

Abstract
For the first time, structurally defined manganese pincer complexes catalyze the dehydrogenation of aqueous methanol to hydrogen and carbon dioxide, which is a transformation of interest with regard to the implementation of a hydrogen and methanol economy. Excellent long‐term stability was demonstrated for the Mn‐PNPiPr catalyst, as a turnover of more than 20 000 was reached. In addition to methanol, other important hydrogen carriers were also successfully dehydrogenated.
Keywords: dehydrogenation, homogeneous catalysis, ligand design, manganese, methanol
The long‐term transition from a fossil‐based energy supply to renewable energies requires the development of efficient technologies for energy storage. In this respect, the focus has been increasingly on hydrogen as an environmentally benign energy carrier.1 Nevertheless, it is a highly volatile gas and features very low gravimetric energy density. Hence, other chemical energy carriers, which can be reversibly (de)hydrogenated are also considered as viable options. Among these, methanol is promising because of its comparably high hydrogen content and its liquid state under ambient conditions, which significantly simplifies transportation and handling.2 In the last decade, a variety of catalysts have been successfully developed for the dehydrogenation of methanol, as well as its use in hydrogen‐borrowing reactions.3 While heterogeneous systems in general require harsh reaction conditions (>200 °C), molecularly defined complexes based on ruthenium and iridium show good activities at temperatures below 100 °C. Besides these precious catalysts, in recent years non‐noble metals have attracted increasing attention. However, so far only a few iron‐containing complexes were reported for this transformation (Scheme 1).4 This year, manganese pincer catalysts were successfully employed for the first time in both hydrogenation and dehydrogenation reactions, though this metal is commonly used in oxidations.5 Clearly, manganese features a range of favorable properties as it is low toxic, earth abundant, and inexpensive because of its use for the iron and steel production.6
Scheme 1.
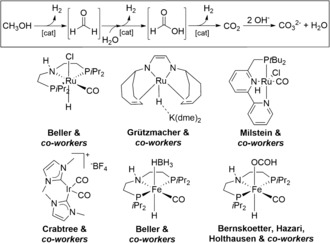
A selection of ruthenium, iridium and iron catalysts reported for the dehydrogenation of aqueous methanol.
Inspired by these recent achievements, herein we report, for the first time, the low‐temperature methanol reforming in the presence of a highly stable manganese PNP‐pincer complex. Initially, a range of different manganese catalysts and precursors were tested following a standard protocol developed for today's state‐of‐the‐art homogenous methanol reforming catalyst (Table 1).5a, 7 Interestingly, the iPr complex 1, the corresponding pincer complex bearing ethyl groups (3), and the precursor [Mn(CO)5Br] with an additional 10 equivalents of the HN(CH2CH2)P(CH(CH3)2)2 ligand (from here on named PNPiPr ligand) showed reasonable activity for methanol dehydrogenation, thus achieving turnover numbers (TONs) of 54, 65, and 68, respectively (entries 1, 3, and 8). During these preliminary tests, high sensitivity to light irradiation was noted in the case of the dissolved manganese complex 1, which caused a rapid decline in its activity. This phenomenon stands in contrast to the reported stability of this complex in its solid state and is ascribed to the light‐triggered cleavage of the PNPiPr ligand (see section SI6.2 in the Supporting Information).5a Hence, all described experiments were carried out under rigorous exclusion of light.
Table 1.
Different manganese catalysts for the dehydrogenation of aqueous methanol.[a]
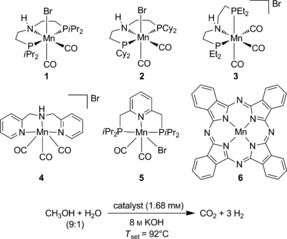
| Entry | Catalyst | V 5h [mL] | TON5h |
|---|---|---|---|
| 1[b] | 1 | 11 | 54 |
| 2 | 2 | – | – |
| 3[b] | 3 | 13 | 65 |
| 4 | 4 | – | – |
| 5 | 5 | 9 | 41 |
| 6 | 6 | – | – |
| 7 | [Mn2(CO)10] | – | – |
| 8[b] | [Mn(CO)5Br] + 10 equiv PNPiPr ligand | 14 | 68 |
[a] Reaction conditions: MeOH/H2O (5 mL, ratio 9:1), 8 m KOH (molarity based on total MeOH + H2O volume), catalyst (8.4 μmol, 1.68 mm); T set: 92 °C. Gas volumes were determined using manual or automatic gas burettes and were analyzed by GC. Each molecule of hydrogen is counted as one turnover. All gas measurements were performed at least twice and corrected by blank values. [b] Standard deviation is less than 15 %.
A variation of Milstein's catalyst (5) was less active and catalyst performance fluctuated significantly. The pincer catalysts 2 and 4, as well as the commercially available manganese porphyrine complex 6 and [Mn2(CO)10] did not show measurable activities and gas evolution did not exceed blank values (Table 1, entries 2, 4, 6 and 7).
Further investigations of critical reaction parameters concentrated on the catalyst 1 and its precursor [Mn(CO)5Br] because of their promising activities and easy availability compared to the manganese ethyl complex 3 (see SI3). Replacing KOH by other bases, such as tBuOK or LiOH, led to either a significant decrease in activity or to complete deactivation of the catalyst, as in the case of the latter. Analogous to the recent findings reported by Boncella and co‐workers, the addition of LiBF4 completely shut down catalytic activity.5d On the other hand, catalytic activity was improved by the addition of 10 equivalents of the PNPiPr ligand. A long‐term stabilizing effect for a reaction time of more than ten hours was achieved by using triglyme as solvent.
To gain better insight into the catalytic systems based on the iPr complex and the precursor [Mn(CO)5Br], and to test their stability, long‐term experiments were performed under optimized reaction conditions (Figure 1). Remarkably, the combination of 1 in the presence of additional PNPiPr ligand proved to be stable for longer than a month, thus reaching a TON of more than 20 000. Notably, this manganese catalyst exceeded the stability of the related Fe‐iPr complex, which was only stable up to five days.4e In contrast, the [Mn(CO)5Br] precursor plus additional PNPiPr ligand showed a decline in activity after 100 hours and the activity ceased entirely after 300 hours (see SI5).
Figure 1.
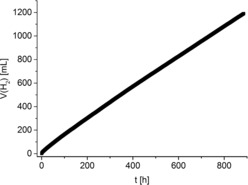
Long‐term experiment. Reaction conditions: MeOH/H2O (20 mL, ratio 9:1), triglyme (20 mL), 8 m KOH, 1 (2.1 μmol, 0.05 mm), 10 equiv. PNPiPr ligand; T set: 92 °C.
For a better understanding of possible catalytic intermediates, ex‐situ IR investigations under reaction conditions were performed. To address the aspect of the role of the base in the mechanistic cycle, KOH was added to a 9:1 MeOH/H2O solution of 1 and samples were measured. Under non‐basic conditions, two bands at 1925 and 1843 cm−1 were detected and assigned to the symmetric and asymmetric C−O frequencies of 1. Already when adding 0.5 equivalents of KOH, additional bands appeared at slightly lower wavenumbers (1910 and 1825 cm−1). At 10 equivalents of KOH full conversion into this new species was reached (Figure 2). To identify this complex, NMR investigations were carried out (see SI6): Dissolving 1 in a basic MeOH/H2O solution lead to the appearance of two species [31P NMR (additional MeOD‐d4): δ=84.7 (bs, major species) and 86.5 ppm (bs, minor species]. The formation of the hydride complex 12 could be excluded as neither a hydride signal was detected in the 1H NMR spectrum nor did the 31P signals match the previously reported shift of δ=109.6 ppm for the hydride complex.5a Next, the amido complex 8 was generated by adding base to the catalyst in benzene‐d6 to prove the occurrence of possible intermediates in the catalytic cycle such as the methoxide 9, hydroxide 7 or formate 11 complexes (Scheme 2).
Figure 2.
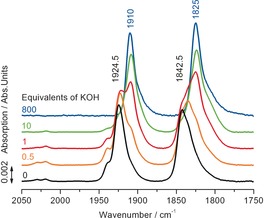
IR measurements of the active catalytic species in dependence on base molarity. Reaction conditions: MeOH/H2O (1 mL, ratio 9:1), 1 (10 μmol, 10 mm), RT.
Scheme 2.
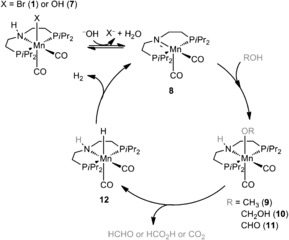
Proposed catalytic cycle for the manganese‐catalyzed aqueous‐phase reforming of methanol.
Addition of water confirmed that the minor peak in the 31P NMR spectra can be attributed to the hydroxide species 7. The analogous experiment of adding methanol and formic acid resulted in the appearance of broad peaks in the 31P NMR spectrum with very similar chemical shifts to the major peak. Hence, it was not possible to unambiguously differentiate between the methoxide and formate complex by NMR experiments.8 However, calculated IR shifts support the assumption that the prevalent species is the methoxide complex 9 and that the corresponding hydroxide complex 7 is present to a minor extent (see SI8 and Figure SI14). The bands of the latter are the reason for the very broad tailing of the bands, which can be observed at 1910 and 1825 cm−1.
To clarify the temperature influence on the catalytic species, the reaction solution containing 10 mm of the catalyst was heated from room temperature to 90 °C (Figure 3). Similar to the previous investigations, both methoxide and hydroxide species were observed. Additionally, formate was detected after five minutes and its concentration gradually increased during the reaction. This latter band was assigned by adding formic acid to the basic reaction solution. Based on all these findings and in agreement with DFT calculations,5a, 9 we propose the mechanistic cycle shown in Scheme 2. Initially, the active amido species 8 is generated by either base‐mediated dehydrobromination of 1 or by dehydration of the hydroxide complex 7. By coordination of methanol, the methoxide species 9 is formed, and it undergoes C−H cleavage to form formaldehyde and 12. In the last step hydrogen is produced and the active catalyst is regenerated.
Figure 3.
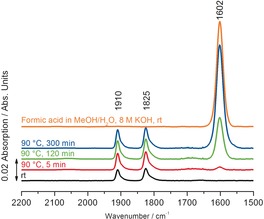
Reaction monitoring by IR. Reaction conditions: MeOH/H2O (10 mL, ratio 9:1), 8 m KOH, 1 (100 μmol, 10 mm), RT −90 °C.
Finally, we were interested in the performance of our novel dehydrogenation catalyst for other important C1 and C2 substrates. Thus, reactions of paraformaldehyde, formic acid and ethanol were investigated in the presence of 1.
To our delight, the catalyst was active for all these substrates and activity was still linear after 14 hours. For EtOH as a substrate a TON of 163 after five hours (Table 2, entry 1) was obtained. Dehydrogenation of paraformaldehyde in a slightly basic tBuOH solution at 81 °C resulted in a TON of 80. Following a formic acid dehydrogenation protocol,10 the manganese catalyst showed significantly higher activity, thus reaching a TON of 283 (entries 2 and 3).
Table 2.
Dehydrogenation of different substrates with 1.
| Entry | Substrate | V 5h [mL] | TON5h |
|---|---|---|---|
| 1[a] | EtOH | 33 | 163 |
| 2[b,d] | paraformaldehyde | 32 | 79 |
| 3[c,d] | formic acid | 73 | 283 |
[a] Reaction conditions: EtOH/H2O (5 mL, ratio 9:1), KOH (0.04 mol, 8 m), 1 (8.4 μmol, 1.68 mm); T set: 92 °C. [b] Reaction conditions: tBuOH (36 mL), H2O (4 mL), KOH (0.05 m), paraformaldehyde (4 mmol, 0.1 m), 1 (8.28 μmol, 0.21 mm); T set: 81 °C. [c] Reaction conditions: PC (5 mL), 11 mol FA/10 mol DMOA (5 mL), 1 (5.3 μmol, 0.53 μm); T set: 60 °C. [d] Concomitant production of CO2 (ratio H2/CO2 1:1, analyzed by GC).
In conclusion, we have demonstrated for the first time that pincer complexes based on nontoxic and easily available manganese successfully promote the selective dehydrogenation of aqueous methanol. Under optimized reaction conditions, an impressive TON of more than 20 000 was attained and 1 was still active after one month. Although for practical purposes the general activity still has to be improved, these promising results show that such molecularly defined non‐noble metal catalysts can be extremely stable and robust, which is also of importance for future applications.
Experimental Section
All reactions were carried out under argon atmosphere with exclusion of air and light using Schlenk techniques. Specific reaction procedures can be found in the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. E. Alberico, for her excellent support and very helpful discussions. We also thank P. Bartels for her outstanding practical support. Furthermore, we would like to thank M. Perez and J. Neumann for kindly providing us with the catalysts 4 and 5. L.K.V. would like to thank the VCI for generous funding (scholarship n. 196241). W.Z. would like to thank the EU fund H2020‐MSCA‐ITN‐2015 in Horizon 2020 as part of the NoNoMeCat.
M. Andérez-Fernández, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig, M. Beller, Angew. Chem. Int. Ed. 2017, 56, 559.
References
- 1.
- 1a. Müller K., Arlt W., Energy Technol. 2013, 1, 501–511; [Google Scholar]
- 1b. Eberle U., Felderhoff M., Schüth F., Angew. Chem. Int. Ed. 2009, 48, 6608–6630; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6732–6757. [Google Scholar]
- 2.
- 2a. Olah G. A., Angew. Chem. Int. Ed. 2005, 44, 2636–2639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 2692–2696; [Google Scholar]
- 2b. Armaroli N., Balzani V., ChemSusChem 2011, 4, 21–36. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Palo D. R., Dagle R. A., Holladay J. D., Chem. Rev. 2007, 107, 3992–4021; [DOI] [PubMed] [Google Scholar]
- 3b. Sá S., Silva H., Brandãoa L., Sousa J. M., Mendes A., Appl. Catal. B 2010, 99, 43–57; [Google Scholar]
- 3c. Alberico E., Nielsen M., Chem. Commun. 2015, 51, 6714–6725; [DOI] [PubMed] [Google Scholar]
- 3d. Ortega N., Richter C., Glorius F., Org. Lett. 2013, 15, 1776–1779; [DOI] [PubMed] [Google Scholar]
- 3e. Chan L. K. M., Poole D. L., Shen D., Healy M. P., Donohoe T. J., Angew. Chem. Int. Ed. 2014, 53, 761–765; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 780–784; [Google Scholar]
- 3f. Shen D., Poole D. L., Shotton C. C., Kornahrens A. F., Healy M. P., Donohoe T. J., Angew. Chem. Int. Ed. 2015, 54, 1642–1645; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1662–1665; [Google Scholar]
- 3g. Quan X., Kerdphon S., Andersson P. G., Chem. Eur. J. 2015, 21, 3576–3579; [DOI] [PubMed] [Google Scholar]
- 3h. Moran J., Preetz A., Mesch R. A., Krische M. J., Nat. Chem. 2011, 3, 287–290; [DOI] [PubMed] [Google Scholar]
- 3i. Nguyen K. D., Herkommer D., Krische M. J., J. Am. Chem. Soc. 2016, 138, 14210–14213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3j. Sam B., Breit B., Krische M. J., Angew. Chem. Int. Ed. 2015, 54, 3267–3274; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3317–3325. [Google Scholar]
- 4.
- 4a. Nielsen M., Alberico E., Baumann W., Drexler H. J., Junge H., Gladiali S., Beller M., Nature 2013, 495, 85–89; [DOI] [PubMed] [Google Scholar]
- 4b. Rodríguez-Lugo R. E., Trincado M., Vogt M., Tewes F., Santiso-Quinones G., Grützmacher H., Nat. Chem. 2013, 5, 342–347; [DOI] [PubMed] [Google Scholar]
- 4c. Hu P., Diskin-Posner Y., Ben-David Y., Milstein D., ACS Catal. 2014, 4, 2649–2652; [Google Scholar]
- 4d. Campos J., Sharninghausen L. S., Manas M. G., Crabtree R. H., Inorg. Chem. 2015, 54, 5079–5084; [DOI] [PubMed] [Google Scholar]
- 4e. Alberico E., Sponholz P., Cordes C., Nielsen M., Drexler H.-J., Baumann W., Junge H., Beller M., Angew. Chem. Int. Ed. 2013, 52, 14162–14166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14412–14416; [Google Scholar]
- 4f. Bielinski E. A., Förster M., Zhang Y., Bernskoetter W. H., Hazari N., Holthausen M. C., ACS Catal. 2015, 5, 2404–2415; [Google Scholar]
- 4g. Fujita K.-I., Kawahara R., Aikawa T., Yamaguchi R., Angew. Chem. Int. Ed. 2015, 54, 9057–9060; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9185–9188; [Google Scholar]
- 4h. Wakizaka M., Matsumoto T., Tanaka R., Chang H.-C., Nat. Commun. 2016, 7, 12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Elangovan S., Topf C., Fischer S., Jiao H., Spannenberg A., Baumann W., Ludwig R., Junge K., Beller M., J. Am. Chem. Soc. 2016, 138, 8809–8814; [DOI] [PubMed] [Google Scholar]
- 5b. Mastalir M., Glatz M., Gorgas N., Stöger B., Pittenauer E., Allmaier G., Veiros L. F., Kirchner K., Chem. Eur. J. 2016, 22, 12316–12320; [DOI] [PubMed] [Google Scholar]
- 5c. Mukherjee A., Nerush A., Leitus G., Shimon L. J. W., Ben David Y., Espinosa Jalapa N. A., Milstein D., J. Am. Chem. Soc. 2016, 138, 4298–4301; [DOI] [PubMed] [Google Scholar]
- 5d. Tondreau A. M., Boncella J. M., Organometallics 2016, 35, 2049–2052; [Google Scholar]
- 5e. Vijjamarri S., Chidara V. K., Rousova J., Du G., Catal. Sci. Technol. 2016, 6, 3886–3892; [Google Scholar]
- 5f. Kallmeier F., Irrgang T., Dietel T., Kempe R., Angew. Chem. Int. Ed. 2016, 55, 11806–11809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11984–11988; [Google Scholar]
- 5g. Elangovan S., Neumann J., Sortais J.-B., Junge K., Darcel C., Beller M., Nat. Commun. 2016, 7, 12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Olsen S. E., Tangstand M., Lindstad T., Production of Manganese Ferroalloys, Tapir Akademisk Forlag, Trondheim, 2007, pp. 12–13; [Google Scholar]
- 6b. Hasan H., Manganese, Vol. 1, The Rosen Publishing Group, New York, 2008, pp. 18–29. [Google Scholar]
- 7.
- 7a. Gonzalez M. A., Lim M. A., Cheng S., Moyes A., Hobbs A. J., Mascharak P. K., Inorg. Chem. 2012, 51, 601–608; [DOI] [PubMed] [Google Scholar]
- 7b.S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge, M. Beller, Angew. Chem. Int. Ed 2016, DOI: 10.1002/anie.201607233; Angew. Chem 2016, DOI: ; [DOI] [PubMed]
- 7c.M. Perez, S. Elangovan, A. Spannenberg, K. Junge, M. Beller, ChemSusChem 2016, DOI: 10.1002/cssc.201601057; [DOI] [PubMed]
- 7d.J. Neumann, S. Elangovan, A. Spannenberg, K. Junge, M. Beller, unpublished results.
- 8. 13C-enriched MeOH was added to the in situ generated amido complex 8, thus leading to the appearance of a carbon signal at δ=26.56 ppm, which can be assigned to the -OCH3-group of the Mn-methoxide complex 9 No signal in the region of δ=170 ppm was detected, which could be attributed to the formation of the Mn-formate complex 11 as reported by Boncella and co-workers.[6d] Unluckily, the poor solubility of 1 in basic MeOH/H2O solution led to a very poor S/N ratio and made it impossible to assign any 13C signals.
- 9. Alberico E., Lennox A. J. J., Vogt L. K., Jiao H., Baumann W., Drexler H.-J., Nielsen M., Spannenberg A., Checinski M. P., Junge H., Beller M., J. Am. Chem. Soc. 2016, 138, 14890–14904. [DOI] [PubMed] [Google Scholar]
- 10. Enthaler S., Brück A., Kammer A., Junge H., Irran E., Gülak S., ChemCatChem 2015, 7, 65–69. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary