

Abstract
Cystic fibrosis (CF) is an autosomal recessive genetic disease caused by variants in the gene encoding the cystic fibrosis transmembrane conduction regulator (CFTR) protein. Loss of CFTR function disrupts chloride, bicarbonate and regulation of sodium transport, producing a cascade of mucus obstruction, inflammation, pulmonary infection, and ultimately damage in numerous organs. Established CF therapies treat the downstream consequences of CFTR dysfunction and have led to steady improvements in patient survival. A class of drugs termed CFTR modulators has recently entered the CF therapeutic landscape. These drugs differ fundamentally from prior therapies in that they aim to improve the function of disease‐causing CFTR variants. This review summarizes the science behind CFTR modulators, including their targets, mechanism of action, clinical benefit, and future directions in the field. CFTR modulators have dramatically changed how CF is treated, validated CFTR as a therapeutic target, and opened the door to truly personalized therapies and treatment regimens.
Keywords: CFTR, cystic fibrosis, ion transport, modulator
1. INTRODUCTION
Cystic fibrosis (CF) is considered a rare and lethal genetic disease, occurring in approximately 1:3500 births in the United States and at a higher incidence in many northern European countries.1, 2 It is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein, an anion channel and regulator of other ion transporters which controls the hydration and viscoelastic properties of mucus in several epithelial tissues.1, 2, 3, 4 Disease‐causing CFTR variants alter ion transport, leading to mucus obstruction. In the lungs, this sets the stage for chronic infection, inflammation, and airway damage that can progress to respiratory failure. There are more than 2000 variants in the CFTR gene that have been described, and a subset of these are clearly disease‐associated (www.genet.sickkids.on.ca/cftr and www.cftr2.org). The vast majority of people with CF have a relatively small number of variants that have been carefully characterized; however, there are numerous uncommon or rare variants that remain poorly characterized (with approximately 1200 variants identified in less than five CF subjects worldwide).
2. ESTABLISHED, SYMPTOM‐BASED THERAPY FOR CF
Symptom‐based therapies have been highly successful in CF, leading to steady increases in patient lifespan and qualitative outcomes over several decades. These established therapies address critical aspects of disease pathology, including exocrine pancreatic insufficiency (pancreatic replacement enzymes), nutritional deficiencies (fat soluble vitamins and high caloric supplements), airway bacterial infection (inhaled and systemic antibiotics), mucus hydration (inhaled osmotic agents), inhaled mucolytics (recombinant human DNAse), and anti‐inflammatories (high‐dose NSAIDs, chronic macrolides).5, 6, 7, 8, 9, 10 Even with these effective therapies, the median predicted survival of subjects born with CF remains well below that of unaffected newborns throughout the developed world.11 This singular fact provides a clear need to develop more effective therapies for CF. There continues to be a great deal of effort directed towards new drug development that targets symptomatic aspects of CF, with numerous drugs in the therapeutic pipeline.12 These include strategies to normalize epithelial sodium transport (eg, inhibitors of the epithelial sodium channel), agents which reduce or help to resolve inflammation, muco‐active compounds, novel anti‐infective therapies, and treatments that address nutritional and GI aspects of CF disease.12, 13, 14, 15, 16, 17, 18, 19 In addition to these more traditional targets, clinical trials testing highly novel strategies are underway, such as allogenic human mesenchymal stem cells (NCT02866721), iron chelators to disrupt microbial biofilms (NCT02354859), and inhaled nitric oxide that target difficult to treat CF infections (NCT02498535, NCT01958944). These approaches focus on important disease pathologies downstream of the genetic defect causing CF and can be applied to CF patients independent of their background genotype.
3. PRINCIPLES OF CFTR MODULATION
There has been a great deal of interest and excitement in the CF care and research community recently regarding a new class of drugs termed CFTR modulators.12, 19, 20, 21, 22, 23, 24, 25, 26, 27 These drugs differ fundamentally from established CF therapeutics in that they aim to increase or potentially restore the function of disease‐causing CFTR variant protein. In general they are matched to different disease‐causing CFTR variants and are specific in their application (as opposed to the established therapies described above in section 2). Clinical trial evidence suggests that highly effective CFTR modulators can be transformational for CF patients with responsive mutations, leading to improvements in multiple relevant endpoints such as lung function, pulmonary exacerbation rates, respiratory symptoms, weight and growth, lung function stability, mucus clearance, intestinal pH, sweat chloride (SC), structural lung disease, inflammatory burden in mucus, and the detection of CF pathogens.19, 20, 21, 22, 23, 24, 25, 26, 27 These clear benefits across numerous endpoints and trials have provided validation of CFTR as an appropriate target for drug development. CFTR modulators have some advantages relative to current nucleotide strategies in development that seek to repair or replace CFTR (eg, mRNA or DNA gene delivery, gene editing). These include absorption and systemic availability of small molecule drugs that allows oral dosing, and treatment of CFTR defects in multiple organ systems. They also have a potential disadvantage, as they do not produce wild‐type CFTR. Thus, there is theoretically a ceiling of benefit that can be achieved by CFTR modulators that depends on the underlying characteristics of a given disease‐causing CFTR variant. However, data from recent studies suggest that the level of CFTR function that can be achieved by CFTR modulators is significant, producing changes in CFTR biomarkers in patients with highly responsive CFTR variants that fall outside of the range for CF diagnosis.20, 21, 22, 23, 24, 25, 26, 27
When discussing modulation of CFTR, it is helpful to consider what the fundamental channel factors are that contribute to CFTR‐dependent ion transport. CFTR is a traffic ATPase that is composed of 1480 amino acids that normally resides in the apical membrane of various epithelial cells (eg, airway, gastrointestinal, vas deferens, pancreatic duct, biliary tree, sweat gland duct).1, 2, 3, 4, 28, 29 Like all members of this protein family, it has two transmembrane domains (TMD‐1 and ‐2) which anchor the protein in the plasma membrane and two nucleotide binding domains (NBD‐1 and ‐2) which are cytoplasmic, bind, and hydrolyze ATP as a heterodimer, and control gating (opening and closing) of the channel.30, 31 CFTR is unique among traffic ATPases in that it has a regulatory domain that imparts phosphorylation dependent regulation (protein kinase A), and that it, functions as a chloride and bicarbonate channel.29, 30, 31, 32 Traffic ATPases typically function as small molecule pumps which move their cargo across the plasma membrane at speeds that are orders of magnitude slower than ion channels.33 New insights into the molecular (human) and atomic (zebrafish) structure of CFTR have recently been published, providing more information regarding relationships between its structure and unique functions.34, 35
There are essentially three ways to impact or improve the activity of CFTR (or other ion channels). These include increasing the number (N) of CFTR channels available in the plasma membrane, increasing the time a given channel is open (open channel probability, or Po), and/or increasing the size of the channel (channel conductance, or G). The product of these three factors (N × Po × G) determines the total CFTR‐dependent ion transport capacity in a given epithelial cell. The activity of CFTR modulators is based on these basic principles, and combinations of modulators can be used to address CFTR variants that have more than one defect. Currently available modulators include potentiators (increase open channel probability) and correctors (improve folding of some CFTR variants and localization to the plasma membrane).
Disease‐causing CFTR variants fall into a number of different classes which can serve as a first step to categorize them by common or similar defects. As the scientific community has learned more about CFTR variants, it has become clear that they often have numerous defects affecting N, Po, and/or G. Thus, this classification is perhaps becoming arbitrary, and CFTR modulators can increase CFTR activity by either directly impacting a defect (eg, restoring gating function to a CFTR variant that is unable to efficiently bind and hydrolyze ATP such as G551D CFTR), or exploiting another aspect of the variant that is still intact (eg, increasing the ion transport capacity of R117H CFTR 5T by increasing its gating function, despite the presence of additional conductance and splicing defects).1, 2, 21, 36 The most common disease‐causing CFTR variant is F508del CFTR, which is caused by the deletion of three base pairs in exon 10 that results in the deletion of phenylalanine from position 508 of the full‐length protein. Its primary defect is in protein folding, with failure of the nascent protein to mature beyond the ER and undergo glycosylation. It is rapidly degraded in the 26S proteosome, and little if any F508del CFTR normally reaches its proper destination in the apical membrane of CFTR‐expressing epithelia (ie, reduced N).28, 37 However, if F508del CFTR is localized to the plasma membrane by improving it's folding in the ER, it subsequently exhibits defects in both channel gating (reduced Po) and protein stability at the plasma membrane (which further reduces N).38, 39 A more recent example is the P67L CFTR variant, which has been described in detail with both maturation and gating defects (that are responsive in vitro to approved CFTR modulators).40 These examples highlight the rationale of combining CFTR modulators to maximize the activity of CFTR variants.
Disease‐causing CFTR variants that severely reduce the number of channels available in the plasma membrane (very low/absent N) include (i) biosynthetic defects (eg, premature termination codons, frameshift and canonical splice variants) and (ii) trafficking and folding defects. These are typically considered severe disease‐causing variants as they have little if any function and are associated with pancreatic insufficiency. Disease‐causing CFTR variants that localize to the plasma membrane in appreciable amounts include those with primary defects in gating (Po), noncanonical splice variants that produce reduced plasma membrane levels of functional CFTR (N), variants with reduced conductance (G) and/or plasma membrane stability (N). Many variants that primarily impact gating have little if any function and are considered severe. The other variants that have appreciable amounts of CFTR at the plasma membrane and have some residual function are often considered mild, as their presence is often associated with pancreatic sufficiency.1, 2, 3 It is important to note that there are many exceptions to these general groupings; CF patients with two severe mutations may have a comparably mild phenotype, and CF patients with variants that are predicted to be mild often have many typical CF disease manifestations. Airway epithelial cells from some F508del CFTR homozygous donors have readily measured CFTR activity without modulation, and some variants with primary conduction defects and normal N have very low/absent function.41, 42 Figure 1 provides examples of common CFTR variants and their described primary defects in N, Po, and G. CFTR modulators that have gained regulatory approval or are in development impact the number of channels present in the epithelial cell membrane or the gating of individual channels. It is not clear if current CFTR modulators can increase the size of a CFTR channel.
Figure 1.
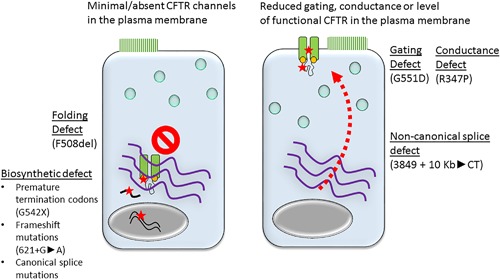
CFTR variant classes discriminated by presence or absence of CFTR at the plasma membrane. Left: CFTR variants that result in minimal or absent CFTR in the plasma membrane. The red stars represent defects in transcription, translation, or protein folding. Examples within each group are listed below in parentheses. Right: CFTR variants with significant levels of CFTR in the plasma membrane. The red arrow represents reduced levels of functional CFTR localizing to the plasma membrane (with a non‐canonical splice defect represented). The red stars represent CFTR variants with normal levels in the plasma membrane, but with defective gating or conduction. Examples within each group are listed below in parentheses
4. POTENTIATION OF CFTR
Potentiators are CFTR modulators which increase the gating of CFTR (Po), leading to more time spent by the channel in the open configuration.43, 44 This results in increased ion transport across the epithelium, which impacts many of the downstream processes that are defective in CF‐affected tissues. Compounds that have been developed into potentiator drugs were identified through high throughput screening (HTS) assays performed in standardized heterologous expression systems.43 One of these (ivacaftor) has advanced through the regulatory process and is available to CF patients with responsive mutations. Ivacaftor has in many ways set the standard for CFTR modulator development, demonstrating significant improvements across numerous clinical endpoints during controlled and observational clinical trials in patients with highly responsive mutations. The compound scaffold that through medicinal chemistry became ivacaftor (VX‐770) was identified through HTS in cells stably transduced with G551D CFTR. This is the third most common disease‐causing CFTR variant, and is found in approximately 4% of CF patients. As part of its development, ivacaftor underwent extensive testing in primary human bronchial epithelial (HBE) cells derived from lung explants of CF patients and grown in planar cultures.43 Using this model system, ivacaftor was shown to increase total G551D CFTR activity to approximately 50% of that observed in HBE cultures from non‐CF donors (eg, wtCFTR). This level of G551D CFTR potentiation was sufficient to improve HBE regulation of airway surface liquid volume, mucociliary clearance, and ciliary beat frequency to near non‐CF levels. Ivacaftor was developed independent of animal model efficacy studies due to limitations of the available animal model (CF mice). Based on this and subsequent experience with studies in primary HBEs, reliance on this model system has become the gold standard for preclinical CFTR modulator evaluation. The model system is an excellent positive predictor of agents that have achieved clinical efficacy.
Ivacaftor monotherapy has demonstrated bioactivity and efficacy in several randomized, double‐blind placebo controlled clinical trials that have enrolled several different CF subpopulations, including subjects with the G551D CFTR and a variety of other gating variants, the R117H CFTR variant, and patients with CFTR variants that localize in appreciable amounts to the plasma membrane and retain some level of residual function.20, 21, 22, 23, 24, 25, 26, 27, 36, 45, 46, 47 Subjects with gating mutations have demonstrated the greatest evidence of bioactivity and clinical benefit, including increases in the forced expiratory volume in one second (FEV1) percent predicted of >10%, reduction in risk of pulmonary exacerbations by >50%, increases in weight and reductions in pulmonary symptoms relative to baseline and placebo treated controls. Mean reductions in SC concentrations have been on the order of 50 mM in participants with CFTR gating variants, with perhaps larger effects observed in younger subjects.46, 47 The studies in toddlers and young children have also shown increases in fecal elastase levels, suggesting improvements in exocrine pancreatic function with open label ivacaftor treatment.46, 47 Mechanistic studies suggest that ivacaftor directly binds to CFTR to improve channel gating, based on its activation of isolated wild‐type and mutant CFTR expressed in proteoliposomes, and its capacity to activate multiple CFTR missense variants expressed in heterologous expression systems.42, 43, 48 Furthermore, ivacaftor monotherapy did not demonstrate clinical efficacy in CF subjects homozygous for the F508del CFTR variant, indicating it's requirement for appreciable CFTR at the plasma membrane for drug effect.49 In addition to these acute effects, prolonged treatment of CF patients with the G551D CFTR mutation reduces the loss of lung function over time relative to untreated controls and reduces mortality in patients with advanced disease.50, 51 Thus, ivacaftor has the capacity to modify the course of CF disease in certain CF populations. Additional observational studies have detected reductions in inflammation, structural lung disease, and possibly detection of CF pathogens, but these data are currently limited in terms of length of follow‐up and number of subjects.23, 25 Based on accumulating evidence of safety and efficacy in several CF genotypic subgroups, coupled with relationships observed between results in preclinical model systems compared to clinical trials, the FDA has expanded the label of ivacaftor‐approved mutations.52 This expansion has included some rare CFTR variants that have not been represented in clinical trials. This approach is an exciting development in the CF field, as patients with very rare variants that are difficult to include in rigorous clinical trials may be able to gain access to ivacaftor based on results in standardized preclinical model systems.
5. MODULATING F508del CFTR
F508del CFTR is the most common disease‐causing CFTR variant and is found in nearly 90% of CF patients.53 The loss of phenylalanine at position 508 of the full‐length protein produces a primary defect in protein maturation and localization to the plasma membrane.28, 54, 55 F508del CFTR has additional defects in channel gating and stability that contribute to its overall limitation of chloride transport.38, 39 VX‐809 is a CFTR modulator that was developed through HTS efforts and improves F508del CFTR trafficking and plasma membrane localization.56 This effect is specific to F508del CFTR as VX‐809 does not impact the maturation of other proteins with folding defects introduced.56 Recent data indicates that VX‐809 has no corrective effect on the second most common CFTR folding variant (N1303K CFTR), and protease digest studies suggest that VX‐809 is able to stabilize an N‐terminal domain in CFTR limited to MSD1 and improve the function of CFTR constructs with missense mutations introduced into MSD1.57, 58, 59 The results suggest that VX‐809 interactions with MSD1 inhibits F508del folding defects by increasing NBD1, MSD1, and MSD2 domain interactions. These data point towards specific binding of VX‐809 to F508del CFTR which drives its corrective effects. In vitro and subsequent in vivo studies have demonstrated that VX‐809 can improve the level of mature Band C F508del CFTR levels and activity, again based primarily on studies performed in primary HBEs derived from F508del CFTR homozygous CF patients undergoing lung transplantation.56 This corrective effect is on the order of 15% of wild‐type CFTR, but there is significant donor to donor variability seen in preclinical studies. Importantly, this effect can be essentially doubled by co‐treatment with VX‐770 in vitro, which provided the rationale for the development of VX‐809 (lumacaftor) in combination with VX‐770 (ivacaftor) in CF patients with two copies of the F508del CFTR variant.
Clinical trials of lumacaftor (and subsequent trials with the chemically similar drug tezacaftor) alone and in combination with ivacaftor in CF subjects with the F508del variant have generally aligned with predictions from preclinical observations in HBEs. Specifically, lumacaftor monotherapy was insufficient to produce measurable clinical benefits in CF adults homozygous for F508del CFTR despite small, but significant dose‐dependent reductions in SC.60 Lumacaftor combined with ivacaftor was insufficient to produce meaningful clinical benefits in CF subjects with one F508del CFTR variant and a second minimal function variant.61 In contrast, lumacaftor or tezacaftor combined with ivacaftor in F508del/F508del CF subjects led to modest increases in FEV1 percent predicted, and moderate reductions in pulmonary exacerbation risk and improvements in weight and pulmonary symptoms compared with placebo.62, 63 The effects of these therapies on SC were relatively small and not substantially different than results with corrector alone.60, 64, 65 As lumacaftor/ivacaftor has been studied in younger CF subjects (Ages 6‐11 years), more robust reductions in SC and improvements in lung function (measured by the nitrogen multiple breath washout test and the lung clearance index or LCI) have been observed compared with studies in older populations.66, 67 It is clear that individual changes in SC are not directly associated with improvements in clinical outcome measures during CFTR modulator therapy, but group changes in SC do directly correlate with changes in FEV1 percent predicted.68, 69 Thus, it is exciting to speculate that CFTR modulators may have higher efficacy in younger CF subjects with early disease, therefore, serving a disease preventive role in CF management. Similar to results with ivacaftor in the G551D CFTR population, lumacaftor/ivacaftor therapy in F508del/F508del CF subjects is associated with reduced decline in lung function over time compared with observational matched controls.70 Moreover, recent results reported in CF subjects with one F508del CFTR variant and one ivacaftor responsive variant demonstrated that tezacaftor/ivacaftor produced greater increases in lung function relative to ivacaftor alone.45 The results of this study, indicate that targeting each CFTR variant (when they differ) can be more efficacious than targeting one, and provides support for the rationale to develop additional CFTR modulator strategies that build upon the current foundation. They also highlight recent thinking in regards to patient categorization for clinical trials and treatment. CF patients with one F508del allele combined with a minimal function second CFTR allele are likely to require new CFTR modulators to obtain measurable clinical benefit. Recent press reports are consistent with this concept, providing preliminary evidence that combining tezacaftor and ivacaftor with a third drug (a “next generation corrector” that further corrects F508del CFTR folding defects) produce large improvements in lung function and SC in CF subjects with two F508del CFTR variants or one F508del CFTR variant and one minimal function variant.71 This latter population is significant, and currently lacks efficacious CFTR modulator therapies. Phase 3 trials of these “next generation” correctors combined with ivacaftor and tezacaftor are currently active, and if successful have the capacity to dramatically expand the number of CF patients who are candidates for highly efficacious CFTR modulators.
6. ADDITIONAL MODULATOR STRATEGIES
In addition to the drugs described above that have gained regulatory approval or are in late phase clinical trials in CF patients, several early phase clinical trials of novel modulators are underway (eg, NCT03173573, NCT02170025, NCT03540524). These agents generally fit into the categories of CFTR potentiators or correctors. Additional CFTR modulator strategies that are under development target other aspects of CFTR biology and offer CF patients with rare and common CFTR variants potential future benefit.
Premature termination codons or PTCs are found in approximately 10% of CF patients, and are much more common in some ethnicities including those of Ashkenazi descent.72, 73, 74 They are caused by base pair substitutions that create an in‐frame PTC (UGA, UAA, UAG), which leads to the cessation of translation and the production of truncated CFTR protein.74 Suppressors of PTCs are compounds which lead to the insertion of a near cognate aminoacyl tRNA in the A site of the eukaryotic ribosome, allowing translation to continue to the authentic termination codon and the production of a full‐length protein. These CFTR protein products following PTC suppression may or may not have full function based on the nature of the amino acid substitution coupled with the site of the substitution within the full‐length protein.74, 75 Drugs that induce PTC read‐through and full length CFTR production have been tested in several contexts, including cell lines, primary airway cells from CF patients with PTCs, transgenic mice, and CF patients.75, 76, 77, 78, 79, 80 Some provocative small clinical trials have provided evidence that certain aminoglycosides, which are known to bind to the eukaryotic ribosome complex, can improve the function of CFTR in CF patients harboring PTC variants in CFTR.72, 73, 77, 78 Ataluren was developed through HTS, and was studied in two phase 3 randomized, double blind placebo controlled clinical trials in CF subjects with PTC‐mediated CF, but ultimately this drug was insufficient to produce measurable benefit in subjects with a harboring a variety of PTCs.80, 81 Since this concept was first described in CF cells nearly two decades ago, accumulating data has helped the CF research community to better understand the many factors that contribute to PTC‐mediated CF.76 These include the importance of nonsense mediated decay impacting CFTR mRNA substrate levels, variable responsiveness of different PTCs to suppression, the role of the mRNA microenvironment (including nearby base pairs) in read‐through susceptibility, and the observation that some CFTR variants with PTCs late in the CFTR open reading frame retain partial function.74, 75, 82, 83, 84, 85 This improved understanding of how cells regulate PTCs and their read‐through has led to the identification of new agents capable of producing more robust and predictable PTC suppression.85, 86, 87 The importance of using patient derived cells to study PTC‐mediated CF, and novel model systems to evaluate putative PTC suppressive candidates has become clear.88 It is hoped that this new understanding will help to rapidly advance promising strategies to the clinic.
A new approach that has the potential to impact many disease‐causing CFTR variants is co‐treatment with a CFTR amplifier. This concept increases the amount of translated CFTR protein substrate, and therefore, could be readily applicable to CF subjects with a variety of CFTR variants.12 These include subjects with reduced levels of functional CFTR at the plasma membrane, or subjects primarily treated with potentiators and/or correctors. A CFTR amplifier (PTI‐148) has recently advanced to early phase clinical trials, and a recent press release and scientific presentation supports safety, tolerability, and pulmonary efficacy in CF patients receiving PTI‐148 added to ivacaftor/lumacaftor therapy.88 Safety and tolerability data is typically a primary goal of early phase clinical trials, and will hopefully provide the necessary data to advance this strategy that has the potential to impact modulator therapies across a variety of CF populations. One published report supports the hypothesis of amplifier bioactivity in cells derived from a CF patient with a rare disease‐causing CFTR variant.89
7. CONCLUSIONS AND FUTURE DIRECTIONS
CFTR modulator science has advanced dramatically since therapeutic CFTR modulators were first described less than 10 years ago. Through steady advancement of our understanding of the biology underpinning CFTR variants and modulation, the number of CF subjects who are candidates for therapy has increased to greater than 50%. The short‐term future appears quite bright in that it is anticipated that highly effective CFTR modulator therapy may soon be approved for greater than 90% of CF patients. Furthermore, additional modulators that potentiate and correct CFTR variants are well into clinical trials, which may ultimately overlap with novel approaches to increase CFTR translation. Successful development of these agents is paramount to the CF population, as a full and diverse pipeline is necessary to ensure that CF subjects have access to those therapies that are best matched to their CFTR variants and their needs. Challenges will undoubtedly be faced, including performance of clinical trials to develop new therapies in the face of a CF population heavily treated with CFTR modulators, and the design of studies that assess the relative benefits of established, symptom‐based therapies in the context of highly effective CFTR modulator treatment. Finally, the cost of these life‐long drugs is extremely high and is a significant barrier to patient access in many countries.90 This issue is likely to grow as more and more CF subjects become candidates for modulators that are currently in development. Equitable solutions are needed that balance the incentive needed for companies to develop these drugs for a limited market, and the capacity of third party payers and national health systems to offer CF patients the best available treatment for their personal CF disease. CF is at the cutting edge of personalized care, and the scientific and health policy lessons that we learn will undoubtedly have important implications well beyond the science of CFTR modulation.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Clancy JP. Rapid therapeutic advances in CFTR modulator science. Pediatric Pulmonology. 2018;53:S4–S11. 10.1002/ppul.24157
REFERENCES
- 1. Spielberg DR, Clancy JP. Cystic fibrosis and its management through established and emerging therapies. Annu Rev Genomics Hum Genet. 2016;17:155–175. [DOI] [PubMed] [Google Scholar]
- 2. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. [DOI] [PubMed] [Google Scholar]
- 3. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. [DOI] [PubMed] [Google Scholar]
- 5. Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637–642. [DOI] [PubMed] [Google Scholar]
- 6. Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long‐term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–240. [DOI] [PubMed] [Google Scholar]
- 7. Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med. 1999;340:23–30. [DOI] [PubMed] [Google Scholar]
- 8. Saiman L, Marshall BC, Mayer‐Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749–1756. [DOI] [PubMed] [Google Scholar]
- 9. Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high‐dose ibuprofen in patients with cystic fibrosis. N Engl J Med. 1995;332:848–854. [DOI] [PubMed] [Google Scholar]
- 10. Konstan MW, Accurso FJ, Nasr SZ, Ahrens RC, Graff GR. Efficacy and safety of a unique enteric‐coated bicarbonate‐buffered pancreatic enzyme replacement therapy in children and adults with cystic fibrosis. Clin Investig (Lond). 2013;3:723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goss CH, Sykes J, Stanojevic S, et al. Comparison of nutrition and lung function outcomes in patients with cystic fibrosis living in Canada and the United States. Am J Respir Crit Care Med. 2018;197:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hudock KM, Clancy JP. An update on new and emerging therapies for cystic fibrosis. Expert Opin Emerg Drugs. 2017;22:331–346. [DOI] [PubMed] [Google Scholar]
- 13. Elborn JS, Horsley A, MacGregor G, et al. Phase I studies of acebilustat: biomarker response and safety in patients with cystic fibrosis. Clin Transl Sci. 2017;10:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haberman R, Ling M, Van Goor F, Higgins M, Jain M. Preclinical evidence for adding ENAC inhibition to corrector/potentiator therapy (lumacaftor‐ivacaftor combination therapy). Pediatr Pulmonol. 2016;51:S274. [Google Scholar]
- 15. Wheeler A, Schaberg A, Scott D, Crowder T, Couroux P. Safety and pharmacokinetics of spx‐101 in healthy human subjects. Am J Respir Crit Care Med. 2017;195:A4736. [Google Scholar]
- 16. Gardiner P, Malmgren A, Ersdal E, Goldwater R, Patel N. ENaC inhibitor AZD5634 first in human trial reveals promising clinical profile for the treatment of cystic fibrosis. Am J Respir Crit Care Med. 2017;195:A7306. [Google Scholar]
- 17. Devereux G, Steele S, Griffiths KJ, et al. An open label investigation of the tolerability and pharmacokinetics of oral cysteamine in adults with cystic fibrosis. J Cyst Fibrosis. 2015;14:S93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sagel SD, Khan U, Jain R, et al. Effects of an antioxidant‐enriched multivitamin in cystic fibrosis: randomized, controlled, multicenter trial. Am J Respir Crit Care Med. 2018; 10.1164/rccm.201801-0105OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chin M, Aaron SD, Bell SC. The treatment of the pulmonary and extrapulmonary manifestations of cystic fibrosis. Presse Med. 2017;46:e139–e164. [DOI] [PubMed] [Google Scholar]
- 20. Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation. N Engl J Med. 2010;363:1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non‐G551D gating mutation. J Cyst Fibros. 2014;13:674–680. [DOI] [PubMed] [Google Scholar]
- 23. Rowe SM, Heltshe SL, Gonska T, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D‐mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hisert KB, Heltshe SL, Pope C, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. 2017;195:1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ronan NJ, Einarsson GG, Twomey M, et al. CORK study in cystic fibrosis: sustained improvements in ultra‐low‐dose chest CT scores after CFTR modulation with ivacaftor. Chest. 2018;153:395–403. [DOI] [PubMed] [Google Scholar]
- 26. Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2‐5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open‐label, single‐arm study. Lancet Respir Med. 2016;4:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Graeber SY, Hug MJ, Sommerburg O, et al. Intestinal current measurements detect activation of mutant CFTR in patients with cystic fibrosis with the G551D mutation treated with ivacaftor. Am J Respir Crit Care Med. 2015;192:1252–125. [DOI] [PubMed] [Google Scholar]
- 28. Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. [DOI] [PubMed] [Google Scholar]
- 29. Anderson MP, Gregory RJ, Thompson S, et al. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. [DOI] [PubMed] [Google Scholar]
- 30. Rich DP, Gregory RJ, Anderson MP, Manavalan P, Smith AE, Welsh MJ. Effect of deleting the R domain on CFTR‐generated chloride channels. Science. 1991;253:205–207. [DOI] [PubMed] [Google Scholar]
- 31. Smith JJ, Welsh MJ. CAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest. 1992;89:1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hug MJ, Clarke LL, Gray MA. How to measure CFTR‐dependent bicarbonate transport: from single channels to the intact epithelium. Methods Mol Biol. 2011;741:489–509. [DOI] [PubMed] [Google Scholar]
- 33. Beis K. Structural basis for the mechanism of ABC transporters. Biochem Soc Trans. 2015;43:889–893. [DOI] [PubMed] [Google Scholar]
- 34. Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J. Molecular structure of the human CFTR ion channel. Cell. 2017;169:85–95. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Z, Chen J. Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell. 2016;167:1586–1597. [DOI] [PubMed] [Google Scholar]
- 36. Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His‐CFTR mutation: a double‐blind, randomised controlled trial. Lancet Respir Med. 2015;3:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rabeh WM, Bossard F, Xu H, et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bebok Z, Collawn JF, Wakefield J, et al. Failure of cAMP agonists to activate rescued ΔF508 CFTR in CFBE41o‐airway epithelial monolayers. J Physiol. 2005;569:601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jurkuvenaite A, Chen L, Bartoszewski R, et al. Functional stability of rescued delta F508 cystic fibrosis transmembrane conductance regulator in airway epithelial cells. Am J Respir Cell Mol Biol. 2010;42:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sabusap CM, Wang W, McNicholas CM, et al. Analysis of cystic fibrosis‐associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases. JCI Insight. 2016;1:e86581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del‐CFTR protein processing defect in vitro by the investigational drug VX‐809. Proc Natl Acad Sci USA. 2011;108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014;13:29–36. [DOI] [PubMed] [Google Scholar]
- 43. Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX‐770. Proc Natl Acad Sci USA. 2009;106:18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kramer EL, Clancy JP. CFTR modulator therapies in pediatric cystic fibrosis: focus on ivacaftor. Expert Opin Orphan Drugs. 2016;4:1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor‐ivacaftor in residual‐function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377:2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenfeld M, Wainwright CE, Higgins M, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single‐arm study. Lancet Respir Med. 2018;6:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX‐770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation‐dependent but ATP‐independent manner. J Biol Chem. 2012;287:36639–36649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del‐CFTR mutation. Chest. 2012;142:718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sawicki GS, McKone EF, Pasta DJ, et al. Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med. 2015;192:836–42. [DOI] [PubMed] [Google Scholar]
- 51. Dilokthornsakul P, Hansen RN, Campbell JD. Forecasting US ivacaftor outcomes and cost in cystic fibrosis patients with the G551D mutation. Eur Respir J. 2016;47:1697–1705. [DOI] [PubMed] [Google Scholar]
- 52. Durmowicz AG, Lim R, Rogers H, Rosebraugh CJ, Chowdhury BA. The US Food and Drug administration's experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in lieu of a clinical trial. Ann Am Thorac Soc. 2018;15:1–2. [DOI] [PubMed] [Google Scholar]
- 53.Cystic Fibrosis Foundation. CFF patient registry report. www.CFF.org; 2016.
- 54. Hudson RP, Chong PA, Protasevich II, et al. Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2012;287:28480–28494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kleizen B, van Vlijmen T, de Jonge HR, Braakman I. Folding of CFTR is predominantly cotranslational. Mol Cell. 2005;20:277–287. [DOI] [PubMed] [Google Scholar]
- 56. Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del‐CFTR protein processing defect in vitro by the investigational drug VX–809. Proc Natl Acad Sci USA. 2011;108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. DeStefano S, Gees M, Hwang TC. Physiological and pharmacological characterization of the N1303K mutant CFTR. J Cyst Fibros. 2018;pii: S1569‐1993:30593–30599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ren HY, Grove DE, De La Rosa O, et al. VX–809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane‐spanning domain 1. Mol Biol Cell. 2013;24:3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thibodeau PH, Richardson JM, 3rd , Wang W, et al. The cystic fibrosis‐causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem. 2010;285:35825–35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX–809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del‐CFTR mutation. Thorax. 2012;67:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rowe SM, McColley SA, Rietschel E, et al. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del‐CFTR. Ann Am Thorac Soc. 2017;14:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor‐Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231. [DOI] [PubMed] [Google Scholar]
- 63. Taylor‐Cousar JL, Munck A, McKone EF, et al. Tezacaftor‐Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe5087del. N Engl J Med. 2017;377:2013–2023. [DOI] [PubMed] [Google Scholar]
- 64. Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2:527–538. [DOI] [PubMed] [Google Scholar]
- 65. Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del‐CFTR or F508del/G551D‐CFTR. Am J Respir Crit Care Med. 2018;197:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del‐CFTR: a randomised, placebo‐controlled phase 3 trial. Lancet Respir Med. 2017;5:557–567. [DOI] [PubMed] [Google Scholar]
- 67. Milla CE, Ratjen F, Marigowda G, et al. Lumacaftor/ivacaftor in patients aged 6–11 years with cystic fibrosis and homozygous for F508del‐CFTR. Am J Respir Crit Care Med. 2017;195:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Durmowicz AG, Witzmann KA, Rosebraugh CJ, Chowdhury BA. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest. 2013;143:14–18. [DOI] [PubMed] [Google Scholar]
- 69. Fidler MC, Beusmans J, Panorchan P, Van Goor F. Correlation of sweat chloride and percent predicted FEV(1) in cystic fibrosis patients treated with ivacaftor. J Cyst Fibros. 2017;16:41–44. [DOI] [PubMed] [Google Scholar]
- 70. Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long‐term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del‐CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5:107–118. [DOI] [PubMed] [Google Scholar]
- 71.Preliminary Safety and Efficacy of Triple Combination CFTR Modulator Regimens in CF. Poster 777, and Oral presentation during Workshop W18–NT: Innovative Approaches to CF Therapy. North American Cystic Fibrosis Conference, November 3, 2017. www.vertex.com.
- 72. Wilschanski M, Famini C, Blau H, et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med. 2000;161:860–865. [DOI] [PubMed] [Google Scholar]
- 73. Wilschanski M, Yahav Y, Yaacov Y, et al. Gentamicin‐induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–1441. [DOI] [PubMed] [Google Scholar]
- 74. Rowe SM, Clancy JP. Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs. 2009;23:165–174. [DOI] [PubMed] [Google Scholar]
- 75. Xue X, Mutyam V, Thakerar A, et al. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum Mol Genet. 2017;26:3116–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. [DOI] [PubMed] [Google Scholar]
- 77. Sermet‐Gaudelus I, Renouil M, Fajac A, et al. In vitro prediction of stop‐codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Clancy JP, Bebök Z, Ruiz F, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med. 2001;163:1683–1692. [DOI] [PubMed] [Google Scholar]
- 79. Du M, Jones JR, Lanier J, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr‐/‐ mouse carrying a human CFTR‐G542X transgene. J Mol Med (Berl). 2002;80:595–604. [DOI] [PubMed] [Google Scholar]
- 80. Elborn JS, Melotti P, Bronsveld I, et al. Ataluren for the treatment of nonsense‐mutation cystic fibrosis: a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.PTC Therapeutics Announces Results from Pivotal Phase 3 Clinical Trial of Ataluren in Patients Living with Nonsense Mutation Cystic Fibrosis, March 2, 2017. www.ptcbio.com.
- 82. Linde L, Boelz S, Nissim‐Rafinia M, et al. Nonsense‐mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context‐dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rowe SM, Varga K, Rab A, et al. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol. 2007;37:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dekkers JF, Berkers G, Kruisselbrink E, et al. Characterizing responses to CFTR‐modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8:344ra84. [DOI] [PubMed] [Google Scholar]
- 86. Mutyam V, Du M, Xue X, et al. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. Am J Respir Crit Care Med. 2016;194:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Xue X, Mutyam V, Tang L, et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am J Respir Cell Mol Biol. 2014;50:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Evaluation of safety, tolerability and pharmacokinetics, and pharmacodynamics data of PTI‐428, a novel CFTR amplifier, in patients with cystic fibrosis. The European Cystic Fibrosis Society Meeting, July 7, 2018. www.proteostasis.com.
- 89. Molinski SV, Ahmadi S, Ip W, et al. Orkambi® and amplifier co‐therapy improves function from a rare CFTR mutation in gene‐edited cells and patient tissue. EMBO Mol Med. 2017;9:1224–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hollin IL, Robinson KA. A scoping review of healthcare costs for patients with cystic fibrosis. Appl Health Econ Health Policy. 2016;14:151–159. [DOI] [PubMed] [Google Scholar]