

Non-amnestic Alzheimer’s disease is associated with hippocampal sparing; however, the progression of this condition is understudied. Phillips and Da Re et al. show that the focus and rate of atrophy differ across non-amnestic phenotypes, and that longitudinal atrophy is associated with both domain-specific cognitive decline and degree of structural connectivity.
Keywords: non-amnestic Alzheimer’s disease, logopenic-variant primary progressive aphasia, posterior cortical atrophy, frontal-variant Alzheimer’s disease, longitudinal neuroimaging
Abstract
Recent models of Alzheimer’s disease progression propose that disease may be transmitted between brain areas either via local diffusion or long-distance transport via white matter fibre pathways. However, it is unclear whether such models are applicable in non-amnestic Alzheimer’s disease, which is associated with domain-specific cognitive deficits and relatively spared episodic memory. To date, the anatomical progression of disease in non-amnestic patients remains understudied. We used longitudinal imaging to differentiate earlier atrophy and later disease spread in three non-amnestic variants, including logopenic-variant primary progressive aphasia (n = 25), posterior cortical atrophy (n = 20), and frontal-variant Alzheimer’s disease (n = 12), as well as 17 amnestic Alzheimer’s disease patients. Patients were compared to 37 matched controls. All patients had autopsy (n = 7) or CSF (n = 67) evidence of Alzheimer’s disease pathology. We first assessed atrophy in suspected sites of disease origin, adjusting for age, sex, and severity of cognitive impairment; we then performed exploratory whole-brain analysis to investigate longitudinal disease spread both within and outside these regions. Additionally, we asked whether each phenotype exhibited more rapid change in its associated disease foci than other phenotypes. Finally, we investigated whether atrophy was related to structural brain connectivity. Each non-amnestic phenotype displayed unique patterns of initial atrophy and subsequent neocortical change that correlated with cognitive decline. Longitudinal atrophy included areas both proximal to and distant from sites of initial atrophy, suggesting heterogeneous mechanisms of disease spread. Moreover, regional rates of neocortical change differed by phenotype. Logopenic-variant patients exhibited greater initial atrophy and more rapid longitudinal change in left lateral temporal areas than other groups. Frontal-variant patients had pronounced atrophy in left insula and middle frontal gyrus, combined with more rapid atrophy of left insula than other non-amnestic patients. In the medial temporal lobes, non-amnestic patients had less atrophy at their initial scan than amnestic patients, but longitudinal rate of change did not differ between patient groups. Medial temporal sparing in non-amnestic Alzheimer’s disease may thus be due in part to later onset of medial temporal degeneration than in amnestic patients rather than different rates of atrophy over time. Finally, the magnitude of longitudinal atrophy was predicted by structural connectivity, measured in terms of node degree; this result provides indirect support for the role of long-distance fibre pathways in the spread of neurodegenerative disease.
Introduction
Recent theories of neurodegenerative disease progression have raised the possibility that pathogenic protein aggregates do not arise spontaneously throughout the brain; rather, they may be transmitted from areas of existing pathology through one or more mechanisms, including local diffusion of pathogenic proteins through the extracellular medium as well as long-distance transmission along white-matter pathways between brain areas (Guo and Lee, 2014). This transmission model of neurodegenerative disease is supported by a diverse body of research ranging from rodent models (Liu et al., 2012; Iba et al., 2013; Ahmed et al., 2014; Mezias et al., 2017) to computational modelling of human neuroimaging data (Raj et al., 2012, 2015; Iturria-Medina et al., 2014; Hu et al., 2016). This research particularly supports the relevance of the transmission model in typical, amnestic Alzheimer’s disease, which spreads from the transentorhinal cortex and hippocampus to the rest of the medial temporal lobes and ultimately to the neocortex (Braak and Braak, 1991). This stereotyped progression provides detailed expectations against which to test models of interregional transmission.
However, it is unclear whether the transmission hypothesis has equal relevance in atypical presentations of Alzheimer’s disease, which demonstrate a number of pathological and clinical differences from amnestic Alzheimer’s disease, including relatively prominent neocortical disease and relative hippocampal sparing (Galton et al., 2000; Murray et al., 2011; Whitwell et al., 2012; Mesulam et al., 2014a). Clinically, atypical Alzheimer’s disease includes at least four non-amnestic phenotypes: logopenic-variant primary progressive aphasia (lvPPA), characterized by primary language deficits (Gorno-Tempini et al., 2011); posterior cortical atrophy (PCA), characterized by visuospatial deficits (Crutch et al., 2017); a variant defined by deficits in executive function and/or social behaviour (Dubois et al., 2014), frequently referred to as frontal-variant Alzheimer’s disease; and corticobasal syndrome (CBS), which can present with a constellation of lateralized motor and cognitive deficits (Medaglia et al., 2017). These syndromes are marked by different rates of clinical progression than in amnestic Alzheimer’s disease (Duara et al., 2013; Byun et al., 2015; Poulakis et al., 2018). Moreover, each phenotype is associated with distinct anatomical distributions of disease, particularly in early stages. Patients with lvPPA have strongly left-lateralized disease with pathological accumulations in left superior temporal and inferior parietal cortex (Mesulam et al., 2014b; Giannini et al., 2017); additional disease is commonly observed in left dorsolateral prefrontal cortex (Rogalski et al., 2016; Giannini et al., 2017). PCA is marked by involvement of the parietal and/or occipital lobes (Tang-Wai et al., 2004; Crutch et al., 2017); depending on the patient, disease in PCA may or may not have a right-hemisphere bias (Crutch et al., 2012; Ossenkoppele et al., 2015a). In frontal-variant Alzheimer’s disease, elevated pathology has been most commonly reported in the frontal lobes (Johnson et al., 1999; Blennerhassett et al., 2014), although a recent report based on a larger sample of these patients also highlights disease in posterior cortical areas (Ossenkoppele et al., 2015b). In CBS due to Alzheimer’s disease, disease is distributed widely and often asymmetrically throughout the temporal and parietal lobes, sometimes extending into the posterior portion of the frontal lobes (Lee et al., 2011; McMillan et al., 2016). Each of these non-amnestic Alzheimer’s disease phenotypes shares clinical similarities to presentations of frontotemporal lobar degeneration (FTLD) spectrum pathology, making it imperative to corroborate clinical diagnosis through Alzheimer’s disease biomarkers.
Non-amnestic syndromes with underlying Alzheimer’s disease pathology may be more prevalent than previously thought (Peter et al., 2014; Dickerson et al., 2017). However, relatively little research has examined the anatomical spread of non-amnestic Alzheimer’s disease. In a previous MRI analysis (Phillips et al., 2018), we inferred patterns of atrophy spread in non-amnestic Alzheimer’s disease phenotypes from cross-sectional MRI data: following the logic of post-mortem pathology studies, we explicitly assumed that the relative frequency of atrophy in different brain areas could be used to infer the anatomical progression of disease over time. These results corroborated the hypothesis that each non-amnestic Alzheimer’s disease phenotype has a distinct neocortical origin with relative sparing of the medial temporal lobes (MTL). Moreover, this study suggested that each non-amnestic Alzheimer’s disease phenotype has a distinct pattern of disease spread that differs from amnestic Alzheimer’s disease.
In the present study, we sought to validate and extend this previous work, using longitudinal MRI to differentiate patterns of earlier atrophy from subsequent disease spread in each phenotype. Additionally, we investigated whether amnestic Alzheimer’s disease and non-amnestic Alzheimer’s disease differ in the anatomic distribution and longitudinal rate of grey matter atrophy over time. We reasoned that such differences could explain phenotype-specific patterns of clinical progression across amnestic and non-amnestic Alzheimer’s disease variants. In a hypothesis-driven analysis based on our previous cross-sectional study (Phillips et al., 2018), we investigated differences in grey matter volume at the time of initial MRI as well as volume change over time in regions of interest associated with lvPPA, PCA, frontal-variant, and amnestic Alzheimer’s disease. Exploratory, whole-brain, voxelwise analysis of cortical thickness was performed to map patterns of disease spread beyond these initial regions of interest. We sought to identify group differences in atrophy distribution and progression independent of age, which has been reported previously to differ between typical and atypical forms of Alzheimer’s disease (Murray et al., 2011). Based on the high neocortical disease burden and domain-specific cognitive deficits that we previously observed in non-amnestic Alzheimer’s disease, we predicted that patients with non-amnestic Alzheimer’s disease would exhibit faster rates of atrophy in phenotype-specific neocortical regions of interest relative to those with amnestic Alzheimer’s disease. Additionally, we tested the hypothesis that patients with non-amnestic Alzheimer’s disease would exhibit slower atrophy than amnestic Alzheimer’s disease patients in the hippocampus and surrounding MTL areas, as a possible explanation for the relative memory sparing associated with these structures in non-amnestic Alzheimer’s disease. Finally, we compared longitudinal atrophy patterns to measures of interregional structural connectivity estimated from a large population of healthy controls; we predicted that connectivity would predict longitudinal atrophy, consistent with the transmission hypothesis.
Materials and methods
Patients
The current study used a longitudinal case-control design based on data retrospectively selected from the Integrated Neurodegenerative Disease Database at the University of Pennsylvania. Participants were recruited through the Penn Frontotemporal Degeneration Center (FTDC) and the Penn Memory Center (PMC). All procedures were approved by the University of Pennsylvania’s Institutional Review Board, and all patients and/or their caregivers gave written informed consent according to the principles established by the Declaration of Helsinki. An initial database query yielded 1897 patients scanned on the same 3 T Siemens MRI scanner at the Hospital of the University of Pennsylvania. Of these, 360 patients had either autopsy or CSF biomarker evidence of underlying Alzheimer’s disease pathology. An additional 58 patients were excluded because of major cerebrovascular disease, stroke, head trauma, or comorbid psychiatric, neurodegenerative, medical, or developmental disorders apart from their primary diagnoses. Of the remainder, 90 patients had longitudinal data available and exhibited clinical phenotypes of interest, as described below. At time of recruitment, MRI scans for all patients were screened for signs of cerebrovascular disease, hydrocephalus, or white matter lesions; those with a Fazekas scale score > 1 were excluded. Additionally, MRI scans were visually inspected by two raters (J.S.P. and F.D.R.), and 16 patients were excluded for poor quality data. The final sample included 181 T1-weighted MRI scans from 74 patients (25 with lvPPA, 20 PCA, 12 with frontal-variant Alzheimer’s disease, and 17 with amnestic Alzheimer’s disease) and 85 scans from 37 demographically-matched controls. A majority of participants (48/74 patients and 29/37 controls) had only two available scans; the remaining participants contributed three to four scans each. We included scans acquired with a minimum interscan interval of 6 months up to 3.5 years from the initial MRI; beyond this window, there were insufficient observations for a valid analysis. Seven patients had primary neuropathological diagnoses and 67 had CSF biomarkers (total tau/amyloid-β ratio > 0.34) indicative of Alzheimer’s disease pathology according to methods previously described (Shaw et al., 2009; Irwin et al., 2012; Toledo et al., 2012). APOE genotyping was performed on 66 of 74 patients. One patient (white male, amnestic Alzheimer’s disease, age 51 at onset) with an APOE ɛ3/ɛ4 genotype was found to have a mutation in the PSEN1 gene; supplementary analyses indicated that excluding this patient did not have substantive effects on the outcome of key analyses. All patients were clinically diagnosed by experienced neurologists (M.G., D.J.I., D.W., and S.V.), and diagnoses were confirmed by consensus after patients’ initial visit by clinicians with expertise in dementia. Clinical criteria for each patient phenotype were as follows: for lvPPA, primary language impairment including deficits in repetition and/or naming (Gorno-Tempini et al., 2011; Giannini et al., 2017); for PCA, visuospatial deficits (e.g. in object/spatial perception, neglect, or oculomotor apraxia) (Crutch et al., 2017); for frontal-variant Alzheimer’s disease, clinical evidence of a behavioural/dysexecutive syndrome per Rascovsky et al.’s (2011) criteria for behavioural-variant frontotemporal dementia; and for amnestic Alzheimer’s disease, primary memory impairment plus deficits in one or more additional cognitive domains (McKhann et al., 2011). Patients with non-amnestic Alzheimer’s disease had relatively preserved episodic memory, as assessed through clinical interviews and detailed mental status examinations; however, we note that the label ‘non-amnestic’ is used throughout this manuscript to denote patients’ initial presentation and does not preclude the development of memory deficits in more advanced disease. Patients with non-amnestic Alzheimer’s disease also had relatively spared abilities in other cognitive domains except their domain of primary impairment at initial presentation. Because of the challenges of clinically differentiating behavioural/dysexecutive syndromes due to Alzheimer’s disease versus FTLD, we performed additional screening on the frontal-variant Alzheimer’s disease group, as detailed in the Supplementary material (‘Patient selection details’). The current study included 54 patients from our previous, cross-sectional study (Phillips et al., 2018) (amnestic Alzheimer’s disease, n = 8; lvPPA, n = 24; PCA, n = 16; and frontal-variant Alzheimer’s disease, n = 6).
Shapiro-Wilks tests indicated non-normal distributions for education and disease duration, age, and Mini Mental Status Examination (MMSE) score at initial MRI (all P < 0.001). Kruskal-Wallis tests of group differences were non-significant, with the exception of MMSE [χ2(4) = 38.5, P < 0.001], reflecting patients’ cognitive deficits relative to controls. Mann-Whitney tests confirmed that all patient groups exhibited significantly lower MMSE scores than controls (all U ≥ 428, P < 0.001); all other pairwise comparisons were non-significant. To corroborate non-amnestic Alzheimer’s disease patients’ domain-specific cognitive impairment, we analysed neuropsychological performance on assessments independent of those used in clinical diagnosis, including performance on specific items of the Philadelphia Brief Assessment of Cognition (PBAC) (Libon et al., 2011b). Only neuropsychological observations acquired within 1 year of an MRI scan were included. Language was assessed in terms of speech features (with lower scores indicating speech and language impairment), forward digit span as a measure of repetition (Giannini et al., 2017), and letter fluency, which is sensitive to deficits in executive-mediated lexical retrieval (Rascovsky et al., 2007; Ramanan et al., 2017). Visuospatial function was assessed by patients’ ability to copy a modified version of the Rey complex figure as well as the judgment of line orientation. Social behaviour was assessed on an 18-point scale evaluating social comportment, apathy, disinhibition, agitation, empathy, and ritualistic behaviours. Executive function was evaluated through an oral version of the trail-making test as well as backward digit span. Finally, episodic memory was assessed by recognition on the Philadelphia Verbal Learning Test (PVLT) (Libon et al., 2011a) or the PBAC verbal memory test, as available. All neuropsychological assessments were acquired within 1 year of the initial MRI scan [PVLT: mean = 0.19 years, standard deviation (SD) = 0.25; letter fluency: mean = 0.14 years, SD = 0.25; PBAC: mean = 0.21 years, SD = 0.27; digit span: mean = 0.11 years, SD = 0.21]. Results were consistent with each phenotype’s primary impairment in all domains except for executive function (Table 1). Post hoc comparisons between patient groups for neuropsychological performance at initial MRI are reported in Supplementary Table 11. The median and maximum follow-up intervals for the MMSE were 1.4 and 3.8 years, respectively; for verbal recognition memory, 1.7 and 4.2 years; for letter fluency, 1.6 and 4.6 years; for forward and reverse digit span, 1.5 and 4.0 years; and for additional measures, which were derived from the PBAC, 1.6 and 4.6 years.
Table 1.
Participant characteristics at time of first scan
| Control | aAD | lvPPA | PCA | fvAD | P-value | |
|---|---|---|---|---|---|---|
| n | 37 (85) | 17 (40) | 25 (66) | 20 (48) | 12 (27) | |
| Male, n (%) | 16 (43.2) | 6 (35.3) | 9 (36.0) | 7 (35.0) | 7 (58.3) | 0.672 |
| Education | 16.0 [16.0, 18.0] | 16.0 [14.0, 18.0] | 16.0 [14.0, 19.0] | 16.0 [12.0, 16.0] | 16.0 [13.5, 18.0] | 0.421 |
| Age at MRI, years | 61.9 [57.9, 65.6] | 59.4 [53.5, 70.3] | 58.5 [56.9, 64.5] | 58.0 [55.1, 61.4] | 63.9 [59.7, 69.5] | 0.137 |
| Inter-scan interval, years | 1.2 [0.9, 1.7] | 1.2 [0.9, 1.5] | 1.1 [0.9, 1.3] | 1.0 [0.9, 1.2] | 1.0 [0.7, 1.1] | 0.162 |
| Disease duration, years | – | 3.0 [1.9, 4.0] | 2.7 [1.7, 3.9] | 2.2 [1.3, 4.0] | 2.2 [1.8, 5.2] | 0.747 |
| MMSE, 0–30 | 29.0 [28.0, 30.0] (20) | 23.0 [20.0, 25.0] (17) | 25.0 [23.0, 28.0] (25) | 24.5 [18.8, 25.2] (20) | 23.0 [17.0, 26.0] (12) | <0.001 |
| Recognition memory, discrimination, 0–1 | 1.0 [0.9, 1.0] (7) | 0.6 [0.5, 0.7] (10) | 0.8 [0.8, 1.0] (25) | 0.7 [0.6, 0.9] (19) | 0.6 [0.6, 0.8] (12) | <0.001 |
| Speech, 0–4 | 4.0 [4.0, 4.0] (3) | 2.5 [2.5, 3.0] (9) | 2.5 [2.0, 3.0] (19) | 3.0 [3.0, 4.0] (15) | 3.5 [2.2, 4.0] (11) | 0.004 |
| Letter fluency, number of words/60 s | 19.0 [17.5, 20.5] (7) | 9.0 [5.0, 13.0] (13) | 8.5 [5.2, 10.8] (22) | 10.0 [6.5, 15.5] (19) | 6.5 [3.0, 11.0] (12) | 0.001 |
| Forward digit span, length correct | 7.0 [7.0, 8.0] (11) | 5.0 [3.0, 6.0] (9) | 5.0 [4.0, 5.0] (25) | 6.0 [5.0, 7.0] (20) | 5.0 [4.0, 6.0] (12) | 0.005 |
| Rey figure copy, 0–12 | 12.0 [12.0, 12.0] (3) | 11.0 [4.0, 12.0] (9) | 12.0 [11.0, 12.0] (19) | 2.5 [0.0, 8.8] (12) | 9.5 [4.5, 11.0] (10) | 0.001 |
| Judgement of line orientation, 0–6 | 6.0 [6.0, 6.0] (3) | 3.0 [0.8, 5.0] (8) | 5.0 [4.0, 6.0] (19) | 2.0 [0.0, 4.0] (13) | 4.0 [3.0, 5.0] (9) | 0.004 |
| Social behaviour, 0–18 | 17.0 [17.0, 17.0] (3) | 17.5 [16.8, 18.0] (8) | 18.0 [17.0, 18.0] (19) | 17.0 [16.0, 18.0] (15) | 13.0 [11.1, 16.5] (11) | 0.004 |
| Oral trail-making test, 0–6 | 6.0 [5.5, 6.0] (3) | 0.0 [0.0, 3.0] (5) | 2.0 [0.2, 3.0] (10) | 0.5 [0.0, 2.8] (10) | 2.0 [0.2, 3.8] (6) | 0.051 |
| Reverse digit span, length correct | 6.0 [4.5, 6.0] (11) | 3.0 [3.0, 3.0] (9) | 3.0 [3.0, 4.0] (25) | 3.0 [2.0, 3.0] (19) | 3.0 [2.0, 3.2] (12) | <0.001 |
Data are presented as median [IQR] for all continuous variables. Education, age at MRI, interscan interval, and disease duration are expressed in years. Sample sizes (n) indicate number of unique individuals per group; total number of scans per group is given in parentheses. For each cognitive score, numbers in parentheses indicate the number of observations per group. P-values reflect the results of a chi-squared test for sex and Kruskal-Wallis tests for all other variables.
aAD= amnestic Alzheimer’s disease; fvAD = frontal-variant Alzheimer’s disease; naAD = non-amnestic Alzheimer’s disease.
Neuroimaging methods
T1-weighted magnetic resonance images were acquired axially with 0.98 mm × 0.98 mm × 1 mm voxels, a 256 × 192 matrix, a repetition time of 1620 ms, an inversion time of 950 ms, and a flip angle of 15°. Scans were visually inspected for quality by two authors (J.P. and F.D.R.). Advanced Normalization Tools (ANTs) (Avants et al., 2014; Tustison et al., 2014) was used to process each image using an a priori-based approach. Images underwent intensity normalization (Tustison et al., 2010) and were spatially normalized to a template based on healthy controls from the Open Access Series of Imaging Studies (OASIS) dataset (Marcus et al., 2007) using a symmetric diffeomorphic algorithm (Klein et al., 2009; Avants et al., 2011). Images were segmented into six tissue classes (cortical grey matter, subcortical grey matter, deep white matter, CSF, brainstem, and cerebellum) using template-based priors; this tissue segmentation was then used to estimate cortical thickness; ANTs cortical thickness measurements have been extensively validated relative to surface-based methods such as FreeSurfer (Tustison et al., 2014; Klein et al., 2017). We used a joint label fusion approach (Wang et al., 2013) to align the Mindboggle-101 labels (based on the Desikan-Killainy-Tourville label scheme) (Klein and Tourville, 2012) with each image using pseudo-geodesic registration (Tustison and Avants, 2013) and calculated the volume of grey matter voxels within each label, normalized by intracranial volume and converted to a z-score relative to controls’ initial scans. To perform voxelwise group analyses, we warped cortical thickness images to the template using the previously-computed spatial transforms; these images were then spatially smoothed with a 2-sigma Gaussian kernel and down-sampled to 2 mm isotropic voxels.
Statistical analysis
In a hypothesis-driven analysis, we analysed grey matter volumes in phenotype-specific regions of interest motivated by our previous study of disease progression in non-amnestic Alzheimer’s disease (Phillips et al., 2018). This study identified the regions most commonly atrophied in each non-amnestic Alzheimer’s disease phenotype, reflecting the likely anatomical origin of disease. These regions of interest included left middle and superior temporal gyri in lvPPA; right precuneus, superior parietal lobule, and angular, supramarginal, and middle temporal gyri in PCA; and left anterior insula and middle frontal gyrus as well as right middle temporal gyrus in frontal-variant Alzheimer’s disease (Table 2). Each region of interest was expected to exhibit lower volume at the time of participants’ initial MRI scan as well as more rapid volume loss over time in its associated patient group(s) relative to other groups. We additionally hypothesized that the amnestic Alzheimer’s disease group would demonstrate selective atrophy in the MTL, including bilateral hippocampi, parahippocampal gyri, and entorhinal cortex. Atrophy at the time of initial MRI was analysed using multiple linear regression models with a factor of group and covariates for age, sex, and MMSE score at the time of initial MRI; controls formed the reference group in these models. Longitudinal atrophy was assessed using linear mixed effects (LME) models with fixed factors of group, time since first scan, and the interaction of group × time. As in the baseline model, covariates included age, sex, and MMSE score at initial MRI. A subject-specific random intercept was included to account for intra-individual correlations in imaging measures. Post hoc comparisons were performed for the effect of group at initial MRI as well as the group × time interaction in longitudinal models; values of P < 0.05, corrected using the false discovery rate (FDR) method, were considered significant.
Table 2.
Differences in grey matter volume at initial MRI and longitudinal atrophy in hypothesis-driven analysis of regional brain volumes, relative to matched controls
| A priori association | Region | F First MRI(4,103) | aAD | lvPPA | PCA | fvAD | F Group×time (4,150) | aAD | lvPPA | PCA | fvAD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| aAD | L entorhinal | 7.2*** | −3.4a | −1.5 | −1.5 | −0.1 | 6.7*** | −2.2 | −3.7b | −3.3b | −3.9b |
| R entorhinal | 5.5*** | −2.6c | 0.7 | −1.9 | −0.1 | 7.4*** | −4.1c | −2.9b | −4.1b | −3.0b | |
| L hippocampus | 8.4*** | −4.6c | −3.2c | −2.1 | −1.6 | 5.4*** | −4.1c | −2.9c | −2.9b | −0.3 | |
| R hippocampus | 7.4*** | −4.3c | −1.7 | −3.1c | −1.8 | 4.7** | −3.5c | −2.3 | −3.4c | −1.2 | |
| L parahippocampal | 2.8** | −1.7 | −2.1 | 0.1 | 0.8 | 5.6*** | −3.5b | −4.0b | −2.6b | −2.1 | |
| R parahippocampal | 1.6* | −2.2 | −0.5 | −1.3 | −0.3 | 5.9*** | −3.7b | −2.5b | −4.0b | −0.5 | |
| lvPPA | L middle temporal | 30.5*** | −3.0c | −7.3c | −3.2c | −3.0c | 34.7*** | −9.6c | −9.2c | −6.4c | −5.3c |
| L superior temporal | 21.5*** | −2.6c | −6.8c | −2.3 | −1.8 | 17.3*** | −4.4c | −8.1c | −3.7b | −2.8b | |
| PCA | R angular | 14.7*** | −2.8a | −1.3 | −3.9c | −3.0a | 2.4 | −1.4 | −2.6b | −2.5c | −1.0 |
| R precuneus | 9.8*** | −0.7 | −0.5 | −4.4c | −1.3 | 13.7*** | −6.5b | −4.8b | −4.5c | −0.9 | |
| R superior parietal lobule | 16.7*** | 0.6 | 0.0 | −5.5a | −1.7 | 4.2** | −3.6b | −2.7b | −2.1 | −0.1 | |
| R supramarginal | 6.4*** | 0.2 | 0.4 | −2.6c | −0.6 | 11.2*** | −6.5b | −3.1b | −2.6c | −0.5 | |
| fvAD | L anterior insula | 10.3*** | −2.1 | −2.6a | 0.6 | −3.4c | 3.5* | −2.0 | −1.6 | −0.4 | −3.3c |
| L middle frontal | 15.3*** | −2.6c | −3.8c | −2.4 | −4.4a | 8.2*** | −3.9c | −4.9c | −3.8b | −2.1 | |
| PCA, fvAD | R middle temporal | 23.6*** | −2.8c | −2.7c | −5.6c | −4.2c | 44.8*** | −11.3c | −9.3c | −7.5c | −6.4c |
| Reference ROI | L precentral | 3.4*** | 0.8 | −1.3 | −1.4 | 1.2 | 3.7** | −3.1b | −3.1b | −2.1 | −1.3 |
| R precentral | 5.4*** | 0.2 | −0.7 | −2.9c | −0.7 | 5.0** | −3.3b | −3.3b | −3.3c | −0.6 |
Hypotheses included selective atrophy of neocortical areas associated with early disease in non-amnestic Alzheimer’s disease (naAD) (Phillips et al., 2018) and of the MTL (hippocampus, entorhinal cortex, and parahippocampal gyrus) in patients with anmesticc Alzheimer’s disease (aAD). The left and right precentral gyri are included to demonstrate the regional specificity of atrophy. F-statistics indicate the main effect of group at initial MRI scan and the group × time interaction across all scans. Additional columns report z-statistics for pairwise contrasts of each patient group versus controls.
aSignificant differences in volume only at initial MRI.
bSignificant differences in longitudinal atrophy rates.
cDifferences in both initial volume and longitudinal atrophy, based on a threshold of P < 0.05, corrected using the FDR method.
fvAD = frontal-variant Alzheimer’s disease; n.s. = non-significant; ROI = region of interest.
*P < 0.05; **P < 0.01; ***P < 0.001.
We used LME models to relate grey matter volume change to neuropsychological performance within 1 year of each imaging session. Because of the limited number of observations, only linear associations between atrophy and time were assessed. The mean interval between test and MRI was 0.30 years (SD = 0.30) for recognition memory; 0.22 years (SD = 0.29) for letter fluency; 0.18 years (SD = 0.29) for digit span; and 0.25 years (SD = 0.33) for all other longitudinal neuropsychological measures. Separate LME models were computed for each measure and change in associated regions of interest. Thus, recognition performance was related to grey matter volume in each of the six MTL regions of interest; language measures were compared to volume change in left middle and superior temporal gyrus; visuospatial measures were related to change in the right superior parietal lobule, precuneus, and angular, supramarginal, and middle temporal gyri; and behavioural and executive measures were related to left anterior insula and middle frontal gyrus as well as right middle temporal gyrus. Neuropsychological performance formed the outcome in each model; predictors treated as fixed effects included regional grey matter volume at initial MRI and subsequent volume change, as well as covariates of sex and education. Additionally, a subject-specific random intercept was included in the LME model. Because of limited neuropsychological data, controls were omitted from these models. The association with regional volume change in each model was assessed at a significance level of P < 0.05, corrected for FDR.
Additionally, we performed exploratory, whole-brain, voxelwise analysis to investigate differences in cortical thickness that were not assessed by a priori regions of interest. Region of interest-based and voxelwise analyses both present distinct advantages and weaknesses. Voxelwise analysis is not constrained by the borders of anatomically-defined regions of interest, and it allows more precise anatomical localization of effects. However, region of interest volume is regarded as a more reliable measure of grey matter atrophy than cortical thickness (Schwarz et al., 2016). Moreover, voxelwise parametric tests depend on patients displaying neurodegeneration at the same precise point within a brain area. Thus, region of interest-based volumetric analysis may be more sensitive to atrophy if the precise focus of atrophy within a region differs across individuals. Voxelwise analysis did not include hippocampus, where cortical thickness is not well estimated (Han et al., 2006; Gronenschild et al., 2012; Schwarz et al., 2016), but did include entorhinal cortex and parahippocampal gyri. As in region of interest-based analysis, we used multiple regression to assess group differences at initial MRI and an LME model to investigate longitudinal atrophy. These voxelwise models used the same regression formulae as region of interest-based models, and the LME was implemented in the 3dLME (Chen et al., 2013) function from the Analysis of Functional NeuroImaging (AFNI) software suite. Multiple comparisons correction was performed by first thresholding voxelwise results at P < 0.001 (uncorrected), then applying a cluster extent threshold corresponding to a cluster-wise alpha value of 0.05. To calculate cluster extent thresholds, we first estimated spatial auto-correlation from the model residuals using AFNI’s 3dFWHMx. We then used the 3dClustSim function, which is based on a Monte Carlo approach (Forman et al., 1995; Cox et al., 2017), to determine the cluster size corresponding to a false-positive rate of 0.05 at a voxelwise threshold of P < 0.001 (uncorrected). These simulations indicated a cluster threshold of 73 voxels (i.e. 584 µl) for the baseline MRI model and a threshold of 75 voxels (600 µl) for the longitudinal LME model. For both the baseline effect of group and the group × time interaction, we performed post hoc contrasts between all groups, which were corrected to cluster-wise P < 0.05 using the same method. In the Supplementary material, we display voxelwise contrasts versus controls at a lenient threshold of P < 0.01, uncorrected for multiple comparisons (Supplementary Fig. 3).
Structural connectivity
To investigate associations between atrophy progression and brain connectivity, we related longitudinal atrophy to structural population-average structural connectivity measures computed by Yeh et al. (2018). The decision to use population-average connectivity measures rather than estimating connectivity from patients was based on both practical and conceptual considerations. First, constraining participant selection by the availability of white-matter imaging data would have further reduced sample sizes. Second, white-matter degeneration in patients’ brains might adversely affect fibre tractography, leading to false negatives in estimating region-to-region brain connectivity.
Yeh et al. (2018) reported a whole-brain connectivity matrix (available at http://brain.labsolver.org/) based on diffusion MRI data from 842 healthy participants in the Human Connectome Project; connectivity values represent average anisotropy values for white matter fibre tracts connecting 65 regions in a modified version of the Automated Anatomical Labeling (AAL) brain parcellation (Tzourio-Mazoyer et al., 2002). Because label boundaries for major cortical structures vary between the AAL and Mindboggle parcellations, we warped the modified AAL atlas into the native acquisition space for each of the T1-weighted scans in the current study and recomputed grey matter volumes based on this parcellation. An example of the anisotropy-based structural connectivity values reported by Yeh et al. (2018) is shown in Supplementary Table 6 for the areas of greatest overlap with Mindboggle regions of interest in the hypothesis-driven analysis described above.
Using the igraph package for R (https://igraph.org/r/), we created an unweighted, undirected graph of structural connectivity from Yeh et al.’s (2018) connectivity matrix, omitting the cerebellum and brainstem to yield a total of 62 nodes (i.e. brain areas). The degree of each node was computed as the number of non-zero white-matter connections with other regions. Self-connections were excluded; thus, the maximum possible degree of a node was 61. As with Mindboggle labels, volumes were normalized by each participants’ intracranial volume and converted to a z-score relative to the region-wise mean and standard deviation of the control sample. We calculated annualized change in grey matter volume over time for each region by subtracting these z-score volume measures from the first and last available scans for each participant and dividing by the time interval. We then computed a linear mixed effects model with annualized change as the outcome and fixed effects of group, node degree, and the group × degree interaction, covarying for the baseline volume of each region, patients’ age at initial MRI, and sex. The average volume of each region (i.e. raw volume divided by intracranial volume) among control participants was also included as a covariate to ensure that variation in node degree did not simply reflect differences in region size. A random intercept was estimated for each participant, and a significance threshold of P < 0.05 was used.
Data availability
Computer code for the current manuscript (including all text, analysis, and visualization of results) is available in the form of Rmarkdown and LaTeX scripts in a public GitHub repository (https://github.com/jeffrey-phillips/naAD-longitudinal.git). Rmarkdown code requires R version 3.4.4 or higher. Investigators who wish to access imaging and clinical data may submit a direct request to the corresponding author.
Results
Hypothesis-driven analysis of region of interest volumes
Areas of earlier atrophy in each phenotype
First, we assessed both regional volume at initial MRI and longitudinal volume change in each group relative to matched controls in regions of interest associated a priori with each phenotype. The purpose of this hypothesis-driven analysis was 2-fold: (i) to dissociate effects of earlier versus later degeneration that are confounded in cross-sectional studies; and (ii) to test hypotheses regarding differential rates of atrophy between phenotypes. This analysis identified multiple regions displaying initial atrophy in each phenotype (Table 2), which reflect atrophy prior to patients’ initial scans. While a subset of these regions continued to degenerate over the follow-up period, others exhibited no further change (Table 2). Additionally, we detected a number of regions that were not atrophied relative to controls at the initial scan but demonstrated progression over the follow-up period (Table 2); these areas are interpreted as areas of later disease spread in each phenotype. In cross-sectional analysis of participants’ initial MRI scans, all regions of interest exhibited a main effect of group, independent of age and MMSE.
Patterns of atrophy at initial MRI corroborated prior cross-sectional studies of non-amnestic Alzheimer’s disease, supporting the accuracy of clinical diagnoses. LvPPA patients exhibited strong lateralization of disease, with early atrophy relative to controls and other patient groups in left superior and middle temporal gyri (Table 2 and Fig. 1). Additionally, they had significant atrophy relative to controls in left anterior insula, hippocampus, and middle frontal gyrus as well right middle temporal gyrus. PCA patients, in turn, had significant atrophy at first MRI in right angular gyrus, precuneus, superior parietal lobule, and hippocampus as well as bilateral middle temporal gyri. Patients with frontal-variant Alzheimer’s disease had significant atrophy in left anterior insula and middle frontal gyrus; right angular gyrus; and bilateral middle temporal gyri. The precentral gyrus, which comprises primary motor areas, exhibited early atrophy only in the PCA group and was restricted to the right hemisphere, consistent with the general right-lateralization of these patients’ atrophy pattern; the relative sparing of these structures is consistent with patients’ preserved motor function and demonstrates the regional specificity of atrophy patterns. The amnestic Alzheimer’s disease patients exhibited initial atrophy relative to controls in bilateral hippocampi and entorhinal cortex, left middle frontal gyrus, bilateral temporal cortex, and right angular gyrus. These temporoparietal areas have been previously characterized as nodes of the posterior default mode network in which different Alzheimer’s disease phenotypes demonstrate convergence of atrophy patterns (Ossenkoppele et al., 2015a). Additionally, amnestic Alzheimer’s disease patients demonstrated more severe atrophy than non-amnestic Alzheimer’s disease groups in bilateral hippocampi and entorhinal cortex (Fig. 1). Non-amnestic patients exhibited characteristic sparing of MTL structures, with initial atrophy limited to left hippocampus in lvPPA and right hippocampus in PCA. In longitudinal models, areas of significant early atrophy tended to demonstrate further progression over the follow-up period relative to controls (Table 2; see also Supplementary Fig. 2). However, a subset of brain areas had a non-significant slope of change over time (Table 2), suggesting a slowing of atrophy. These areas included left entorhinal and right angular gyrus in amnestic Alzheimer’s disease; left anterior insula in lvPPA; right superior parietal lobule in PCA; and left middle frontal gyrus in frontal-variant Alzheimer’s disease. Importantly, variation in sample sizes should be taken into consideration when interpreting results for different patient groups. For example, the frontal-variant Alzheimer’s disease group (the smallest sample) may be more susceptible to false negatives than larger groups. We caution against drawing conclusions about group differences by visual comparisons of each group’s results versus controls (Table 2 and Fig. 3). Rather, group differences in atrophy are assessed directly in Figs 1–2 and 4–5.
Figure 1.

Patient group differences at time of initial MRI in normalized volumes for a priori regions of interest. Box-and-whisker plots represent the distribution of regional grey matter volumes, expressed in z-score units relative to the healthy control reference group and adjusted for age, sex, and global cognition. More negative values on the x-axis indicate greater atrophy. The vertical bar in each box indicates the median volume; the edges of the box represent the IQR, i.e. the difference between the first and third quartiles. The whiskers extend to the most extreme point within 1.5 × IQR from the left or right edge of the box; observations falling outside this range are plotted individually. The notches in each box extend 1.58 × IQR / √n, displaying a ∼95% confidence interval for the median. Black brackets indicate significant pairwise group differences (P < 0.05 after FDR correction). Group is indicated by colour as well as by the shape centred over the median line in each box. aAD = amnestic Alzheimer’s disease; fvAD = frontal variant Alzheimer’s disease.
Figure 3.

Voxelwise differences in cortical thickness relative to matched controls. Image overlays are binarized t-statistic maps for simple contrasts of controls minus each patient group. Blue: simple effect of group (patients < controls) from cross-sectional analysis of participants’ initial MRI scans; red: group × time interaction from longitudinal mixed effects models, indicating where patients have more rapid cortical thinning than controls; green: overlap between group and group × time effects. All results were thresholded at voxelwise P < 0.001 with a minimum cluster volume of 584 µl for results at initial MRI and 600 µl for longitudinal results, corresponding to a corrected cluster-wise threshold of P < 0.05. aAD = amnestic Alzheimer’s disease; fvAD = frontal variant Alzheimer’s disease.
Figure 2.

Patient group differences in the effect of time for a priori regions of interest. The plot displays annualized change in regional grey matter volume in each group, expressed in z-score units relative to the healthy control reference group and adjusted for sex as well as age and global cognition at initial MRI. More negative values on the x-axis indicate more rapid atrophy over time. The vertical bar in each box indicates the median change in regional volume; the edges of the box represent the IQR, i.e. the difference between the first and third quartiles. The whiskers extend to the most extreme point within 1.5 × IQR from the left or right edge of the box; observations falling outside this range are plotted individually. The notches in each box extend 1.58 × IQR / √n, displaying an ∼95% confidence interval for the median. Black brackets indicate significant pairwise group differences (P < 0.05 after FDR correction). Group is indicated by colour as well as by the shape centred over the median line in each box. aAD = amnestic Alzheimer’s disease; fvAD = frontal variant Alzheimer’s disease.
Figure 4.

Voxelwise differences between patient groups in cortical thickness at time of initial MRI scan. Results are thresholded at voxelwise P < 0.001 with a minimum cluster volume of 584 µl, corresponding to a corrected cluster-wise threshold of P < 0.05. Warm colours indicate thinner cortical grey matter in the second group than the first; cool colours indicate thinner cortical grey matter in the first group than the second. aAD = amnestic Alzheimer’s disease; fvAD = frontal variant Alzheimer’s disease.
Figure 5.

Voxelwise differences between patient groups in rates of cortical thinning over time. Image overlays are t-statistic maps for the interaction of each group with time, calculated from LME models and thresholded at voxelwise P < 0.001 with a minimum cluster volume of 600 µl, corresponding to a corrected cluster-wise threshold of P < 0.05. Warm colours indicate that cortical thinning over time is more rapid in the second group than the first; cool colours indicate that cortical thinning is more rapid in the first group than the second. aAD = amnestic Alzheimer’s disease; fvAD = frontal variant Alzheimer’s disease.
Longitudinal analysis identifies areas of later change in each phenotype
Additionally, multiple brain areas in each phenotype demonstrated significant change over time despite an absence of atrophy at initial MRI; these areas appear to represent disease spread in later stages. In the neocortex, lvPPA patients exhibited longitudinal atrophy in right temporoparietal areas, while PCA patients exhibited new left-hemisphere atrophy in superior temporal and middle frontal gyrus. Patients with frontal-variant Alzheimer’s disease exhibited new atrophy in left superior temporal gyrus, marking lateral temporal cortex as one of the most consistent areas of longitudinal change across patient groups. In the MTL, amnestic Alzheimer’s disease, lvPPA, and PCA patients all exhibited later atrophy in bilateral parahippocampal gyri; and all three non-amnestic Alzheimer’s disease groups demonstrated later atrophy in bilateral entorhinal cortex. Additionally, PCA patients exhibited later-stage atrophy in right hippocampus. Because a subset of PCA patients have a disease focus in the ventral visual processing stream (Crutch et al., 2017), we evaluated longitudinal atrophy in bilateral inferior occipital gyri (Supplementary Tables 7–10); while we observed significant atrophy across patient groups, there were no between-group differences in either mean atrophy or its rate of change. Finally, in precentral gyrus reference regions, all patient groups except frontal-variant Alzheimer’s disease exhibited longitudinal change relative to controls, consistent with their more advanced disease status; however, in the PCA group this change remained restricted to the right hemisphere.
Group differences in regional rates of change
The longitudinal design allowed us to test the hypothesis that each non-amnestic Alzheimer’s disease phenotype would exhibit faster atrophy in its associated neocortical regions of interest than other patient groups, consistent with phenotype-specific disease patterns. Additionally, we predicted that patients with non-amnestic Alzheimer’s disease would exhibit more gradual rates of change in MTL structures than those with amnestic Alzheimer’s disease, providing a dynamic correlate of MTL sparing in non-amnestic Alzheimer’s disease. These hypotheses were tested through pairwise contrasts of group × time interaction terms from linear mixed effects models of grey matter volume change. Consistent with hypotheses, patients with lvPPA had more rapid atrophy than patients with PCA in left superior temporal gyrus (z = 2.8, P < 0.02) as well as marginally more rapid change than patients with amnestic Alzheimer’s disease (z = 2.1, P < 0.09). Similarly, the frontal-variant Alzheimer’s disease group exhibited significantly greater atrophy rates in left anterior insula than lvPPA (z = 2.5, P < 0.04) and PCA patients (z = 2.9, P < 0.02). Contrary to hypotheses, patients with PCA did not exhibit faster neurodegeneration during the follow-up period than other phenotypes. Because PCA is associated with heterogeneous disease distributions including both dorsal and ventral occipito-temporal variants (Crutch et al., 2017), we performed supplementary analyses of longitudinal atrophy in ventrolateral occipital cortex (i.e. bilateral inferior occipital gyri). While PCA patients exhibited significantly lower grey matter volumes than controls in both left and right inferior occipital gyri, there were no significant differences in either mean volumes or rates of longitudinal change with other patient groups (Supplementary Tables 7–10).
In addition, amnestic Alzheimer’s disease patients had more rapid atrophy in right middle temporal gyrus than lvPPA (z = 3.5, P < 0.01) and PCA patients (z = 3.2, P < 0.01); in left middle temporal gyrus relative to lvPPA (z = 2.7, P < 0.03); in right precuneus relative to lvPPA (z = 2.5, P < 0.04) and frontal-variant Alzheimer’s disease (z = 3.0, P < 0.02); and in right supramarginal gyrus relative to all three non-amnestic Alzheimer’s disease groups (all z > 3.3, P < 0.01). We had predicted that patients with non-amnestic Alzheimer’s disease would exhibit more gradual atrophy than patients with amnestic Alzheimer’s disease in MTL structures. However, all patient groups demonstrated significant atrophy relative to controls in one or more MTL structures (Table 2), and we found no significant differences between patient groups in atrophy rates for bilateral hippocampi, entorhinal cortex, or parahippocampal gyri. To address limitations in statistical power, we performed a supplementary analysis on MTL regions of interest in which all non-amnestic Alzheimer’s disease phenotypes were combined into a single group; while both the non-amnestic and amnestic Alzheimer’s disease groups had significantly faster atrophy than controls in all six MTL regions, we again observed no difference in atrophy rates between non-amnestic and amnestic Alzheimer’s disease (Supplementary Table 3).
Exploratory whole-brain analysis
Exploratory whole-brain analysis of cortical thickness was performed to identify areas of early atrophy and later spread that were not captured by a priori regions of interest. As in the region of interest analysis (Table 2), areas were categorized by whether they exhibited significant atrophy at patients’ first MRI and whether they exhibited significant longitudinal change during the follow-up period relative to controls. As mentioned above, the hippocampi were excluded from voxelwise analysis because of the difficulty of reliably segmenting and estimating cortical thickness for this structure (Han et al., 2006; Gronenschild et al., 2012; Schwarz et al., 2016).
Voxelwise cortical thickness differences at initial MRI
Whole-brain atrophy patterns at initial MRI corroborated region of interest-based analyses and indicated areas of earlier neurodegeneration that fell outside of a priori regions of interest. At initial MRI, the lvPPA group exhibited lower cortical thickness versus controls in left middle and superior temporal gyri, our hypothesized disease focus for lvPPA, corroborating region of interest volume analysis (Fig. 3A). In addition to these regions, patients with lvPPA exhibited early atrophy in multiple left hemisphere temporal, parietal, and frontal areas including central and parietal opercula, planum temporale, planum polare, and inferior temporal, fusiform, supramarginal, angular, inferior occipital, and middle occipital gyri (Fig. 3). In prefrontal cortex, patients with lvPPA had cortical thinning in left anterior insula and frontal operculum as well as bilateral middle and superior frontal gyri. Moreover, nearly all of these areas continued to exhibit longitudinal change during the follow-up period (Fig. 3, green areas). Peak t-statistics and cluster volumes for these regions are reported in Supplementary Table 4. Voxelwise analysis of the PCA group not only demonstrated expected atrophy in right parietal, occipital, and posterior temporal areas, but also in their left hemisphere homologues (Fig. 3B). Additionally, PCA patients’ baseline atrophy extended into right precentral, middle frontal, and superior frontal gyri. Among these areas, the bilateral precuneus/posterior cingulate gyrus and middle temporal gyrus continued to demonstrate change during the follow-up period. Overall, baseline results thus indicated that despite some right lateralization of disease, PCA patients in the current sample had bilateral cortical involvement consistent with recent consensus criteria for PCA (Crutch et al., 2017). As in the region of interest-based analysis, patients with frontal-variant Alzheimer’s disease exhibited initial atrophy relative to controls in left anterior insula and middle frontal gyrus, right angular gyrus, and bilateral middle temporal gyri. However, areas of early atrophy extended far beyond these regions to include right insula and middle frontal gyrus as well as bilateral medial and ventral prefrontal cortex, inferior and superior frontal gyri, temporal poles, and opercular cortex (Fig. 3C). The frontal-variant Alzheimer’s disease group also had initial atrophy relative to controls in the anterior and dorsal portion of right entorhinal cortex, a finding that was not captured by region of interest-based analysis. In apparent contrast to the findings of Ossenkoppele et al. (2015b), posterior atrophy was limited, most notably including the right precuneus. Among areas of initial atrophy in frontal-variant Alzheimer’s disease, only right anterior insula and bilateral central opercula displayed significant cortical thinning over the follow-up period. The amnestic Alzheimer’s disease group exhibited expected atrophy in right entorhinal cortex as well as bilateral middle and superior temporal gyri, partially replicating region of interest-based findings (Fig. 3D). Outside a priori regions of interest, patients with amnestic Alzheimer’s disease also exhibited early atrophy in bilateral parietal areas including the precunei and middle cingulate, posterior cingulate, angular, and supramarginal gyri; right insula; and right frontal lobe areas including anterior orbital, middle frontal, superior frontal, and medial precentral gyri (Supplementary Table 4). Of these areas, only the right insula demonstrated continued atrophy throughout the follow-up period. Figure 4 presents contrasts between patient groups of initial cortical thickness. Consistent with expectations from previous cross-sectional studies, these results indicate left lateralized atrophy in lvPPA (Fig. 4A); parietal and occipitotemporal disease in PCA that exhibits some right-hemisphere bias (Fig. 4B, D and F), and greater frontal lobe involvement in frontal-variant Alzheimer’s disease than in other phenotypes (Fig. 4C, E and F). Collectively, these results replicate initial volume differences from region of interest-based analysis and highlight additional phenotype-specific areas of atrophy reported in prior studies of lvPPA (Rogalski et al., 2016), PCA (Lehmann et al., 2012), and frontal-variant Alzheimer’s disease (Whitwell et al., 2011).
Voxelwise whole-brain analysis of longitudinal disease spread
Longitudinal whole-brain analysis also allowed us to identify brain areas that were not significantly atrophied at baseline but demonstrated progressive atrophy over the follow-up period. As in region of interest-based analysis, we interpret these effects to indicate the spread of disease to brain areas that were relatively spared in early disease stages. The lvPPA group showed extensive new atrophy in right temporoparietal areas and throughout bilateral prefrontal, medial parietal, and anterior temporal cortex (Fig. 3A, red regions), suggesting spread of disease to these areas following patients’ initial scans. In PCA, progressive atrophy was observed in several areas unaffected at initial MRI, including the temporal poles, bilateral superior frontal gyri, and bilateral perisylvian cortex (Fig. 3B and Supplementary Table 5). In contrast to lvPPA and PCA patients, areas of newer atrophy progression were sparse among patients with frontal-variant Alzheimer’s disease, limited to portions of right anterior insula as well as left opercular and perisylvian cortex (Fig. 3C). Because the small sample size of this group might have limited statistical sensitivity, we also present voxelwise contrasts versus controls at a liberal statistical threshold of P < 0.01, without cluster-wise correction for multiple comparisons (Supplementary Fig. 3). While these results must be interpreted with caution because of the potential for false positive results, they suggest more extensive disease spread to left posterior insula, left dorsolateral prefrontal cortex, and bilateral anterior prefrontal areas. Finally, patients with amnestic Alzheimer’s disease showed new longitudinal change during the follow-up period in bilateral parietal cortex as well as right posterior temporal, anterior temporal, opercular, and prefrontal areas (Fig. 3D and Supplementary Table 5).
Voxelwise whole-brain differences in regional rates of change
As in region of interest-based analysis, we assessed group differences in the regional pace of cortical thinning over time. Consistent with region of interest-based analysis (Fig. 2), patients with lvPPA had significantly more rapid atrophy than patients with amnestic Alzheimer’s disease in left anterior and posterior superior/middle temporal gyri (Fig. 5A). Additionally, patients with amnestic Alzheimer’s disease exhibited faster atrophy progression than patients with frontal-variant Alzheimer’s disease in right middle occipital gyrus and superior parietal lobule (Fig. 5B), consistent with parietal differences observed between these groups in region of interest-based analysis. Similarly, patients with lvPPA exhibited more rapid atrophy than patients with frontal-variant Alzheimer’s disease in left precuneus and bilateral middle occipital gyri (Fig. 5C). These results corroborate region of interest-based findings that suggest neocortical rates of atrophy may vary by region according to patient phenotype.
Degree of structural connectivity predicts longitudinal atrophy
Nodes in the AAL region graph had a median degree of 19.5 [interquartile range (IQR) = 17–25.75]. Nodes in the top quartile corresponded to several a priori regions of interest, including right superior parietal lobule; bilateral inferior, middle, and superior temporal gyri; and right angular gyrus. Both left and right hippocampus labels (which encompassed proximal MTL structures) had degrees of 17; left insula, 23; left middle frontal gyrus, 21; right supramarginal gyrus, 21; right precuneus, 22; and left and right precentral gyri, 21 and 18, respectively. Node degree was positively associated with regions’ average volume among controls (Pearson’s R = 0.46, P < 0.001); to account for this potential confound, average control volume for each region was included as a covariate. Linear mixed effects modelling showed that higher node degree predicted greater annualized grey matter volume loss in each patient group relative to controls (Fig. 6), as evidenced by group × degree interaction terms: for lvPPA, β = −0.011, t(6703) = −6.4, P < 0.001; for PCA, β = −0.0059, t(6703) = −3.1, P < 0.001; for frontal-variant Alzheimer’s disease, β = −0.0049, t(6703) = −2.2, P < 0.03; and for amnestic Alzheimer’s disease, β = −0.010, t(6703) = −5.3, P < 0.001. The main effect of degree was marginally significant [β = −0.0020, t(6703) = 1.7, P < 0.09], reflecting the lack of substantial grey matter volume loss in the control group (Fig. 6). Among covariates, volume at participants’ first MRI was significantly associated with annualized change [β = −0.024, t(6703) = −6.2, P < 0.001], as was average region size among controls [β = −13.2, t(6703) = −8.3, P < 0.001]. Simple effects of group were not significant, although the frontal-variant Alzheimer’s disease group had marginally lower volumes relative to controls [β = −0.15, t(103) = −1.9, P < 0.06]. Effects of age and sex were also non-significant (both P > 0.12). In pairwise post hoc contrasts, the amnestic Alzheimer’s disease group had significantly greater overall volume loss than the lvPPA group (z = 2.3, P < 0.02). Additionally, the amnestic Alzheimer’s disease group had a more negative slope of association between degree and longitudinal change than the PCA group (z = 2.0, P < 0.05) and the frontal-variant Alzheimer’s disease group (z = 2.1, P < 0.04), suggesting that brain connectivity predicted greater atrophy in amnestic Alzheimer’s disease than these other phenotypes. Other contrasts of group and group × degree interaction terms were non-significant (all z < 0.6, P > 0.5). Importantly, these associations were based on grey matter volumes estimated for AAL labels and are thus unaffected by differences in the AAL and Mindboggle parcellation schemes. Furthermore, because the present analysis relies on population-averaged connectivity values for all groups, it does not address potential connectivity differences between non-amnestic and amnestic Alzheimer’s disease in networks associated with language, visuospatial function, social behaviour, executive control, and memory.
Figure 6.
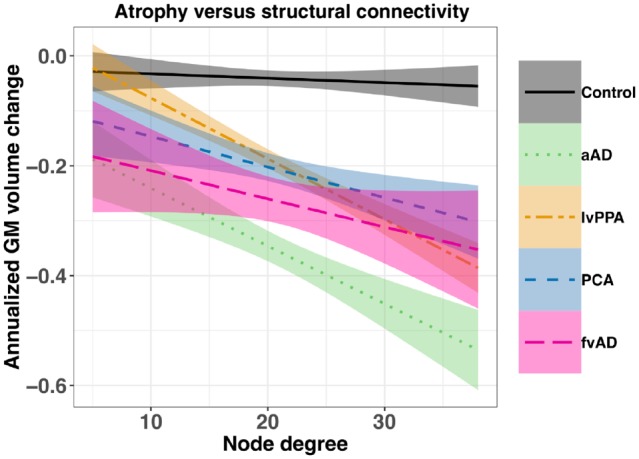
Node degree predicts annualized grey matter volume decline among study participants in regions defined by the AAL atlas. Node degree is based on structural connectivity measures computed by Yeh et al. (2018) and reflects the number of white matter connections that each AAL region has with other regions. Shaded areas show the pointwise 95% confidence interval for each regression line. aAD = amnestic Alzheimer’s disease; fvAD = frontal variant Alzheimer’s disease.
Effects of global cognition and age
Initial MMSE score (which was included as a measure of global cognitive impairment) was positively associated with grey matter volume in the majority of regions of interest [all t(103) ≥ 2.3, P < 0.03], with the exception of bilateral hippocampi and parahippocampal gyri [all t(103) < 1.0, P > 0.3]. In contrast, age at initial MRI was inversely associated with volume in all six MTL regions investigated, including left and right hippocampus [t(103) = −2.5, P < 0.02 and t(103) = −3.2, P < 0.02, respectively, after FDR correction], left and right parahippocampal gyri [t(103) = −3.7, P < 0.001 and t(103) = −4.9, P < 0.001], and left and right entorhinal cortex [t(103) = −3.2, P < 0.002 and t(103) = −3.8, P < 0.001]. In addition, age effects were observed in bilateral precentral gyri [both t(103) < −2.9, P < 0.01], suggesting age-related atrophy in motor cortex. No other regions of interest displayed an effect of age. To determine whether this age effect differed by group, we performed secondary analyses on MTL volumes at the time of first scan using multiple regression models with predictors of group, age, and their interaction, covarying for MMSE score and the interval between MMSE and MRI. After FDR correction, no MTL regions showed a significant group × age interaction [all F(4,99) < 2.3, P > 0.2], suggesting that the association of increased age with MTL atrophy was similar across groups. Age and MMSE effects for the exploratory voxelwise analysis are shown in Supplementary Fig. 1. Consistent with region of interest-based results, voxelwise associations with baseline MMSE score were distributed throughout all lobes of the brain (Supplementary Fig. 1, bottom). Voxelwise analysis further showed robust age effects in the MTL as well as the precentral gyri, anterior temporal lobes, and ventral prefrontal cortex. Conversely, age was positively associated with cortical thickness in the precuneus, which exhibits greater atrophy in early-onset than late-onset Alzheimer’s disease (Möller et al., 2013). No significant effects of sex were observed in either region of interest-based or voxelwise analysis.
Effects of APOE genotype
We assessed the distribution of APOE genotypes among amnestic and non-amnestic Alzheimer’s disease patients. Genotyping data were unavailable for one lvPPA, one PCA, one frontal-variant Alzheimer’s disease, and two amnestic Alzheimer’s disease patients. One amnestic Alzheimer’s disease, three PCA, and three frontal-variant Alzheimer’s disease patients each carried one copy of the ɛ2 allele, which is associated with lower risk for Alzheimer’s disease (Corder et al., 1994). The proportions of lvPPA, PCA, frontal-variant Alzheimer’s disease, and amnestic Alzheimer’s disease patients carrying one to two copies of the APOE ɛ4 allele were 29.2%, 36.8%, 63.6%, and 73.3%, respectively. The frequency of individuals carrying zero, one, or two copies of the ɛ4 allele significantly differed across patient groups [χ2(6) = 14.9, P < 0.02]. In post hoc comparisons, these frequencies differed between the lvPPA and PCA groups [χ2(1) = 8.3, P < 0.02] and between the PCA and frontal-variant Alzheimer’s disease groups [χ2(1) = 6.1, P < 0.05]. Because APOE genotypes were unavailable for control participants, we recomputed LMEs for a priori regions of interest using lvPPA (the largest group) as the reference group and number of ɛ4 alleles as a covariate. No regions of interest exhibited an association with number of ɛ4 alleles independent of group (all P > 0.2).
Longitudinal associations between neuropsychological performance and grey matter volume
Associations between longitudinal neuropsychological performance and concurrent grey matter volume loss were evaluated in patients that had two or more assessments, each within 1 year of a structural MRI scan. This longitudinal analysis contrasts with previous studies that have inferred associations by correlating brain imaging data from a single time point with cognitive change. For recognition memory, this yielded 121 observations from 51 patients, including 21 lvPPA, 13 PCA, eight amnestic Alzheimer’s disease, and nine frontal-variant Alzheimer’s disease patients. For letter fluency, 129 observations were available from 20 lvPPA, 14 PCA, 12 amnestic Alzheimer’s disease, and nine frontal-variant Alzheimer’s disease patients. For forward and reverse digit span, 117 observations were available from 21 lvPPA, 13 PCA, eight amnestic Alzheimer’s disease, and eight frontal-variant Alzheimer’s disease patients. Ninety PBAC observations, from which all other test measures were obtained, were available for 17 lvPPA, 11 PCA, seven amnestic Alzheimer’s disease, and seven frontal-variant Alzheimer’s disease patients. In all cognitive domains except for social behaviour, longitudinal cognition was directly associated with grey matter volume change in one or more associated brain areas, independent of volume at the time of initial MRI (Supplementary Table 12). In the memory domain, volume loss in bilateral hippocampi and left entorhinal predicted declines in recognition discrimination. In the language domain, volume loss in left middle and superior temporal gyri was associated with decreases in letter fluency and forward digit span. In the visuospatial domain, Rey figure copy performance over time was associated with volume loss in right angular, middle temporal, and supramarginal gyrus as well as right precuneus. Judgment of line orientations was likewise associated with right precuneus volume change. No significant associations were found for social behaviour; however, left middle frontal gyrus, left anterior insula, and right middle temporal gyrus predicted reverse digit span, a measure of working memory and executive function (Kramer et al., 2003).
Discussion
Our previous work (Phillips et al., 2018) used cross-sectional analyses to identify areas of frequent atrophy in several Alzheimer’s disease phenotypes, which we hypothesized to be likely regions of disease onset. This approach was inspired by pathological staging studies that have inferred disease progression over time in Alzheimer’s disease (Braak and Braak, 1991), Parkinson’s disease (Braak et al., 2003), and amyotrophic lateral sclerosis (Brettschneider et al., 2013) based on post-mortem pathology. However, this cross-sectional design did not allow us to directly observe within-patient effects of earlier versus later disease progression in each phenotype. The current study compared longitudinal disease progression in multiple clinically-defined non-amnestic Alzheimer’s disease phenotypes with autopsy or CSF evidence of Alzheimer’s disease pathology. We differentiated earlier and later disease stages through a two-part approach. Region of interest-based analysis allowed us to focus on grey matter volume change in the most likely sites of disease onset for each phenotype (Phillips et al., 2018). A second, exploratory analysis of whole-brain cortical thickness values allowed us to examine disease spread outside this cluster of a priori regions of interest. In each phenotype, we observed a combination of local spread surrounding areas of early atrophy and distal spread to brain areas that were not significantly atrophied at the beginning of the follow-up period. Both patterns of initial atrophy and subsequent progression differed between phenotypes. Further, we found that longitudinal rates of neurodegeneration differed across patient groups in phenotype-specific neocortical disease foci, a result which could at least partially account for each phenotype’s characteristic disease distribution. In contrast, we observed no evidence of phenotype-dependent differences in atrophy rates within the MTL, although MTL atrophy appeared to begin later in non-amnestic than in amnestic Alzheimer’s disease. Finally, we found that structural connectivity, assessed by node degree, was a significant predictor of grey matter volume loss over time in both amnestic and non-amnestic Alzheimer’s disease; this result supports brain connectivity as a general factor mediating atrophy progression in Alzheimer’s disease.
Atrophy at initial MRI indicates possible sites of early disease
We hypothesized that each non-amnestic Alzheimer’s disease phenotype would be characterized by a distinct pattern of early atrophy, observed through cross-sectional contrasts of patients’ first MRI scans. We consider significant baseline atrophy an expected and necessary marker for identifying potential sites of disease onset, although early atrophy alone is not sufficient to determine these onset sites. A priori regions of interest for each phenotype (including left temporal cortex in lvPPA, posterior temporoparietal cortex in PCA, prefrontal cortex in frontal-variant Alzheimer’s disease, and the MTL in amnestic Alzheimer’s disease) demonstrated significant initial atrophy, consistent with hypotheses. However, the lvPPA and PCA groups also exhibited lateralized hippocampal atrophy versus controls at initial MRI; although this atrophy was mild relative to the amnestic Alzheimer’s disease group (Fig. 1), we cannot rule out early, lateralized hippocampal disease in these phenotypes. Longitudinal imaging of patients from earlier disease stages, when atrophy will presumably be more focal than in the current sample, is thus necessary to conclusively determine whether focal neocortical disease precedes, follows, or arises concurrently with MTL disease in these phenotypes. Nevertheless, the current study narrows the field of brain areas where disease is likely to originate in each non-amnestic Alzheimer’s disease phenotype, providing a valuable prior constraint on future hypothesis testing. Overall, we propose that the current results are more consistent with the prevailing hypothesis that non-amnestic Alzheimer’s disease patients have disease originating in the neocortex, as inferred by cross-sectional or single-group longitudinal imaging studies (Rogalski et al., 2011; Lehmann et al., 2012; Rohrer et al., 2013; Ossenkoppele et al., 2015a; Xia et al., 2017; Phillips et al., 2018) as well as autopsy studies of hippocampal-sparing Alzheimer’s disease (Giannakopoulos et al., 1994; Murray et al., 2011; Ferreira et al., 2017). Phenotypic variability in initial atrophy patterns (Table 2), including sparing of primary motor cortex at the time of initial MRI, supports the regional specificity of atrophy in non-amnestic Alzheimer’s disease patients.
Interestingly, some areas of initial atrophy continued to change over time, while others did not. From the data at our disposal, we cannot say with certainty what differentiates these regions. One statistical explanation is simply that variability prevented reliable detection of longitudinal atrophy in some regions and phenotypes. An alternative, biological explanation is that areas that failed to exhibit further change over the follow-up period (Table 2 and Fig. 3) had already undergone massive atrophy by the time of patients’ first MRI, reaching a plateau determined by the limited amount of remaining grey matter tissue (Sabuncu et al., 2011; Schuff et al., 2012). The right superior parietal lobule in PCA and left middle frontal gyrus in frontal-variant Alzheimer’s disease may exemplify such slowing: in region of interest-based analysis, both regions were severely atrophied at initial MRI and did not significantly progress over time in their respective phenotypes. Further research is needed to determine why the pace of atrophy changes in some areas of early degeneration but not others.
Differences between phenotypic groups in the neocortical spread of atrophy
Areas that exhibited longitudinal atrophy in the absence of initial cross-sectional differences provide a window onto disease spread in each phenotype. In region of interest-based analysis, the lvPPA group showed strong left lateralization of atrophy at baseline, consistent with prior studies (Rogalski et al., 2016; Phillips et al., 2018). This pattern included left lateral temporal cortex, an area specifically associated with language deficits in lvPPA (Gorno-Tempini et al., 2011). Region of interest-based analysis also indicated early atrophy in left prefrontal cortex, anterior insula, and hippocampus; and right lateral temporal areas. Voxelwise analysis indicated atrophy in left precuneus and right prefrontal cortex as well. Over the follow-up period, we observed new progressive atrophy in brain areas both adjacent to and distal from these areas of initial atrophy. Proximal disease spread was observed throughout the left temporal and parietal lobes as well as bilateral frontal lobes. This proximal atrophy may indicate diffusive spread of pathology through the extracellular medium or along short-distance axonal connections between neighbouring cells in cortex (Guo and Lee, 2014). However, we also observed progression through parts of bilateral frontal lobes and right temporoparietal cortex distal from foci of initial atrophy (Fig. 3); diffusive spread from adjacent disease areas appears insufficient to account for this progression. Two possible explanations may account for new, distal atrophy progression. First, pathology may have arisen independently in these areas. Second, pathogenic proteins may have been transmitted to these areas via long-distance white matter projections, according to the transmission hypothesis of neurodegenerative disease (Guo and Lee, 2014). It is particularly interesting to consider these two possibilities with respect to cross-sectional reports of right temporal atrophy in lvPPA, which—if observed—tends to be much milder than left temporal atrophy. In such cases, it is tempting to infer that right temporal atrophy results from the spread of disease from left to right hemispheres via callosal projections. However, this apparent ‘progression’ may result from a subset of patients having bilateral disease. The current study cannot rule out this possibility, as region of interest-based analysis indicated right temporal atrophy that predated lvPPA patients’ first MRI (Table 2). Earlier recruitment and longitudinal imaging of patients with language disturbances is thus necessary to conclusively demonstrate interhemispheric disease spread in lvPPA.
The PCA group also exhibited a combination of proximal and distal disease spread. Initial atrophy was observed in bilateral precunei and temporoparietal regions (Fig. 3) as well as right hippocampus (Table 2). These parietal areas, in particular, are important to visuospatial processing (Astafiev et al., 2003; Greenberg et al., 2010; Gmeindl et al., 2016) and are consistent with early disease patterns observed in prior studies of PCA (Tang-Wai et al., 2004; Lehmann et al., 2012). In voxelwise analysis, patients with PCA had newer atrophy extending from areas of early disease into inferior parietal, posterior temporal, and insular/opercular cortex; they also exhibited spread proximal to areas of prefrontal atrophy observed at baseline. In addition, however, the PCA group exhibited atrophy progression in the anterior temporal lobes distal from any cluster of existing atrophy (Fig. 3). This finding suggests testable hypotheses regarding the diffusion of disease-causing agents along fibre pathways that terminate in anterior temporal cortex. These pathways include projections from MTL areas as well as more distal connections via the inferior longitudinal fasciculus to striate and prestriate cortex, which may in turn connect with parietal cortex (Nieuwenhuys et al., 2008).
In the frontal-variant Alzheimer’s disease group, region of interest-based and voxelwise analysis collectively indicated grey matter volume loss at initial MRI in bilateral prefrontal, temporal, and anterior insular cortex as well as right middle cingulate and angular gyri. The involvement of the insula is particularly interesting given this group’s behavioural dysfunction, as anterior insula is crucially implicated in primates’ emotion (Phan et al., 2002) as well as in empathy and social life (Singer, 2006). The anterior insula is also implicated in behavioural-variant frontotemporal dementia (bvFTD) (Seeley, 2010), and Ossenkoppele et al. (2015a) found that insula was one of the few regions of atrophy specific to behavioural-variant Alzheimer’s disease patients who were initially misdiagnosed as bvFTD. While our findings suggest early involvement of frontal, temporal, and limbic regions, previous studies of behavioural/dysexecutive Alzheimer’s disease have shown either predominantly frontal (Blennerhassett et al., 2014) or predominantly temporal (Ossenkoppele et al., 2015a) disease. In region of interest-based analysis, the frontal-variant Alzheimer’s disease group demonstrated new atrophy progression only in left perisylvian cortex; voxelwise analysis indicated additional disease progression in bilateral insular/opercular cortex. These findings are located proximally to atrophy clusters observed at first MRI and thus may reflect local, diffusive spread of disease. Although more distal atrophy progression was not observed, we emphasize that null results in this group should be interpreted with extreme caution due to the small sample size; while the reported foci may represent the areas of most robust atrophy in the current sample, true disease progression may be missed due to type II statistical error and may be more anatomically widespread than reported here.
In the amnestic Alzheimer’s disease group, region of interest-based analysis showed new neocortical atrophy in bilateral precentral gyri as well as right temporoparietal cortex (Table 2). Voxelwise analysis similarly indicated neocortical atrophy progression throughout the right temporal lobe as well as in bilateral parietal cortex and right prefrontal cortex. The slight lateralization of disease progression (right hemisphere > left) may be incidental to the current sample, and we do not propose that it is characteristic of amnestic Alzheimer’s disease generally. However, the results are broadly consistent with spreading neocortical disease in later Braak stages (Braak and Braak, 1991). Clusters of newer atrophy in right temporal cortex may indicate local, diffusive spread from right angular gyrus, which was atrophied at initial MRI in the amnestic Alzheimer’s disease group. However, other areas of new progression observed in the voxelwise analysis (Fig. 3) are distal from sites of early atrophy and may result from either white-matter-mediated disease spread or de novo accumulation of pathology. Notably, structural connectivity data from healthy adults indicate that the superior parietal lobule is connected to the hippocampus and angular gyrus (Supplementary Table 6), both of which exhibited baseline atrophy in the amnestic Alzheimer’s disease group; newer areas of superior parietal atrophy in amnestic Alzheimer’s disease may thus result from disease transmission along white-matter pathways connecting these areas.
MTL atrophy in amnestic and non-amnestic phenotypes
At initial MRI, patients with amnestic Alzheimer’s disease demonstrated significant atrophy in bilateral entorhinal cortex and hippocampi, as expected from Braak staging (Braak and Braak, 1991). Bilateral parahippocampal gyri were not atrophied, but they demonstrated significant change over the follow-up period; this pattern of results is consistent with progression from approximate Braak stages IV–V (Whitwell et al., 2008) among our amnestic Alzheimer’s disease sample. Based on well-characterized patterns of disease spread in amnestic Alzheimer’s disease, these MTL foci may be the source of disease spread to the neocortex. The hippocampus has well-characterized white matter connections to posterior cortical areas via the posterior cingulate (Nieuwenhuys et al., 2008; Teipel et al., 2010); these pathways thus represent tracts of interest for investigating the spread of pathogenic proteins to the neocortex.
Patients with non-amnestic Alzheimer’s disease, in turn, demonstrated relative sparing of MTL structures at baseline. In region of interest-based analysis, atrophy was limited to left hippocampus in lvPPA and right hippocampus in PCA; these patterns of lateralization were consistent with the general hemispheric bias observed in both phenotypes. Over the follow-up period, patients with non-amnestic Alzheimer’s disease demonstrated significant atrophy progression in the MTL (Table 2); in the frontal-variant Alzheimer’s disease group, these changes were limited to bilateral entorhinal cortex, although null findings in other MTL structures may reflect the small size of this group. MTL progression in patients with non-amnestic Alzheimer’s disease suggests that sparing of the hippocampus and surrounding MTL (a set of clinically-defined syndromes) is a graded rather than an absolute phenomenon, and that patients with non-amnestic Alzheimer’s disease may become increasingly susceptible to hippocampal degeneration at older ages and in more advanced disease. Indeed, age was a strong predictor of MTL atrophy, as evidenced by both region of interest-based results (see above, ‘Effects of global cognition and age’) and voxelwise results (Supplementary Fig. 1). We note that while patients with non-amnestic Alzheimer’s disease tend to be younger than typical amnestic Alzheimer’s disease patients, the current study controlled for this potential confound both by demographic balancing of groups and by covarying for age in statistical models. Thus, baseline differences between amnestic and non-amnestic Alzheimer’s disease patients in grey matter volume within the MTL (Fig. 1) were not attributable to age differences between these patient groups. Seminal studies of hippocampal sparing in Alzheimer’s disease (Giannakopoulos et al., 1994; Murray et al., 2011; Whitwell et al., 2012) grouped patients based on post-mortem pathology findings; these studies may not have included patients who initially presented with non-amnestic syndromes but developed hippocampal pathology in later disease.
Differences between phenotypic groups in rates of atrophy progression
The design of the current study not only allowed us to investigate differences between phenotypic groups in the topographical distribution of atrophy but also differences in the rate of atrophy within each region. We reasoned that each phenotype might exhibit more rapid degeneration within its associated disease foci, reflecting phenotype-specific susceptibility to disease (Bergeron et al., 2016; Mattsson et al., 2016) in that area. Among neocortical areas associated with non-amnestic Alzheimer’s disease phenotypes, we found evidence to support this reasoning. Patients with lvPPA demonstrated more rapid atrophy in the left temporal cortex than the PCA group (region of interest-based analysis, Fig. 2) and the amnestic Alzheimer’s disease group (voxelwise analysis, Fig. 5). Patients with frontal variant Alzheimer’s disease, in turn, had more rapid atrophy in left anterior insula than both PCA and lvPPA patients in region of interest-based analysis (Fig. 2); and voxelwise analysis indicated additional prefrontal, temporal, and insular differences between frontal-variant Alzheimer’s disease and lvPPA (Fig. 5C). Contrary to our initial hypotheses, we saw no difference in MTL atrophy rates between amnestic and non-amnestic Alzheimer’s disease patients, even when all three non-amnestic Alzheimer’s disease variants were combined to enhance statistical power. Considered together with amnestic Alzheimer’s disease patients’ significant MTL atrophy at initial MRI, this result suggests that relative MTL sparing in non-amnestic Alzheimer’s disease may result from a delayed onset of degeneration in these structures; but that once neurodegeneration has begun, it proceeds at a similar rate as in amnestic Alzheimer’s disease. However, we caution that these findings warrant replication in longitudinal studies involving larger sample sizes.
Associations between longitudinal atrophy and brain connectivity
To investigate the possible role of brain connectivity in mediating disease spread, we related patients’ atrophy patterns to population-average structural connectivity, as estimated from Human Connectome Project white-matter imaging data (Yeh et al., 2018). Several a priori regions of interest in the current study corresponded to hubs in Yeh et al.’s structural connectivity matrix, as evidenced by their high node degree. These findings replicate established functional connectivity results that have related the neuroanatomy of Alzheimer’s disease to brain network hubs including bilateral middle temporal, inferior parietal, and superior parietal cortex (Buckner et al., 2009; Crossley et al., 2014). Moreover, we found that node degree was a significant predictor of regional grey matter volume loss over time in each of the patient groups. We caution that this result is correlative in nature and does not demonstrate long-distance disease spread along white-matter pathways. Indeed, network influences on neurodegeneration need not be limited to physical transport of pathogenic proteins along white-matter tracts; rather, they may reflect effects such as diaschisis (Chételat, 2018), with disease in one area leading to metabolic and functional disruptions in its network neighbors. Computational analysis of atrophy patterns using network models such as those of Raj and colleagues (2012, 2015), Iturria-Medina et al. (2014), and Hu et al. (2016) offers a more rigorous approach for testing hypotheses regarding disease spread in brain networks. Nevertheless, associations between atrophy and node degree provide supporting evidence for the hypothesis that atrophy progression is mediated by structural connectivity. Importantly, the structural connectivity analysis reported here does not address potential hypotheses regarding connectivity differences in non-amnestic and amnestic Alzheimer’s disease. Indeed, initial research on this question suggests that the connectivity of specific brain networks may differ between typical and atypical presentations of Alzheimer’s disease (Lehmann et al., 2015; Whitwell et al., 2015). Because the current study relied on population-averaged structural connectivity values, it was limited to showing a general relationship between degree of connectivity and magnitude of longitudinal change; future analysis of patients’ specific connectivity patterns remains a high priority.
Convergence of atrophy in advanced disease
The spread of disease along white-matter pathways may help explain the reported convergence of atrophy patterns across Alzheimer’s disease phenotypes. For example, Ossenkoppele et al. (2015a) proposed that the common temporoparietal atrophy observed among lvPPA, PCA, early-onset Alzheimer’s disease, and late-onset Alzheimer’s disease patients could result from convergent disease in nodes of the posterior default mode network. In support of the convergence hypothesis, we note that amnestic and non-amnestic Alzheimer’s disease variants alike had common early atrophy and subsequent progression in bilateral temporal cortex (Table 2). These results replicate findings from our previous study (Phillips et al., 2018) of substantial overlap in temporoparietal areas among typical and atypical patients with Alzheimer’s disease. Indeed, this study reported that spatial distributions of atrophy become more similar across phenotypes in later disease phases. At the same time, however, we found that simple logistic regression models could effectively discriminate non-amnestic and Alzheimer’s disease phenotypes from one another based on atrophy patterns, even in advanced disease (Phillips et al., 2018). Moreover, post-mortem studies of Alzheimer’s disease variants show that regional differences in pathology burden persist among Alzheimer’s disease variants even until the end of life. One proposal for resolving these apparently conflicting results is the proposal of Warren et al. (2012) that different clinical presentations of Alzheimer’s disease involve a common temporal, parietal, and frontal network, but that genetic variation or other factors cause the nodes of these networks to be differentially engaged across syndromes.
Conclusions and limitations
Strengths of the current study include a novel comparison of longitudinal anatomical changes in multiple clinically-defined non-amnestic Alzheimer’s disease phenotypes using both a priori region of interest-based and whole-brain voxelwise analyses. The longitudinal study design allowed us to differentiate areas of earlier and later atrophy and to compare these patterns of disease progression across phenotypes. Moreover, we sought to ensure the comparability of the heterogeneous patient groups included here by controlling for demographic and clinical characteristics both during sample selection and in statistical analysis. The relevance of a priori regions of interest is supported by analyses showing that longitudinal anatomical change is associated with concurrent domain-specific cognitive decline.
However, one major limitation was the inability to evaluate non-linear atrophy progression in Alzheimer’s disease: prior evidence suggests that an initial acceleration due to spreading cumulative damage is followed by a deceleration due to the reduction of intact tissue (Sabuncu et al., 2011; Schuff et al., 2012). Such non-linearities complicate study design and interpretation in ways that may not be fully addressed by equating patient groups for chronological age and estimated disease duration: for example, in the current study, it is possible that areas of early atrophy in each phenotype (i.e. those exhibiting atrophy at initial MRI) have entered the deceleration phase, while for other phenotypes the same regions may have been imaged during the acceleration phase. Investigating longitudinal change in earlier-stage patients may allow us to observe a more complete trajectory of neurodegeneration, and including a minimum of three to four imaging time points may allow us to discriminate between linear, quadratic, and sigmoid models of neurodegeneration. Another possible limitation in our findings is statistical power, which is likely to have affected voxelwise analysis more severely than region of interest-based analysis due to the much stricter multiple-comparisons correction of the former. Power limitations may thus have resulted in underestimation of disease spread in the relatively small frontal-variant and amnestic Alzheimer’s disease groups; we thus emphasize the importance of further longitudinal study, particularly of patients for whom post-mortem pathological diagnoses are available to rule out the possibility of co-morbid FTLD or other pathologies. Relatedly, the PBAC behavioural scale did not demonstrate expected worsening of behavioural symptoms over time in the frontal-variant Alzheimer’s disease group; while it is possible that this null result stems from successful treatment of behavioural symptoms through psychiatric medications, future research should strive to include more sensitive measures of behavioural dysfunction. An additional limitation is that patients in the current sample were not selected based on availability of white matter imaging data, preventing us from performing a structured white matter analysis to support interpretations of disease spread along white matter pathways. The current study was also not designed to investigate associations with the APOE genotype or other genetic risk modifiers for Alzheimer’s disease. We found that APOE ɛ4 allele counts added little predictive power to our imaging models after accounting for group effects; however, continued study of the APOE genotype and other genetic risk modifiers in non-amnestic Alzheimer’s disease remains an important research aim. Finally, future studies should include patients with CBS due to underlying Alzheimer’s disease pathology; insufficient longitudinal data prevented us from including this uncommon non-amnestic Alzheimer’s disease phenotype in the current study.
Understanding the neuropathological and clinical heterogeneity of Alzheimer’s disease is crucial to understanding the mechanisms of its progression. The current study not only corroborated probable areas of early disease for lvPPA, PCA, and frontal-variant Alzheimer’s disease but also showed that each phenotype has a different pattern of atrophy progression across the cortex. Moreover, we report novel evidence that the longitudinal rate of neocortical atrophy varies by region and phenotype in non-amnestic Alzheimer’s disease, reflecting phenotype-specific cognitive decline. In contrast, the rate of MTL atrophy in non-amnestic Alzheimer’s disease was similar to that found in amnestic Alzheimer’s disease, suggesting that early sparing of these structures results from a later onset of MTL atrophy in non-amnestic Alzheimer’s disease. Finally, we observed associations between longitudinal atrophy and structural brain connectivity, providing indirect support for models of interregional disease spread in association with white-matter fibre pathways in non-amnestic Alzheimer’s disease.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Valeria Isella and Dr Carlo Ferrarese for their valuable feedback on this project; and Dr Ilya Nasrallah for assistance with visual reads of PET images. Additionally, the authors thank Dr Fang-Cheng Yeh for making structural connectivity results available at http://brain.labsolver.org/.
Glossary
Abbreviations
- LME
linear mixed effects
- lvPPA
logopenic-variant primary progressive aphasia
- MMSE
Mini Mental Status Examination
- MTL
medial temporal lobe
- PBAC
Philadelphia Brief Assessment of Cognition
- PCA
posterior cortical atrophy
Funding
This work was supported by grants from the Alzheimer’s Association (AARF-16–443681), National Institutes of Health (AG061277, AG054519, AG017586, AG010124, AG043503, and NS088341), BrightFocus Foundation (A2016244S), Dana Foundation, Newhouse Foundation, Wyncote Foundation, Arking Family Foundation, and the Italian Ministry of Education, University, and Research.
Competing interests
All authors report that they have no competing interests to disclose.
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 2014; 127: 667–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astafiev SV, Shulman GL, Stanley CM, Snyder AZ, Van Essen DC, Corbetta M. Functional Organization of human intraparietal and frontal cortex for attending, looking, and pointing. J Neurosci 2003; 23: 4689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC. A reproducible evaluation of ANTs similarity metric performance in brain image registration. NeuroImage 2011; 54: 2033–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Stauffer M, Song G, Wu B, Gee JC. The Insight ToolKit image registration framework. Front Neuroinform 2014; 8: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron D, Bensaïdane R, Laforce R. Untangling Alzheimer’s disease clinicoanatomical heterogeneity through selective network vulnerability - an effort to understand a complex disease. Curr Alzheimer Res 2016; 13: 589–96. [DOI] [PubMed] [Google Scholar]
- Blennerhassett R, Lillo P, Halliday GM, Hodges JR, Kril JJ. Distribution of pathology in frontal variant Alzheimer’s disease. J Alzheimer’s Dis: JAD 2014; 39: 63–70. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24: 197–211. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013; 74: 20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci 2009; 29: 1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun MS, Kim SE, Park J, Yi D, Choe YM, Sohn BK et al. Heterogeneity of regional brain atrophy patterns associated with distinct progression rates in Alzheimer’s disease. PLoS One 2015; 10: e0142756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Saad ZS, Britton JC, Pine DS, Cox RW. Linear mixed-effects modeling approach to FMRI group analysis. NeuroImage 2013; 73: 176–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G. Multimodal neuroimaging in Alzheimer’s disease: early diagnosis, physiopathological mechanisms, and impact of lifestyle. J Alzheimer’s Dis 2018; 64: S199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nature Genet 1994; 7: 180–84. [DOI] [PubMed] [Google Scholar]
- Cox RW, Chen G, Glen DR, Reynolds RC, Taylor PA. FMRI clustering in AFNI: false-positive rates redux. Brain Connect 2017; 7: 152–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley NA, Mechelli A, Scott J, Carletti F, Fox PT, McGuire P et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain 2014; 137: 2382–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. The Lancet Neurology 2012; 11: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, van der Flier WM et al. Consensus classification of posterior cortical atrophy. Alzheimer’s & Dement 2017; 13: 870–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, McGinnis SM, Xia C, Price BH, Atri A, Murray ME et al. Approach to atypical Alzheimer’s disease and case studies of the major subtypes. CNS Spectr 2017; 22: 439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duara R, Loewenstein DA, Shen Q, Barker W, Greig MT, Varon D et al. Regional patterns of atrophy on MRI in Alzheimer’s disease: neuropsychological features and progression rates in the ADNI cohort. Adv Alzheimer’s Dis 2013; 02: 135–47. [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 2014; 13: 614–29. [DOI] [PubMed] [Google Scholar]
- Ferreira D, Verhagen C, Hernández-Cabrera JA, Cavallin L, Guo C-J, Ekman U et al. Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci Rep 2017; 7: 46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman SD, Cohen JD, Fitzgerald M, Eddy WF, Mintun MA, Noll DC. Improved assessment of significant activation in functional magnetic resonance imaging (fMRI): use of a cluster-size threshold. Magnet Resonan Med 1995; 33: 636–47. [DOI] [PubMed] [Google Scholar]
- Galton CJ, Patterson K, Xuereb JH, Hodges JR. Atypical and typical presentations of Alzheimer’s disease: A clinical neuropsychological, neuroimaging and pathological study of 13 cases. Brain 2000; 123: 484–98. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Hof PR, Bouras C. Alzheimer’s disease with asymmetric atrophy of the cerebral hemispheres: Morphometric analysis of four cases. Acta Neuropathol 1994; 88: 440–7. [DOI] [PubMed] [Google Scholar]
- Giannini LAA, Irwin DJ, McMillan CT, Ash S, Rascovsky K, Wolk DA et al. Clinical marker for Alzheimer disease pathology in logopenic primary progressive aphasia. Neurology 2017; 88: 2276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmeindl L, Chiu Y-C, Esterman MS, Greenberg AS, Courtney SM, Yantis S. Tracking the will to attend: cortical activity indexes self-generated, voluntary shifts of attention. Atten Percept Psychophys 2016; 78: 2176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76: 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AS, Esterman M, Wilson D, Serences JT, Yantis S. Control of spatial and feature-based attention in frontoparietal cortex. J Neurosci 2010; 30: 14330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronenschild EHBM, Habets P, Jacobs HIL, Mengelers R, Rozendaal N, van Os J et al. The effects of FreeSurfer version, workstation type, and Macintosh operating system version on anatomical volume and cortical thickness measurements. PLos One 2012; 7: e38234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VMY. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nature Medicine 2014; 20: 130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Jovicich J, Salat D, van der Kouwe A, Quinn B, Czanner S et al. Reliability of MRI-derived measurements of human cerebral cortical thickness: the effects of field strength, scanner upgrade and manufacturer. NeuroImage 2006; 32: 180–94. [DOI] [PubMed] [Google Scholar]
- Hu C, Hua X, Ying J, Thompson PM, Fakhri GE, Li Q. Localizing sources of brain disease progression with network diffusion model. IEEE J Sel Top Sig Process 2016; 10: 1214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM-Y. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 2013; 33: 1024–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, McMillan CT, Toledo JB, Arnold SE, Shaw LM, Wang L-S et al. Comparison of cerebrospinal fluid levels of tau and Aβ 1–42 in Alzheimer disease and frontotemporal degeneration using 2 analytical platforms. Arch Neurol 2012; 69: 1018–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Evans AC, Alzheimer’s Disease Neuroimaging Initiative. Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput Biol 2014; 10: e1003956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 1999; 56: 1233–9. [DOI] [PubMed] [Google Scholar]
- Klein A, Andersson J, Ardekani BA, Ashburner J, Avants B, Chiang M-C et al. Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage 2009; 46: 786–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Ghosh SS, Bao FS, Giard J, Häme Y, Stavsky E et al. Mindboggling morphometry of human brains. PLos Comput Biol 2017; 13: e1005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Tourville J. 101 labeled brain images and a consistent human cortical labeling protocol. Front Neurosci 2012; 6: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 2003; 16: 211–8. [DOI] [PubMed] [Google Scholar]
- Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011; 70: 327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Barnes J, Ridgway GR, Ryan NS, Warrington EK, Crutch SJ et al. Global gray matter changes in posterior cortical atrophy: a serial imaging study. Alzheimer’s Dement 2012; 8: 502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Madison C, Ghosh PM, Miller ZA, Greicius MD, Kramer JH et al. Loss of functional connectivity is greater outside the default mode network in nonfamilial early-onset Alzheimer’s disease variants. Neurobiol Aging 2015; 36: 2678–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libon DJ, Bondi MW, Price CC, Lamar M, Eppig J, Wambach DM et al. Verbal serial list learning in mild cognitive impairment: a profile analysis of interference, forgetting, and errors. J Int Neuropsychol Soc 2011a; 17: 905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libon DJ, Rascovsky K, Gross RG, White MT, Xie SX, Dreyfuss M et al. The Philadelphia brief assessment of cognition (PBAC): a validated screening measure for dementia. Clin Neuropsychol 2011b; 25: 1314–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012; 7: e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DS, Wang TH, Parker J, Csernansky JG, Morris JC, Buckner RL. Open Access Series of Imaging Studies (OASIS): cross-sectional MRI data in young, middle aged, nondemented, and demented older adults. J Cogn Neurosci 2007; 19: 1498–507. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Schott JM, Hardy J, Turner MR, Zetterberg H. Selective vulnerability in neurodegeneration: Insights from clinical variants of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2016; 87: 1000–4. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack Jr CR, Kawas CH et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement 2011; 7: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan CT, Irwin DJ, Nasrallah I, Phillips JS, Spindler M, Rascovsky K et al. Multimodal evaluation demonstrates in vivo 18F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016; 132: 935–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medaglia JD, Huang W, Segarra S, Olm C, Gee J, Grossman M et al. Brain network efficiency is influenced by the pathologic source of corticobasal syndrome. Neurology 2017; 89: 1373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M-M, Rogalski EJ, Wieneke C, Hurley RS, Geula C, Bigio EH et al. Primary progressive aphasia and the evolving neurology of the language network. Nat Rev Neurol 2014a; 10: 554–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M-M, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain 2014b; 137: 1176–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezias C, LoCastro E, Xia C, Raj A. Connectivity, not region-intrinsic properties, predicts regional vulnerability to progressive tau pathology in mouse models of disease. Acta Neuropathol Commun 2017; 5: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller C, Vrenken H, Jiskoot L, Versteeg A, Barkhof F, Scheltens P et al. Different patterns of gray matter atrophy in early- and late-onset Alzheimer’s disease. Neurobiol Aging 2013; 34: 2014–22. [DOI] [PubMed] [Google Scholar]
- Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011; 10: 785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuys R, Voogd J, Voogd J, van Huijzen C, van Huijzen C. The human central nervous system. 4th edn. Berlin: Springer; 2008. [Google Scholar]
- Ossenkoppele R, Cohn-Sheehy BI, La Joie R, Vogel JW, Möller C, Lehmann M et al. Atrophy patterns in early clinical stages across distinct phenotypes of Alzheimer’s disease. Hum Brain Mapp 2015a; 36: 4421–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Pijnenburg YAL, Perry DC, Cohn-Sheehy BI, Scheltens NME, Vogel JW et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 2015b; 138: 2732–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, Abdulkadir A, Kaller C, Kümmerer D, Hüll M, Vach W et al. Subgroups of Alzheimer’s disease: stability of empirical clusters over time. J Alzheimer’s Dis 2014; 42: 651–61. [DOI] [PubMed] [Google Scholar]
- Phan KL, Wager T, Taylor SF, Liberzon I. Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. NeuroImage 2002; 16: 331–48. [DOI] [PubMed] [Google Scholar]
- Phillips JS, Da Re F, Dratch L, Xie SX, Irwin DJ, McMillan CT et al. Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer’s disease. Neurobiol Aging 2018; 63: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulakis K, Pereira JB, Mecocci P, Vellas B, Tsolaki M, Kłoszewska I et al. Heterogeneous patterns of brain atrophy in Alzheimer’s disease. Neurobiol Aging 2018; 65: 98–108. [DOI] [PubMed] [Google Scholar]
- Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron 2012; 73: 1204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, LoCastro E, Kuceyeski A, Tosun D, Relkin N, Weiner M. Network diffusion model of progression predicts longitudinal patterns of atrophy and metabolism in Alzheimer’s disease. Cell Rep 2015; 10: 359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan S, Bertoux M, Flanagan E, Irish M, Piguet O, Hodges JR et al. Longitudinal executive function and episodic memory profiles in behavioral-variant frontotemporal dementia and Alzheimer’s disease. J Int Neuropsychol Soc 2017; 23: 34–43. [DOI] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134: 2456–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Salmon DP, Hansen LA, Thal LJ, Galasko D. Disparate letter and semantic category fluency deficits in autopsy-confirmed frontotemporal dementia and Alzheimer’s disease. Neuropsychology 2007; 21: 20–30. [DOI] [PubMed] [Google Scholar]
- Rogalski E, Cobia D, Harrison TM, Wieneke C, Weintraub S, Mesulam M-M. Progression of language decline and cortical atrophy in subtypes of primary progressive aphasia. Neurology 2011; 76: 1804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski E, Sridhar J, Rader B, Martersteck A, Chen K, Cobia D et al. Aphasic variant of Alzheimer disease: clinical, anatomic, and genetic features. Neurology 2016; 87: 1337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Caso F, Mahoney C, Henry M, Rosen HJ, Rabinovici G et al. Patterns of longitudinal brain atrophy in the logopenic variant of primary progressive aphasia. Brain Lang 2013; 127: 121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabuncu MR, Desikan RS, Sepulcre J, Yeo BTT, Liu H, Schmansky NJ et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol 2011; 68: 1040–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Tosun D, Insel PS, Chiang GC, Truran D, Aisen PS et al. Nonlinear time course of brain volume loss in cognitively normal and impaired elders. Neurobiol Aging 2012; 33: 845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz CG, Gunter JL, Wiste HJ, Przybelski SA, Weigand SD, Ward CP et al. A large-scale comparison of cortical thickness and volume methods for measuring Alzheimer’s disease severity. NeuroImage Clin 2016; 11: 802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW. Anterior insula degeneration in frontotemporal dementia. Brain Struct Funct 2010; 214: 465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 2009; 65: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer T. The neuronal basis and ontogeny of empathy and mind reading: review of literature and implications for future research. Neurosci Biobehav Rev 2006; 30: 855–63. [DOI] [PubMed] [Google Scholar]
- Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, Crook R et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004; 63: 1168–74. [DOI] [PubMed] [Google Scholar]
- Teipel SJ, Bokde ALW, Meindl T, Amaro E, Soldner J, Reiser MF et al. White matter microstructure underlying default mode network connectivity in the human brain. NeuroImage 2010; 49: 2021–32. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Brettschneider J, Grossman M, Arnold SE, Hu WT, Xie SX et al. CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol 2012; 124: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA et al. N4ITK: Improved N3 bias correction. IEEE Trans Med Imaging 2010; 29: 1310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB. Explicit B-spline regularization in diffeomorphic image registration. Front Neuroinform 2013; 7: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Cook PA, Klein A, Song G, Das SR, Duda JT et al. Large-scale evaluation of ANTs and FreeSurfer cortical thickness measurements. NeuroImage 2014; 99: 166–79. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 2002; 15: 273–89. [DOI] [PubMed] [Google Scholar]
- Wang H, Suh JW, Das SR, Pluta J, Craige C, Yushkevich PA. Multi-Atlas Segmentation with Joint Label Fusion. IEEE Trans Pattern Anal Mach Intell 2013; 35: 611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JD, Fletcher PD, Golden HL. The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol 2012; 8: 451–64. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: A case-control study. Lancet Neurol 2012; 11: 868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Przybelski SA, Parisi JE, Senjem ML, Boeve BF et al. Temporoparietal atrophy: A marker of AD pathology independent of clinical diagnosis. Neurobiol Aging 2011; 32: 1531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jones DT, Duffy JR, Strand EA, Machulda MM, Przybelski SA et al. Working memory and language network dysfunction in logopenic aphasia: A task-free fMRI comparison to Alzheimer’s dementia. Neurobiology of aging 2015; 36: 1245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD et al. MRI correlates of neurofibrillary tangle pathology at autopsy. Neurology 2008; 71: 743–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Makaretz SJ, Caso C, McGinnis S, Gomperts SN, Sepulcre J et al. Association of In Vivo [18F]AV-1451 tau pet imaging results with cortical atrophy and symptoms in typical and atypical alzheimer disease. JAMA Neurol 2017; 74: 427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh F-C, Panesar S, Fernandes D, Meola A, Yoshino M, Fernandez-Miranda JC et al. Population-averaged atlas of the macroscale human structural connectome and its network topology. NeuroImage 2018; 178: 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Computer code for the current manuscript (including all text, analysis, and visualization of results) is available in the form of Rmarkdown and LaTeX scripts in a public GitHub repository (https://github.com/jeffrey-phillips/naAD-longitudinal.git). Rmarkdown code requires R version 3.4.4 or higher. Investigators who wish to access imaging and clinical data may submit a direct request to the corresponding author.