

Abstract
Amenamevir (formerly ASP2151) is a helicase‐primase inhibitor being developed for the treatment of herpesvirus infection. Amenamevir is both a substrate and inducer of cytochrome P450 (CYP) 3A4. Three studies were done in healthy volunteers to investigate potential CYP3A pharmacokinetic interactions with the following drugs: (1) Midazolam (probe substrate for CYP3A): After 10 days’ pretreatment with amenamevir 400 mg daily, geometric mean maximum concentration of drug in blood plasma (Cmax) and area under the plasma drug concentration‐time curve from time zero to infinity (AUC0‐∞) of midazolam 7.5 mg were about 68% and 51%, respectively, of those after midazolam alone. (2) Cyclosporine (substrate and inhibitor of CYP3A): After 5 days’ pretreatment with cyclosporine 100 mg twice daily, geometric mean Cmax of amenamevir after 400‐mg and 1200‐mg single doses was, respectively, about 66% and 69%, and AUC0‐∞ about 82% and 79%, of those after amenamevir alone. (3) Ritonavir (inhibitor of CYP3A): When given with single doses of ritonavir 600 mg, geometric mean Cmax of amenamevir after 400‐mg and 1200‐mg single doses was, respectively, about 1.4 and 1.6 times higher, and geometric mean AUC0‐∞ about 2.6 and 3.3 times higher, than after amenamevir alone. Amenamevir has the potential to be involved in CYP3A‐mediated pharmacokinetic interactions in clinical practice.
Keywords: amenamevir, cyclosporine, drug‐drug interaction, midazolam, ritonavir
Amenamevir, an oxadiazolephenyl derivative (see Figure 1),1 is a nonnucleoside helicase‐primase inhibitor being developed for the treatment of herpes simplex virus types 1 and 2 (HSV‐1 and HSV‐2), and varicella zoster virus (VZV) infection. The viral helicase‐primase complex is essential for viral DNA replication.
Figure 1.
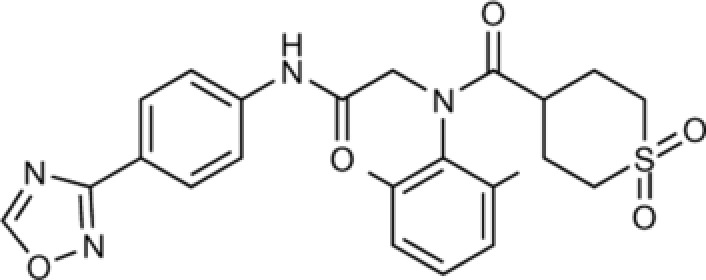
Molecular structure of amenamevir.1
Nucleoside analogues such as acyclovir and valacyclovir are currently first‐line treatment for HSV and VZV.2 Drug‐resistant HSV infection is unusual in immunocompetent patients but is more common in immunocompromised individuals, in whom it is more likely to be clinically important.3 Amenamevir has good activity against acyclovir‐resistant VZV strains in vitro1 and therefore might be effective in the treatment of infection with virus resistant to first‐line antiviral drugs.
The pharmacokinetics and metabolic profile of amenamevir have previously been studied (unpublished data held by Maruho). In healthy volunteers, amenamevir was rapidly absorbed after single, oral administration, with time of peak concentration of 1.33 to 2.5 hours. Plasma protein binding of 14C‐amenamevir in humans was about 75%. Elimination half‐life was about 7 to 8 hours and was independent of dose over the range of 5 to 2400 mg. After a single oral dose of 200 mg 14C‐amenamevir, 74.6% of the 14C radioactivity was recovered in feces; the remainder was recovered in urine.
Studies of 14C‐amenamevir using cells expressing the human multidrug resistance transporter P‐glycoprotein (P‐gp) and the human multidrug resistance‐associated protein‐2 showed that amenamevir is a substrate of the human multidrug resistance P‐gp transporter but not of the human multidrug resistance‐associated protein‐2 transporter. There was no uptake of 14C‐amenamevir into organic anion transporting polypeptide (OAT) P1B1‐, OATP1B3‐, OAT1‐ and OAT3‐, and organic cation uptake transporter‐2–expressing cells, which suggests that amenamevir is not a substrate of those transporters. In vitro studies using human liver microsomes show that cytochrome P450 (CYP) 3A4/5 are the main isozymes involved in the metabolism of amenamevir. A major metabolite of amenamevir in human plasma was the monohydroxy metabolite AS1955888‐00. The metabolite/parent compound ratio was ∼0.1. The antiherpetic potency of amenamevir, as judged by half maximal effective concentration values, was much greater than that of AS1955888‐00: up to 12‐fold greater for HSV‐1, up to 8‐fold for HSV‐2, and up to 4‐fold for VZV. Amenamevir 1 to 100 μM showed the potential to induce CYP3A4 in human hepatocytes but had very weak or no inhibitory activity against CYP3A4/5 (half maximal inhibitory concentration >100 μM).
Thus, amenamevir is both a substrate and inducer of CYP3A. CYP3A comprises over 30% of hepatic enzymes and is involved in the metabolism of more than 50% of medicines that undergo metabolic elimination.4 CYP3A interactions involving amenamevir could therefore be clinically relevant.
In this paper, we describe 3 studies that investigated, in healthy volunteers, potential pharmacokinetic interactions mediated by CYP3A. The rationale for our choice of drugs was as follows:
Midazolam. In this study, we tested the ability of amenamevir to induce CYP3A, using midazolam (a sedative) as a “probe” substrate.5 We used a single oral dose of midazolam 7.5 mg, which is within the recommended therapeutic range and is generally well tolerated, albeit somewhat sedative.6
Cyclosporine. In this study, we tested the susceptibility of amenamevir to inhibition of CYP3A by cyclosporine, an immunosuppressant that is both a substrate and an inhibitor of CYP3A.7 Immunocompromised status increases the frequency of reactivation and the severity of symptoms of herpesvirus infection.3 Thus, many patients taking immunomodulator therapy and patients infected with HIV (see ritonavir section below) are likely to be treated with amenamevir. We preferred cyclosporine to tacrolimus because cyclosporine is a stronger CYP3A inhibitor. In clinical practice, the usual oral dose of cyclosporine is 2.5 to 15 mg/kg/day,8 equivalent to about 175 to 1050 mg daily in a 70‐kg subject. We chose to give cyclosporine 100 mg twice daily, which we considered low enough to be safe but high enough to allow detection of drug interactions mediated by CYP3A.
Ritonavir. In this study, we tested the susceptibility of amenamevir to CYP3A inhibition by ritonavir, an antiretroviral drug that is a powerful inhibitor of CYP3A.9 Ritonavir is often used to “boost” the circulating concentrations of other protease inhibitors, most of which are substrates for CYP3A.10 We chose a single dose of 600 mg because repeated doses of that size are poorly tolerated,11 and Kempf et al12 showed that a single concomitant dose of 600‐mg ritonavir increased the plasma concentrations of saquinavir >50‐fold.
Subjects and Methods
All Studies
All 3 studies were done at Hammersmith Medicines Research (HMR), London, after approval by the Medicines and Healthcare Products Regulatory Agency (MHRA) and London‐Brent Research Ethics Committee. The studies were conducted in accordance with the ethical principles outlined in the Declaration of Helsinki. All subjects gave written, informed consent before the start of the study.
Subjects
The subjects comprised healthy men aged 18 to 45 years who had a body mass index of 18.0 to 30.9 kg/m2 and were capable of giving informed consent. Subjects were deemed healthy on the basis of clinical history, medical examination, electrocardiogram, vital signs, and clinical laboratory tests of routine hematology, biochemistry, and urinalysis. Subjects were excluded if, during the 28 days before dosing, they had used any prescription or nonprescription medicine or herbal remedy known to interfere with CYP3A activity.
Midazolam Study
This was a phase 1, open‐label, single‐center, drug‐drug interaction study in 18 healthy men aged 20 to 43 years (see Table 1).
Table 1.
Summary of Demographic Details of Subjects in All 3 Studies
| Ritonavir Study (N = 48) | ||||||
|---|---|---|---|---|---|---|
| Midazolam Study (N = 18) | Cyclosporine Study (N = 26)b | Group 1 (N = 24) | Group 2 (N = 24) | |||
| Age (y) | Mean (SD) | 30.7 (7.53) | 32.7 (7.2) | 28.2 (6.4) | 27.0 (4.9) | |
| Range | 20‐43 | 20‐45 | 19‐42 | 18‐36 | ||
| Race | ||||||
| Asian | n (%) | 2 (11.1) | 0 (0) | 1 (4.2) | 3 (12.5) | |
| Black | 4 (22.2) | 0 (0) | 4 (16.7) | 5 (20.8) | ||
| White | 11 (61.1) | 26 (100) | 17 (70.8) | 15 (62.5) | ||
| Other | 1 (5.6) | 0 (0) | 2 (8.3) | 1 (4.2) | ||
| Height (cm) | Mean (SD) | 176.7 (4.8) | 182.4 (6.5) | 179.8 (6.8) | 178.2 (7.2) | |
| Range | 166‐184 | 173‐202 | 165‐196 | 167‐190 | ||
| Weight (kg) | Mean (SD) | 75.4 (9.8) | 81.4 (8.2) | 79.1 (7.8) | 75.5 (11.3) | |
| Range | 56.7‐91.5 | 67.9‐96.1 | 62.0‐92.1 | 58.2‐104.9 | ||
| BMI | Mean (SD) | 24.1 (2.7) | 24.5 (2.2) | 24.5 (2.5) | 23.7 (2.9) | |
| kg/m2 | Range | 18.9‐28.5 | 20.6‐29.0 | 19.9‐30.1 | 19.2‐29.1 | |
| Smoker | n (%) | 0 | 2 (7.7) | 6 (25.0) | 3 (12.5) | |
| Cigarettes1 (daily) | Mean (SD) | 2 (1.4) | 3.3 (1.5) | 3.7 (2.3) | ||
| Range | 1‐3 | 2‐5 | 2‐5 | |||
| Consumes alcohol | n (%) | 11 (61.1) | 2 (1.4) | 20 (83.3) | 12 (50) | |
| Units/weeka | Mean (SD) | 4.5 (3.1) | 5.8 (4.8) | 7.4 (4.0) | 6.3 (5.4) | |
| Range | 2‐11 | 1‐20 | 2‐16 | 1‐16 | ||
BMI, body mass index; SD, standard deviation.
Includes only those subjects who smoke/drink alcohol.
Includes 2 replacement subjects.
Each subject received 4 single oral doses of 7.5‐mg midazolam on days 1, 12, 19, and 26, and 10 daily doses of 400‐mg amenamevir on days 3 through 12 (see Figure 2A). On day 12, the doses of midazolam and amenamevir were administered at the same time. We chose a dose level of 400‐mg instead of 1200‐mg amenamevir because 1200‐mg amenamevir is being developed only as a single dose, and repeated doses are needed to assess enzyme induction. Blood samples for midazolam and 1‐hydroxymidazolam assay were taken before and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 16, and 24 hours after each dose of midazolam.
Figure 2.
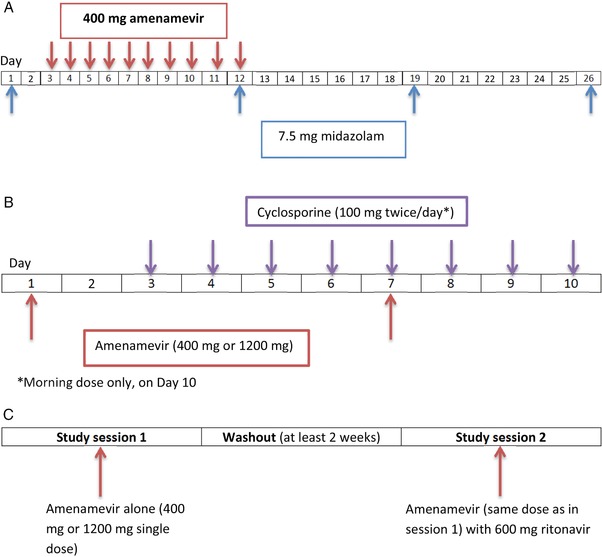
(A) Midazolam study schematic; (B) cyclosporine study schematic showing 1 treatment session (each subject underwent 2 such sessions, separated by a minimum 2‐week washout); (C) ritonavir study schematic.
To assess the return of CYP3A activity to baseline, the effect of amenamevir on midazolam and 1‐hydroxymidazolam was assessed on days 19 and 26 using the analysis of variance model described above. The 95% confidence interval (CI) for the difference in means (day 19 vs day 1 and day 26 vs day 1) were used to determine how long it took for the CYP3A levels to return to normal.
Cyclosporine Study
This was a randomized, phase 1, open‐label, single‐center, balanced 2‐way crossover, drug‐drug interaction study in 24 healthy men aged 20 to 45 years (see Table 1).
Each subject underwent 2 treatment sessions in which they received single oral doses of amenamevir (400 mg in one session, 1200 mg in the other) on days 1 and 7, and cyclosporine 100 mg twice daily on days 3 through 9, and once in the morning of day 10 (see Figure 2B). On day 7, the doses of amenamevir and cyclosporine were administered at the same time. Twelve subjects were randomized to receive the 400‐mg dose of amenamevir in treatment session 1 and the 1200‐mg dose in treatment session 2, and 12 were randomized to receive the 1200‐mg dose in treatment session 1 followed by the 400‐mg dose in treatment session 2. There was a washout of at least 2 weeks between treatment sessions.
Blood samples for assay of amenamevir and its metabolite AS1955888‐00 were taken before and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48 hours on days 1 and 7 after each dose of amenamevir, and at 72 hours after each dose of amenamevir on day 7. Blood samples for assay of cyclosporine were taken before, and at 0.5, 1, 2, 3, 4, 6, 8, 10, and 12 hours after, morning doses of cyclosporine on days 6 and 7 only.
Ritonavir Study
A randomized, phase 1, open‐label, balanced 2‐way crossover, drug‐drug interaction study in 48 healthy men aged 18 to 42 years (see Table 1).
The subjects were randomized to receive single doses of amenamevir 400 mg (n = 24) or 1200 mg (n = 24) in 2 treatment sessions. Each subject was further randomized to receive amenamevir alone in the first session, followed by amenamevir with a single dose of ritonavir 600 mg (administered at the same time) in the second session, or vice versa (see Figure 2C). Blood samples for assay of amenamevir and its metabolite AS1955888‐00 were taken before and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, and 72 hours after each dose. There was a washout of at least 2 weeks between treatment sessions.
Blood samples for ritonavir assay were taken before and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 hours after each dose.
Safety and Tolerability Assessments
Safety and tolerability assessments included adverse events, vital signs, 12‐lead electrocardiogram, physical examination, and clinical laboratory tests.
Assay of Amenamevir and Its Metabolite AS1955888‐00
Blood samples for assay of amenamevir and AS1955888‐00 (2 mL) were collected in sodium heparin tubes. Plasma was separated by centrifugation at ∼1500 G for 10 min at 4°C, then stored at –20°C or below until analysis by Shin Nippon Biomedical Laboratories Ltd, using a validated liquid chromatography tandem mass spectrometry (LC‐MS/MS) method as follows.
Internal Standard
YM‐385482 and AS2357699‐00 (both 100 μg/mL in acetonitrile) were diluted with 50% acetonitrile solution to yield a final concentration of 400 ng/mL of each analyte.
Calibrators
Deuterated amenamevir (2.5 mg/mL in acetonitrile) and deuterated AS1955888‐00 (1.25 mg/mL in 50% acetonitrile) were diluted with 50% acetonitrile to yield final concentrations of 25 to 1 000 000 ng/mL amenamevir and 10 to 400 000 ng/mL AS1955888‐00.
Analytical Method
Samples were extracted using the organic solvent t–butyl methyl ether (MTBE).
20 μL of 50% acetonitrile solution and 50 μL of internal standard were added to 100‐μL plasma samples and mixed; 100 μL of 5% ammonia solution and 2.5 mL of t‐MTBE were then added and the samples centrifuged. The samples were placed in a dry ice/acetone bath to freeze the aqueous layer. The organic layer was then removed and evaporated to dryness under a stream of nitrogen at 40°C. The residue was reconstituted in 1 mL of acetonitrile/ultrapure water/formic acid (98%) 50:50:0.1 v/v/v for analysis by LC‐MS/MS.
High‐Performance Liquid Chromatography (HPLC) Conditions
| Column | Chromolith SpeedROD RP–18e (50 mm × 4.6 mm i.d., Phenomenex Inc.) |
| Mobile phase | Methanol/ultrapure water/formic acid, 60:40:0.1, v/v/v |
| Elution | Isocratic |
| Flow rate | 0.5 mL/min |
| Injection volume | 5 μL |
Mass Spectrometry Settings
| MS type | Multiple Reaction Monitoring (MRM) |
|---|---|
| Ionisation type | ESI (TurboIonSpray) |
| Ionisation polarity | Positive |
| Compound | Q1 (m/z) | Q3 (m/z) |
|---|---|---|
| Amenamevir | 483 | 322 |
| AS1955888–00 | 499 | 338 |
| YM‐385482 (IS substance‐1) | 489 | 328 |
| AS2357699‐00 (IS substance‐2) | 504 | 338 |
Variability
| Compound | Concentration (ng/mL) | Mean (ng/mL) | Precision (%CV) | Accuracy (Relative Error) (%) |
|---|---|---|---|---|
| Amenamevir | 5 | 5.28 | 4.5 | 105.6 |
| 10 | 10.12 | 3.9 | 101.2 | |
| 500 | 510.4 | 2.6 | 102.1 | |
| 4000 | 4017 | 2.1 | 100.4 | |
| AS1955888‐00 | 2 | 2.07 | 5.2 | 103.7 |
| 4 | 4.04 | 6.2 | 101.1 | |
| 200 | 202.8 | 4.0 | 101.4 | |
| 1600 | 1613 | 3.8 | 100.8 |
CV, coefficient of variation.
The lower limit of reliable quantification was 5 ng/mL for amenamevir and 2 ng/mL for AS1955888‐00.
Assay of Other Drugs
Blood samples for assay of midazolam, its metabolite 1‐hydroxymidazolam (3 mL), and ritonavir (2 mL) were collected in lithium heparin tubes. Blood samples for assay of cyclosporine (2 mL) were collected in ethylenediaminetetraacetic acid tubes. Plasma was separated using the same method as for amenamevir and AS1955888‐00 and stored at –20°C until analysis by Analytical Services International using validated LC‐MS/MS as follows.
Midazolam
Internal Standard
Midazolam D4 maleate and alpha‐hydroxymidazolam‐D4 (both 100 μg/mL in methanol) were diluted with 0.1% formic acid to a final concentration of 50 ng/mL of each analyte.
Calibrators
The calibrators were 0.1 to 100.0 ng/mL midazolam and α‐hydroxymidazolam in 0.1% formic acid.
Analytical Method
Samples were extracted using the organic solvent MTBE.
The following were added to a polypropylene tube:
250 μL plasma sample (or 250 μL blank plasma, for calibration samples)
250 μL 0.1% formic acid (or 250 μL calibrator, for calibration samples)
100 μL internal standard
500 μL 2 M Tris (containing Orange G)
2 mL MTBE
The organic phase supernatant was separated by centrifugation, then evaporated to dryness in a vacuum. The residue was reconstituted with 250 μL of 10% methanol and briefly vortexed before analysis by LC‐MS/MS.
HPLC Conditions
| Column | Chromolith RP‐18e column (100 mm × 3 mm) |
|---|---|
| Mobile phase | Methanol |
| Formic acid 0.1% solution in deionized water | |
| Elution | Isocratic |
| Flow rate | 0.30 mL/min |
| Injection volume | 5 μL |
MS Settings
| MS type | Sciex API4000 |
| Ionization type | TurboIonSpray |
| Ionisation polarity | Positive |
| Midazolam mass ratio | 326.1/291.0 amu |
| Midazolam standard mass ratio | 330.2/295.0 amu |
| Hydroxymidazolam mass ratio | 342.1/323.9 amu |
| Hydroxymidazolam standard mass ratio | 346.1/328.0 amu |
Variability
| Concentration | Mean | Precision | Accuracy (Relative Error) | |
|---|---|---|---|---|
| Midazolam | (ng/mL) | (ng/mL) | (%CV) | (%) |
| QC1 | 0.300 | 0.29 | 9.50 | 97.77 |
| QC2 | 30.00 | 29.78 | 4.86 | 99.26 |
| QC3 | 80.00 | 75.78 | 5.06 | 94.72 |
| α‐Hydroxymidazolam | Concentration (ng/mL) | Mean (ng/mL) | Precision (%CV) | Accuracy (Relative Error) (%) |
|---|---|---|---|---|
| QC1 | 0.300 | 0.29 | 9.03 | 97.04 |
| QC2 | 30.00 | 31.57 | 3.97 | 105.25 |
| QC3 | 80.00 | 80.29 | 4.63 | 100.37 |
CV, coefficient of variation.
Cyclosporine
Internal Standard
Cyclosporine‐D12 (0.504 mg/mL cyclosporine‐D12 in methanol) was diluted with 25% ethanol:25% ethanediol:50% deionized water to a final concentration of 250 ng/mL.
Calibrators
Cyclosporine (987.97 μg/mL in methanol) was diluted with whole blood to yield final concentrations of 24.7 to 1976.0 ng/mL.
Analytical Method
Samples were prepared by protein precipitation and liquid‐liquid extraction.
The following were added to a polypropylene tube:
50 μL plasma sample, calibrator or quality control
50 μL internal standard
500 μL 5% zinc sulphate
500 μL acetone
After centrifugation the supernatant was added to 0.1 mL of 0.1‐M sodium hydroxide and 2 mL of MTBE, then mixed and centrifuged. The resulting solvent layer was evaporated to dryness in a vacuum. The residue was reconstituted in 250 μL of 80% methanol, centrifuged, and the resulting supernatant analyzed by LC‐MS/MS.
HPLC Conditions
| Column | Alltima C18 (150 mm × 2.1 mm, 5 μm) |
| Mobile phase | Ammonium acetate 0.01‐M solution in 85% methanol |
| Elution | Isocratic |
| Flow rate | 0.40 mL/min |
| Injection volume | 5–30 μL |
MS Settings
| MS type | MRM |
| Ionisation type | TurboIonSpray |
| Ionisation polarity | Positive |
| Cyclosporine mass ratio | 1220.0/1202.8 amu |
| Cyclosporine standard mass ratio | 1232.1/1215.0 amu |
Variability
| Concentration | Mean | Precision | Accuracy (Relative Error) | |
|---|---|---|---|---|
| Cyclosporine | (μg/L) | (μg/L) | (%CV) | (%) |
| QC1 | 1482 | 1344.57 | 3.66 | 90.73 |
| QC2 | 741 | 698.27 | 3.63 | 94.23 |
| QC3 | 74.1 | 73.28 | 3.50 | 98.90 |
CV, coefficient of variation.
Ritonavir
Internal Standard
Ritonavir‐d6 (0.05 mg/mL in methanol) was diluted with methanol/deionized water (50/50 v/v) to a final concentration of 10 μg/mL.
Calibrators
Ritonavir (100.2 μg/mL in methanol) was diluted with plasma to yield final concentrations of 0.5 to 25.0 μg/mL.
Analytical Method
Samples were extracted by protein precipitation.
The following were added to a polypropylene tube:
50 μL plasma sample, calibrator or quality control
50 μL internal standard
1 mL acetonitrile
After centrifugation, the supernatant was mixed with 5 mL of deionized water and analysed by LC‐MS/MS.
HPLC Conditions
| Column | Alltima C18 (150 mm × 2.1 mm; 5 μm) |
| Mobile phase | Deionized water supplemented with 2 mM of ammonium acetate and 0.1% formic acid, about 15% |
| Methanol, deionized water, 2 mM of ammonium acetate and 0.1% formic acid, about 85% | |
| Elution | Isocratic |
| Flow rate | 0.40 mL/min |
| Injection volume | 5 μL |
MS Settings
| MS type | MRM |
| Ionisation type | TurboIonSpray |
| Ionisation polarity | Positive |
| Ritonavir mass ratio | 721.2/296.1 amu |
| Ritonavir standard mass ratio | 727.1/302.0 amu |
Variability
| Ritonavir | Concentration (μg/L) | Mean (μg/L) | Precision (%CV) | Accuracy (Relative Error)(%) |
|---|---|---|---|---|
| QC1 | 1.5 | 1.53 | 8.26 | 101.96 |
| QC2 | 7.5 | 7.32 | 5.02 | 97.55 |
| QC3 | 20.0 | 21.07 | 4.09 | 105.34 |
CV, coefficient of variation.
The lower limit of reliable quantification was 0.1 ng/mL for midazolam and 1‐hydroxymidazolam, 25 ng/mL for cyclosporine, and 500 ng/mL for ritonavir.
Pharmacokinetic and Statistical Analysis
Pharmacokinetic parameters were calculated using WinNonlin version 6.3. Lower limit of reliable quantification values were taken as zero, unless they fell between 2 quantifiable concentrations, in which case they were treated as missing. Interactions were tested using an equivalence analysis as follows: area under the plasma drug concentration‐time curve from time zero to infinity (AUC0‐∞) and maximum concentration of drug in blood plasma (Cmax) of the substrate drug were logarithmically transformed and subjected to analysis of variance with treatment dose, group, sequence, session, and dose by treatment interaction as fixed effects, and subject as a random effect. The least square mean of the treatment difference and its 90%CI were transformed to the original scale, to obtain mean AUC0‐∞ and Cmax ratios. Absence of a clinically significant drug‐drug interaction was concluded if the 90%CI for both AUC0‐∞ and Cmax ratios fell within the prespecified interval of 80% to 125%.
Results
Midazolam Study
Plasma concentrations and AUC0‐∞ of midazolam were significantly reduced when midazolam was administered with the last of 10 daily doses of 400‐mg amenamevir, compared with midazolam given before treatment with amenamevir (see Figure 3 and Table 2). Elimination half‐life of midazolam was also shortened by amenamevir from 4.2 to 3.3 hours.
Figure 3.
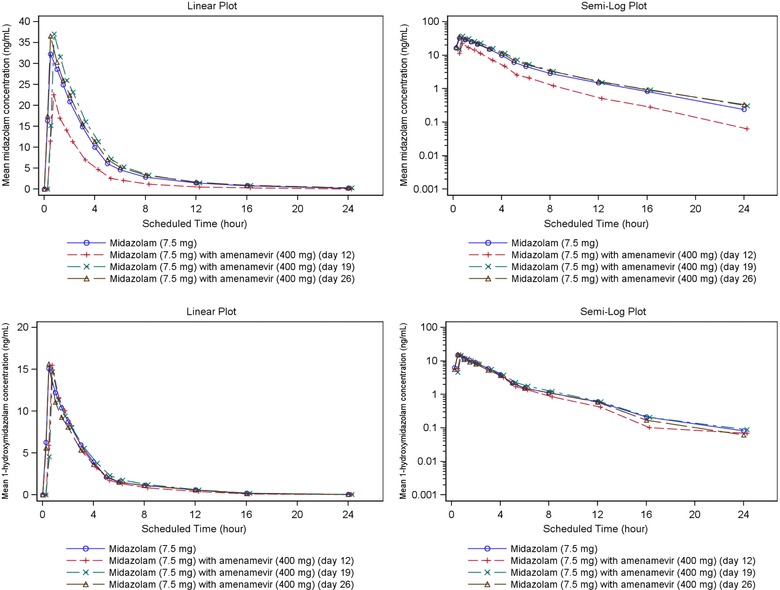
Mean midazolam and 1‐hydroxymidazolam plasma concentration‐time plots (linear and semilog) with and without amenamevir (n = 18).
Table 2.
Summary of Midazolam Pharmacokinetic Parameters (n = 18)
| Day 1 Midazolam 7.5 mg Alone (Control) | Day 12 Midazolam 7.5 mg With Amenamevir 400 mg | Day 19 Midazolam 7.5 mg Alone | Day 26 Midazolam 7.5 mg Alone | ||
|---|---|---|---|---|---|
| Cmax (ng/mL) | Arithmetic mean | 35.2 | 24.5 | 39.7 | 39.5 |
| SD | 8.1 | 7.7 | 8.3 | 15.5 | |
| Geometric mean | 34.5 | 23.4 | 38.8 | 36.8 | |
| %CVb | 21.2 | 32.5 | 23.1 | 41.4 | |
| LS mean | 34 | 23 | 39 | 37 | |
| LS mean ratio vs control (90%CI) (%) | N/A | 68 (59, 78) | 113 (96, 132)a | 107 (91, 125)a | |
| AUC0–tn (ng ⋅ h/mL) | Arithmetic mean | 115.3 | 59 | 127.3 | 126.2 |
| SD | 35.4 | 17.9 | 39.5 | 45.8 | |
| Geometric mean | 111 | 56.7 | 121.9 | 118.9 | |
| %CVb | 28 | 28.8 | 30.7 | 36.2 | |
| AUC0–∞ (ng ⋅ h/mL) | Arithmetic mean | 117.5 | 60.3 | 130.3 | 130 |
| SD | 36.2 | 18.6 | 42.6 | 49.2 | |
| Geometric | |||||
| mean | 113.1 | 58 | 124.4 | 122.1 | |
| %CVb | 28.1 | 29.2 | 31.7 | 37.3 | |
| LS mean | 113 | 58 | 124 | 122 | |
| LS mean ratio vs control (90%CI) (%) | N/A | 51 (47, 56) | 110 | 108 | |
| Tmax (h) | Median | 0.5 | 0.5 | 0.5 | 0.5 |
| Range | 0.50‐1.52 | 0.25‐1.50 | 0.50‐1.50 | 0.28‐2.00 | |
| t1/2 (h) | Arithmetic mean | 4.5 | 3.7 | 4.6 | 4.6 |
| SD | 1.5 | 1.8 | 1.8 | 2 | |
| Geometric | |||||
| mean | 4.2 | 3.3 | 4.3 | 4.1 | |
| %CVb | 37 | 52.7 | 43.7 | 52.3 | |
| CL/F (L/h) | Arithmetic mean | 68.5 | 134.3 | 63 | 65.2 |
| SD | 16.9 | 36.4 | 18.9 | 22.3 |
AUC 0–∞, area under concentration‐time curve extrapolated to infinite time; AUC 0–tn, area under concentration–time curve up to last nonzero value; CI, confidence interval; CL/F, apparent total body clearance from plasma; Cmax, peak concentration; %CVb, between‐subject coefficient of variance; LS, least squares; SD, standard deviation; t1/2, half‐life; Tmax, time of peak concentration.
95%CI.
Exposure to midazolam 7 and 14 days after the last dose of amenamevir was similar to that before amenamevir dosing (see Table 2).
After midazolam with amenamevir, geometric mean Cmax and AUC0‐∞ of midazolam were about 68% and 51%, respectively, of that after midazolam alone (see Table 2). Results failed to exclude a significant drug‐drug effect (the 90%CI of the log ratio midazolam with amenamevir:midazolam did not fall within the range 80%‐125% for Cmax or AUC0‐∞).
Plasma concentrations of 1‐hydroxymidazolam were not influenced by coadministration of amenamevir (see Figure 3 and Table S1): They remained about half those of unchanged midazolam.
Somnolence was the most common adverse event and was reported by every subject after 1 or more of their 4 doses of midazolam. No subject reported somnolence while receiving amenamevir alone.
Cyclosporine Study
Plasma concentrations (1‐12 hours after dose) and AUC0‐∞ of amenamevir were reduced after coadministration with cyclosporine compared with amenamevir alone (see Figure 4 and Table 3).
Figure 4.
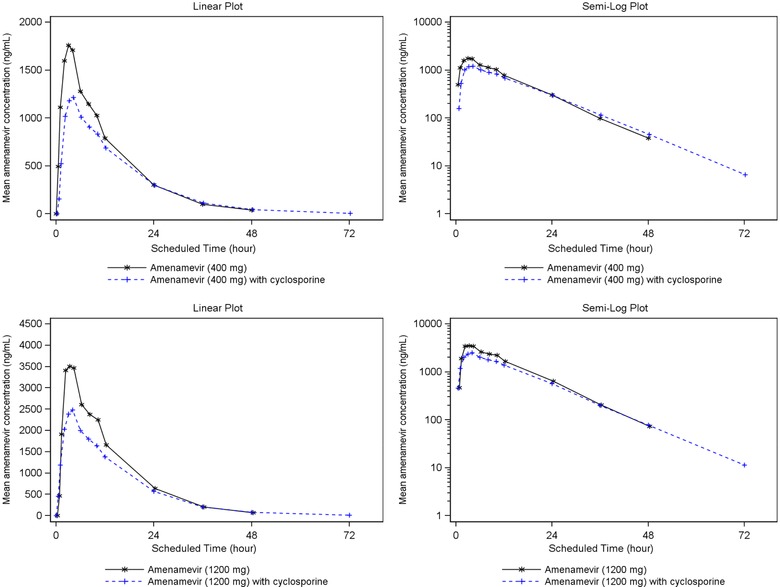
Mean amenamevir plasma concentration‐time plots (linear and semilog) with and without cyclosporine (n = 24).
Table 3.
Summary of Amenamevir Pharmacokinetic Parameters From Cyclosporine Study (n = 24)
| Amenamevir 400 mg | Amenamevir 1200 mg | ||||
|---|---|---|---|---|---|
| Amenamevir Alone (Control) | Amenamevir With Cyclosporine | Amenamevir Alone (Control) | Amenamevir With Cyclosporine | ||
| Cmax (ng/mL) | Arithmetic mean | 1875.3 | 1260.6 | 3681.8 | 2563.0 |
| SD | 475.6 | 418.3 | 886.5 | 782.0 | |
| Geometric mean | 1818.0 | 1202.3 | 3573.1 | 2452.1 | |
| %CVb | 26.1 | 31.5 | 26.1 | 31.4 | |
| LS mean | 2568 | 1730 | 2568 | 1730 | |
| LS mean ratio vs control (90%CI) (%) | N/A | 67 (63, 72) | N/A | 67 (63, 72) | |
| AUC0‐tn (ng ⋅ h/mL) | Arithmetic mean | 23 670.0 | 19 755.6 | 48 655.9 | 38 867.0 |
| SD | 6221.1 | 7094.7 | 12 353.3 | 9143.8 | |
| Geometric mean | 22 903.1 | 18 784.4 | 47 282.0 | 37 734.2 | |
| %CVb | 26.7 | 31.8 | 24.6 | 26.1 | |
| AUC0‐∞ (ng ⋅ h/mL) | Arithmetic mean | 24 111.7 | 19 973.7 | 49 496.4 | 39 066.6 |
| SD | 6430.1 | 7116.8 | 12876.8 | 9176.1 | |
| Geometric mean | 23 307.4 | 19 013.5 | 48 051.2 | 37 940.8 | |
| %CVb | 27.1 | 31.4 | 25.0 | 25.9 | |
| LS mean | 33 764 | 27 098 | 33 764 | 27 098 | |
| LS mean ratio vs control (90%CI) (%) | N/A | 80 (75, 86) | N/A | 80 (75, 86) | |
| Tmax (h) | Median | 3.00 | 4.00 | 3.00 | 4.00 |
| Range | 0.50‐4.02 | 1.00‐8.00 | 2.00‐4.00 | 2.00‐8.00 | |
| t1/2 (h) | Arithmetic mean | 7.8 | 8.7 | 7.5 | 8.4 |
| SD | 0.9 | 1.4 | 1.0 | 1.1 | |
| Geometric mean | 7.8 | 8.6 | 7.5 | 8.3 | |
| %CVb | 12.1 | 16.4 | 13.2 | 14.1 | |
| CL/F (L/h) | Arithmetic mean | 17.7 | 21.9 | 25.7 | 32.7 |
| SD | 4.6 | 5.9 | 6.3 | 9.3 | |
AUC 0–∞, area under concentration–time curve extrapolated to infinite time; AUC 0–tn, area under concentration‐time curve up to last nonzero value; CI, confidence interval; CL/F, apparent total body clearance from plasma; Cmax, peak concentration; %CVb, between‐subject coefficient of variance; LS, least squares; SD, standard deviation; t1/2, half‐life; Tmax, time of peak concentration.
After 400 mg of amenamevir with cyclosporine, geometric mean (90%CI) Cmax and AUC0‐∞ of amenamevir were about 66(59, 74)% and 82(73, 91)%, respectively, of those after amenamevir alone. After 1200 mg of amenamevir with cyclosporine, geometric mean Cmax and AUC0‐∞ were about 69(63, 74)% and 79(73, 85)%, respectively, of those after amenamevir alone. To assess the effect of cyclosporine on amenamevir, an equivalence analysis was done. Because the dose by treatment interaction was not significant at the 20% level, it was not included in the model. The geometric mean ratio amenamevir with cyclosporine:amenamevir for Cmax of amenamevir was 67%, ie, below the prespecified interval 80% to 125%, and the corresponding figure for AUC0‐∞ was 80%, ie, just within the prespecified interval 80% to 125%. For both Cmax and AUC0‐∞ the 90%CIs excluded unity. Thus, the difference between the doses of amenamevir with and without cyclosporine with respect to pharmacokinetic parameters was modest in the case of Cmax and minor in the case of AUC0‐∞, even though the differences achieved a conventional level of statistical significance.
To assess the effect of cyclosporine on the pharmacokinetics of the metabolite AS1955888‐00, a further equivalence analysis was done. Because the dose by treatment interaction was not significant at the 20% level, it was not included in the model; the resulting estimates presented in Table 4 are for both 400‐mg and 1200‐mg amenamevir. After amenamevir with cyclosporine, Cmax and AUC0‐∞ of AS1955888‐00 were about 78% and 92%, respectively, of those after amenamevir alone. The 90%CI of the log ratio amenamevir alone vs amenamevir with cyclosporine, did not fall within the prespecified interval 80% to 125% for Cmax, but did for AUC0‐∞. Therefore, a significant drug‐drug effect on plasma concentrations of AS1955888‐00 could not be ruled out, as both ratios fell outside the prespecified interval. However, given that the AUC0‐∞ ratio was close to 100%, any interaction is likely to be small and of no consequence.
Table 4.
Least Squares Means and Ratios of AUC0‐∞ and Cmax for AS1955888‐00 Before Cyclosporine Treatment and on the Fifth Day of Dosing With Cyclosporine 100 mg Twice Daily (n = 24)
| LS Means | Amenamevir With Cyclosporine vs Amenamevir Alone | |||
|---|---|---|---|---|
| Amenamevir Alone | Amenamevir With Cyclosporine | Ratio (%) | 90%CI | |
| Cmax (ng/mL) | 256 | 200 | 78 | 72, 84 |
| AUC0‐∞ (ng ⋅ h/mL) | 3987 | 3672 | 92 | 86, 99 |
AUC0‐∞, area under concentration‐time curve extrapolated to infinite time; CI, confidence interval; Cmax, peak concentration; LS, least squares.
Cyclosporine plasma concentrations were not affected by coadministration with amenamevir (see Table 5).
Table 5.
Least Squares Means and Ratios of AUC0‐t and Cmax for Cyclosporine Before (Day 6) and After a Single Dose of Amenamevir (n = 24)
| LS Means | ||||
|---|---|---|---|---|
| Cyclosporine (Day 6) | Amenamevir With Cyclosporine (Day 7) | Ratio (%) Day 7/Day 6 | 90%CI | |
| Cmax (μg/mL) | 480 | 462 | 96 | 92, 101 |
| AUC0‐τ (ng ⋅ h/mL) | 1931 | 1960 | 102 | 99, 105 |
AUC0–τ, area under concentration‐time curve over the dosing interval; CI, confidence interval; Cmax, peak concentration; LS, least squares.
The most frequently reported treatment‐emergent adverse events were headache (12.5% in treatment sessions 1 and 2) and oropharyngeal pain (8.3% in treatment session 1, 16.7% in treatment session 2).
Ritonavir Study
Plasma concentrations and AUC0‐∞ of amenamevir were significantly increased after coadministration with 600 mg of ritonavir compared with administration of amenamevir alone (see Figure 5 and Table 6).
Figure 5.
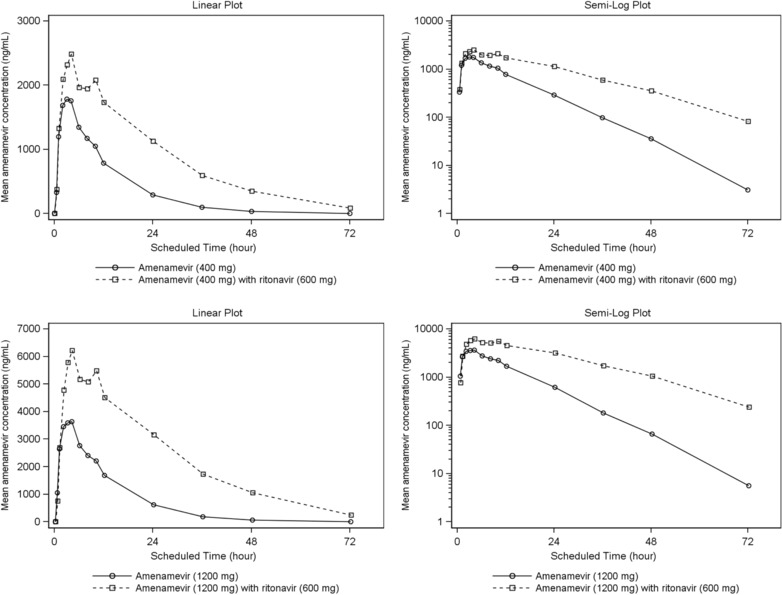
Mean amenamevir plasma concentration‐time plots (linear and semilog) with and without ritonavir (n = 24).
Table 6.
Summary of Amenamevir Pharmacokinetic Parameters From Ritonavir Study (n = 24)
| Amenamevir 400 mg | Amenamevir 1200 mg | ||||
|---|---|---|---|---|---|
| Amenamevir Alone (N = 24) | Amenamevir With Ritonavir 600 mg (N = 24) | Amenamevir Alone (N = 24) | Amenamevir With Ritonavir 600 mg (N = 24) | ||
| Cmax (ng/mL) | Arithmetic mean | 1896.7 | 2568.3 | 3948.2 | 6457.4 |
| SD | 418.5 | 459.8 | 1096.5 | 1855.0 | |
| Geometric mean | 1845.7 | 2518.7 | 3804.0 | 6211.6 | |
| %CVb | 25.4 | 21.9 | 28.8 | 29.2 | |
| AUC0‐tn (ng ⋅ h/mL) | Arithmetic mean | 24 113.8 | 59 926.3 | 50 141.0 | 161 853.7 |
| SD | 7029.9 | 12 482.1 | 13 944.6 | 38 163.6 | |
| Geometric mean | 22 987.3 | 58 616.1 | 48 245.8 | 157 352.7 | |
| % CVb | 34.2 | 22.2 | 29.3 | 25.1 | |
| AUC0‐∞ (ng ⋅ h/mL) | Arithmetic mean | 24 296.0 | 61 678.1 | 50 440.3 | 166 855.3 |
| SD | 7072.3 | 13 384.7 | 14 021.9 | 39 814.1 | |
| Geometric mean | 23 162.4 | 60 225.3 | 48 532.3 | 162 131.6 | |
| %CVb | 34.2 | 23.0 | 29.3 | 25.3 | |
| Tmax (h) | Median | 3 | 4 | 3 | 4 |
| Range | 1.00‐4.00 | 1.00‐10.00 | 1.00‐4 | 2.00‐12.00 | |
| t1/2 (h) | Arithmetic mean | 7.5 | 12.7 | 7.3 | 12.7 |
| SD | 1.2 | 2.2 | 1.4 | 2.5 | |
| Geometric mean | 7.4 | 12.6 | 7.2 | 12.5 | |
| %CVb | 14.9 | 15.9 | 17.5 | 19.6 | |
| CL/F (L/h) | Arithmetic mean | 18.3 | 6.8 | 25.7 | 7.6 |
| SD | 7.3 | 1.7 | 7.6 | 2.0 | |
AUC0–∞, area under concentration‐time curve extrapolated to infinite time; AUC0‐tn, area under concentration‐time curve up to last nonzero value; CL/F, apparent total body clearance from plasma; Cmax, peak concentration; %CVb: between‐subject coefficient of variance; SD, standard deviation; t1/2, half‐life; tmax, time of peak concentration.
After amenamevir 400 mg with ritonavir, geometric mean Cmax and AUC0‐∞ of amenamevir were, respectively, about 1.4 and 2.6 times higher than after amenamevir 400 mg alone. After amenamevir 1200 mg with ritonavir, geometric mean Cmax and AUC0‐∞ of amenamevir were, respectively, about 1.6 and 3.3 times higher than after amenamevir 1200 mg alone (see Table 7). After 400 mg and 1200 mg of amenamevir, the 90%CI of the log ratio amenamevir with ritonavir:amenamevir did not fall within the range 80% to 125% for either Cmax or AUC0‐∞, and therefore failed to exclude a significant drug‐drug effect of ritonavir on those parameters.
Table 7.
Least Squares Means and Ratios of AUC0‐∞ and Cmax for 400‐ and 1200‐mg Amenamevir With and Without Coadministration of 600‐mg Ritonavir (n = 24)
| LS Means | Amenamevir With Ritonavir vs Amenamevir Alone | ||||
|---|---|---|---|---|---|
| Amenamevir Dose | Amenamevir Alone | Amenamevir With Ritonavir | Ratio (%) | 90%CI | |
| 400 mg | Cmax (ng/mL) | 1846 | 2519 | 136 | 124, 151 |
| AUC0‐∞ (ng ⋅ h/mL) | 23 162 | 60 225 | 260 | 234, 289 | |
| 1200 mg | Cmax (ng/mL) | 3804 | 6212 | 163 | 146, 182 |
| AUC0‐∞ (ng ⋅ h/mL) | 48 532 | 162 132 | 334 | 305, 366 | |
AUC0–∞, area under concentration‐time curve extrapolated to infinite time; CI: confidence interval; Cmax, peak concentration; LS, least squares.
Ritonavir increased the geometric mean elimination half‐life of amenamevir from about 7 hours to about 12 to 13 hours, irrespective of the dose of amenamevir.
After dosing of amenamevir with ritonavir, plasma concentrations of AS1955888‐00 were much lower than after amenamevir monotherapy. Geometric mean Cmax and AUC0‐∞ after amenamevir and ritonavir coadministration were about 11% and 28%, respectively, of those after 400‐mg amenamevir monotherapy, and about 13% and 26%, respectively, of those after 1200‐mg amenamevir monotherapy (see Table 8).
Table 8.
Least Squares Means and Ratios of AUC0‐∞ and Cmax for AS1955888‐00 With and Without Coadministration of 600‐mg Ritonavir (n = 24)
| LS Mean | Amenamevir With Ritonavir vs Amenamevir Alone | ||||
|---|---|---|---|---|---|
| Amenamevir Dose | AS1955888‐00 Parameter | Amenamevir Alone | Amenamevir With Ritonavir | Ratio (%) | 90%CI |
| 400 mg | Cmax (ng/mL) | 190 | 22 | 11 | 9, 14 |
| AUC0‐∞ (ng ⋅ h/mL) | 2636 | 739 | 28 | 24, 33 | |
| 1200 mg | Cmax (ng/mL) | 421 | 56 | 13 | 10, 17 |
| AUC0‐∞ (ng ⋅ h/mL) | 6023 | 1587 | 26 | 22, 32 | |
AUC0–∞, area under concentration–time curve extrapolated to infinite time; CI: confidence interval; Cmax, peak concentration; LS, least squares.
Plasma concentrations and AUC0‐tn of ritonavir were similar when ritonavir was given with either 400 mg or 1200 mg of amenamevir (see Figure 6 and Table 9).
Figure 6.
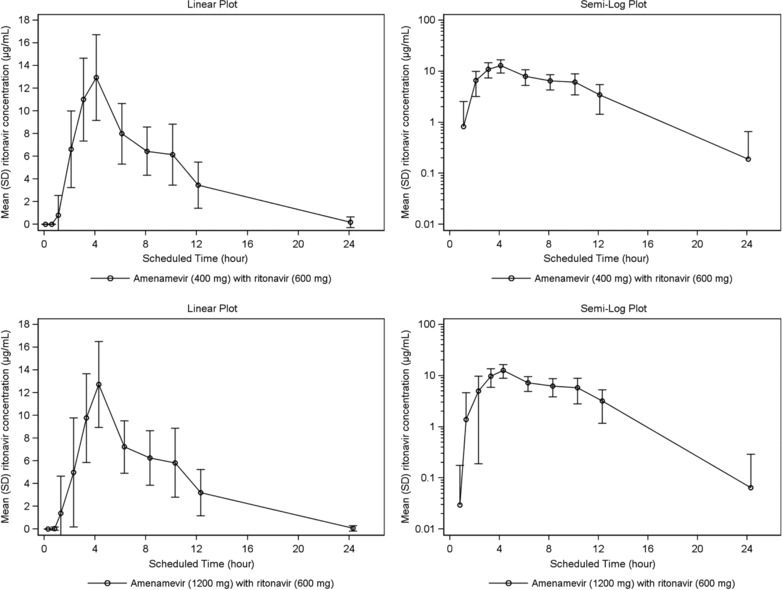
Mean (SD) ritonavir plasma concentration versus time (linear and semilog); (n = 24*) *at the 24‐hour time point, n = 4 for 400 mg; and n = 2 for 1200‐mg amenamevir.
Table 9.
Summary of Ritonavir Pharmacokinetic Parameters (n = 24)
| Amenamevir 400 mg and Ritonavir 600 mg (N = 24) | Amenamevir 1200 mg and Ritonavir 600 mg (N = 24) | ||
|---|---|---|---|
| Cmax (μg/mL) | Arithmetic mean | 13.6 | 12.9 |
| SD | 3.7 | 3.7 | |
| Geometric mean | 13.2 | 12.4 | |
| %CVb | 26.6 | 29.8 | |
| AUC0‐tn (μg ⋅ h/mL) | n | 24 | 24 |
| Arithmetic mean | 87.3 | 78.1 | |
| SD | 31.4 | 29.9 | |
| Geometric mean | 82.5 | 72.6 | |
| %CVb | 34.9 | 41.3 | |
| AUC0‐∞ (μg ⋅ h /mL) | n | 14 | 13 |
| Arithmetic mean | 98.1 | 77.7 | |
| SD | 37.5 | 32.2 | |
| Geometric mean | 92.1 | 71.8 | |
| %CVb | 37.7 | 44.0 | |
| Tmax (h) | n | 24 | 24 |
| Median | 4.0 | 4.0 | |
| Range | 3.00‐6.00 | 2.00‐4.22 | |
| t1/2 (h) | n | 24 | 24 |
| Arithmetic mean | 5.0 | 4.5 | |
| SD | 2.9 | 2.1 | |
| Geometric mean | 4.5 | 4.1 | |
| %CVb | 47.7 | 47.9 |
AUC0–∞, area under concentration‐time curve extrapolated to infinite time; AUC0‐tn, area under concentration‐time curve up to last nonzero value; Cmax, peak concentration; %CVb, between‐subject coefficient of variance; t1/2, half‐life; tmax, time of peak concentration.
The most frequently reported treatment‐emergent adverse event was headache (16.7% with 400‐mg amenamevir, 29.2% with 1200‐mg amenamevir).
Discussion
Midazolam
Treatment with amenamevir 400 mg once daily for 10 days reduced Cmax of midazolam by about 32%, and average concentrations (AUC) by about 51%. Mean elimination half‐life was also reduced from 4.2 to 3.3 hours. The reduction in Cmax suggests that amenamevir increased first‐pass intestinal and hepatic extraction of midazolam to a modest degree. The effect on AUC was greater, probably because amenamevir treatment increased the clearance and reduced the half‐life of midazolam, thereby reducing plasma concentrations of midazolam beyond the 32% reduction that would be predicted from the effect on Cmax alone. The effect of amenamevir was minor compared with that of a powerful enzyme inducer such as rifampicin: A study of similar design to ours, but using rifampicin as an inducer instead of amenamevir, showed that rifampicin reduced midazolam Cmax 27‐fold and AUC 64‐fold, and shortened mean elimination half‐life from 3.2 to 1.8 hours.13
Our results show that 10 days’ treatment with amenamevir 400 mg once daily induced the activity of CYP3A in the intestine and liver, but the effect was only moderate and was no longer evident at 7 and 14 days after the end of amenamevir treatment. The reversal of amenamevir's enzyme‐inducing effect was much more rapid than has been reported after rifampicin, whose effect on the pharmacokinetics of midazolam may take 3 weeks to reverse,13 presumably because the inducing effect of rifampicin is so much greater than that of amenamevir.
Plasma concentrations of 1‐hydroxymidazolam, the main metabolite of midazolam, were not affected by coadministration of amenamevir with midazolam. Concentrations of 1‐hydroxymidazolam might be expected to increase because of induction of the metabolism of midazolam by amenamevir, but that expectation is incorrect: 1‐hydroxymidazolam is cleared by glucuronidation, and hepatic enzyme induction increases the activity of glucuronyltransferases. It has been shown convincingly that hepatic enzyme induction by rifampicin leads to a reduction in plasma concentrations of 1‐hydroxymidazolam that is greater than the corresponding reduction in parent midazolam.13
Cyclosporine
After single doses of amenamevir 400 mg or 1200 mg given on day 5 of 7 days’ treatment with cyclosporine 100 mg twice daily, plasma concentrations of amenamevir were lower than when the same doses of amenamevir had been given before the start of cyclosporine treatment. The interval between doses of amenamevir was 6 days. Results were similar after both the 400‐mg and 1200‐mg dose levels of amenamevir: Taking the average result for the 2 dose levels, Cmax and AUC of amenamevir given with cyclosporine were about 67% and 80%, respectively, of values obtained after the dose of amenamevir given 6 days previously. We were surprised by that result: cyclosporine is an inhibitor of CYP3A7 and therefore might have been expected to reduce first‐pass metabolism and clearance of amenamevir, leading to increased plasma concentrations. However, amenamevir is not only a substrate of CYP3A but also an inducer; see the results of our study of the effect of amenamevir on the pharmacokinetics of midazolam above. Although it is theoretically possible that our paradoxical result could have been due to amenamevir inducing its own metabolism, that seems very unlikely because the results of our midazolam study show that any induction of CYP3A by repeated doses of amenamevir was fully reversed by 7 days after the last dose (see Table 2).
Cyclosporine is an inhibitor of P‐gp5: Because amenamevir is a P‐gp substrate, we would again expect inhibition of P‐gp by cyclosporine to increase amenamevir plasma concentrations rather than decrease them, as occurred in this study. Our findings with respect to unchanged amenamevir are consistent with the report of Hesselink et al14 on the effects of cyclosporine on the pharmacokinetics of mycophenolate mofetil: Coadministration of mycophenolate mofetil with cyclosporine to solid‐organ transplant recipients reduced mycophenolic acid concentrations and increased exposure to its metabolite, mycophenolic acid–glucuronide (MPAG), possibly by interfering with the biliary excretion of MPAG.14 The authors of that report suggested a role for multidrug resistance‐associated protein‐2 in that interaction, as it was not observed in multidrug resistance‐associated protein‐2–deficient rats.
In contrast with the results of Hesselink et al14 with respect to the MPAG metabolite of mycophenolate mofetil, we found that plasma concentrations of amenamevir's main metabolite, AS1955888‐00, were lower after the second dose of amenamevir (coadministered with cyclosporine) than after the first dose of amenamevir, which was given alone. Although plasma concentrations of AS1955888‐00 might have been expected to be higher, because of increased hepatic metabolism of amenamevir after the second dose, experience with the metabolites of midazolam13 (see above) shows that the reverse may be the case in practice.
An important limitation of the design of our study is that it did not include a control arm in which single doses of amenamevir were given on days 1 and 7 in the absence of any treatment with cyclosporine. As a result of that limitation, we cannot determine whether cyclosporine treatment had any effect on the plasma concentrations of amenamevir. A further study would be needed to elucidate the matter fully. For the present, however, we can safely conclude that cyclosporine treatment does not increase plasma concentrations of amenamevir.
Two single doses of amenamevir 400 mg and 1200 mg, given 7 days apart, had no effect on the pharmacokinetics of cyclosporine. Because amenamevir is both a substrate and an inducer of CYP3A, it has the potential to produce opposing effects on cyclosporine concentrations that might cancel each other out.
Ritonavir
A single 600‐mg dose of ritonavir substantially increased plasma concentrations of amenamevir after coadministration with either 400‐mg or 1200‐mg doses. The increase in Cmax was about 1.5‐fold, which is consistent with a modest decrease in first‐pass extraction of amenamevir. In contrast, AUC increased >3‐fold, reflecting the dual contributions of decreased first‐pass extraction and reduced clearance. These findings are similar to those of Kusawake et al,15 who coadministered amenamevir with ketoconazole, another CYP3A inhibitor. In that study, the geometric mean ratio of amenamevir plus ketoconazole vs amenamevir alone for Cmax and AUCinf was 1.30 (90%CI, 1.17‐1.45) and 2.58 (90%CI, 2.32‐2.87), respectively.15 Elimination half‐life of amenamevir after both 400‐mg and 1200‐mg doses was increased from about 7 hours to about 12 to 13 hours by a single dose of ritonavir 600 mg. Plasma concentrations of the metabolite AS1955888‐00 were much lower when amenamevir was given with ritonavir compared with amenamevir alone, reflecting decreased hepatic metabolism of amenamevir.
Amenamevir is a substrate of P‐gp in vitro, and ritonavir inhibits P‐gp as well as CYP3A.16 Inhibition of P‐gp by ritonavir in the intestinal epithelium could have contributed to the observed increases in Cmax and AUC of amenamevir, and could also have contributed to the prolongation of elimination half‐life by reducing transport into bile.
Single doses of amenamevir 400 mg and 1200 mg did not affect the pharmacokinetics of ritonavir.
Conclusions
As predicted from in vitro metabolic studies of amenamevir, our 3 clinical studies confirm the existence of interactions between amenamevir and midazolam (a probe substrate for CYP3A), cyclosporine (a substrate and inhibitor of CYP3A), and ritonavir (an inhibitor of CYP3A). In our healthy volunteers, amenamevir induced the metabolism and enhanced the clearance of midazolam. Cyclosporine decreased Cmax and AUC of amenamevir; further studies would be needed to identify the mechanism of that interaction. Ritonavir, even when given as a single dose, substantially inhibited the clearance of amenamevir.
The CYP3A‐inducing effect of amenamevir caused a 52% reduction in plasma concentrations of the probe drug midazolam, and might be expected to have a similar effect on the plasma concentrations of other drugs that are substrates of CYP3A. Halving plasma concentrations might have important consequences in the case of drugs with a narrow therapeutic window but would probably be of no consequence for others. The prescriber would have to make an informed decision in each case. However, if a prescriber were to double the dose of a drug to maintain plasma concentrations during amenamevir treatment, it would be important to reduce the dose of that drug within a few days of the end of amenamevir treatment, as the enzyme‐inducing effect clearly wanes rapidly.
Although our study has not fully defined the potential interactions between cyclosporine and amenamevir, the results are reassuring in that they show that cyclosporine does not increase plasma concentrations of amenamevir, and any reduction by cyclosporine is unlikely to be greater than that attributable to autoinduction of metabolism of amenamevir. Single doses of amenamevir up to 1200 mg have no effect on the pharmacokinetics of cyclosporine.
As demonstrated by the ritonavir study, concentrations of amenamevir are likely to be substantially increased by coadministration of inhibitors of CYP3A, such as erythromycin, itraconazole, or ritonavir.
Amenamevir clearly has the potential to be involved in CYP3A‐mediated pharmacokinetic interactions in clinical practice. Prescribers should therefore consider the need for dose adjustment when amenamevir is coadministered with other CYP3A substrates, inhibitors, or inducers.
Declaration of Conflicting Interests
The authors are employees of either Maruho or the clinical unit. The authors declare that they have no conflicts of interest to disclose.
Supporting information
Table S1: Summary of Pharmacokinetic Parameters for 1‐Hydroxymidazolam (n = 18)
Funding
The studies were sponsored by Maruho.
References
- 1. Chono K, Katsumata K, Kontani T, et al. ASP2151, a novel helicase–primase inhibitor, possesses antiviral activity against varicella–zoster virus and herpes simplex virus types 1 and 2. J Antimicrob Chemother. 2010;65:1733–1741. [DOI] [PubMed] [Google Scholar]
- 2. Perry CM, Faulds D. Valaciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections. Drugs. 1996;52:754‐772. [DOI] [PubMed] [Google Scholar]
- 3. Levin MJ, Bacon TH, Leary JJ. Resistance of herpes simplex virus infections to nucleoside analogues in HIV‐infected patients. Clin Infect Dis. 2004;39(suppl 5):S248‐S257. [DOI] [PubMed] [Google Scholar]
- 4. Topletz AR, Dennison JB, Barbuch RJ, et al. The relative contributions of CYP3A4 and CYP3A5 to the metabolism of vinorelbine. Drug Metab Dispos. 2013;41:1651‐1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. 2012:46‐47.
- 6. Whai T, Easaw Thomas, Choo S. Premedication with midazolam is more effective by the sublingual than oral route. Can J Anaesth. 1997;44:723‐726. [DOI] [PubMed] [Google Scholar]
- 7. Amundsen R, Asberg A, Ohm IK, Christensen H. Cyclosporine A‐ and tacrolimus‐mediated inhibition of CYP3A4 and CYP3A5 in vitro. Drug Metab Dispos. 2012;40:655‐661. [DOI] [PubMed] [Google Scholar]
- 8. Tilney NL, Chang A, Milford EL, et al. Ten‐year experience with cyclosporine as primary immunosuppression in recipients of renal allografts. Ann Surg. 1991;214:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Greenblatt DJ, Harmatz JS. Ritonavir is the best alternative to ketoconazole as an index inhibitor of cytochrome P450‐3A in drug‐drug interaction studies. Br J Clin Pharmacol. 2015;80:342‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hill A, van der Lugt J, Sawyer W, et al. How much ritonavir is needed to boost protease inhibitors? Systematic review of 17 dose‐ranging pharmacokinetic trials. AIDS. 2009;23:2237‐2245. [DOI] [PubMed] [Google Scholar]
- 11. Park J, Vousden M, Brittain C, et al. Dose‐related reduction in bupropion plasma concentration by ritonavir. J Clin Pharmacol. 2010;50:1180‐1187. [DOI] [PubMed] [Google Scholar]
- 12. Kempf DJ, Marsh KC, Kumar G, et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob Agents Chemother. 1997;41:654‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Link B, Haschke M, Grignaschi N, et al. Pharmacokinetics of intravenous and oral midazolam in plasma and saliva in humans: usefulness of saliva as matrix for CYP3A phenotyping. Br J Clin Pharmacol. 2008;66:473‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hesselink DA, van Hest RM, Mathot RAA, et al. Cyclosporine interacts with mycophenolic acid by inhibiting the multidrug resistance‐associated protein 2. Am J Transplant. 2005;5:987‐994. [DOI] [PubMed] [Google Scholar]
- 15. Kusawake T, den Adel M, Groenendaal‐van de Meent D, et al. Pharmacokinetic evaluation of the interactions of amenamevir (ASP2151) with ketoconazole, rifampicin, midazolam, and warfarin in healthy adults. Adv Ther. 2017;34:2466‐2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Profit L, Eagling VA, Back DJ. Modulation of P‐glycoprotein function in human lymphocytes and Caco‐2 cell monolayers by HIV‐1 protease inhibitors. AIDS. 1999;13:1623‐1627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Summary of Pharmacokinetic Parameters for 1‐Hydroxymidazolam (n = 18)