following heavy, chronic cannabis use. J Anal Toxicol. 2009;33(9):610‐614. 10.1093/jat/33.9.610 [DOI] [PubMed] [Google Scholar]
- 15. Toennes SW, Geraths A, Pogoda W, et al. Excretion of metabolites of the synthetic cannabinoid JWH‐018 in urine after controlled inhalation. J Pharm Biomed Anal. 2018;150:162‐168. 10.1016/j.jpba.2017.12.016 [DOI] [PubMed] [Google Scholar]
- 16. Castaneto MS, Wohlfarth A, Desrosiers NA, Hartman RL, Gorelick DA, Huestis MA. Synthetic cannabinoids pharmacokinetics and detection methods in biological matrices. Drug Metab Rev. 2015;47(2):124‐174. 10.3109/03602532.2015.1029635 [DOI] [PubMed] [Google Scholar]
- 17. Hutter M, Moosmann B, Kneisel S, Auwärter V. Characteristics of the designer drug and synthetic cannabinoid receptor agonist AM‐2201 regarding its chemistry and metabolism. J Mass Spectrom. 2013;48(7):885‐894. 10.1002/jms.3229 [DOI] [PubMed] [Google Scholar]
- 18. Carlier J, Scheidweiler KB, Wohlfarth A, Salmeron BD, Baumann MH, Huestis MA. Quantification of [1‐(5‐fluoropentyl)‐1H‐indol‐3‐yl](naphthalene‐1‐yl)methanone (AM‐2201) and 13 metabolites in human and rat plasma by liquid chromatography‐tandem mass spectrometry. J Chromatogr A. 2016;1451:97‐106. 10.1016/j.chroma.2016.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, Gerona R. "Zombie" outbreak caused by the synthetic cannabinoid AMB‐FUBINACA in New York. N Engl J Med. 2017;376(3):235‐242. 10.1056/NEJMoa1610300 [DOI] [PubMed] [Google Scholar]
- 20. Vikingsson S, Gréen H, Brinkhagen L, Mukhtar S, Josefsson M. Identification of AB‐FUBINACA metabolites in authentic urine samples suitable as urinary markers of drug intake using liquid chromatography quadrupole tandem time of flight mass spectrometry. Drug Test Anal. 2016;8(9):950‐956. 10.1002/dta.1896 [DOI] [PubMed] [Google Scholar]
- 21. Holm NB, Pedersen AJ, Dalsgaard PW, Linnet K. Metabolites of 5F‐AKB‐48, a synthetic cannabinoid receptor agonist, identified in human urine and liver microsomal preparations using liquid chromatography high‐resolution mass spectrometry. Drug Test Anal. 2015;7(3):199‐206. 10.1002/dta.1663 [DOI] [PubMed] [Google Scholar]
- 22. Wohlfarth A, Gandhi AS, Pang S, Zhu M, Scheidweiler KB, Huestis MA. Metabolism of synthetic cannabinoids PB‐22 and its 5‐fluoro analog, 5F‐PB‐22, by human hepatocyte incubation and high‐resolution mass spectrometry. Anal Bioanal Chem. 2014;406(6):1763‐1780. 10.1007/s00216-014-7668-0 [DOI] [PubMed] [Google Scholar]
- 23. Broecker S, Herre S, Wüst B, Zweigenbaum J, Pragst F. Development and practical application of a library of CID accurate mass spectra of more than 2,500 toxic compounds for systematic toxicological analysis by LC‐QTOF‐MS with data‐dependent acquisition. Anal Bioanal Chem. 2011;400(1):101‐117. 10.1007/s00216-010-4450-9 [DOI] [PubMed] [Google Scholar]
- 24. Peters FT, Drummer OH, Musshoff F. Validation of new methods. Forensic Sci Int. 2007;165(2–3):216‐224. 10.1016/j.forsciint.2006.05.021 [DOI] [PubMed] [Google Scholar]
- 25. Andersson M, Diao X, Wohlfarth A, Scheidweiler KB, Huestis MA. Metabolic profiling of new synthetic cannabinoids AMB and 5F‐AMB by human hepatocyte and liver microsome incubations and high‐resolution mass spectrometry. Rapid Commun Mass Spectrom. 2016;30(8):1067‐1078. 10.1002/rcm.7538 [DOI] [PubMed] [Google Scholar]
- 26. Castaneto MS, Scheidweiler KB, Gandhi A, et al. Quantitative urine confirmatory testing for synthetic cannabinoids in randomly collected urine specimens. Drug Test Anal. 2015;7(6):483‐493. 10.1002/dta.1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simoes SS, Silva I, Ajenjo AC, Dias MJ. Validation and application of a UPLC‐MS/MS method for the quantification of synthetic cannabinoids in urine samples and analysis of seized materials from the Portuguese market. Forensic Sci Int. 2014;243C:117‐125. 10.1016/j.forsciint.2014.07.022 [DOI] [PubMed] [Google Scholar]
- 28. Neifeld JR, Regester LE, Holler JM, et al. Ultrafast screening of synthetic cannabinoids and synthetic cathinones in urine by RapidFire‐tandem mass spectrometry. J Anal Toxicol. 2016;40(5):379‐387. 10.1093/jat/bkw025 [DOI] [PubMed] [Google Scholar]
- 29. Freijo TD Jr, Harris SE, Kala SV. A rapid quantitative method for the analysis of synthetic cannabinoids by liquid chromatography‐tandem mass spectrometry. J Anal Toxicol. 2014;38(8):466‐478. 10.1093/jat/bku092 [DOI] [PubMed] [Google Scholar]
- 30. de Jager AD, Warner JV, Henman M, Ferguson W, Hall A. LC‐MS/MS method for the quantitation of metabolites of eight commonly‐used synthetic cannabinoids in human urine‐‐an Australian perspective. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;897:22‐31. 10.1016/j.jchromb.2012.04.002 [DOI] [PubMed] [Google Scholar]
- 31. Jang M, Yang W, Choi H, et al. Monitoring of urinary metabolites of JWH‐018 and JWH‐073 in legal cases. Forensic Sci Int. 2013;231(1–3):13‐19. 10.1016/j.forsciint.2013.03.053 [DOI] [PubMed] [Google Scholar]
- 32. Jamerson MH, McCue JJ, Klette KL. Urine pH, container composition, and exposure time influence adsorptive loss of 11‐nor‐Δ9‐tetrahydrocannabinol‐9‐carboxylic acid. J Anal Toxicol. 2005;29(7):627‐631. 10.1093/jat/29.7.627 [DOI] [PubMed] [Google Scholar]
- 33. Moody DE, Monti KM, Spanbauer AC, Hsu JP. Long‐term stability of abused drugs and antiabuse chemotherapeutical agents stored at −20°C. J Anal Toxicol. 1999;23(6):535‐540. 10.1093/jat/23.6.535 [DOI] [PubMed] [Google Scholar]
- 34. Golding Fraga S, Diaz‐Flores Estevez J, Diaz Romero C. Stability of cannabinoids in urine in three storage temperatures. Ann Clin Lab Sci. 1998;28(3):160‐162. [PubMed] [Google Scholar]
- 35. de Jager AD, Bailey NL. Online extraction LC–MS/MS method for the simultaneous quantitative confirmation of urine drugs of abuse and metabolites: Amphetamines, opiates, cocaine, cannabis, benzodiazepines and methadone. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(25):2642‐2652. 10.1016/j.jchromb.2011.07.029 [DOI] [PubMed] [Google Scholar]
- 36. Vikingsson S, Josefsson M, Gréen H. Identification of AKB‐48 and 5F‐AKB‐48 metabolites in authentic human urine samples using human liver microsomes and time of flight mass spectrometry. J Anal Toxicol. 2015;39(6):426‐435. 10.1093/jat/bkv045 [DOI] [PubMed] [Google Scholar]
- 37. Castaneto MS, Wohlfarth A, Pang SK, et al. Identification of AB‐FUBINACA metabolites in human hepatocytes and urine using high‐resolution mass spectrometry. Forensic Toxicol. 2015;33(2):295‐310. 10.1007/s11419-015-0275-8 [DOI] [Google Scholar]
- 38. Mogler L, Franz F, Rentsch D, et al. Detection of the recently emerged synthetic cannabinoid 5F‐MDMB‐PICA in 'legal high' products and human urine samples. Drug Test Anal. 2018;10(1):196‐205. 10.1002/dta.2201 [DOI] [PubMed] [Google Scholar]
- 39. Franz F, Angerer V, Moosmann B, Auwärter V. Phase I metabolism of the highly potent synthetic cannabinoid MDMB‐CHMICA and detection in human urine samples. Drug Test Anal. 2017;9(5):744‐753. 10.1002/dta.2049 [DOI] [PubMed] [Google Scholar]
- 40. Carlier J, Diao X, Huestis MA. Synthetic cannabinoid BB‐22 (QUCHIC): Human hepatocytes metabolism with liquid chromatography‐high resolution mass spectrometry detection. J Pharm Biomed Anal. 2018;157:27‐35. 10.1016/j.jpba.2018.05.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Acquired CID of AB‐FUBINACA M3 in A, positive sample and C, the library CID of AB‐FUBINACA M3 B is a comparison of the two
Figure S2‐A1 to S2‐C3. 1H, 13C, and 19F‐NMR spectra were recorded on a Varian Mercury 300 MHz instrument at 25°C in CDCl3, MeOH‐d4 or acetone‐d6.[1]
Table S1. The name, formula, accurate mass, CAS‐number, IUPAC name, and structure of 35 metabolites of synthetic cannabinoids and three deuterium‐labeled internal standards in the method
Table S2. Concentration levels of the 35 metabolites in the six calibration levels and two quality control levels distributed in five groups of working solutions
Table S3. The 35 metabolites of synthetic cannabinoids in urine and the lowest concentration detected using the different ID criteria where (+) or (‐) means detected/not detected at the correspondent concentration. Criterion I means identified by mass accuracy and RT using MassHunter Quantitative, criterion II means identified using Find by formula algorithm with a minimum score of 80 and criterion III means confirmed by MS/MS library spectra both using MassHunter Qualitative