

Abstract
Alzheimer’s disease (AD) is the most common (60% to 80%) age‐related disease associated with dementia and is characterized by a deterioration of behavioral and cognitive capacities leading to death in few years after diagnosis, mainly due to complications from chronic illness. The characteristic hallmarks of the disease are extracellular senile plaques (SPs) and intracellular neurofibrillary tangles (NFTs) with neuropil threads, which are a direct result of amyloid precursor protein (APP) processing to Aβ, and τ hyperphosphorylation. However, many indirect underlying processes play a role in this event. One of these underlying mechanisms leading to these histological hallmarks is the uncontrolled hyperactivation of a family of cysteine proteases called calpains. Under normal physiological condition calpains participate in many processes of cells’ life and their activation is tightly controlled. However, with an increase in age, increased oxidative stress and other excitotoxicity assaults, this regulatory system becomes impaired and result in increased activation of these proteases involving them in the pathogenesis of various diseases including neurodegeneration like AD. Reviewed here is a pool of data on the implication of calpains in the pathogenesis of AD, the underlying molecular mechanism, and the potential of targeting these enzymes for AD therapeutics.
Keywords: Alzheimer’s disease (AD); calpain; τ, amyloid beta; therapeutics
1. INTRODUCTION
Alzheimer’s disease (AD) is currently the most widely prevalent neurodegenerative disease affecting global health, mostly affecting aged individuals. The disease begins with a gradual loss in short‐term memory leading to a progressive impairment of cognition and behavior and finally a compromised ability of thinking, planning, judgment, and social skills.1 AD constitutes about 60 to 80% of overall dementia cases and is inevitably ending with the death of the affected individual in about 5 to 10 years after clinical diagnosis, mainly due to complications from chronic illness.2 The disease is marked histopathologically by extracellular deposit of amyloid beta (Aβ) surrounded by dystrophic neuritis forming senile plaques (SPs), and intracellular hyperphosphorylated τ made neurofibrillary tangles (NFTs).3, 4 The Aβ peptides are the result of proteolysis of the amyloid precursor protein (APP) by two proteases, the beta APP cleaving enzyme 1 (BACE1), also called β‐secretase, and the γ‐secretase,5, 6 and the hyperphosphorylated τ protein in the NFTs results from the dysregulation of kinase/phosphatase system, involving mainly glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A) among other.7 Other key players in AD pathogenesis include oxidative stress seen as lipids; proteins and nucleic acid oxidations, neuroinflammation, calcium homeostasis disturbance. These inexorably lead to a decrease in PP2A and increase in GSK3β activities8, 9 which ultimately result in τ hyperphosphorylation and an increase Aβ production through increasing BACE1 activity via cyclin‐dependent kinase 5 (CDK5) activation by calpain.10 There is also an impaired autophagy/lysosomal clearance which results in the buildup and aggregation of harmful proteins including Aβ and τ.11, 12
Calpains are a family of cytoplasmic nonlysosomal cysteine proteases that are ubiquitously expressed in cells of human and other organisms. Calpains play a myriad of roles in calcium (Ca2+)‐regulated cellular functions.13, 14 Their enzymatic activity is under the regulation of Ca2+ and calpastatin (CAST) that activates and inhibits them, respectively.14, 15 The activation of calpains is controlled within smaller range, however, the stability of this regulating system tends to be impaired with increase in age resulting in an uncontrolled calpain activation,16, 17 implicating these enzymes in many disease conditions including neurodegenerative diseases like AD. Calpains are involved in the neuropathogenesis of AD where factors like oxidative stress and increased activation of N‐methyl‐d‐aspartate (NMDA) receptor play a role in intracellular calcium influx thus hyperactivating these enzymes. Upon activation, calpain mediates the cleavage of p35 to p25 (the most potent form of these CDK5 activators), GSK3β, dopamine‐regulated and cyclic adenosine monophosphate (cAMP)‐regulated phosphoprotein 32 kDa (DARPP‐32), promotes BACE1 synthesis, and APP truncation.18, 19, 20 In turn, the calpain‐mediated truncation of DARPP‐32 induced the dephosphorylation of a key protein involved in memory and learning and conversion of short‐term to long‐term memory21 known as cAMP‐response element‐binding protein (CREB).18 Calpain also cleaved lysosomal stabilizing protein like heat shock protein (Hsp) 70 and lysosome‐associated membrane protein 2a (LAMP2a).22, 23, 24 Together, these lead to Aβ plaques formation, τ phosphorylation, synaptic abnormalities, and memory and learning impairments observed in AD. In the present review, we first considered the types and functions of these proteases and how they are regulated in normal physiological conditions. We then reviewed data on the causes and consequences of the implication of calpains upregulation in AD pathogenesis. Lastly, we summarized available data on how calpain is targeted as potential AD therapeutics and the future direction.
2. TYPES AND BIOLOGICAL FUNCTION OF CALPAIN
Calpains are a family of widely distributed with highly conserved homology among species, Ca2+‐dependent proteases which were first discovered in 1964.25 It was given the name “calpain” to show its similarity with two other enzymes, the Ca2+‐regulated signaling protein, calmodulin “cal,” and the cysteine protease from papaya, papain “pain” (“cal” + “pain” = “calpain”). Although about 15 human genes (Table 1) of calpain family members are known to date,13, 26 only a few of them have attracted researchers’ attention. These mainly include the two isoforms, μ‐calpain and m‐calpain, reflecting their µM and mM Ca2+ requirement for activation, also known as calpain‐1 and calpain‐2, respectively, or conventional calpains, and their only and specific endogenous inhibitor CAST. Each of the μ‐calpain and m‐calpain form heterodimers composed of large and small subunits (Figure 1A). The large protease core subunit (80 kDa molecular weight) is encoded by the CAPN1 gene for calpain‐1 and CAPN2 gene for calpain‐2, with about 60% homology, while the shared small regulatory (28 kDa molecular weight) subunit is encoded by CAPNS1 gene.27 These conventional calpains have a ubiquitous distribution, in contrast to some of other unconventional calpains which expression is limited to particular tissues. Characteristic examples are the expression of calpain‐3 in skeletal, calpain‐6 in embryonic muscles and placenta, calpains 8 and 9 in the gastrointestinal tract smooth muscles, calpain‐11 in the testes, calpain‐12 in the hair follicles, and calpain‐13 mostly in the lung and skin while others unconventional calpains, like calpain 5, 7, 9, and 10 have more ubiquitous distribution (Table 1).13, 14, 28
Table 1.
Calpain family genes for human
| Gene | Chromosome | Name | Distribution | Deficiency |
|---|---|---|---|---|
| CAPN1 | 11q13 | CAPN1 | Ubiquitous | Platelet dysfunction |
| CAPN2 | 1q41‐q42 | CAPN2 | Ubiquitous except erythrocytes | Embryonic lethality |
| CAPN3 | 15q15.1‐q21.1 | CAPN3 | Skeletal muscle | Muscular dystrophy |
| CAPN5 | 11q14 | CAPN5 | Ubiquitous | Vitreoretinopathy |
| CAPN6 | Xq23 | CAPN6 | Embryonic muscles, placenta | Hypergenesis |
| CAPN7 | 3p24 | CAPN7 | Ubiquitous | ⋯ |
| CAPN8 | 1q41 | CAPN8 | Gastrointestinal tracts | Gastric ulcer |
| CAPN9 | 1q42.11‐q42.3 | CAPN9 | Gastrointestinal tracts | Gastric ulcer |
| CAPN10 | 2q37.3 | CAPN10 | Ubiquitous | Type 2 diabetes |
| CAPN11 | 6p12 | CAPN11 | Testis | ⋯ |
| CAPN12 | 19q13.2 | CAPN12 | Hair follicle | ⋯ |
| CAPN13 | 2p22‐p21 | CAPN13 | Ubiquitous | ⋯ |
| CAPN14 | 2p23.1‐p21 | CAPN14 | Ubiquitous | Eosinophilic Esophagitis |
| CAPN15 | 16p13.3 | CAPN15 | Ubiquitous | ⋯ |
| CAPN16 | 6q24.3 | CAPN16 | Ubiquitous | ⋯ |
| CAPNS1 | 19q13.1 | CAPNS1 | Ubiquitous | ⋯ |
| CAPNS2 | 16q12.2 | CAPNS2 | Ubiquitous | ⋯ |
| CAST | 5q15 | Calpastatin | Ubiquitous | ⋯ |
Figure 1.
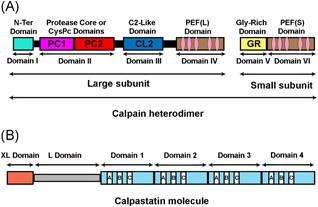
Structure of conventional calpains and calpastatin. A, Schematic representation of conventional calpains. Conventional calpains are heterodimer made up of a large catalytic subunit and a small regulatory subunit. The large subunit contains four domains which include: the anchor helix at the N‐terminus also called the N‐terminal domain or domain I, the catalytic CysPc domain made up of two protease core (PC1 and PC2) domains together constituting the domain II, the C2‐like domain and the penta‐EF‐hand (PEF[L]) domain, which contain the calcium‐binding sites play a regulatory role and constitute the domains III and IV, respectively. The small subunit contains a glycine‐rich (GR) domain and a PEF(S) domain similar to the large subunit, and together they make up the domains V and VI of the complete calpain heterodimer molecule. B, Schematic representation of the longest isoform of human calpastatin: the XL and L domains have no inhibitory activity, while the four inhibitory domains (1 to 4), each of them is capable of binding and inhibiting one calpain molecule. They are made up of A, B, and C subdomains or regions. While the subdomain B mediates calpain inhibitory effect of this molecule, the subdomains A and C interact with the PEF(L) and PEF(S) on the large and small subunits, respectively, and are required for the inhibitory effect of the B subdomain [Color figure can be viewed at wileyonlinelibrary.com]
The large subunit of the conventional calpains is divided into four domains (Figure 1A). Domain I (the N‐terminus) undergoes autolysis when Ca2+ binds to this molecule, domain II (the protease core or CysPc domain) forms the catalytic unit, domain III (the C2‐like domain) has a regulatory role, and domain IV (penta‐EF‐hand [PEF] domain) contains the Ca2+‐binding site.29, 30, 31 The shared small regulatory subunit has two domains a glycine‐rich (GR) domain and a calmodulin‐like PEF domain32, respectively, representing the (domain V) and (domain VI) of the calpain heterodimer (Figure 1A). CAST, the third most commonly studied member of the calpain system, is a heat‐stable unstructured protein which according to the type of the cell has a molecular weight ranging from ∼70 to ∼140 kDa with a ubiquitous widespread expression and is conserved (>70% identical) among mammalian species.33 The longest CAST molecule has six domains, four inhibitory homology units (CAST1‐4), and two N‐terminal domains, called XL domain and L domain (Figure 1B). Each of the four domains can specifically bind and effectively inhibit one calpain molecule, while the XL domain and L domain are devoid of any inhibitory potential.34, 35
Calpains have optimum activity at around neutral pH and are regulatory rather than digestive proteases. In normal physiological cell conditions, the concentration of Ca2+ is lower than 0.05 µM, a condition where calpains act as a biomodulator for Ca2+‐regulated processes, including among other signal transduction, cell proliferation, cell cycle progression, differentiation, apoptosis, learning, and memory and long‐term potentiation.13, 14, 32 It was previously thought that calpain cleavage sites have a Leu or a Val residue in the P2 position,36 but subsequent studies have shaded light on the fact that calpain‐mediated truncation of its substrates is not amino acid sequence‐specific but rather it is determined by the protein conformation.37, 38 Alterations in these proteases have been linked to some human diseases collectively known as calpainopathies, thus providing evidence of the physiological functions of calpains and the indispensable role they play. Supporting this are data that highlighted that deficiencies of calpain‐3 is associated with limb‐girdle muscular dystrophy 2A (LGMD‐2A),39, 40 calpain‐10 linked with diabetes mellites41, while recently calpain 5 mutation has been found to be associated with autoimmune uveitis and photoreceptor degeneration (vitreoretinopathy)42 and calpain‐14 has been associated with eosinophilic esophagitis.43
3. CALPAIN ACTIVATION
The enzymatic activity of calpains is under the regulatory control of Ca2+ that activates them and the endogenous protein CAST that inhibits them through a high‐affinity Ca2+‐dependent binding to the calpain.14, 15 When Ca2+ binds to the calpains it drives a conformational change that initiates their proteolytic properties. Calpain activity is also a measure of its autolyzed fragments.44 Besides induction of conformational changes, Ca2+ also induces calpains’ autoproteolytic cleavage that increases its protease activity. Most of the native µ‐calpain was found to be inactive until it undergoes autolysis and most of its activity is conferred to the 76 kDa fragment.45 In the presence of Ca2+, incubation of purified µ‐calpain with protein kinase A (PKA) or alkaline phosphatase (AP) increased its autolysis and therefore its activity46, 47 compared with control. A 6‐month chronic alcohol consumption in rats resulted in increased μ‐calpain autolytic activation in skeletal muscles.48 In another study in aging muscle, S‐nitrosylation of cysteine residues of μ‐calpain were observed and were suspected to be responsible for negatively regulating the autolytic activation of μ‐calpain as in the absence of nitrosylation agent the μ‐calpain is rapidly autolyzed due to the release of Ca2+ from sarcoplasmic reticulum.49, 50 Recently we have also reported an increase in calpain activity as seen by the increase in its autolyzed fragments in a hyperhomocysteinemia AD rat model.51 The presence of phosphatidylinositol was also reported to decrease the Ca2+ requirement for both µ‐calpain and m‐calpain autolysis in vitro.52 However, autolysis was also shown to lead to the loss of proteolytic activity of both µ‐ calpain and m‐calpain in high salt condition as they form aggregates into dimers and trimers.53 The half in vitro maximal Ca2+ requirement for activation of µ‐ calpain and m‐calpain, respectively, was reported to be around 3 to 50 µM and 0.4 to 0.8 mM which are significantly higher than the cytosolic concentration found in the living cells (<0.05 µM),13 suggesting that other mechanisms (interaction with phosphatidylinositol) are involved in the activation of these proteases. But still, the phospholipid/calpain molar ratio required for lowering the concentration of Ca2+ required for their activation is difficult to be achieved in the living cells.54, 55 Phosphorylation also plays a role as both conventional calpains were reported to be phosphorylated at multiple sites by kinases including PKA, protein kinase C, protein kinase G, calmodulin kinase II, and casein kinase I.13 Phosphorylation by extracellular signal‐regulated kinase (ERK) was believed to activate these proteases, specially calpain‐2 is activated via direct phosphorylation at its Ser50 by ERK.56 Supporting this were the findings that both epidermal growth factor (EGF) and brain‐derived neurotrophic factor (BDNF) could activate m‐calpain in hippocampal neurons dendrites by ERK‐mediated phosphorylation.57 Even though phosphorylation by PKA was demonstrated to increase µ‐calpain activity,46, 47 however, others reported a decrease calpains activity58 following PKA phosphorylation. It should also be noted that the dimerization of the two subunits is also important for the calpain activity as CAPNS1 −/−mice are embryonically lethal because both conventional calpains cannot dimerize with the small subunit indicating the crucial role of the small subunit in calpain stability.59
Calpains have more than 100 various substrates, however, their activation in normal physiological conditions is controlled within smaller range to prevent the devastating consequences of massive proteolysis of these numerous substrates. Unfortunately, these regulatory mechanisms decrease with increase in age leading to an upregulated calpains activity implicating them in multiple conditions ranging from muscular dystrophy, diabetes, traumatic brain injury (TBI) to neurodegenerative diseases including AD.
4. CALPAIN IN ALZHEIMER’S DISEASE
4.1. Overview
Elevated calpain activity as a result of disturbances in cellular Ca2+ homeostasis has been related to neuronal death in various neurotoxic conditions spanning from ischemia to AD.60 Aberrant uncontrolled calpain‐mediated proteolysis has been associated with various neurodegenerative diseases including AD.13, 14, 61 τ Hyperphosphorylation and Aβ accumulation are the main markers of AD. These were found to be promoted in neuronal cells through Ca2+ dysregulation.62 These Ca2+‐regulated proteases “calpains” were abnormally activated and abnormally high in AD patients’ brains.63 Other reports have shown evidence of calpain involvement in the AD pathogenesis whereby the amount of m‐calpain in the cytosolic fraction of brain from AD patients is increased,13 the ratio of autolyzed to unautolyzed µ‐calpain threefold higher, and amount of CAST lowered in AD prefrontal cortex than in normal brains.61, 64 This decrease in CAST, which results from calpain and caspase proteolysis64 led to a subsequent decrease in the CAST/calpain ratio therefore increasing calpain activation.44, 65 Using APP overexpressing neurons, a study demonstrated an increased activation of caspase‐3 and calpain as shown by the calpain‐cleaved high levels of α‐fodrin and autolyzed µ‐calpain fragments while these two proteases were not activated in APP mutant neurons. Furthermore, E64d a membrane permeable calpain inhibitor almost completely inhibited the neuronal caspase‐3 activation and neuronal death induced by APP, an indication that high amount of wild‐type APP might induce neuronal apoptosis through calpain‐mediated caspase‐3 activation in postmitotic neurons.66
Upregulation of calpain activation in the brain of AD activates CDK5 through cleavage of p35 to p2567 and its homolog p39 to p2960 by excitotoxicity‐activated calpain and CDK5 activates BACE1 promoter by binding to its target STAT3, therefore increasing BACE1 expression, Aβ40 and Aβ42 production in transgenic mice.68 Thus, calpain overactivation leads to the two AD hallmarks, Aβ aggregates leading to SP and τ hyperphosphorylation leading to NFTs. The high amount of calpain‐cleaved spectrin detected in AD patients’ cerebrospinal fluid (CSF) is also suggestive of the hyperactivated calpains in the brains of these patients.63 Abnormal calpain‐2 but not calpain‐1 activation was shown to be increased in NFTs and induces degradation of nicotinic acetylcholine receptor α4 thus impairing cholinergic system in AD.69, 70
With age the central nervous system (CNS) becomes more and more susceptible to oxidative stress with widespread consequences on proteins, lipids, and nucleic acids.71, 72 As glutamate is the main excitatory neurotransmitter in the CNS, oxidative stress‐induced glutamatergic excitotoxicity lead to uncontrolled hyperactivation of NMDA receptors which increased neurons permeability to Ca2+ and therefore injuring them. Interestingly NMDA receptor antagonists’ treatment reduced the glutamatergic excitotoxicity associated damage.73 The resulting Ca2+ influx in the neurons induced an increase calpain activation which through increase production of Aβ74, 75 also increases the oxidative stress thus making a circle damaging more and more neurons resulting in observed neurodegeneration in the late stage of AD.
Inflammation seen as reactive astrocytes is also a marker of AD. In line with this a study found αΙΙ spectrin breakdown product (BDP) positive astrocytes in progressive activation stages, and this spectrin BDP persists till fully reactive astrocytes. This is suggestive of the likelihood of calpains and their spectrin proteolytic activity involvement in the physiological and morphologic transformation of astrocytes from resting to reactive fibrous astrocytes.76 In support of this overexpressing CAST in a mouse model of PD resulted in reduced calpain activity and decreased gliosis77 and calpain inhibition by A‐705053 decreased the astrocytic and microglial reactivity associated with AD‐like pathology in aged 3xTg‐AD mice.78 Furthermore, Aβ deposit was found to be in association with neuronal and astrocytic Ca2+ dysregulation.79, 80 Treatment of cortical neurons with Aβ oligomers led to Ca2+ influx resulting in calpain activation.67 All these highlighted the role that calpain plays in the neuroinflammation associated with neurodegenerative diseases.
4.2. Calpain in Aβ pathology
Aβ aggregates is one of the hallmarks of AD and Aβ oligomers aggregation process which was reported to adversely affect synaptic structure and plasticity81, 82 highlighting the observed cognitive impairments in AD. Aβ aggregation was found to be in association with neuronal and astrocytic Ca2+ disturbances.79, 80 Many studies have provided evidence of the increased level and/or activity of BACE1 upon a variety of assaults like oxidative stress,83 hypoxia,84 ischemia85, and TBI.86 Likewise, the level and activity of BACE1 were reported to be upregulated in AD brain,87, 88 and this might initiate or at least worsen AD progression. On another hand the level of BACE1 and the corresponding β‐carboxy terminal fragment (β‐CTF) of APP were decreased in CAST overexpressing APP/PS1 mice, meaning that this could be a mechanism via which calpain activation might promote APP processing through BACE1 synthesis.20 In support of this, is the observation that BACE1 protein levels were increased by m‐calpain overexpression in HEK293 cells expressing APP20, and also calpain activation affected APP processing as shown by increased β‐CTF production.89 However, various calpain inhibitors led to opposite effects.20 This indicates that downregulation of calpain suppressed the levels of BACE1, which ultimately decreased APP proteolysis and Aβ generation. BACE1 upregulation could be mediated by CDK5, which is calpain regulated through truncation of p35 to p25.74 CDK5 activates BACE1 promoter by binding to its target STAT3, therefore increasing BACE1 level and activity, Aβ40 and Aβ42 production in transgenic mice.68 Another experiment also reported that upregulation of calpain and p25/CDK5 resulted in phosphorylation of STAT3 (pSTAT3) which led to increase in both BACE1 and presenilin‐1 (component of γ‐secretase) transcription both of which are implicated in Aβ production.90 Of particular interest is the observation that p25, a product resulting from p35 cleavage by calpain, induces the production and accumulation of Aβ in vivo,75 presumably by increasing BACE1 transcription.68 The phosphorylation of APP at Thr668 is believed to play a critical role in its proteolytic cleavage into soluble Aβ producing fragment. The main kinase to phosphorylate APP at this site was shown to be CDK5,91 suggesting that the calpain‐mediated activation of CDK5 also leads to the amyloidogenic APP processing both via BACE1 expression and APP Thr668 phosphorylation. Thus, calpain activation triggered by various stressors including oxidative stress, excitotoxicity, and Aβ among other might increase the level of BACE1, which in turn promotes processing of APP and more Aβ production. In turn the upregulated Aβ levels lead to increased Ca2+ influx and therefore increasing calpain activation.63
It has been reported that extracellular Ca2+ influx can be triggered by Aβ through stimulation of membrane ion channels or receptors, such as ionotropic glutamate receptors.92 Aβ could also impair the distribution of intracellular Ca2+ by increasing membrane permeability to Ca2+ via oxidative stress.93, 94 It was also found that in familial AD, some presenilin mutations, linked to increased Aβ levels, impaired Ca2+ homeostasis by deregulating voltage‐gated internal Ca2+ channels ryanodine receptor,95, 96 inositol 1,4,5‐triphosphate (IP3) channel,97 or sarcoendoplasmic reticulum Ca2+ ATPase (SERCA).96, 98 Increased Ca2+ in hippocampal neurons has been observed and thought to originate from endoplasmic reticulum leading to calpain hyperactivation99 and BAPTA a Ca2+ chelators was shown to inhibit calpain activation.100 Aβ was reported to enhance calpain activation by inducing extracellular Ca2+ influx99 and NMDA receptors has been found to play a role as shown by studies where M7K801 a specific NMDA receptor inhibitor101 and memantine an NMDA receptor antagonist102 were reported to attenuate the Aβ‐induced Ca2+ influx thereby blocking calpain activation in hippocampal cultured neurons.99 Cholesterol a risk factor for AD103 might also play a role in Aβ‐mediated calcium influx through influencing NMDA receptors components and their membrane microdomain localization at synaptic sites.104 Aβ was also found to activate NR2B‐containing NMDA receptors.105
Another way Aβ affect Ca2+ is through the βAPP intracellular domain (AICD) which was shown to be involved in Ca2+ signaling106 or homeostasis in different cell culture models.107 Advance glycation end products (AGEs) influence Aβ generation through induction of βAPP expression via oxidative stress in SHSY5Y cells108 while injection of receptors for AGEs (RAGE) in AD transgenic mice model increases Aβ accumulation and SP formation.109 The same study revealed an increased BACE1 expression and activity by RAGE which was mediated by AGEs or Aβ stimulation of RAGE thus increasing cytosolic Ca2+ concentration and finally activating the transcription factor NFAT1 which leads to increased transcription of BACE1 in SHSY5Y cells.109 The increased cytosolic Ca2+ might also activate calpain.
Together, the data reviewed here suggest that in AD calpain and Aβ form a closed circle whereby Aβ leads to the activation of calpain, which in turn leads to a feedforward process to produce more Aβ and therefore more calpain activation.
4.3. Calpain in τ pathology
Calpain is also involved in AD by favoring τ pathology including indirect‐mediated phosphorylation and direct‐cleavage favoring aggregation. Potential candidate τ protein kinases include CDK5110 and GSK3.111 Both CDK5 and GSK3β were shown to be activated by calpain. Studies have shown that, τ phosphorylation is achieved through increased activation of both CDK5 and GSK3β, the main kinases of τ, and decreasing PP2A activity, the main phosphatase of τ. Several kinases induce τ phosphorylation at over 38 Ser/Thr residues, these include ERK, CDK5,112 GSK3β,113 calcium/calmodulin‐dependent protein kinase II (CaMKII),114 PKA,115 the dual‐specificity tyrosine phosphorylation‐regulated kinase‐1A (Dyrk1A),116 mitogen‐activated protein (MAP) kinase117, and stress‐activated protein kinases (SAPKs)118 among others.
The most elucidated pathway of calpains involvement in AD is the calpain‐mediated proteolysis of p35 to a 25 kDa form p25. Calpain was reported to cleave p35, an activator of CDK5, and generates p2520, 67, 74, 78 which has longer half‐life than p35, and therefore is a more potent CDK5 activator, leading to a sustained activation of CDK5 and causes phosphorylation of many of its substrates. Since τ is a substrate of CDK5, it is not surprising that the p25‐mediated activation of CDK5 led to τ hyperphosphorylation and the resulting NFTs formation. Furthermore, this p25 was found to accumulate in brains of AD patients,67 suggesting that calpain‐cleaved p25 is at least in part responsible for the hyperphosphorylation of τ in the intracellular NFTs observed in AD brains.119 Moreover, the production of p25 resulted in an increased Aβ level in forebrain before any sign of neuropathology in APP transgenic mice75 probably through upregulation of BACE1 expression.68 Abnormal τ phosphorylation was observed in the APP/PS1120 and this was prevented by overexpressing CAST20 also suggesting calpain intervention.
GSK3β the other main τ kinase was also reported to be activated by calpain.19, 121, 122 Incubation of mouse brain tissue homogenates with CaCl2 was found to trigger the cleavage of GSK3β in two fragments of 40 and 30 kDa in a time‐dependent way, while these cleaved fragments disappeared in the absence of Ca2+.19 Compared with control, this Ca2+‐mediated proteolysis increased the GSK3β kinase activity. Furthermore, this cleavage and activation of GSK3β were inhibited by calpeptin, a calpain inhibitor, not other protease inhibitors, suggesting that this breakdown and increased activity may be due to Ca2+‐dependent calpain activation.19 It should be noted also that phosphorylation of GSK3β at Ser9 abolished the calpain‐mediated cleavage at both C and N‐termini while phosphorylation at Ser‐389 only inhibit N‐terminal cleavage.122
CaMKII can undergo autophosphorylation (activation) in the presence of Ca2+,123 was shown to be a τ kinase and has a role in memory and neuroplasticity.114 However, its redistribution or prolonged activation could implicate it in AD. In support, a correlation with cognitive dysfunction in MCI and AD subjects was found with the reduction of T286‐autophosphorylation in apical dendrites of granule cells of the DG.124 Resveratrol was shown to decrease τ hyperphosphorylation via suppression of GSK3β and CaMKII activities in N2A cells exposed to formaldehyde.125 Strong and sustained stress‐like acute severe CO poisoning could lead to both calpain and CaMKII expression and activation.126 Calpain might also induce Ca2+ as revealed by studies where increased expression of µ‐calpain resulted in mitochondrial damage127 with consequent production of reactive oxygen species (ROS) which in turn resulted in a significant Ca2+ increase128 and thus activation of CaMKII. Via membrane skeleton protein degradation calpain was also shown to be upstream activator of CaMKII resulting in ischemia/reperfusion (IR) injury.129 Interestingly CaMKs family are shown to be substrates of calpain. More specially calpain‐mediated CaMKIV activation is characterized in the apoptosis of SHSY5Y cells130 and cerebellar granule cells,131 while m‐calpain was shown to activate CaMKII‐gamma which in turn led to cAMP/PKA‐mediated ERK1/2 phosphorylation resulting in caspase‐3–mediated apoptosis in head kidney macrophages.132 It should also be noted that CaMKII plays an antiapoptotic.
PKA is also reported to be a τ kinase as revealed by the study where cAMP‐dependent PKA phosphorylation of τ at serine 214 was observed in monkey133 and SHSY5Y cells.134 PKA activator forskolin was shown to induce τ phosphorylation at Thr231.115 In addition, an in vitro study showed that in the presence of Ca2+ incubation of μ‐calpain with PKA resulted in more autolysis than control, but lower than the AP group. Furthermore, PKA was found to catalyze μ‐calpain phosphorylation at serine 255, 256, and 476, and upregulated μ‐calpain activity.46, 47 Together this could lead to the downstream of calpain including Aβ and τ pathologies. However, another study showed that the degradation of calpain substrate proteins was higher in AP‐treated sample and less in PKA‐treated ones.135
Both APP and τ were reported to be substrates of Dyrk1A, therefore Dyrk1A is linked to the two main hallmarks of AD. Dyrk1 inhibition rescued the observed cognitive deficits in 3xTg‐AD mice with a concomitant reduction in Aβ and τ pathology136 via decreasing phosphorylation of both APP and insoluble τ, thereby increasing APP metabolism and decreased Aβ levels.137 DYRK1A phosphorylates τ on 11 different Ser/Thr residues many of which are found in NFTs of AD,116 and its overexpression led to increased 3R‐τ and decreased 4R‐τ.138 This is mediated through influence on the alternative slicing of τ exon 10.139, 140 Inhibition of Dyrk1A by Leucettine L41141 or EHT 5372142 mitigated both τ and Aβ pathologies. Interestingly Dyrk1A was reported to be truncated at the C‐terminus and this was associated with increased µ‐calpain activity in AD brain. Moreover, in vitro studies revealed that µ‐calpain proteolyzed Dyrk1A at both C and N termini and enhanced its τ kinase activity, with C‐terminus truncation producing Dyrk1A with a stronger activity than its full‐length protein in promoting τ phosphorylation and exon 10 exclusion.143
Creatine kinase (CK) is found in many tissues including brain, cardiac, and skeletal muscles. The creatine/phosphocreatine system which is under the regulation of CK, plays an important role in energy metabolism therefore very important for neuron. It is well known that energy metabolism and the CK function are affected in AD brain and in cells exposed to the Aβ peptide.144 Creatine was also focally detected in hippocampi from APP transgenic mice and postmortem AD patients brain and was suggested to be a marker of the disease,145 probably due to a decreased CK activity. Supporting this, other studies also found a decrease in this enzyme and proposed that this could due to oxidative damage as CK is sensitive to oxidative stress.146, 147 Moreover, CK was also found to be a calpain substrate.148 Accordingly, another study reported a decrease in CK activity with increased calpain activity149 suggesting the degradation of this enzyme by calpain. Furthermore, calpain inhibition was shown to prevent cell death in IR, via decreasing DNA strand breaks and CK release, suggesting calpain is an upstream of CK.150, 151
Calpains also activates ERK/MAP kinases152 and other kinases that are also involved in τ phosphorylation. Both calpain and ERK1,2 were found to be activated along with concomitant neurofilament phosphorylation and interestingly the inhibition of calpain by calpeptin abolished both neurofilament phosphorylation and ERK1,2 activation, while inhibiting ERK1,2 by a specific inhibitor Meck1,2 had no effect on calpain activation suggesting calpain to be an upstream activator of the ERK/MAP pathway.152
To induce τ phosphorylation, calpain could also directly or indirectly affect phosphatases. PP2A account for most of the dephosphorylation activity of τ, however protein phosphatase 1 (PP1) also play a part. Therefore, even though it was shown to be neuroprotective, the degradation of PP1 mediated by µ‐calpain153 might increase τ phosphorylation as seen in AD. Calpain was also shown to mediate the cleavage of the PP2A regulatory subunit α4 leading to the proteasomal degradation of PP2Ac resulting in τ hyperphosphorylation.154 GSK3β one of the main kinase of τ is known to be truncated by calpain leading to its increased activity. The underlying mechanism is reported to be its translocation to the nucleus and its enhanced interaction with PP2A upon truncation, promoting its Ser9 dephosphorylation and thus increasing its activity155 highlighting an indirect intervention of calpain on PP2A‐mediated activation of this kinase and involvement in AD. Also, extra‐synaptic N‐methyl‐d‐aspartate receptor (NMDAR) activation led to neuronal death as a result of degradation of striatal‐enriched protein tyrosine phosphatase (STEP) mediated by calpain.153 Tyrosine phosphatase (PTPN13) is reported to dephosphorylates and inhibits c‐Abl (τ kinase) but also to be a substrate of m‐calpain which inactivates its phosphatase activity.156 Calpain was also reported to induce the degradation of inositol polyphosphate‐4‐phosphatase (INPP4),157 a negative regulator of the phosphoinositide 3‐kinase (PI3K) signaling pathway.158 PI3K is known to phosphorylate GSK3β at Ser9 inhibiting its kinase activity.159 Thus, by degrading INPP4 and PTPN13, calpain might respectively increase the kinase activities of GSK3β and c‐Abl, therefore increasing τ phosphorylation and accumulation.
Besides τ phosphorylation, another way calpain activation play a role in τ‐mediated neurodegeneration is through inducing τ cleavage.160, 161, 162, 163, 164 But this cleavage might be at earlier stage of the disease since hyperphosphorylation makes τ, which normally is rapidly cleaved by the calpains,13, 165 highly resistant to calpain truncation,166, 167 so the NFTs in AD are resistant to calpain168 suggesting phosphorylation might regulate τ susceptibility to calpain. Hence, because neurofilament proteins are good substrates of calpains, it may play an important role in the AD observed necrotic neuronal death. Therefore, small fragments of τ, which were believed to be generated by calpain‐1 proteolysis, have been observed in Aβ‐treated hippocampal neurons and these fragments of around 17 kDa (τ 45 to 230) are neurotoxic.162, 163 Another study also reported the presence of these types of fragments in the neocortex of AD brains as well as in brain areas affected by other tauopathies and their levels correlated with increased calpain activity.167 Furthermore, these 17 kDa τ fragments induced neuronal cell death even in the absence of Aβ oligomers following their expression in neuronal and nonneuronal cell types or in an in vivo drosophila model system.162, 163, 169 Fragmented τ has also been found in mitochondria present in synaptosomal fraction from AD brain and the level of these fragments were found to be reduced in culture neurons treated with calpain inhibitors.160 These types of fragments were reported to induce mitochondrial dysfunction.164 By the way it could be also noted that calpain‐2 activation generates smaller τ fragments with nonobservable neurotoxic effects in CNS neurons.161
Collectively, these data strongly provided evidence of the deleterious role of calpain in τ hyperphosphorylation and generation of neurotoxic τ fragments in neurodegeneration.
4.4. Calpain in autophagy/lysosomal impairment
Impaired autophagy/lysosomal pathway has been implicated in AD resulting in the buildup of vacuoles containing harmful proteins11, 12 seen as granulovascular degeneration. Oxidative stress is clearly connected to autophagic pathway dysregulation as a result of ROS‐mediated carbonylation of proteins and eventually lead to neuronal dysfunction, degeneration, and cell death that is seen in many neuropathological conditions including AD. Hsp70.1 is one of the major component of the human Hsp70 proteins family which are chaperones that help the cell get rid of harmful aggregated denatured proteins in various environmental insults such as heat, oxidative stresses, and ischemia.170, 171 AD brains were reported to show progressive ischemic and oxidative assaults that lead to Ca2+ homeostasis dysregulation and calpain activation. Another way Hsp70s confer their cellular protection is via inhibition of rupture/permeabilization of lysosomal membrane,172, 173 thus maintaining lysosomal membrane integrity and are essential for both endosomal and lysosomal autophagy. Elevated levels of Hsp70 carbonylation was reported in the brain of mild cognitive impairment which is an early phase of AD174 and in hippocampal CA1 in IR brain.175 Interestingly Hsp70.1 is found to be an in vivo22 and in vitro23 substrate of calpain. Hsp70.1 has been demonstrated to be more prone to the calpain cleavage following carbonylation by oxidative stressors, such as HNE or H2O2.176 Therefore calpain‐induced Hsp70.1 truncation together with ROS‐mediated Hsp70.1 carbonylation, lead not only to impaired autophagy and buildup of harmful proteins but also to lysosomal rupture/permeabilization leading to release of lysosomal enzymes and cell death in both IR and AD brains.23, 176, 177 Moreover, LAMP2a a molecular marker for lysosome stability and an essential component of chaperone‐mediated autophagy,178 was also reported to be cleaved by calpain leading to lysosomal rupture and cell death.24 Also, downregulation of LAMP2a via RNAi, led to impaired lysosomal degradation of long‐lived proteins and chaperone‐mediated autophagy substrates in neuronal systems.179 Hsp70.1 was also shown to attenuate τ toxicity via maintaining τ in its soluble nonaggregated nature and by facilitating its degradation.180, 181, 182 Thus, impaired Hsp70.1 function as a result of carbonylation and/or cleavage, as a result of calpain activation, could lead not only to lysosomal destabilization but also τ aggregation and NFTs formation.182, 183
Together these suggested that cleavage of Hsp70.1 and LAMP2a is another way that calpain contributes to neurodegenerative changes in AD and related conditions.
4.5. Calpain in synaptic dysfunction
Among calpain substrates are included both presynaptic and postsynaptic proteins, so it is not surprising that calpain is implicated in synaptic dysregulation. Accumulating evidence have been provided by many studies as per the abnormal activation of calpains not only favors Aβ accumulation and τ hyperphosphorylation in neurons but also is implicated in the development of synaptic pathology in neurodegenerative diseases including AD.77, 78, 184 Overactivation of calpain in the brain of transgenic mice, induced a myriad of AD pathological markers including βAPP processing, amyloid deposits, τ phosphorylation, activation of astrocytes, synapse loss, and cognitive impairment.20, 63, 77 These synaptic dysfunctions seen as synaptic loss seems to significantly correlate with the functional and cognitive deficits observed in different stages of AD.185 Supporting this is a report from a study, where the calpain involvement in synaptic loss is highlighted by its cleavage of proteins in the presynaptic termini and/or the postsynaptic elements, thus leading to functional changes in affected brain areas.186 Using cell culture and animal models of AD Aβ was recently shown to induce a significantly reduced level of dynamin‐1, a neuron‐specific GTPase highly enriched in presynaptic terminals which function is to detach synaptic vesicles from the membrane so that they can reenter the synaptic vesicle pool and be refilled for future release.187 This decrease in dynamin protein lead to the depletion of synaptic vesicles and the accumulations of empty pouches at presynaptic termini, thus leading to synaptic dysfunction. This Aβ‐mediated dynamin‐1 reduction was believed to be in part due to calpain‐mediated proteolysis.99, 188 At the postsynaptic element, calpain hyperactivation resulted in the degradation of several proteins changing the ultrastructure of the postsynaptic density (PSD). Following this event are the accelerated cleavage of PSD95 and the NMDA receptor subunits NR1 and NR2A and 2B.189, 190 Calpain was shown to also exacerbate NMDA neurotoxicity by cleaving mGluR1.191, 192 Additionally, calpain activation induces the cleavage of PKA leading to a decrease in both its regulatory and the catalytic subunits thus decreasing the activity of CREB, an important memory‐related protein, aggravating synaptic dysfunction193 as seen by the resulting memory impairment. In a mouse model of PD overexpressing CAST the observed decreased calpain activity was found to be accompanied by a concomitant reduction of the cleaved synaptic protein α‐synuclein ameliorating the observed neurodegeneration.77 In support, using calpain inhibitor A‐705053 treatment in 3xTg‐AD mice model significantly increased the levels of synapsin‐1 and PSD95 to nontransgenic animal level thereby ameliorating the synaptic loss observed in these AD mice model.78
All of these revealed the undeniable role that calpain played in synaptic pathologies.
4.6. Calpain in learning and memory
Many studies have provided data concerning the calpain‐mediated dysregulation of learning and memory‐related molecules in AD including ERK, CREB, and DARPP‐32 among others. CREB, a key player in learning and memory and synaptic plasticity, and a core component involved in converting short‐term to long‐term memory,21 has been found to be dysregulated in various neurological disorders, including AD, Huntington’s disease, Parkinson’s disease, ischemia, and addiction.194, 195 In line with this ERK and CREB phosphorylation (activation) were reported to be decreased in transgenic AD mouse model,20, 184 while overexpression of the endogenous calpain inhibitor CAST prevented this decrease with concomitant improvement in learning and memory ability in these animals.20 Also, selectively inhibiting calpain‐2 by C2‐I resulted in an improved state of learning and memory through increasing timely activation of ERK.196 DARPP‐32 is a key player involved in the regulation of the PKA‐CREB signaling pathway.197 An increase in the cAMP level leads to the activation of PKA and phosphorylation of DARPP‐32 at Thr34, which inhibits PP1 a key phosphatase of CREB, therefore increasing CREB phosphorylation.198 Interestingly, in AD brains DARPP‐32 protein level was found to be about 20% lower compared with control brains, however two fragmented forms, about 28 and 4 kDa, of DARPP‐32 were found in high amount in AD brains.18 Cleavage of DARPP‐32 protein was also reported in both OA or Aβ peptides treated SHSY5Y cells and primary neurons along with a simultaneous decrease in CREB phosphorylation ultimately resulting in synaptic abnormalities.18 Consistently, calpain inhibition reversed the DARPP‐32 truncation in the OA‐treated neurons while recombinant DARPP‐32 was cleaved by calpain in an in vitro assay.18 In support of this, restoring CREB phosphorylation through calpain inhibition has been reported to ameliorate synaptic transmission and memory in an AD mouse model.184 This is in accordance with the findings that calpain affects CREB kinases such as PKA and CaMKII.
These data together suggested that in AD, calpain‐mediated DARPP‐32 truncation and activation of PP1 might be involved in the impairment of the PKA‐CREB signaling pathway and therefore impairing learning and memory ability in the affected subject. It is worth noting that, in normal physiological conditions, calpain also play an important role in synaptic plasticity and thus a positive regulator of learning and memory.199, 200 Recently it was reported that calpain‐1 and calpain‐2 are implicated in learning and memory via two opposing effects where calpain‐1 was found to positively affect LTP and learning and memory while calpain‐2 restricted the extent of learning and memory.196, 201 Thus, calpains are essential for life but their uncontrolled hyperactivation is destructive and implicate them in the etiopathology of many diseases conditions including AD.
5. CALPAIN INHIBITORS AS AD THERAPY
The undeniable role that calpains play in AD progression and pathogenesis made it a potential target for therapeutics, and thus has stimulated the development of many calpain inhibitors for AD treatment. Various insults to the CNS activate calpain, leading to Aβ production, τ hyperphosphorylation, synaptic dysfunction, and neuronal degeneration.13 Upregulated calpain activity due to Ca2+ influx or decrease of CAST causes the proteolysis of many functional proteins in the brain.84, 202, 203 Together, this implies that calpain play a multistage role in AD progression from the earlier APP processing via BACE1 activation to the later CDK5 and GSK3β activation, the resulting τ hyperphosphorylation, autophagy impairments, synaptic and neuronal dysfunction, and the finally observed neurodegeneration (Figure 2). Thus, calpain inhibition could be a potential option in preventing neuronal degeneration induced by multiple neurotoxic factors.
Figure 2.
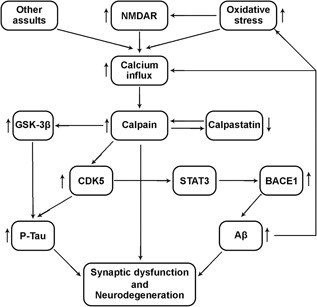
Summary of calpain involvement in AD pathology. Intracellular calcium influx resulting from increase oxidative stress, increased NMDAR activation and other assaults together lead to increased and uncontrolled calpain activation. This calpain hyperactivation could also result from impaired regulatory mechanisms like decreased calpastatin level which could also be a result of calpain proteolysis. The resulting calpain upregulation then lead to truncation and increased GSK3β activation, cleavage of p35 to p25 with resulting prolonged CDK5 activation. On one hand these two (GSK3β and CDK5) lead to τ hyperphosphorylation, while on the other hand, the prolonged CDK5 activation lead to increase BACE1 expression and activation via STAT3 with consequent increased Aβ production. The τ hyperphosphorylation and Aβ deposit together lead to synaptic dysfunction and neurodegeneration. The Aβ peptides could also exert a positive feedback leading to increased oxidative stress and/or increased calcium influx and more calpain activation and the cycle continues. AD, Alzheimer’s disease; Aβ, amyloid beta; BACE1, beta APP cleaving enzyme 1; GSK3β, glycogen synthase kinase 3β; NMDAR, N‐methyl‐d‐aspartate receptor
A study by Trinchese et al184 showed that synthetic calpain inhibitors improved memory and synaptic transmission without significant effects on Aβ deposit in APP/PS1 mice. Considering that synthetic calpain inhibitors are not highly specific, they might affect other proteases and therefore may influence neurodegeneration processes. Thus, inhibiting calpain by overexpressing its endogenous inhibitor CAST would be a better way to study isolated calpain effect. Interestingly, CAST overexpression in APP/PS1 (APP/PS1/CAST),20 APP23 (APP23/CAST),204 APP (APP/CAST),63 and JNPL3 (CAST/JNPL3)205 mice led to a significant decrease in Aβ level and/or τ phosphorylation and this effect appeared to be mediated through decreasing BACE1 and β‐CTF levels. ThisCAST‐mediated inhibition of calpain in the nervous system is more responsive in pathological conditions.204 Similar effects with CAST overexpression have also been demonstrated with synthetic inhibitors as they also suppressed calpain and reduced BACE1 levels, which attenuated APP processing and therefore Aβ generation.20 However, stimulation and activation of calpain by various stressors20 or CAST knock‐out63 caused the opposite effects. Additionally, the CAST overexpression also prevented astrocyte activation, microgliosis, and synapse loss.20, 63 APP/PS1 mice showed an increase in AT8‐positive phosphorylated τ protein in their brain in comparison with wild‐type control mice, while overexpressing CAST abolished it20 and synthetic calpain inhibitors were reported to improve synaptic transmission and memory in AD mouse model.184 Synthetic calpain inhibitors also showed neuroprotective effect in neuronal injuries like ischemia and excitotoxicity‐induced neuronal death.64, 206 To date many calpain inhibitors have been synthesized owing to the role played by this enzyme in AD pathogenesis. These inhibitors include compounds like SNJ‐1945, a compound with good metabolic stability and permeability,207 and has been shown to decrease in vitro and in vivo death of retinal cell.208, 209 MDL28170, another calpain inhibitor, has been reported to have some protective effects against excitotoxicity.191, 210 E64, BDA‐410, and leupeptin calpain inhibitors have also showed protective effects in AD cell culture models.78, 162, 211, 212 Other inhibitors include SJA6017, ALLN,207 and a recently produced A‐705053.213 This novel calpain inhibitor A‐705053, which can easily be taken orally, has improved pharmacokinetics and has provided interesting results as it was shown to prevent calpain‐mediated truncation of dynamin‐1 and τ in primary hippocampal neurons culture.188 It was also effective when used as preventive, simultaneous, or curative treatment against Aβ‐triggered neurodegenerative process188 and prevented neurodegeneration of the nucleus basalis magnocellularis induced by Aβ oligomer.214 A‐705053 was reported to reduce both Aβ40 and Aβ42 and also decreased the thioflavin‐s positive Aβ aggregates through decreasing BACE1 and increasing ATP‐binding cassette transporter A1 (ABCA1) expression, which respectively decrease the production and increase the clearance of Aβ78 and also inhibited Aβ‐induced synaptic deficits214, 215 and prevented in vitro and in vivo stress‐induced hyperphosphorylation of τ.216 Unfortunately, this calpain inhibitor also has limited selectivity against other cysteine protease cathepsins B, K, L, S, and C thus preventing further investigation on this compound.217 Moreover, inhibition of cathepsins C, L, and S might induce serious side effects like immunosuppression while inhibition of cathepsins B and K is not detrimental.218, 219, 220, 221 This idea led to the very recent discovery of new calpain inhibitors derived from A‐705053 which retain the calpain inhibitory effect and with more selectivity. These compounds, A‐933548 and A‐953227, more specially the later, have more preclinical safety with in vivo anti‐AD profiles.217 But still further experiments are needed before considering the use of this calpain inhibitor as potential therapeutic treatment. Other recent developments result from another study that showed that inhibiting calpain‐2 by its selective inhibitor C2‐I enhanced learning and memory in both normal and genetically memory impaired mice by prolonging ERK activation.196 Another report revealed the downregulatory effect of both τ hyperphosphorylation and Aβ peptide production of synthetic chalcone derivatives222 conferring them a neuroprotective effect via decreasing p‐MAPK and BACE1 protein levels. Two other calpain inhibitors NYC438 and NYC488, have also been developed and showed promising results and had proven efficacy, potency, and safety.223
Collectively this suggested the promising potential of inhibiting calpain in AD therapeutics.
6. FUTURE DIRECTIONS
The two main hallmarks of AD are well established to be Aβ aggregate into SP and τ hyperphosphorylation forming NFTs. These are accompanied by a decline in cognitive functions including impaired memory and behavioral deficits. With Aβ deposit as the trigger of the disease, many of the primary AD therapies have focus on reducing Aβ load in the affected brain areas. Many of these strategies including monoclonal antibodies treatment against Aβ, vaccination with Aβ, Aβ aggregation inhibitors, β‐secretase interference, nerve growth factors, just to mention these, have succeeded in reducing the Aβ burden but often failed to improve the cognitive deficit and the resulting neurodegeneration. Considering this and also considering the fact that NFTs correlate more with the degree of the disease, therapeutic strategies that target both Aβ and τ hyperphosphorylation would be more efficient in treating the disease. From the summarized data above, it is clear that calpain hyperactivation is a big player in the generation and/or exacerbating these two AD hallmarks. Thus, targeting calpain either through downregulation or inhibition could be a way out. As reviewed earlier inhibiting calpain through overexpression of its endogenous inhibitor CAST and through the use of different synthetic inhibitors have provided promising results in neurodegenerative animal and cell models for the treatment of this conditions.
Calpain gene deletion has been proven to be lethal224 however suppression of calpain‐1 expression using specific probes prevented cell injury in human hepatic cancer cell lines induced by oxidative stress.225 In AD cell model other calpain inhibitors leupeptin, BDA‐410 and E64, have shown protective effects against cell death,78, 162, 211, 212 while MDL28170 had protective effect against excitotoxicity.191, 210 It is unfortunate to note that most of the currently used calpain inhibitors in AD therapeutics are limited due to their physical and/or chemical properties resulting in inefficient cellular penetration, selectivity, and kinetics. However, hope in this quest occurs from recent development of newly synthetic inhibitors, like A‐705053, A‐933548, A‐953227, NYC438, NYC488, and chalcone derivatives which have proven efficacy.78, 188, 213, 214, 217, 222, 223 Moreover, preliminary studies using these inhibitors in mouse and rat models of AD provided interesting results recovering cognitive functions when treatment was administered at an early age in these animals.18, 212, 214
Taken together, the data reviewed in this literature highlighted the undeniable involvement of calpain upregulation in AD pathology and the potential hope that reside in the development of calpain inhibitors as AD therapeutic tool and related neurodegenerative diseases. But there is still a long way to go in establishing the specificity, efficacy, safety, availability, and adequacy in the inhibitory effect of these compounds for the use in clinical trials and for the subsequent consideration for long‐term AD treatment, since calpains also play a normal physiological role as mentioned above. Supporting this is also a study where calpain inhibition led to the impairment of insulin release.226 It should also be noted that recent developments have provided evidence on the differential involvement of calpains in learning and memory and neurodegeneration where calpain‐1 has been reported to enhance learning and memory and neuroprotective while opposite effects have been attributed to calpain‐2.196, 201 Thus, specific targeting of these isoforms should be considered under different pathological conditions.
Clear and palpable evidence are provided from innumerable studies ranging from cell models to AD patients passing through animal models on the uncontestable role that upregulation of calpain plays in the pathogenesis of numerous neurodegenerative diseases among AD. This triggered a quest for the development of strategies aiming in decreasing calpain activity as treatment of AD and other neuronal injuries involving calpain hyperactivation. The result of this is the synthesis of numerous calpain inhibitors. However, many of these inhibitors are limited because of their insufficient cellular penetration, selectivity, and kinetics. Non‐the‐less, the idea that calpain inhibition could be a potential therapeutic strategy in neurodegenerative diseases still remain one of the promising option as supported by evidence from in vitro, in culture, and animal model studies using calpain inhibitors like MDL28170, A‐705053, A‐953227, NYC438, and NYC488 among others. However, it should be noted that these experiments were conducted in controlled environments with less or minimal influence of age‐related chronic diseases and in less complex organisms than humans.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
ACKNOWLEDGMENTS
This study was supported in parts by grants from National Natural Science Foundation of China (81571255, 31771114, and 31528010), Innovative Research Groups of the National Natural Science Foundation of China (81721005), grant from the Ministry of Science and Technology of China (2016YFC1305800), and the Academic Frontier Youth Team Project to Xiaochuan Wang from Huazhong University of Science and Technology.
Mahaman YAR, Huang F, Henok KA, Maibouge TMS, Ghose B, Wang X. Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med Res Rev. 2019;39:608‐630. 10.1002/med.21534
[Correction added after publication 16 October 2018: in the author group, author name is corrected from “Kessete Afewerky Henok” to “Henok Kessete Afewerky”]
References
REFERENCES
- 1. Iulita MF, Cuello AC. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol Sci. 2014;35(7):338‐348. [DOI] [PubMed] [Google Scholar]
- 2. Alzheimer’s Association . 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12(4):459‐509. [DOI] [PubMed] [Google Scholar]
- 3. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61(3):378‐384. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Braak E, Grundke‐Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65(3):351‐355. [DOI] [PubMed] [Google Scholar]
- 5. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li YM, Xu M, Lai MT, et al. Photoactivated gamma‐secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405(6787):689‐694. [DOI] [PubMed] [Google Scholar]
- 7. Wang JZ, Wang ZH, Tian Q. Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci Bull. 2014;30(2):359‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng Y, Xia Y, Yu G, et al. Cleavage of GSK‐3beta by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK‐3beta activity induced by H2O2 . J Neurochem. 2013;126(2):234‐242. [DOI] [PubMed] [Google Scholar]
- 9. Mondragón‐Rodríguez S, et al. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxid Med Cell Longevity. 2013;2013:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chami L, Checler F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta‐amyloid production in Alzheimer’s disease. Mol Neurodegener. 2012;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Funderburk SF, Marcellino BK, Yue Z. Cell “self‐eating” (autophagy) mechanism in Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goll DE, Thompson VF, LI H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83(3):731‐801. [DOI] [PubMed] [Google Scholar]
- 14. Ono Y, Sorimachi H. Calpains: an elaborate proteolytic system. Biochim Biophys Acta. 2012;1824(1):224‐236. [DOI] [PubMed] [Google Scholar]
- 15. Saido TC, Nagao S, Shiramine M, et al. Autolytic transition of mu‐calpain upon activation as resolved by antibodies distinguishing between the pre‐ and post‐autolysis forms. J Biochem. 1992;111(1):81‐86. [DOI] [PubMed] [Google Scholar]
- 16. Averna M, De Tullio R, Salamino F, Minafra R, Pontremoli S, Melloni E. Age‐dependent degradation of calpastatin in kidney of hypertensive rats. J Biol Chem. 2001;276(42):38426‐38432. [DOI] [PubMed] [Google Scholar]
- 17. Benuck M, Banay‐Schwartz M, DeGuzman T, Lajtha A. Changes in brain protease activity in aging. J Neurochem. 1996;67(5):2019‐2029. [DOI] [PubMed] [Google Scholar]
- 18. Cho K, Cho MH, Seo JH, et al. Calpain‐mediated cleavage of DARPP‐32 in Alzheimer’s disease. Aging cell. 2015;14(5):878‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goñi‐Oliver P, Lucas JJ, Avila J, Hernández F. N‐terminal cleavage of GSK‐3 by calpain: a new form of GSK‐3 regulation. J Biol Chem. 2007;282(31):22406‐22413. [DOI] [PubMed] [Google Scholar]
- 20. Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2010;285(36):27737‐27744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets. 2003;7(1):101‐114. [DOI] [PubMed] [Google Scholar]
- 22. Nakajima E, David LL, Bystrom C, Shearer TR, Azuma M. Calpain‐specific proteolysis in primate retina: Contribution of calpains in cell death. Invest Ophthalmol Vis Sci. 2006;47(12):5469‐5475. [DOI] [PubMed] [Google Scholar]
- 23. Yamashima T. Can ’calpain‐cathepsin hypothesis’ explain Alzheimer neuronal death? Ageing Res Rev. 2016;32:169‐179. [DOI] [PubMed] [Google Scholar]
- 24. Arnandis T, Ferrer‐Vicens I, García‐Trevijano ER, et al. Calpains mediate epithelial‐cell death during mammary gland involution: mitochondria and lysosomal destabilization. Cell Death Differ. 2012;19(9):1536‐1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guroff G. A neutral, calcium‐activated proteinase from the soluble fraction of rat brain. J Biol Chem. 1964;239:149‐155. [PubMed] [Google Scholar]
- 26. Sorimachi H, Ono Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc Res. 2012;96(1):11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sorimachi H, Hata S, Ono Y. Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp Anim. 2010;59(5):549‐566. [DOI] [PubMed] [Google Scholar]
- 28. Ono Y, Ojima K, Shinkai‐Ouchi F, Hata S, Sorimachi H. An eccentric calpain, CAPN3/p94/calpain‐3. Biochimie. 2016;122:169‐187. [DOI] [PubMed] [Google Scholar]
- 29. Sorimachi H, Suzuki K. The structure of calpain. J Biochem. 2001;129(5):653‐664. [DOI] [PubMed] [Google Scholar]
- 30. Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca(2+) switch aligns the active site of calpain. Cell. 2002;108(5):649‐660. [DOI] [PubMed] [Google Scholar]
- 31. Tompa P, Emori Y, Sorimachi H, Suzuki K, Friedrich P. Domain III of calpain is a ca2+‐regulated phospholipid‐binding domain. Biochem Biophys Res Commun. 2001;280(5):1333‐1339. [DOI] [PubMed] [Google Scholar]
- 32. Carafoli E, Molinari M. Calpain: a protease in search of a function? Biochem Biophys Res Commun. 1998;247(2):193‐203. [DOI] [PubMed] [Google Scholar]
- 33. Kiss R, Kovács D, Tompa P, Perczel A. Local structural preferences of calpastatin, the intrinsically unstructured protein inhibitor of calpain. Biochemistry. 2008;47(26):6936‐6945. [DOI] [PubMed] [Google Scholar]
- 34. Maki M, Takano E, Mori H, Sato A, Murachi T, Hatanaka M. All four internally repetitive domains of pig calpastatin possess inhibitory activities against calpains I and II. FEBS Lett. 1987;223(1):174‐180. [DOI] [PubMed] [Google Scholar]
- 35. Hanna RA, Garcia‐Diaz BE, Davies PL. Calpastatin simultaneously binds four calpains with different kinetic constants. FEBS Lett. 2007;581(16):2894‐2898. [DOI] [PubMed] [Google Scholar]
- 36. Murachi T, Hatanaka M, Hamakubo T. Calpains and neuropeptide metabolism In: Chichester TE, ed. Neuropeptides and Their Peptidases. Chichester, UK: Ellis Horwood; 1987:pp. 202‐228. [Google Scholar]
- 37. Fischer S, Vandekerckhove J, Ampe C, Traub P, Weber K. Protein‐chemical identification of the major cleavage sites of the Ca2+ proteinase on murine vimentin, the mesenchymal intermediate filament protein. Biol Chem Hoppe Seyler. 1986;367(11):1147‐1152. [DOI] [PubMed] [Google Scholar]
- 38. Stabach PR, Cianci CD, Glantz SB, Zhang Z, Morrow JS. Site‐directed mutagenesis of alpha II spectrin at codon 1175 modulates its mu‐calpain susceptibility. Biochemistry. 1997;36(1):57‐65. [DOI] [PubMed] [Google Scholar]
- 39. Beckmann JS, Bushby KMD. Advances in the molecular genetics of the limb‐girdle type of autosomal recessive progressive muscular dystrophy. Curr Opin Neurol. 1996;9(5):389‐393. [DOI] [PubMed] [Google Scholar]
- 40. Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb‐girdle muscular dystrophy type 2A. Cell. 1995;81(1):27‐40. [DOI] [PubMed] [Google Scholar]
- 41. Harris F, Biswas S, Singh J, Dennison S, Phoenix DA. Calpains and their multiple roles in diabetes mellitus. Ann N Y Acad Sci. 2006;1084:452‐480. [DOI] [PubMed] [Google Scholar]
- 42. Mahajan VB, Skeie JM, Bassuk AG, et al. Calpain‐5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLOS Genet. 2012;8(10):e1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davis BP, Stucke EM, Khorki ME, et al. Eosinophilic esophagitis‐linked calpain 14 is an IL‐13‐induced protease that mediates esophageal epithelial barrier impairment. JCI Insight. 2016;1(4):e86355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vaisid T, Kosower NS, Katzav A, Chapman J, Barnoy S. Calpastatin levels affect calpain activation and calpain proteolytic activity in APP transgenic mouse model of Alzheimer’s disease. Neurochem Int. 2007;51(6‐7):391‐397. [DOI] [PubMed] [Google Scholar]
- 45. Inomata M, Imahori K, Kawashima S. Autolytic activation of calcium‐activated neutral protease. Biochem Biophys Res Commun. 1986;138(2):638‐643. [DOI] [PubMed] [Google Scholar]
- 46. Du M, Li X, Li Z, et al. Phosphorylation regulated by protein kinase A and alkaline phosphatase play positive roles in mu‐calpain activity. Food Chem. 2018;252:33‐39. [DOI] [PubMed] [Google Scholar]
- 47. Du M, Li X, Li Z, et al. Effects of phosphorylation on mu‐calpain activity at different incubation temperature. Food Res Int. 2017;100(Pt 2):318‐324. [DOI] [PubMed] [Google Scholar]
- 48. Gritsyna YV, Salmov NN, Bobylev AG, et al. Increased autolysis of mu‐calpain in skeletal muscles of chronic alcohol‐fed rats. Alcohol Clin Exp Res. 2017;41(10):1686‐1694. [DOI] [PubMed] [Google Scholar]
- 49. Liu R, Li Y, Wang M, Zhou G, Zhang W. Effect of protein S‐nitrosylation on autolysis and catalytic ability of mu‐calpain. Food Chem. 2016;213:470‐477. [DOI] [PubMed] [Google Scholar]
- 50. Zhang C, Liu R, Wang A, Kang D, Zhou G, Zhang W. Regulation of calpain‐1 activity and protein proteolysis by protein nitrosylation in postmortem beef. Meat Sci. 2018;141:44‐49. [DOI] [PubMed] [Google Scholar]
- 51. Mahaman YAR, Huang F, Wu M, et al. Moringa oleifera alleviates homocysteine‐induced Alzheimer’s disease‐like pathology and cognitive impairments. J Alzheimers Dis. 2018;63(3):1141‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cong J, Goll DE, Peterson AM, Kapprell HP. The role of autolysis in activity of the Ca2+‐dependent proteinases (mu‐calpain and m‐calpain). J Biol Chem. 1989;264(17):10096‐10103. [PubMed] [Google Scholar]
- 53. Li H, Thompson VF, Goll DE. Effects of autolysis on properties of mu‐ and m‐calpain. Biochim Biophys Acta. 2004;1691(2‐3):91‐103. [DOI] [PubMed] [Google Scholar]
- 54. Coolican SA, Hathaway DR. Effect of L‐alpha‐phosphatidylinositol on a vascular smooth muscle Ca2+‐dependent protease. Reduction of the Ca2+requirement for autolysis. J Biol Chem. 1984;259(19):11627‐11630. [PubMed] [Google Scholar]
- 55. Saido TC, Mizuno K, Suzuki K. Proteolysis of protein kinase C by calpain: effect of acidic phospholipids. Biomed Biochim Acta. 1991;50(4‐6):485‐489. [PubMed] [Google Scholar]
- 56. Glading A, Bodnar RJ, Reynolds IJ, et al. Epidermal growth factor activates m‐calpain (calpain II), at least in part, by extracellular signal‐regulated kinase‐mediated phosphorylation. Mol Cell Biol. 2004;24(6):2499‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zadran S, Jourdi H, Rostamiani K, Qin Q, Bi X, Baudry M. Brain‐derived neurotrophic factor and epidermal growth factor activate neuronal m‐calpain via mitogen‐activated protein kinase‐dependent phosphorylation. J Neurosci. 2010;30(3):1086‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shiraha H, Glading A, Chou J, Jia Z, Wells A. Activation of m‐calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m‐calpain. Mol Cell Biol. 2002;22(8):2716‐2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. P. zimmerman, landin boring, jhang UJ, et al. The calpain small subunit gene is essential: its inactivation results in embryonic lethality. IUBMB Life. 2000;50(1):63‐68. [DOI] [PubMed] [Google Scholar]
- 60. Patzke H, Tsai LH. Calpain‐mediated cleavage of the cyclin‐dependent kinase‐5 activator p39 to p29. J Biol Chem. 2002;277(10):8054‐8060. [DOI] [PubMed] [Google Scholar]
- 61. Nixon RA. The calpains in aging and aging‐related diseases. Ageing Res Rev. 2003;2(4):407‐418. [DOI] [PubMed] [Google Scholar]
- 62. Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. Calcium‐mediated transient phosphorylation of τ and amyloid precursor protein followed by intraneuronal amyloid‐beta accumulation. J Biol Chem. 2006;281(52):39907‐39914. [DOI] [PubMed] [Google Scholar]
- 63. Higuchi M, Iwata N, Matsuba Y, et al. Mechanistic involvement of the calpain‐calpastatin system in Alzheimer neuropathology. FASEB J. 2012;26(3):1204‐1217. [DOI] [PubMed] [Google Scholar]
- 64. Rao MV, Mohan PS, Peterhoff CM, et al. Marked calpastatin (CAST) depletion in Alzheimer’s disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J Neurosci. 2008;28(47):12241‐12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Higuchi M, Tomioka M, Takano J, et al. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280(15):15229‐15237. [DOI] [PubMed] [Google Scholar]
- 66. Kuwako K, Nishimura I, Uetsuki T, Saido TC, Yoshikawa K. Activation of calpain in cultured neurons overexpressing Alzheimer amyloid precursor protein. Brain Res Mol Brain Res. 2002;107(2):166‐175. [DOI] [PubMed] [Google Scholar]
- 67. Lee M, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405(6784):360‐364. [DOI] [PubMed] [Google Scholar]
- 68. Wen Y, Yu WH, Maloney B, et al. Transcriptional regulation of beta‐secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57(5):680‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Adamec E, Mohan P, Vonsattel JP, Nixon RA. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol. 2002;104(1):92‐104. [DOI] [PubMed] [Google Scholar]
- 70. Yin Y, Wang Y, Gao D, et al. Accumulation of human full‐length τ induces degradation of nicotinic acetylcholine receptor alpha4 via activating calpain‐2. Sci Rep. 2016;6:27283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Floyd R, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23(5):795‐807. [DOI] [PubMed] [Google Scholar]
- 72. Çakatay U, Telci A, Kayalì R, Tekeli F, Akçay T, Sivas A. Relation of oxidative protein damage and nitrotyrosine levels in the aging rat brain. Exp Gerontol. 2001;36(2):221‐229. [DOI] [PubMed] [Google Scholar]
- 73. Osborne NN, Ugarte M, Chao M, et al. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol. 1999;43(Suppl 1):S102‐S128. [DOI] [PubMed] [Google Scholar]
- 74. Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain‐dependent proteolytic cleavage of the p35 cyclin‐dependent kinase 5 activator to p25. J Biol Chem. 2000;275(22):17166‐17172. [DOI] [PubMed] [Google Scholar]
- 75. Cruz JC, Kim D, Moy LY, et al. p25/cyclin‐dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006;26(41):10536‐10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim JH, Kwon SJ, Stankewich MC, Huh GY, Glantz SB, Morrow JS. Reactive protoplasmic and fibrous astrocytes contain high levels of calpain‐cleaved alpha 2 spectrin. Exp Mol Pathol. 2016;100(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 77. Diepenbroek M, Casadei N, Esmer H, et al. Overexpression of the calpain‐specific inhibitor calpastatin reduces human alpha‐Synuclein processing, aggregation and synaptic impairment in [A30P]alphaSyn transgenic mice. Hum Mol Genet. 2014;23(15):3975‐3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Medeiros R, Kitazawa M, Chabrier MA, et al. Calpain inhibitor A‐705253 mitigates Alzheimer’s disease‐like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol. 2012;181(2):616‐625. [DOI] [PubMed] [Google Scholar]
- 79. Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59(2):214‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323(5918):1211‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Koffie RM, Meyer‐Luehmann M, Hashimoto T, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106(10):4012‐4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Stern EA, et al. Cortical synaptic integration in vivo is disrupted by amyloid‐beta plaques. J Neurosci. 2004;24(19):4535‐4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tamagno E, et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10(3):279‐288. [DOI] [PubMed] [Google Scholar]
- 84. Zhang X, Zhou K, Wang R, et al. Hypoxia‐inducible factor 1alpha (HIF‐1alpha)‐mediated hypoxia increases BACE1 expression and beta‐amyloid generation. J Biol Chem. 2007;282(15):10873‐10880. [DOI] [PubMed] [Google Scholar]
- 85. Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Increased beta‐secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004;1009(1‐2):1‐8. [DOI] [PubMed] [Google Scholar]
- 86. Blasko I, Beer R, Bigl M, et al. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta‐secretase (BACE‐1). J Neural Transm (Vienna). 2004;111(4):523‐536. [DOI] [PubMed] [Google Scholar]
- 87. Fukumoto H, et al. Beta‐secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59(9):1381‐1389. [DOI] [PubMed] [Google Scholar]
- 88. Yang LB, Lindholm K, Yan R, et al. Elevated beta‐secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9(1):3‐4. [DOI] [PubMed] [Google Scholar]
- 89. Chen M, Fernandez HL. Stimulation of beta‐amyloid precursor protein alpha‐processing by phorbol ester involves calcium and calpain activation. Biochem Biophys Res Commun. 2004;316(2):332‐340. [DOI] [PubMed] [Google Scholar]
- 90. Liu L, Martin R, Kohler G, Chan C. Palmitate induces transcriptional regulation of BACE1 and presenilin by STAT3 in neurons mediated by astrocytes. Exp Neurol. 2013;248:482‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Iijima K, Ando K, Takeda S, et al. Neuron‐specific phosphorylation of Alzheimer’s beta‐amyloid precursor protein by cyclin‐dependent kinase 5. J Neurochem. 2000;75(3):1085‐1091. [DOI] [PubMed] [Google Scholar]
- 92. Alberdi E, Sánchez‐Gómez V, Cavaliere F, et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47(3):264‐272. [DOI] [PubMed] [Google Scholar]
- 93. Small DH, Gasperini R, Vincent AJ, Hung AC, Foa L. The role of Abeta‐induced calcium dysregulation in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2009;16(2):225‐233. [DOI] [PubMed] [Google Scholar]
- 94. Sepulveda FJ, Parodi J, Peoples RW, Opazo C, Aguayo LG. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLOS One. 2010;5(7):e11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin‐1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275(24):18195‐18200. [DOI] [PubMed] [Google Scholar]
- 96. Oules B, Del Prete D, Greco B, et al. Ryanodine receptor blockade reduces amyloid‐beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32(34):11820‐11834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cheung KH, Shineman D, Müller M, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58(6):871‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Green KN, Demuro A, Akbari Y, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181(7):1107‐1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kelly BL, Ferreira A. beta‐Amyloid‐induced dynamin 1 degradation is mediated by N‐methyl‐d‐aspartate receptors in hippocampal neurons. J Biol Chem. 2006;281(38):28079‐28089. [DOI] [PubMed] [Google Scholar]
- 100. Abdel‐Hamid KM, Baimbridge KG. The effects of artificial calcium buffers on calcium responses and glutamate‐mediated excitotoxicity in cultured hippocampal neurons. Neuroscience. 1997;81(3):673‐687. [DOI] [PubMed] [Google Scholar]
- 101. Huettner JE, Bean BP. Block of N‐methyl‐d‐aspartate‐activated current by the anticonvulsant MK‐801: selective binding to open channels. Proc Natl Acad Sci U S A. 1988;85(4):1307‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bullock R. Efficacy and safety of memantine in moderate‐to‐severe Alzheimer disease: the evidence to date. Alzheimer Dis Assoc Disord. 2006;20(1):23‐29. [DOI] [PubMed] [Google Scholar]
- 103. Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement Geriatr Cogn Disord. 2009;28(1):75‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nicholson AM, Methner DNR, Ferreira A. Membrane cholesterol modulates {beta}‐amyloid‐dependent tau cleavage by inducing changes in the membrane content and localization of N‐methyl‐d‐aspartic acid receptors. J Biol Chem. 2011;286(2):976‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rönicke R, Mikhaylova M, Rönicke S, et al. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B‐containing NMDA receptors. Neurobiol Aging. 2011;32(12):2219‐2228. [DOI] [PubMed] [Google Scholar]
- 106. Leissring MA, Murphy MP, Mead TR, et al. A physiologic signaling role for the γ‐secretase‐derived intracellular fragment of APP. Proc Natl Acad Sci U S A. 2002;99(7):4697‐4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hamid R, Kilger E, Willem M, et al. Amyloid precursor protein intracellular domain modulates cellular calcium homeostasis and ATP content. J Neurochem. 2007;102(4):1264‐1275. [DOI] [PubMed] [Google Scholar]
- 108. Ko SY, Lin YP, Lin YS, Chang SS. Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic Biol Med. 2010;49(3):474‐480. [DOI] [PubMed] [Google Scholar]
- 109. Cho HJ, Son SM, Jin SM, et al. RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J. 2009;23(8):2639‐2649. [DOI] [PubMed] [Google Scholar]
- 110. Paudel HK, Lew J, Ali Z, Wang JH. Brain proline‐directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in τ associated with Alzheimer’s paired helical filaments. J Biol Chem. 1993;268(31):23512‐23518. [PubMed] [Google Scholar]
- 111. Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase‐3 induces Alzheimer’s disease‐like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147(1):58‐62. [DOI] [PubMed] [Google Scholar]
- 112. Shelton SB, Johnson GVW. Cyclin‐dependent kinase‐5 in neurodegeneration. J Neurochem. 2004;88(6):1313‐1326. [DOI] [PubMed] [Google Scholar]
- 113. Lucas JJ, et al. Decreased nuclear beta‐catenin, tau hyperphosphorylation and neurodegeneration in GSK‐3beta conditional transgenic mice. EMBO J. 2001;20(1‐2):27‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ghosh A, Giese KP. Calcium/calmodulin‐dependent kinase II and Alzheimer’s disease. Mol Brain. 2015;8(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sanchez‐Mut JV, Aso E, Heyn H, et al. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA‐dependent tau phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus. 2014;24(4):363‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Stotani S, Giordanetto F, Medda F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med Chem. 2016;8(6):681‐696. [DOI] [PubMed] [Google Scholar]
- 117. Drewes G, Lichtenberg‐Kraag B, Döring F, et al. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer‐like state. EMBO J. 1992;11(6):2131‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Reynolds CH, Utton MA, Gibb GM, Yates A, Anderton BH. Stress‐activated protein kinase/c‐jun N‐terminal kinase phosphorylates tau protein. J Neurochem. 1997;68(4):1736‐1744. [DOI] [PubMed] [Google Scholar]
- 119. Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741‐766. [DOI] [PubMed] [Google Scholar]
- 120. Kurt M, et al. Hyperphosphorylated tau and paired helical filament‐like structures in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin‐1 transgenes. Neurobiol Dis. 2003;14(1):89‐97. [DOI] [PubMed] [Google Scholar]
- 121. Jin N, Yin X, Yu D, et al. Truncation and activation of GSK‐3beta by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep. 2015;5:8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ma S, Liu S, Huang Q, et al. Site‐specific phosphorylation protects glycogen synthase kinase‐3beta from calpain‐mediated truncation of its N and C termini. J Biol Chem. 2012;287(27):22521‐22532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lučić V, Greif GJ, Kennedy MB. Detailed state model of CaMKII activation and autophosphorylation. Eur Biophys J. 2008;38(1):83‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Reese LC, Laezza F, Woltjer R, Taglialatela G. Dysregulated phosphorylation of Ca(2+)/calmodulin‐dependent protein kinase II‐alpha in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J Neurochem. 2011;119(4):791‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. He X, Li Z, Rizak JD, et al. Resveratrol attenuates formaldehyde induced hyperphosphorylation of tau protein and cytotoxicity in N2a cells. Front Neurosci. 2016;10:598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bi MJ, Sun XN, Zou Y, et al. N‐butylphthalide improves cognitive function in rats after carbon monoxide poisoning. Front Pharmacol. 2017;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Thompson J, Hu Y, Lesnefsky EJ, Chen Q. Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release. Am J Physiol Heart Circ Physiol. 2016;310(3):H376‐H384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wang N, Kang HS, Ahmmed G, et al. Calpain activation by ROS mediates human ether‐a‐go‐go‐related gene protein degradation by intermittent hypoxia. Am J Physiol Cell Physiol. 2016;310(5):C329‐C336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kong LH, Gu XM, Wu F, Jin ZX, Zhou JJ. CaMKII inhibition mitigates ischemia/reperfusion‐elicited calpain activation and the damage to membrane skeleton proteins in isolated rat hearts. Biochem Biophys Res Commun. 2017;491(3):687‐692. [DOI] [PubMed] [Google Scholar]
- 130. McGinnis KM, Whitton MM, Gnegy ME, Wang KKW. Calcium/calmodulin‐dependent protein kinase IV is cleaved by caspase‐3 and calpain in SH‐SY5Y human neuroblastoma cells undergoing apoptosis. J Biol Chem. 1998;273(32):19993‐20000. [DOI] [PubMed] [Google Scholar]
- 131. Tremper‐Wells B, Vallano ML. Nuclear calpain regulates Ca2+‐dependent signaling via proteolysis of nuclear Ca2+/calmodulin‐dependent protein kinase type IV in cultured neurons. J Biol Chem. 2005;280(3):2165‐2175. [DOI] [PubMed] [Google Scholar]
- 132. Banerjee C, Khatri P, Raman R, Bhatia H, Datta M, Mazumder S. Role of calmodulin‐calmodulin kinase II, cAMP/protein kinase A and ERK 1/2 on Aeromonas hydrophila‐induced apoptosis of head kidney macrophages. PLOS Pathog. 2014;10(4):e1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Carlyle BC, Nairn AC, Wang M, et al. cAMP‐PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A. 2014;111(13):5036‐5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Liu Y, Su Y, Wang J, et al. Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP‐dependent kinase. Neurochem Int. 2013;62(4):458‐467. [DOI] [PubMed] [Google Scholar]
- 135. Li Z, Li X, Gao X, Shen QW, Du M, Zhang D. Phosphorylation prevents in vitro myofibrillar proteins degradation by mu‐calpain. Food Chem. 2017;218:455‐462. [DOI] [PubMed] [Google Scholar]
- 136. Ryoo SR, Jeong HK, Radnaabazar C, et al. DYRK1A‐mediated hyperphosphorylation of tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;282(48):34850‐34857. [DOI] [PubMed] [Google Scholar]
- 137. Branca C, Shaw DM, Belfiore R, et al. Dyrk1 inhibition improves Alzheimer’s disease‐like pathology. Aging cell. 2017;16(5):1146‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yin X, Jin N, Shi J, et al. Dyrk1A overexpression leads to increase of 3R‐tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci Rep. 2017;7(1):619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ding S, Shi J, Qian W, et al. Regulation of alternative splicing of tau exon 10 by 9G8 and Dyrk1A. Neurobiol Aging. 2012;33(7):1389‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wegiel J, Kaczmarski W, Barua M, et al. Link between DYRK1A overexpression and several‐fold enhancement of neurofibrillary degeneration with 3‐repeat tau protein in Down syndrome. J Neuropathol Exp Neurol. 2011;70(1):36‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Naert G, Ferré V, Meunier J, et al. Leucettine L41, a DYRK1A‐preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Abeta25‐35 peptide administration in mice. Eur Neuropsychopharmacol. 2015;25(11):2170‐2182. [DOI] [PubMed] [Google Scholar]
- 142. Coutadeur S, Benyamine H, Delalonde L, et al. A novel DYRK1A (dual specificity tyrosine phosphorylation‐regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: effect on tau and amyloid pathologies in vitro. J Neurochem. 2015;133(3):440‐451. [DOI] [PubMed] [Google Scholar]
- 143. Jin N, Yin X, Gu J, et al. Truncation and activation of dual specificity tyrosine phosphorylation‐regulated kinase 1A by calpain I: A molecular mechanism linked to tau pathology in Alzheimer disease. J Biol Chem. 2015;290(24):15219‐15237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Aksenov MY, Aksenova MV, Markesbery WR, Butterfield DA. Amyloid beta‐peptide (1‐40)‐mediated oxidative stress in cultured hippocampal neurons. Protein carbonyl formation, CK BB expression, and the level of Cu, Zn, and Mn SOD mRNA. J Mol Neurosci. 1998;10(3):181‐192. [DOI] [PubMed] [Google Scholar]
- 145. Gallant M, Rak M, Szeghalmi A, et al. Focally elevated creatine detected in amyloid precursor protein (APP) transgenic mice and Alzheimer disease brain tissue. J Biol Chem. 2006;281(1):5‐8. [DOI] [PubMed] [Google Scholar]
- 146. Aksenov MY, Aksenova MV, Payne RM, Smith CD, Markesbery WR, Carney JM. The expression of creatine kinase isoenzymes in neocortex of patients with neurodegenerative disorders: Alzheimer’s and Pick’s disease. Exp Neurol. 1997;146(2):458‐465. [DOI] [PubMed] [Google Scholar]
- 147. Aksenov M, Aksenova M, Butterfield DA, Markesbery WR. Oxidative modification of creatine kinase BB in Alzheimer’s disease brain. J Neurochem. 2000;74(6):2520‐2527. [DOI] [PubMed] [Google Scholar]
- 148. Purintrapiban J, Wang M, Forsberg NE. Identification of glycogen phosphorylase and creatine kinase as calpain substrates in skeletal muscle. Int J Biochem Cell Biol. 2001;33(5):531‐540. [DOI] [PubMed] [Google Scholar]
- 149. Zhang ZW, Qin XY, Che FY, Xie G, Shen L, Bai YY. Effects of beta 2 adrenergic agonists on axonal injury and mitochondrial metabolism in experimental autoimmune encephalomyelitis rats. Genet Mol Res. 2015;14(4):13572‐13581. [DOI] [PubMed] [Google Scholar]
- 150. Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277(32):29181‐29186. [DOI] [PubMed] [Google Scholar]
- 151. Neuhof C, Götte O, Trumbeckaite S, et al. A novel water‐soluble and cell‐permeable calpain inhibitor protects myocardial and mitochondrial function in postischemic reperfusion. Biol Chem. 2003;384(12):1597‐1603. [DOI] [PubMed] [Google Scholar]
- 152. Veeranna, Kaji T, Boland B, et al. Calpain mediates calcium‐induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer’s disease. Am J Pathol. 2004;165(3):795‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wang Y, Briz V, Chishti A, Bi X, Baudry M. Distinct roles for mu‐calpain and m‐calpain in synaptic NMDAR‐mediated neuroprotection and extrasynaptic NMDAR‐mediated neurodegeneration. J Neurosci. 2013;33(48):18880‐18892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Watkins GR, Wang N, Mazalouskas MD, et al. Monoubiquitination promotes calpain cleavage of the protein phosphatase 2A (PP2A) regulatory subunit alpha4, altering PP2A stability and microtubule‐associated protein phosphorylation. J Biol Chem. 2012;287(29):24207‐24215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Jin N, Wu Y, Xu W, Gong CX, Iqbal K, Liu F. C‐terminal truncation of GSK‐3beta enhances its dephosphorylation by PP2A. FEBS Lett. 2017;591(7):1053‐1063. [DOI] [PubMed] [Google Scholar]
- 156. Wang Y, Hall RA, Lee M, Kamgar‐Parsi A, Bi X, Baudry M. The tyrosine phosphatase PTPN13/FAP‐1 links calpain‐2, TBI and tau tyrosine phosphorylation. Sci Rep. 2017;7(1):11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Aich J, Mabalirajan U, Ahmad T, Agrawal A, Ghosh B. Loss‐of‐function of inositol polyphosphate‐4‐phosphatase reversibly increases the severity of allergic airway inflammation. Nat Commun. 2012;3:877. [DOI] [PubMed] [Google Scholar]
- 158. Fedele CG, Ooms LM, Ho M, et al. Inositol polyphosphate 4‐phosphatase II regulates PI3K/Akt signaling and is lost in human basal‐like breast cancers. Proc Natl Acad Sci U S A. 2010;107(51):22231‐22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Jiang J, Wang ZH, Qu M, et al. Stimulation of EphB2 attenuates tau phosphorylation through PI3K/Akt‐mediated inactivation of glycogen synthase kinase‐3beta. Sci Rep. 2015;5:11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Amadoro G, Corsetti V, Stringaro A, et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis. 2010;21(2):445‐470. [DOI] [PubMed] [Google Scholar]
- 161. Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y. Cleavage of tau by calpain in Alzheimer’s disease: the quest for the toxic 17 kD fragment. Neurobiol Aging. 2011;32(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 162. Park SY. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta‐amyloid‐induced neurodegeneration. J Neurosci. 2005;25(22):5365‐5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Park SY, Tournell C, Sinjoanu RC, Ferreira A. Caspase‐3‐ and calpain‐mediated tau cleavage are differentially prevented by estrogen and testosterone in beta‐amyloid‐treated hippocampal neurons. Neuroscience. 2007;144(1):119‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Atlante A, Amadoro G, Bobba A, et al. A peptide containing residues 26‐44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim Biophys Acta. 2008;1777(10):1289‐1300. [DOI] [PubMed] [Google Scholar]
- 165. Guillozet‐Bongaarts AL, Garcia‐Sierra F, Reynolds MR, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer’s disease. Neurobiol Aging. 2005;26(7):1015‐1022. [DOI] [PubMed] [Google Scholar]
- 166. Litersky JM, Johnson GV. Phosphorylation by cAMP‐dependent protein kinase inhibits the degradation of tau by calpain. J Biol Chem. 1992;267(3):1563‐1568. [PubMed] [Google Scholar]
- 167. Ferreira A, Bigio E. Calpain‐mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. 2011;17(7‐8):676‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK. Dual vulnerability of tau to calpains and caspase‐3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro. 2011;3(1):e00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Reinecke JB, DeVos SL, McGrath JP, et al. Implicating calpain in tau‐mediated toxicity in vivo. PLOS One. 2011;6(8):e23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381(6583):571‐579. [DOI] [PubMed] [Google Scholar]
- 171. Mosser DD, Caron AW, Bourget L, et al. The chaperone function of hsp70 is required for protection against stress‐induced apoptosis. Mol Cell Biol. 2000;20(19):7146‐7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Nylandsted J, Gyrd‐Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200(4):425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Figueiredo C, Pais TF, Gomes JR, Chatterjee S. Neuron‐microglia crosstalk up‐regulates neuronal FGF‐2 expression which mediates neuroprotection against excitotoxicity via JNK1/2. J Neurochem. 2008;107(1):73‐85. [DOI] [PubMed] [Google Scholar]
- 174. Sultana R, Perluigi M, Newman SF, et al. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease. Antioxid Redox Signal. 2010;12(3):327‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89(4):343‐358. [DOI] [PubMed] [Google Scholar]
- 176. Sahara S, Yamashima T. Calpain‐mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem Biophys Res Commun. 2010;393(4):806‐811. [DOI] [PubMed] [Google Scholar]
- 177. Tofighi R, Johansson C, Goldoni M, et al. Hippocampal neurons exposed to the environmental contaminants methylmercury and polychlorinated biphenyls undergo cell death via parallel activation of calpains and lysosomal proteases. Neurotox Res. 2011;19(1):183‐194. [DOI] [PubMed] [Google Scholar]
- 178. Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000;113(Pt 24):4441‐4450. [DOI] [PubMed] [Google Scholar]
- 179. Alvarez‐Erviti L, Rodriguez‐Oroz MC, Cooper JM, et al. Chaperone‐mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67(12):1464‐1472. [DOI] [PubMed] [Google Scholar]
- 180. Petrucelli L, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13(7):703‐714. [DOI] [PubMed] [Google Scholar]
- 181. Dou F, Netzer WJ, Tanemura K, et al. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100(2):721‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Patterson KR, Ward SM, Combs B, et al. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry. 2011;50(47):10300‐10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Yamashima T. Reconsider Alzheimer’s disease by the ‘calpain‐cathepsin hypothesis’—a perspective review. Prog Neurobiol. 2013;105:1‐23. [DOI] [PubMed] [Google Scholar]
- 184. Trinchese F, Fa’ M, Liu S, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118(8):2796‐2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572‐580. [DOI] [PubMed] [Google Scholar]
- 186. Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal. 2008;1(14):re1. [DOI] [PubMed] [Google Scholar]
- 187. Clark SG, Shurland DL, Meyerowitz EM, Bargmann CI, van der Bliek AM. A dynamin GTPase mutation causes a rapid and reversible temperature‐inducible locomotion defect in C. elegans. Proc Natl Acad Sci U S A. 1997;94(19):10438‐10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Sinjoanu RC, Kleinschmidt S, Bitner RS, Brioni JD, Moeller A, Ferreira A. The novel calpain inhibitor A‐705253 potently inhibits oligomeric beta‐amyloid‐induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochem Int. 2008;53(3‐4):79‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Bi R, Bi X, Baudry M. Phosphorylation regulates calpain‐mediated truncation of glutamate ionotropic receptors. Brain Res. 1998;797(1):154‐158. [DOI] [PubMed] [Google Scholar]
- 190. Vinade L, Petersen JD, Do K, Dosemeci A, Reese TS. Activation of calpain may alter the postsynaptic density structure and modulate anchoring of NMDA receptors. Synapse. 2001;40(4):302‐309. [DOI] [PubMed] [Google Scholar]
- 191. Calo L, et al. Interactions between ephrin‐B and metabotropic glutamate 1 receptors in brain tissue and cultured neurons. J Neurosci. 2005;25(9):2245‐2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain‐mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53(3):399‐412. [DOI] [PubMed] [Google Scholar]
- 193. Liang Z, Liu F, Grundke‐Iqbal I, Iqbal K, Gong CX. Down‐regulation of cAMP‐dependent protein kinase by over‐activated calpain in Alzheimer disease brain. J Neurochem. 2007;103(6):2462‐2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Walton MR, Dragunow M. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23(2):48‐53. [DOI] [PubMed] [Google Scholar]
- 195. Nucifora Jr. FC, Jr. , et al. Interference by huntingtin and atrophin‐1 with cbp‐mediated transcription leading to cellular toxicity. Science. 2001;291(5512):2423‐2428. [DOI] [PubMed] [Google Scholar]
- 196. Liu Y, Wang Y, Zhu G, Sun J, Bi X, Baudry M. A calpain‐2 selective inhibitor enhances learning & memory by prolonging ERK activation. Neuropharmacology. 2016;105:471‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP‐32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269‐296. [DOI] [PubMed] [Google Scholar]
- 198. Hemmings HC, Jr. , Nairn AC, Elliott JI, Greengard P. Synthetic peptide analogs of DARPP‐32 (Mr 32,000 dopamine‐ and cAMP‐regulated phosphoprotein), an inhibitor of protein phosphatase‐1. Phosphorylation, dephosphorylation, and inhibitory activity. J Biol Chem. 1990;265(33):20369‐20376. [PubMed] [Google Scholar]
- 199.Briz V, Baudry M. Calpains: master regulators of synaptic plasticity. Neuroscientist. 2017;23(2):221–231. [DOI] [PubMed] [Google Scholar]
- 200. Karpenko MN, Tikhomirova MS. The role of calpains in regulating synaptic transmission. Neurosci Behav Physiol. 2015;45(8):952‐956. [Google Scholar]
- 201. Baudry M, Bi X. Calpain‐1 and calpain‐2: the yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci. 2016;39(4):235‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Abe K, Takeichi M. NMDA‐receptor activation induces calpain‐mediated beta‐catenin cleavages for triggering gene expression. Neuron. 2007;53(3):387‐397. [DOI] [PubMed] [Google Scholar]
- 203. Jang YN, Jung YS, Lee SH, Moon CH, Kim CH, Baik EJ. Calpain‐mediated N‐cadherin proteolytic processing in brain injury. J Neurosci. 2009;29(18):5974‐5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Morales‐Corraliza J, Berger JD, Mazzella MJ, et al. Calpastatin modulates APP processing in the brains of beta‐amyloid depositing but not wild‐type mice. Neurobiol Aging. 2012;33(6):1125. e9‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Rao MV, McBrayer MK, Campbell J, et al. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J Neurosci. 2014;34(28):9222‐9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Cao G, Xing J, Xiao X, et al. Critical role of calpain I in mitochondrial release of apoptosis‐inducing factor in ischemic neuronal injury. J Neurosci. 2007;27(35):9278‐9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Shirasaki Y, Miyashita H, Yamaguchi M, Inoue J, Nakamura M. Exploration of orally available calpain inhibitors: peptidyl alpha‐ketoamides containing an amphiphile at P3 site. Bioorg Med Chem. 2005;13(14):4473‐4484. [DOI] [PubMed] [Google Scholar]
- 208. Shimazawa M, Suemori S, Inokuchi Y, et al. A novel calpain inhibitor, ((1S)‐1‐((((1S)‐1‐benzyl‐3‐cyclopropylamino‐2,3‐di‐oxopropyl)amino)carbonyl)‐3‐me thylbutyl)carbamic acid 5‐methoxy‐3‐oxapentyl ester (SNJ‐1945), reduces murine retinal cell death in vitro and in vivo. J Pharmacol Exp Ther. 2010;332(2):380‐387. [DOI] [PubMed] [Google Scholar]
- 209. Ryu M, Yasuda M, Shi D, et al. Critical role of calpain in axonal damage‐induced retinal ganglion cell death. J Neurosci Res. 2012;90(4):802‐815. [DOI] [PubMed] [Google Scholar]
- 210. Kunz S, Niederberger E, Ehnert C, et al. The calpain inhibitor MDL 28170 prevents inflammation‐induced neurofilament light chain breakdown in the spinal cord and reduces thermal hyperalgesia. Pain. 2004;110(1‐2):409‐418. [DOI] [PubMed] [Google Scholar]
- 211. Di Rosa G, Odrlijn T, Nixon RA, Arancio O. Calpain inhibitors: a treatment for Alzheimer’s disease. J Mol Neurosci. 2002;19(1‐2):135‐141. [DOI] [PubMed] [Google Scholar]
- 212. Battaglia F, Trinchese F, Liu S, Walter S, Nixon RA, Arancio O. Calpain inhibitors, a treatment for Alzheimer’s disease: position paper. J Mol Neurosci. 2003;20(3):357‐362. [DOI] [PubMed] [Google Scholar]
- 213. Lubisch W, Beckenbach E, Bopp S, et al. Benzoylalanine‐derived ketoamides carrying vinylbenzyl amino residues: discovery of potent water‐soluble calpain inhibitors with oral bioavailability. J Med Chem. 2003;46(12):2404‐2412. [DOI] [PubMed] [Google Scholar]
- 214. Granic I, Nyakas C, Luiten PGM, et al. Calpain inhibition prevents amyloid‐beta‐induced neurodegeneration and associated behavioral dysfunction in rats. Neuropharmacology. 2010;59(4‐5):334‐342. [DOI] [PubMed] [Google Scholar]
- 215. Nimmrich V, Reymann K, Strassburger M, et al. Inhibition of calpain prevents NMDA‐induced cell death and beta‐amyloid‐induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol. 2010;159(7):1523‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Nikkel AL, Martino B, Markosyan S, et al. The novel calpain inhibitor A‐705253 prevents stress‐induced tau hyperphosphorylation in vitro and in vivo. Neuropharmacology. 2012;63(4):606‐612. [DOI] [PubMed] [Google Scholar]
- 217. Kling A, Jantos K, Mack H, et al. Discovery of novel and highly selective inhibitors of calpain for the treatment of Alzheimer’s disease: 2‐(3‐phenyl‐1H‐pyrazol‐1‐yl)‐nicotinamides. J Med Chem. 2017;60(16):7123‐7138. [DOI] [PubMed] [Google Scholar]
- 218. Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001;382(5):735‐741. [DOI] [PubMed] [Google Scholar]
- 219. Mueller‐Steiner S, Zhou Y, Arai H, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron. 2006;51(6):703‐714. [DOI] [PubMed] [Google Scholar]
- 220. Brömme D, Lecaille F. Cathepsin K inhibitors for osteoporosis and potential off‐target effects. Expert Opin Investig Drugs. 2009;18(5):585‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest. 2010;120(10):3421‐3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Jeon KH, Lee E, Jun KY, et al. Neuroprotective effect of synthetic chalcone derivatives as competitive dual inhibitors against mu‐calpain and cathepsin B through the downregulation of tau phosphorylation and insoluble Abeta peptide formation. Eur J Med Chem. 2016;121:433‐444. [DOI] [PubMed] [Google Scholar]
- 223. Fà M, Zhang H, Staniszewski A, et al. Novel selective calpain 1 inhibitors as potential therapeutics in Alzheimer’s disease. J Alzheimers Dis. 2016;49(3):707‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Arthur JSC, Elce JS, Hegadorn C, Williams K, Greer PA. Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Mol Cell Biol. 2000;20(12):4474‐4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Taniguchi K, Umeshita K, Sakon M, et al. Suppression of oxidative stress‐induced hepatocyte injury by calpain antisense. J Surg Res. 2003;111(1):23‐27. [DOI] [PubMed] [Google Scholar]
- 226. Johnson JD, Otani K, Bell GI, Polonsky KS. Impaired insulin secretion in transgenic mice over‐expressing calpastatin in pancreatic beta‐cells. Islets. 2009;1(3):242‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]