

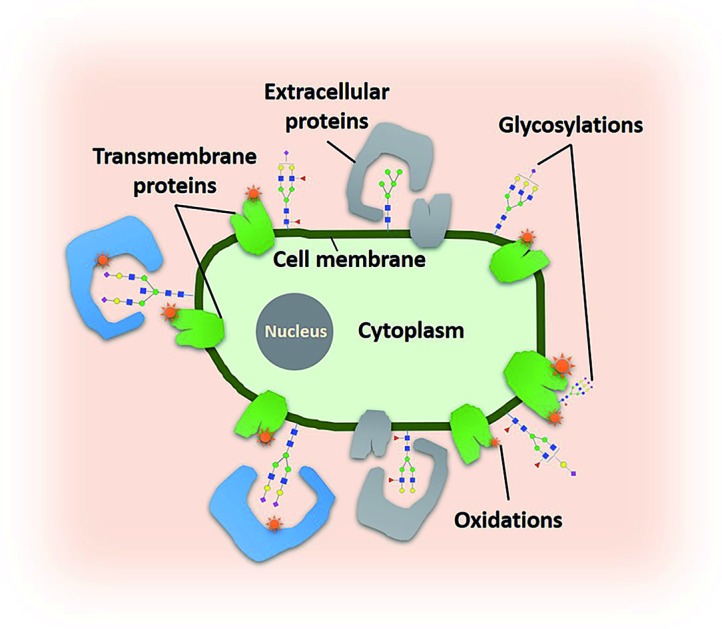
 A “protein oxidation of sialic acid environments” (POSE) mapping tool is developed for sialic acid binding protein discovery.
A “protein oxidation of sialic acid environments” (POSE) mapping tool is developed for sialic acid binding protein discovery.
Abstract
The cell membrane contains a highly interactive glycan surface on a scaffold of proteins and lipids. Sialic acids are negatively charged monosaccharides, and the proteins that bind to sialic acids play an important role in maintaining the integrity and collective functions of this interactive space. Sialic acid binding proteins are not readily identified and have nearly all been discovered empirically. In this research, we developed a proximity labeling method to characterize proteins with oxidation by localized radicals produced in situ. The sites of oxidation were identified and quantified using a standard proteomic workflow. In this method, a clickable probe was synthesized and attached to modified sialic acids on the cell membrane, which functioned as a catalyst for the localized formation of radicals from hydrogen peroxide. The proteins in the sialic acid environment were labeled through amino acid oxidation, and were categorized into three groups including sialylated proteins, non-sialylated proteins with transmembrane domains, and proteins that are associated with the membrane with neither sialylated nor transmembrane domains. The analysis of the last group of proteins showed that they were associated with binding functions including carbohydrate binding, anion binding, and cation binding, thereby revealing the nature of the sialic acid–protein interaction. This new tool identified potential sialic acid-binding proteins in the extracellular space and proteins that were organized around sialylated glycans in cells.
Introduction
Cell membranes are covered with a matrix of glycans on a scaffold of lipids and proteins. This glycocalyx creates a highly interactive environment that governs many cellular functions. At the terminus of many glycans are negatively charged sialic acids, which are involved in a host of glycan–protein interactions. Sialic acid-binding proteins include Factor H and selectins, which are known to recognize negatively charged sialic acids through anion-binding domains.1 Another group includes siglecs (sialic acid-recognizing Ig-superfamily lectins), which belong to the immunoglobulin superfamily and recognize sialylated ligands.2 With few exceptions, sialic acid-binding proteins are discovered empirically, and there are few examples that identify specific sialic acid protein partners. There have been no general methods for identifying sialic acid-binding proteins in a broad scope.
There are currently numerous methods to characterize protein–protein interactions;3 however, there are very few for glycan–protein interactions. For protein–protein interactions, crosslinking methods are used to produce covalent bonds between interacting proteins.4 These methods, while complicated and difficult to elucidate, have become more available with new reagents and automated proteomic workflows. Other covalent labeling techniques including hydroxyl radical oxidation and stable isotope labeling such as hydrogen deuterium exchange (HDX) are also used to distinguish interacting domains.5,6 Oxidative labeling is a useful tool because it covalently distinguishes potential interacting sites. An early approach was developed by Lee and co-workers who applied a tethered metal chelate for footprinting protein complexes.7 However, the method required the isolation of the protein complexes and could not be performed in situ. Branon and co-workers demonstrated enzyme-catalyzed proximity labeling for identifying protein–protein interactions in cell lines.8 However, the required concentration of hydrogen peroxide made the method cytotoxic to the cells in cultures.
Glycan mediated protein–protein binding is typically probed using glycan arrays.9 The complementary lectin arrays are also used but neither can be performed in situ.10 Due to the heterogeneities of cell surface glycosylation and the limitations of existing analytical methodologies, there are only a few examples involving characterization of sialic acid–protein interactions. A crosslinking-based approach using photocleavable linkers that produce reactive carbene was used to capture sialic acid-interacting partners in situ.11 A similar approach was used to determine the protein partner of sialylated glycosphingolipids,12 and the cis-ligands13 and trans-ligands14 binding to CD22 through their sialic acids. Although these studies illustrated that sialic acid binding proteins can be obtained from cross-linking experiments, they were focused on the discovery of binding partners of a specific protein.
We report a method termed “protein oxidation of sialic acid environments” (POSE) to map the protein environment of sialic acids in situ. In this method, azido groups are first metabolically incorporated into cell surfaces as N-azidoacetyl sialic acid (SiaNAz) on sialylated glycoconjugates (Fig. 1a).15 A synthesized catalytic iron (S)-1-(p-bromoacetamidobenzyl)ethylenediaminetetraacetate (FeBABE) probe is then specifically conjugated to the azide group through click chemistry (Fig. 1b). To determine the extent of the incorporation of SiaNAz and the conjugation of the metal complexes, we performed glycomic analysis on released cell surface glycans using liquid chromatography-mass spectrometry (LC-MS). Subsequently, cells were treated with hydrogen peroxide and hydroxyl radicals were produced at the FeBABE probe spreading to nearby proteins (Fig. 1c). Specific amino acids were oxidized, and the proteins were analyzed with LC-MS using standard proteomic workflows. By varying the concentration of hydrogen peroxide and the reaction times, the extent of oxidation of nearby proteins was similarly altered. The oxidized proteins were profiled, and the extent of oxidation was determined using quantitative proteomics. The functional analysis of specifically oxidized proteins provided potential sialic acid-binding candidates on cell surfaces.
Fig. 1. The schematic representation of the POSE concept. (a) The metabolic incorporation of ManNAz into cell surface glycoproteins as SiaNAz. (b) The bio-orthogonal reaction of the DBCO-FeBABE probe with the SiaNAz group through copper-free click chemistry. (c) The oxidation of proteins in the sialic acid environment by hydroxyl (and other) radicals generated from the reaction between the FeBABE probe and hydrogen peroxide.

Methods
Synthesis of dibenzocyclooctyne-functionalized p-bromoacetamidobenzyl-EDTA, iron(iii) chelate (DBCO-FeBABE)
FeBABE (1 mg) and dibenzocyclooctyne amine (DBCO–NH2) (0.726 mg) were mixed in 500 μL of dimethyl sulfoxide (DMSO). The reaction was carried out at room temperature for 48 hours with shaking, and the structure of the product and yield were determined by LC-MS analysis in negative ionization mode.
Synthesis of profluorescent rhodamine
Detailed procedures for synthesizing profluorescent rhodamine was described previously by Cao et al.16 Briefly, rhodamine B (479.1 mg, 1 mmol) and 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (172.2 mg, 1 mmol) were mixed in 10 mL of anhydrous dichloromethane (DCM), followed by the addition of 12.3 mg of 4-dimethylamiopryidine (DMAP, 0.1 mmol) and 206.3 mg of dicyclohexylcarbodiimide (DCC, 1 mmol). The reaction was conducted under an argon atmosphere while stirring at room temperature overnight. The product was purified by column chromatography (5% methanol in DCM, Rf = 0.3) to obtain the solid in dark red color (159.9 mg, 26.8%). The structure of profluorescent rhodamine was confirmed with LC-MS in positive ionization mode.
Cell culture
Human colon adenocarcinoma Caco-2 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown in Eagle's Minimum Essential Medium (EMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U mL–1 penicillin and streptomycin, non-essential amino acids, and 2 mM l-glutamine. Human lung carcinoma A549 cells and human normal prostate epithelial (immortalized with SV40) PNT2 cells were obtained from ATCC and grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U mL–1 penicillin and streptomycin, non-essential amino acids and 2 mM l-glutamine. Cells were maintained in a humidified incubator at 37 °C with 5% CO2 and subcultured at 80% confluency.
Confocal imaging of DBCO conjugation
The PNT2 cells were seeded into FluoroDish™ cell culture dishes (WPI, FL) coated with poly-d-lysine with appropriate density using RPMI 1640 cell culture media. At 40% confluency, cells were treated with 100 μM N-azidoacetyl mannosamine (ManNAz) supplemented growing media for 72 hours. Afterwards, both the control cells without ManNAz treatment and the treated cells were rinsed with phosphate-buffered saline (PBS) and treated with 50 μM dibenzocyclooctyne–cyanine3 (DBCO–Cy3) in PBS at room temperature for 1 hour, followed by the fixation with 4% paraformaldehyde (Affymetrix OH). Cells were then stained for the nucleus with 1.6 μM Hoechst 33342 (Thermo Fisher Scientific, MA) followed by the staining for the plasma membrane with 1000-fold diluted CellMask™ Deep Red Plasma Membrane Stain (Thermo Fisher Scientific, MA), respectively at 37 °C for 10 min. Fluorescence images were captured using a Leica TCS SP8 STED 3X Super-Resolution Confocal Microscope (Wetzlar, Germany).
Visualization of hydroxyl radicals via profluorescent rhodamine
Cells were split and treated with ManNAz for 72 hours on FluoroDish™ cell culture dishes. Three days after ManNAz treatment cells were rinsed with PBS and then subjected to 50 μM DBCO-FeBABE in PBS at 37 °C for 1 h. Subsequently, the cells were rinsed with PBS and stained for the nucleus and plasma membrane as described previously. Live-cell confocal imaging was performed immediately after adding 1 μM profluorescent rhodamine, 2 μL of DMSO, and 100 μM hydrogen peroxide to 1 mL of PBS, with fluorescence images captured every 1 minute using a Leica TCS SP8 STED 3X Super-Resolution Confocal Microscope (Wetzlar, Germany).
Oxidation of cell plasma membrane proteins
For oxidative mapping of the sialic-acid environment on the cell surface, cells were split into T75 flasks and treated with regular media supplemented with 100 μM ManNAz after reaching a 30–40% confluency for 3 days at 37 °C in a humidified incubator with 5% CO2. The medium was further supplemented with 50 μM DBCO-FeBABE for 1 h at 37 °C followed by the treatment with 100 or 300 μM hydrogen peroxide for a 30 minute-reaction. After the reaction was quenched with 10 mM methionine amide hydrochloride in PBS, cells were harvested and resuspended in a homogenization buffer containing 1 : 100 protease inhibitor (EMD Millipore, CA), 0.25 M sucrose, and 20 mM HEPES–KOH (pH 7.4). For control, cells were cultured with the same media and harvested together with treated cells with identical procedures.
Cell membrane extraction
The procedures for cell membrane extraction were described previously and performed here with slight modifications. Cells were lysed on ice with five alternating on and off pulses in 5 and 10 second intervals using a probe sonicator (Qsonica, CT). Nuclear and mitochondrial fractions and cellular debris were pelleted and isolated by centrifugation at 2000 × g for 10 min. The supernatants were then ultra-centrifuged at 200 000 × g for 45 min at 4 °C to extract the plasma membrane. The pellets of the cell membrane were resuspended in 500 μL of 0.2 M Na2CO3 solution and 500 μL of water followed by two more ultracentrifugation treatments at 200 000 × g for 45 min to wash off the endoplasmic reticulum (ER) and cytoplasmic fraction.
Protein digestion
The extracted cell membrane pellets were reconstituted with 60 μL of 8 M urea and sonicated for 15 minutes for denaturing. Dithiothreitol (DTT) was then added to the samples and incubated for 50 minutes at 55 °C. Thereafter, 4 μL of iodoacetamide (IAA) was added to alkylate the free thiol groups for 25 minutes at room temperature in the dark. After diluting the urea concentration and adjusting the pH value of the samples by adding 420 μL of ammonium bicarbonate (NH4HCO3) solution, 3 μg of trypsin was added to the mixture and digestion was conducted at 37 °C for 18 hours. The resulting peptides were desalted by solid-phase extraction with C18 cartridges containing 500 mg materials. The glycopeptides were enriched by solid-phase extraction using iSPE®-HILIC cartridges (The Nest Group, MA). The samples were dried in vacuo using miVac (SP Scientific, PA).
Glycoproteomic analysis with LC-MS/MS
The samples were reconstituted with 2% (v/v) acetonitrile (ACN) and 0.1% (v/v) trifluoroacetic acid (TFA) in water and separated on an EZ-1200 LC system using a C18 column with 15 cm length and 75 μm inner diameter (ID). LC separation was performed with a binary gradient using solvent A with 0.1% (v/v) formic acid (FA) in water and solvent B with 80% ACN and 0.1% FA in water at a flow rate of 300 nL min–1. After the separation, the peptides were analyzed on a Thermo Q-Exactive Plus Orbitrap with the full MS scanned from 700 to 2000 in positive ionization mode. The MS/MS spectra were collected for fragments with m/z values starting from 120.
Glycoproteomic data analysis
Proteins were identified using Byonic software (Protein Metrics, CA). Raw files were searched against a human protein FASTA database acquired from UniProt. C-Terminals of lysine and arginine were used for specific cleavage sites, and missed cleavages were restricted to 2. Precursor mass tolerance was limited to 10 ppm, and CID & HCD fragmentation with a mass tolerance of 20 ppm was applied. Carbamidomethylation at cysteine was assigned as the fixed modification. Deamidation of asparagine and glutamine, methylation of lysine and arginine, and acetylation of the protein N-terminus were selected as the variable modification. For oxidized samples, common oxidized modifications of 13 amino acids (M, C, W, Y, F, H, L, I, R, V, T, P, and K) were selected as variable modifications. For glycoprotein samples, an in-house human database was applied for N-glycosylation at asparagine. MzIdentML files were exported during the search to further quantitate for oxidation. Identifications with high confidence were achieved through filtering 1% false discovery rate (FDR), posterior error probability (PEP) smaller than 0.01, and DeltaMod larger than 10.
Quantitation of oxidation
The extent of oxidation was determined with the software Skyline by quantifying the peptide precursor peak area in the MS1 spectrum. The peptide results in the form of mzid files were generated by searching the proteomics raw data with the software Byonic, followed by establishing a MS spectra library in the software Skyline for the overall cell surface proteomics. The peptides were further matched to proteins by searching them against the human proteome database. The extracted ion chromatograms (EIC) of peptide precursors were obtained and integrated for quantitation. To calculate the oxidation extent (OE), the EIC peak areas of oxidized peptides were normalized to the EIC peak areas of corresponding non-oxidized peptides.
Enzymatic release and purification of N-glycans
Plasma membrane pellets were resuspended with 100 μL of 5 mM DTT in 100 mM NH4HCO3 and heated in a 100 °C water bath for 1 min to thermally denature the proteins. The cleavage of N-glycans was performed by adding 2 μL of peptide N-glycosidase F (PNGase F, New England Biolabs, MA) followed by the incubation at 37 °C in a microwave reactor (CEM Corporation, NC) for 10 min at 20 watts. To convert the N-glycans from amines to aldehydes, the sample was allowed to stand in a 37 °C water bath overnight. To separate the released N-glycans from the membrane fractions, residual deglycosylated proteins and lipids, 350 μL of nanopure water was added and ultra-centrifugation at 200 000 × g was conducted for 45 min. The released N-glycans contained in the supernatant were purified using porous graphitic carbon (PGC) on an SPE plate (Grace, IL) and eluted with 40% (v/v) ACN and 0.5% (v/v) TFA in water. The eluted samples were dried in vacuo and stored at –20 °C until analysis.
Glycomic analysis with LC-MS/MS
The glycan samples were reconstituted in nanopure water, and 5 μL of the sample was injected and analyzed with an Agilent 6520 Accurate Mass Q-TOF LC/MS (Agilent, CA) equipped with a PGC micro-fluidic chip, which incorporates an enrichment column, an analytical column, and a nano-electrospray tip in a single assembly. A binary gradient using solvent A with 3% (v/v) ACN and 0.1% (v/v) FA in water and solvent B with 90% (v/v) ACN and 1% (v/v) FA in water was applied to separate samples at a flow rate of 300 nL min–1. MS spectra were collected with a mass range of m/z 600–2000 at a rate of 1.5 s per spectrum in positive ionization mode. Six of the most abundant precursor ions in each MS spectrum were subjected to fragmentation through collision-induced dissociation (CID) with nitrogen gas.
Glycomic data analysis
Compound chromatographs of glycans with a proton adduct were extracted with the MassHunter Qualitative Analysis B. 07 software (Agilent, CA) using the function ‘Find by Molecular Features’. A mass tolerance of 20 ppm was applied for signal extraction, and signals above 1% S/N ratio (signal-to-noise ratio) were filtered. N-Glycan compounds were identified with an in-house library that contains the accurate mass of common N-glycans, SiaNAz modified N-glycans, and SiaNAz–DBCO–NH2 modified N-glycans. The N-glycan structures were further confirmed through tandem MS fragmentation spectra. Relative abundances of each N-glycan subtype were calculated after normalizing the integrated peak areas to the total peak areas of all glycans detected.
Results
Incorporation of functionalized sialic acids on the cell membrane and conjugation of oxidative catalytic probe
We first determined the extent of SiaNAz incorporation into N-glycans. Although sialic acids are present in O-glycans and in glycolipids, we examined N-glycans as a surrogate for all sialic acid-containing glycoforms. The extent of SiaNAz incorporation was monitored by releasing the glycans followed by LC-MS analysis as described previously.17 The extracted compound chromatogram (ECC) and MS/MS data are provided in Fig. S1 and S2.† The incorporation of SiaNAz varied for three cell lines and increased with time (Fig. S3†). The highest incorporation where SiaNAz replaced 87% of all sialic acids was in the PNT2 cell line. The undifferentiated Caco-2 cell line had the lowest incorporation at 18% incorporation. Notably, the SiaNAz incorporation in the differentiated Caco-2 cell line, which has a greater amount of sialylated glycans compared to the undifferentiated, had a greater value at 26%. The cell line A549 had a maximum incorporation of 45%.
DBCO–NH2 was conjugated to SiaNAz on the cell membrane (Fig. 1b), and glycomic analysis was again conducted on the released N-glycans with LC-MS. As shown in Fig. S1c,† most of the SiaNAz-containing glycans (dark blue peaks) (Fig. S1b†) were converted to DBCO–NH2–conjugated glycans (black peaks) including the most abundant composition Hex5HexNAc4Fuc1SiaNAz1. Tandem MS further confirmed structures of DBCO–NH2–conjugated glycans (Fig. S2c†). To calculate the conjugation efficiency, we divided the total abundances of all the DBCO–NH2–bearing glycans by the total abundances of all the SiaNAzylated glycans. Consequently, the efficiency of the conjugation was estimated to be above 86%.
The localizations of DBCO–conjugated glycans on the cell surface were determined by labeling cells with DBCO–Cy3 and visualization using confocal microscopy. In this method, PNT2 cells were grown on a glass-bottom dish coated with poly-l-lysine and treated with ManNAz for three days followed by the treatment with DBCO–Cy3 for 1 hour. Labeling of the cells using Hoechst 33342 (cell nuclei) and CellMask™ Deep Red (cell plasma membrane) was performed before confocal imaging. As shown in Fig. 2a, PNT2 cells showed strong intensities (red color) after treatment with ManNAz and reaction with DBCO–Cy3. This Cy3 stain overlapped with the cell plasma membrane stain, indicating that the conjugation of the DBCO group was efficient and localized.
Fig. 2. Confocal microscopy of cell lines. (a) Fluorescence imaging of PNT2 cells with localization of SiaNAz-modified glycans. Cells without ManNAz treatment (control) and with ManNAz treatment were stained with Hoechst 33342 (blue), CellMask™ Deep Red (green), and DBCO–Cy3 (red). (b) Detection of produced hydroxyl radicals in PNT2 cells through live cell imaging. After treating the cells with ManNAz and attaching the FeBABE probe to the sialylated glycans, cells were stained with Hoechst 33342 (blue) and CellMask™ Deep Red (green). Pro-fluorescent rhodamine was then added along with hydrogen peroxide and DMSO. The produced hydroxyl radicals (rhodamine, red) at different time points including t = 0 min (starting point), t = 15 min (middle point), and t = 30 min (endpoint) are shown in the middle panel. The combined photos are shown in the lower right panel. The intensities of the stains varying over time are shown in Fig. S4.† .

In situ production of reactive radicals for oxidative protein labeling
To ensure that oxidation was occurring on the cell membrane, we used confocal microscopy with a profluorescent dye to detect hydroxyl radicals.16 After treating the cells with ManNAz and attaching the FeBABE probe to the sialylated glycans, the mixture of rhodamine, hydrogen peroxide, and DMSO was added, and live cell images were taken every 30 seconds for a period of 30 minutes as shown for PNT2 (Fig. 2b). Immediately after addition, at the time point of 0 minutes, the rhodamine dye was just detectable. After 15 minutes, red pigments were noticeable and colocalized with the cell membrane stain, indicating that hydroxyl radicals were produced. At 30 minutes, the dye was observed to diffuse through the membrane. Quantitation of all fluorescence intensities is provided in Fig. S4.† The intensities of rhodamine emission in five membrane and five nuclear regions were quantified and averaged over time (Fig. S4a and b†). The quantitation results showed that at the onset rhodamine intensities on the cell membrane regions were high and increased to a maximum at 700 seconds. The onset at time zero was high because it took at least a few minutes to add the reagent, transfer the cells to the microscope, and prepare the conditions for optimal observation. During this period, the rhodamine intensity in the nuclear regions began at zero and increased even after 2500 seconds due to the movement of hydroxy radicals to the nucleus. Proteomic analysis was performed on the samples collected at various reaction times, and the reaction conditions were selected based on the optimal proteomic results. At 30 minutes, the same cell membrane proteins were oxidized as those in 15 minutes but more extensively (Fig. S5b†), which is consistent with the quantitation results of rhodamine emission intensity illustrated in Fig. S4b.†
The reaction conditions for cell surface protein mapping were optimized based on protein oxidation. To determine the optimal treatment conditions, we measured enhanced oxidation quantitatively using Skyline software (Fig. S5a–g†).18 In general, with the increasing ratio of H2O2 to Fe(iii), the oxidation increased more extensively. The treatment was conducted on four types of cell lines, PNT2, A549, undifferentiated Caco-2 and differentiated Caco-2. These cell lines were chosen because they represented different tissues including prostate, lung, and colon, respectively. In addition, Caco-2 was selected because it represented cells at different states. Caco-2 differentiates at approximately 14 days to express features of intestinal enterocytes.19
The optimal conditions for oxidation were found to vary only slightly among the cell lines. The optimal conditions for PNT2 and A549 were found to be 50 μM DBCO-FeBABE and 100 μM hydrogen peroxide with a reaction time of 30 minutes at 37 °C. Caco-2 was optimized to yield slightly different conditions, namely 50 μM DBCO-FeBABE and 300 μM hydrogen peroxide. To account for background protein oxidation, we used a number of controls including treatment with PBS only, DBCO-FeBABE only, and hydrogen peroxide only to determine baseline oxidation. For the proteins oxidized in both control sample sets and the POSE sample sets, the oxidation in the POSE samples yielded higher degrees of oxidation. We also monitored oxidation in the nuclear proteins and found that their levels were constant and low throughout the reaction period (Fig. S5i†) indicating that the oxidation was primarily occurring in the cell membrane. However, when the ratio of Fe(iii) to H2O2 was changed from 1 : 2 to 1 : 6, an increase of oxidation in the nuclear proteins was observed (Fig. S5h and i†).
The proteomic analysis yielded 200 to 350 oxidized peptides for all cell lines, corresponding to 150 to 200 oxidized proteins for each of the three cell lines (Fig. S6 and ESI 1–4†). The most commonly oxidized amino acid was methionine (M) (over 80%) followed by cysteine (C) and tryptophan (W). Other oxidized amino acids including proline (P), histidine (H), and tyrosine (Y) were in much smaller abundances. Similar observations regarding the relative ease of oxidation were reported previously.20 The functions of these oxidized proteins were annotated using PANTHER (Fig. S7†),21 and the details of the oxidation sites for all cell lines categorized to different functions are summarized in ESI 7.†
Identification of sialic acid binding proteins through oxidative mapping
We identified the proteins that were oxidized using a standard proteomic workflow. The oxidized proteins for each cell line were categorized into three groups: (1) sialylated glycoproteins that contain transmembrane domains, (2) membrane-bound proteins with predicted transmembrane domains and (3) extracellular proteins without transmembrane domains. The first group (Group 1) was oxidized sialylated glycoproteins. The second group (Group 2) contained non-sialylated proteins with transmembrane domains residing in close proximity to sialylated proteins. Proteins in Group 3 were those with no transmembrane domains but reside in the extracellular space and kept in place due potentially to sialic acid interactions (ESI 5†). Potential sialic acid-binding proteins belonging to both Group 1 (protein L1CAM) and 3 (protein LAMA5) were found and their transmembrane locations as well as binding domain locations are shown in Fig. 4a and b.
Fig. 4. Transmembrane location of oxidized proteins. (a) The locations of the sialic-acid-recognizing domain and oxidation sites of protein L1CAM. The oxidation sites are found in the sialic acid-binding domain, which is predicted by THMMH to be extracellular. (b) The location of the sialic-acid-recognizing domain and oxidation sites of protein LAMA5. The oxidation sites are found in the sialic acid-binding domain, which is predicted to be located extracellularly. (c) Predicted transmembrane locations of three oxidation sites after the POSE treatment of the protein VAPA. (d) Predicted transmembrane locations of three oxidation sites after the POSE treatment of protein GTR3.

The glycoproteins with their oxidation sites and glycosylation sites (Group 1) were provided in ESI 1–4.† The distance between the site of sialylation and the site of oxidation was determined for these glycoproteins. We calculated the nearest distance between the two sites for selected proteins using Visual Molecular Dynamics (VMD) software.22 The distances between glycosylation sites and oxidation sites for sialylated glycoproteins from all three cell lines showed that the optimum distance was 20–30 Å (Fig. 3a). This distance matched well with the length of the glycan plus the probe (19 Å, Fig. 3b). For example, the protein DPP4 (dipeptidyl peptidase 4) had four glycosylation sites with sialylated glycans including N150, N321, N520, and N685 (Fig. 3). There were more sialylated glycans than non-sialylated species in this protein (Fig. 3c and d). The proteomic data yielded two oxidation sites M348 and M591. The distances between the oxidation sites and four glycosylation sites were calculated based on the crystal structure, and an average value of 20–25 Å was obtained (Fig. S8†).
Fig. 3. Self-oxidation of glycoproteins. (a) The distribution of distances between oxidation sites and glycosylation sites summarized for over 50 oxidized glycoproteins. (b) The distances between the FeBABE probe and SiaNAz. (c) The number of glycopeptides for each glycosylation site of protein DPP4. The sites N150 and N685 have more than 80% of glycopeptides as sialylated. (d) Approximately 63% of the glycans are sialylated. (e) The glycans associated with the four glycosylation sites of protein DPP4. Putative structures of N-glycans are assigned for each site. The glycosylation and oxidation sites of protein DPP4 are shown.

The second group of oxidized proteins (Group 2) were those that were bound to the membrane through transmembrane domains but did not contain sialylation sites. Membrane proteins weave through the lipid bilayer with segments in the cytosol, the lipid bilayer, and the extracellular space. Software has been developed elsewhere that predicts the location of various protein domains. One of the most commonly used is TMHMM,23 which is based on estimating the hydrophobic character of protein domains. Oxidation of proteins provided indications that the domain was in the extracellular space. An example protein, VAPA (vesicle-associated membrane protein-associated protein A), was oxidized in the PNT2 cell line after treatment. Fig. 4c shows the results of TMHMM with the probability of the location for each amino acid. The protein was oxidized at three amino acids including M132, M142, and M159, suggesting that these amino acids were extracellular. For this protein, the TMHMM results matched well with the oxidation products so that the experiment supported the theoretical results.
With more complicated protein arrangements, we found that the oxidation did not necessarily match the prediction. The protein GTR3 (solute carrier family 2, facilitated glucose transporter member 3) (Fig. 4d) from Caco-2 yielded four oxidation sites including C345, M242, M481, and M483. As predicted by TMHMM, the protein GTR3 had 12 transmembrane regions with the oxidation site M242 being extracellular, however, with only 55% probability. Nonetheless, the oxidation of the M242 site supported the theoretical prediction. Conversely, although the amino acids within the region 449 to 496 were predicted to be in the cytosol with a probability of 71%, the oxidation of M481 and M483 suggested that the domain was external, which contrasted the theoretical predictions. Similar detailed predictions and results of other proteins with glycosylation sites for three cell lines are provided in the Fig. S9–S11.†
The oxidized proteins that associate with the extracellular space but have no membrane domains (Group 3) were consistent in their function as sialic acid-binding proteins. Group 3 contained 165 proteins over three cell lines. In this group 36 were found in at least two cell lines and sometimes all three. The vast majority of these proteins were functionally characterized as either binding or catalytic proteins (>80% of oxidized proteins) according to STRING.24 While the “catalytic” proteins could also be sialic acid-binding proteins, the “binding” proteins were those that were more likely associated with sialylated glycans. However, most of them have not been previously characterized as specifically sialic acid binding proteins. The number of these proteins varied. For Caco-2 (both differentiated and undifferentiated) these proteins numbered as many as 127 and fell into three categories: carbohydrate derivative binding (46), anion binding (56) and cation binding (13). With PNT2 there were 33 proteins, and for A549 there were 40. These proteins and the associated cell lines are summarized in ESI 6.†
Discussion
We showed that the production of oxidizing reagents at a point source provided a comprehensive method for labeling interacting components of sialic acids. SiaNAzylated glycans were produced and could be distinguished from other structures by diagnostic ions originating from the different functional groups with LC-MS. We demonstrated that the efficiency of azido sialic acid incorporation varied according to the cell type, the glycan type, and even the individual structures. The employment of bioorthogonal chemistry and standard proteomic workflow makes this method readily employable to any cell lines and potentially tissues. The results showed that radicals were produced locally, at sialic acids, and oxidized the proteins in the vicinity. Based on the analysis of sialylated glycoproteins that underwent self-oxidization, we concluded that the distances between the sites of radical formation and amino acid oxidization were consistent with an approximate reaction distance at an average of 20 Å. This distance may be varied due to the spatial arrangement of the oxidizable amino acids and the modified sialic acid.
The oxidative labeling of proteins surrounding sialic acids provides a tool for the systematic identification of sialic acid binding proteins. It is difficult to predict a priori sialic acid binding proteins. Indeed, most sialic acid binding proteins have been discovered empirically. The difficulty stems from the variations in the mode of binding between sialic acids and proteins. Sialic acids may bind on specific sialic acid-recognizing motifs, anionic receptors, or even cation-mediated domains in proteins.
Indeed, proteins that contain known sialic acid-recognizing domains were found to be oxidized by POSE. In A549 and PNT2, we found L1CAM (L1 cell adhesion molecule), which was known to interact with sialic acids via its fibronectin protein type-III domain corresponding to sequence V615 to A1015.25 As shown in Fig. 4a, the L1CAM oxidation sites, including W740 in PNT2 cells and W985 in A549 cells, were consistently located in the sialic acid-recognizing domain. Another type of motif that may also interact with sialic acids was the laminin G-like domain.26 The protein LAMA5 (laminin subunit alpha-5) could therefore potentially interact with sialic acid through its laminin-G-like domain (P2736 to C3692). Indeed, we found F2781 (in Caco-2) and C3601 (in PNT2) of the protein oxidized in the respective cell lines (Fig. 4b). Other proteins containing these domains that were oxidized were also found. Additionally, the collagen-like domain containing proteins may also have sialic acid binding function as reported by Bellis et al.27 They showed the reduction of collagen binding after the removal of sialic acids from cell surface integrins. Similarly, the protein COL11A2 (collagen alpha-2(XI) chain) in A549 was oxidized on P488, which was located on its collagen-like domain. Interestingly, the laminin-G-like domain (D57-C228) of this protein had no amino acids oxidized with POSE, suggesting a different form of sialic acid-binding. Three other proteins in other cell lines exhibited similar behavior with oxidation located outside the laminin-G-like domain. Other known sialic acid-binding proteins including siglecs and selectins were not observed due to the low expression levels of these proteins.
The oxidation of anion binding proteins and carbohydrate binding proteins was more consistent with glycan-mediated interactions and the anionic nature of sialic acids.28 Shaikh et al. demonstrated that the sialylated integrins exhibited a greater degree of association with talin, which is a type of carbohydrate protein.29 Indeed, M1407 of TLN 1 (talin-1) was oxidized in the A549 cell line. GTP/GDP interacting proteins such as ras-related proteins have been shown to be involved in cell surface sialic acid regulations.27 The oxidation of these proteins suggested that the GTP/GDP interacting proteins may potentially associate with sialic acids.
Cation binding proteins suggested that sialic acid–protein interactions could also be mediated by cations, especially the calcium ion. Three annexins (ANXA2, ANXA3, and ANXA4) were oxidized, which suggests that they were bound to sialic acids. Previously, Kundranda et al. reported that Lewis lung carcinoma cells could interact with the sialic acid residues on fetuin-A in a calcium-mediated fashion, where the annexins behave like sialic acid-binding lectins that have high affinity for sialic acids.30 With their binding also mediated by calcium ions, cadherin proteins, which are heavily sialylated transmembrane glycoproteins, play essential roles in cell–cell adhesion and tissue differentiation.31 It is known that the alteration of cadherin N-glycosylation varies the protein binding kinetics.32 Specifically, the sialylation of cadherin was shown to be necessary in maintaining cell–cell adhesion.33 In our study, cadherin-binding proteins such as protein SPTB2 (spectrin beta chain, non-erythrocytic 1) and protein EF1G (elongation factor 1-gamma) were oxidized. Although they have not been reported as sialic acid-binding proteins specifically, their oxidation here demonstrated that sialic acids were involved in their binding to cadherin and mediated by calcium ions.
There are potential limitations to the method. The most obvious is that the ligated metal and the linker may prevent closer proximity to the sialic acid. However, the binding of proteins to glycans is generally neither as specific nor strong as binding to small molecule ligands. Additionally, sialic acids are generally found in patches so that binding occurs with multiple sialic acids simultaneously. The replacement of natural sialic acids with modified analogs was generally low. In glycans where there was more than one sialic acid, only one was replaced and in rare cases two. In any case, this method provides potential sialic acid-binding candidates that would need further validation using other approaches such as glycan arrays, which on their own also require validation.
We have shown that cis-sialic acid interactions can be mapped; however, the trans-sialic acid interaction can also be probed. For example, POSE can be used to identify the proteins involved in host–microbe interactions, where bacteria and virus are known to bind to host cells through sialic acid–protein interactions. Similarly, immune cells can be probed as they interact with cancer cells. In these types of cell–cell interactions, either immune cells or cancer cells can be modified and probed. As SiaNAz can also be incorporated into tissues of animals, sialic acid interactions may be performed even with in vivo models. Finally, the method has general utility and can be used with other modified monosaccharides. As these modified monosaccharides can also be incorporated into glycans, there will be opportunities to examine other types of glycan–protein binding interactions.
Author contributions
Q. L. and Y. X. designed and performed experiments, analyzed data, created the figures and co-wrote the manuscript. G. X. designed and performed glycomic experiments. C. B. L. conceived the idea, supervised the study, and co-wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Funding provided by the National Institutes of Health (GMRO1R01 and GM049077) is gratefully acknowledged. The authors thank Professor Claude Meares for providing the inspiration for this project.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01360a
References
- Malhotra R., Ward M., Sim R. B., Bird M. I. Biochem. J. 1999;341:61. [PMC free article] [PubMed] [Google Scholar]
- Crocker P. R., Paulson J. C., Varki A. Nat. Rev. Immunol. 2007;7:255. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- Phizicky E. M., Fields S. Microbiol. Rev. 1995;59:94. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A. Mass Spectrom. Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- Xu G., Chance M. R. Chem. Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- Smith D. L., Deng Y., Zhang Z. J. Mass Spectrom. 1998;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lee S. J. Proteome Res. 2006;5:539–547. doi: 10.1021/pr050299q. [DOI] [PubMed] [Google Scholar]
- Branon T. C. Nat. Biotechnol. 2018;36:880. doi: 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt O. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17033. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X. J. Biol. Chem. 2011;286:31610–31622. doi: 10.1074/jbc.M111.274217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler J., Yu S.-H., Rodriguez A. C. FASEB J. 2016;30:378. [Google Scholar]
- Bond M. R., Whitman C. M., Kohler J. J. Mol. BioSyst. 2010;6:1796–1799. doi: 10.1039/c0mb00069h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Collins B. E., Bengtson P., Paulson J. C. Nat. Chem. Biol. 2005;1:93. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- Ramya T. N. C. Mol. Cell. Proteomics. 2010;9:1339. doi: 10.1074/mcp.M900461-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky S. J., et al., Methods in Enzymology, Academic Press, 2003, vol. 362, pp. 249–272. [DOI] [PubMed] [Google Scholar]
- Cao L., Wu Q., Li Q., Shao S., Guo Y. J. Fluoresc. 2014;24:313–318. doi: 10.1007/s10895-013-1329-0. [DOI] [PubMed] [Google Scholar]
- Park D. D. Chem. Sci. 2018;9:6271–6285. doi: 10.1039/c8sc01875h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo I. J., Raub T. J., Borchardt R. T. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- Xu G., Chance M. R. Anal. Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- Mi H. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Krogh A., Larsson B., Von Heijne G., Sonnhammer E. L. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Franceschini A., von Mering C., Lin J., Jensen L. J. Bioinformatics. 2015;32:1085–1087. doi: 10.1093/bioinformatics/btv696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene R., Yang H., Kutsche M., Schachner M. J. Biol. Chem. 2001;276:21656–21663. doi: 10.1074/jbc.M101790200. [DOI] [PubMed] [Google Scholar]
- Hohenester E., Tisi D., Talts J. F., Timpl R. Mol. Cell. 1999;4:783–792. doi: 10.1016/s1097-2765(00)80388-3. [DOI] [PubMed] [Google Scholar]
- Seales E. C., Jurado G. A., Singhal A., Bellis S. L. Oncogene. 2003;22:7137. doi: 10.1038/sj.onc.1206834. [DOI] [PubMed] [Google Scholar]
- Varki A. Nature. 2007;446:1023. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- Shaikh F. M. Exp. Cell Res. 2008;314:2941–2950. doi: 10.1016/j.yexcr.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundranda M. N. Cancer Res. 2005;65:499–506. [PubMed] [Google Scholar]
- Lodish H., Berk A. and Zipursky S. L., et al., in Molecular Cell Biology, ed. W. H. Freeman, New York, 4th edn, 2000. [Google Scholar]
- Langer M. D., Guo H., Shashikanth N., Pierce J. M., Leckband D. E. J. Cell Sci. 2012;125:2478–2485. doi: 10.1242/jcs.101147. [DOI] [PubMed] [Google Scholar]
- Deman J. In Vitro Cell. Dev. Biol.: Anim. 1995;31:633. doi: 10.1007/BF02634317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.