

Abstract
Aims
To evaluate the specific role of the endocannabinoid/cannabinoid type‐1 (CB1R) system in modulating mitochondrial dynamics in the metabolically active renal proximal tubular cells (RPTCs).
Materials and methods
We utilized mitochondrially‐targeted GFP in live cells (wild‐type and null for the CB1R) and electron microscopy in kidney sections of RPTC‐CB1R‐/‐ mice and their littermate controls. In both in vitro and in vivo conditions, we assessed the ability of CB1R agonism or fatty acid flux to modulate mitochondrial architecture and function.
Results
Direct stimulation of CB1R resulted in mitochondrial fragmentation in RPTCs. This process was mediated, at least in part, by modulating the phosphorylation levels of the canonical fission protein dynamin‐related protein 1 on both S637 and S616 residues. CB1R‐induced mitochondrial fission was associated with mitochondrial dysfunction, as documented by reduced oxygen consumption and ATP production, increased reactive oxygen species and cellular lactate levels, as well as a decline in mitochondrial biogenesis. Likewise, we documented that exposure of RPTCs to a fatty acid flux induced CB1R‐dependent mitochondrial fission, lipotoxicity and cellular dysfunction.
Conclusions
CB1R plays a key role in inducing mitochondrial fragmentation in RPTCs, leading to a decline in the organelle's function and contributing to the renal tubular injury associated with lipotoxicity and other metabolic diseases.
Keywords: cannabinoids, cellular research, experimental pharmacology
1. INTRODUCTION
Mitochondrial morphology is highly dynamic over time, changing rapidly and significantly in response to cellular stress or to altered metabolic demand.1 Mitochondria are shaped by ongoing fusion and fission events, and the lack of balance between these opposing processes is often detrimental to maintaining mitochondrial homeostasis. In fact, a breach in this balance has been implicated in the onset of many pathological conditions, such as neurodegenerative diseases and aging, as well as obesity and its related kidney injuries (reviewed in References 2 and 3).
The endocannabinoid (eCB) system plays an important role in energy balance and homeostasis.4, 5 The main eCBs, N‐arachidonoylethanolamine (AEA) and 2‐arachidonoylglycerol (2‐AG), evoke many cellular pathways by binding and activating the two G‐protein‐coupled receptors, cannabinoid type‐1 (CB1R) and type‐2 (CB2R). The eCB/CB1R system is highly overactive during obesity,6, 7 and both central and peripheral stimulations of this system have been suggested to mediate obesity‐related comorbidities, contributing to the development of the metabolic syndrome.6, 8 Recently, we have demonstrated that over‐activated CB1R mediates obesity‐induced kidney injury, dysfunction, inflammation and fibrosis by compromising the function of renal proximal tubular cells (RPTCs).9 RPTCs are metabolically active and reabsorb up to 80% of the solute and water passing through the nephron via specialized ATP‐dependent membrane transporters.10 To meet this high energy demand, RPTCs rely on mitochondrial oxidative phosphorylation (OXPHOS) to produce ATP.11 Thus, an impairment in mitochondrial bioenergetics may result in renal dysfunction. Indeed, accumulating evidence links mitochondrial dysfunction to the development and progression of diseases such as acute kidney injury, diabetic nephropathy and chronic kidney disease.11, 12, 13, 14, 15, 16, 17 This is further supported by our recent finding that RPTC dysfunction is accompanied by decreased utilization of fatty acids by mitochondria and impaired ATP production. Notably, these effects are rescued by a peripheral blockade of CB1R,9 in agreement with reports by others who describe a role for the eCB/CB1R system in modulating different aspects of mitochondrial physiology.18, 19, 20, 21 However, direct modulation of mitochondrial dynamics by eCBs has not yet been described. Here, we show that directly stimulating CB1R modulates mitochondrial dynamics and function in RPTCs. Specifically, activating CB1R promotes excessive mitochondrial fragmentation by regulating the phosphorylation of dynamin‐related protein 1 (DRP1), leading to mitochondrial dysfunction in the kidney. Interestingly, an acute fatty acid overload also induces CB1R‐dependent mitochondrial fragmentation in RPTCs, suggesting a role for the eCB system in mediating the mitochondrial damage associated with renal lipotoxicity.
2. MATERIALS AND METHODS
2.1. Animals and experimental protocol
All animal studies were approved by the Institutional Animal Care and Use Committee of the Hebrew University of Jerusalem (AAALAC accreditation #1285). Male, 8–10‐week‐old RPTC‐CB1R‐/‐ mice9, 22 and their wild‐type (WT) littermate controls were used for the in vivo experiments. To activate CB1R, mice received a single intraperitoneal injection of 10 mg/kg AEA or arachidonyl‐2′‐chloroethylamide (ACEA) (90050 or 91054, respectively; Cayman Chemicals, Ann Arbor, Michigan). To test the effect of fatty acid flux‐induced CB1R activation on the kidney, mice received a standard diet (STD) (14% fat, 24% protein, 62% carbohydrates; NIH‐31 rodent diet) or a high‐fat diet (HFD) (60% fat, 20% protein and 20% carbohydrates; Research Diet, D12492) for 7 days. Mice were euthanized by a cervical dislocation under anesthesia, and blood and kidneys were harvested for further analyses.
2.2. Multi‐parameter metabolic assessment
Mice were metabolically assessed using the Promethion High‐Definition Behavioral Phenotyping System (Sable Instruments, Inc., Las Vegas, Nevada) as described previously.9
2.3. Cell culture
WT or CB1R‐/‐‐HK‐2 cells (human immortalized RPTCs) were maintained in a low glucose DMEM (01‐050‐1A; Biological Industries, Beit Haemek, Israel) supplemented with 5% FCS, 100 mM glutamine, 100 mM Na‐pyruvate and Pen/Strep (Thermo Fisher Scientific, UK). Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2/95% air. To test the effect of CB1R activation, cells were seeded in 6‐well plates (25 × 104 cells/well) for 24 hours. Then, growth medium was replaced with serum‐free medium (SFM) for an additional 12 hours. On the morning of the experiment, the medium was replaced with fresh SFM containing either vehicle (EtOH), 5 μM AEA, 5 μM ACEA or 250 nM JZL195 for 6 hours. To mimic fatty conditions, cells were overloaded with a mixture of fatty acids containing 0.1 mM sodium Oleate (O7501; Sigma Aldrich, St. Louis, Missouri) and sodium Palmitate (P9767; Sigma Aldrich) in a ratio of 2:1 (O:P, respectively), dissolved in 11% free‐fatty acid BSA solution (A7030; Sigma Aldrich). At indicated time points, cells were harvested for further analyses as described below.
2.4. Stable transfection of RPTCs for mitochondrial overexpression of GFP
HK‐2 cells were transfected with pLYS1‐FLAG‐MitoGFP‐HA (#50057; Addgene, Cambridge, Massachusetts), using Lipofectamin3000 (L3000‐001; Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol. Transfected cells were selected with puromycin (1 μg/μL; P8833, Sigma Aldrich). Antibiotic‐resistant cells were then sorted for high GFP expression using FACSAria II (Becton Dickinson, Franklin Lakes, New Jersey), were plated, and MitoGFP expression was validated using a IX‐73 fluorescent microscope (OLYMPUS).
2.5. Genetic deletion of CB1R in RPTCs
HK‐2‐MitoGFP cells were transfected with CRISPR‐CAS9 vector containing a sgRNA sequence to target all human isoforms of CNR1 (GeneCopeia CS‐HCP263432‐CG01‐01‐B) using Lipofectamine3000 (L3000‐001, Invitrogen), and were selected with hygromycin (200 μM; H3274, Sigma Aldrich). Antibiotic‐resistant cells were then seeded in a single‐cell density and were allowed to grow. Single‐cell clones were analysed by DNA sequencing and mRNA analysis to confirm the deletion of CB1R (Figure S1).
2.6. Electron microscopy
Kidney slices (3 mm) were fixed overnight in 2% paraformaldehyde and 2.5% glutaraldeyde in 0.1 M cacodylate buffer (pH 7.4) at room temperature, and then washed four times in cacodylate buffer. Tissue slices were stained with 1% osmium tetroxide, 1.5% potassium ferricyanide in 0.1 M cacodylate buffer for 1 hour, were washed four times in cacodylate buffer and were dehydrated. Following dehydration, slices were infiltrated with increasing concentrations of Agar 100 resin in propylene oxide, consisting of 25%, 50%, 75% and 100% resin, for 16 hours each, and were then embedded in fresh resin and allowed to polymerize at 60 °C for 48 hours. Embedded tissues in blocks were sectioned with a diamond knife on a Leica Reichert Ultracut S microtome, and ultrathin sections (80 nm) were collected onto 200 Mesh, carbon–formvar‐coated copper grids. The sections on grids were sequentially stained with uranyl acetate and lead citrate for 10 minutes each and were viewed with Tecnai 12 TEM 100 kV (Phillips, Eindhoven, The Netherlands) equipped with a MegaView II CCD camera and Analysis version 3.0 software (SoftImaging System GmbH, Münstar, Germany).
2.7. Extracellular flux analysis
The cellular oxygen consumption rate (OCR) was measured using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies, Billerica, Massachusetts). Briefly, WT‐MitoGFP and CB1R‐/‐‐MitoGFP HK‐2 cells were seeded in an XF24 cell culture plate (25 × 103 cells/well) and were incubated overnight in a 37 °C humidified incubator with 5% CO2. Then, vehicle, AEA or O:P was added to the cells in fresh SFM for the indicated times. Basal OCR was measured at the end of the incubation period. Data were calculated from five consecutive measurements, post instrument calibration.
2.8. Total DNA extraction and mtDNA/nDNA measurement
Cells were harvested using trypsin, and DNA was extracted with a DNeasy Blood & Tissue Kit (69056, QIAGEN) according to the manufacturer's protocol. Isolated DNA served as a template for a qPCR reaction using primer sets for β2‐microglobulin (β2m) to amplify nuclear DNA (nDNA) and a D‐loop region to amplify mitochondrial DNA (mtDNA). Primers are listed in Table S1.
2.9. Quantification of mitochondrial morphology
Fluorescence images in .vsi format were processed in ImageJ software using the OlympusViewer Plugin, and were then subjected to analysis with the publicly available ImageJ macro for mitochondrial morphology, Mito‐Morphology, designed by Ruben K. Dagda.23 The macro returns the circularity value for each mitochondrion in the cell and the average circularity value of each measured cell. The average value ± SEM was plotted. The number of replicates is indicated in each figure legend. The perimeter of each mitochondria and the total mitochondrial area were also measured using the same macro. Interconnectivity was calculated as AVG area/perimeter ratio.
Electron microscope images in .tiff format were analysed using Adobe Photoshop C3S software. Images were analysed in RGB mode, and the measurement scale was set according to the image scale bar. Each mitochondrion was marked using the magnetic laso tool, and measurements of area and perimeter were recorded. Circularity was calculated by the software as 2, and the value of 1 represents a perfect circle. The length of mitochondria was measured by manually drawing a line along the mitochondrial major axis, using a ruler, and recording the value.
2.10. Endocannabinoid measurements by LC‐MS/MS
eCBs were extracted, purified and quantified in kidney and cells, as described previously.9 LC–MS/MS was analysed on an AB Sciex (Framingham, Massachusetts) Triple Quad 5500 mass spectrometer coupled with a Shimadzu (Kyoto, Japan) UHPLC System. eCBs were detected in a positive ion mode using electron spray ionization (ESI) and the multiple reaction monitoring (MRM) mode of acquisition.
The levels of each compound were analysed by monitoring multiple reactions. The molecular ion and fragment for each compound were measured as follows: m/z 348.3→62.1 (quantifier) and 91.1 (qualifier) for AEA, m/z 379.3→287.3 (quantifier) and 91.1 (qualifier) for 2‐AG, m/z 305.2→91.1 (quantifier) and 77.1 (qualifier) for AA, and m/z 352.3→66.1 (quantifier) and 91.1 (qualifier) for [2H4] AEA. The levels of AEA, 2‐AG and AA in the kidney and cells were measured against standard curves.
2.11. Statistical analysis
Data are presented as mean ± SEM. Unpaired two‐tailed Student's t‐test was used to determine variations between groups (GraphPad Prism v6 for Windows). Statistical significance was set at P < 0.05. Complete Materials and Methods may be found in the Supporting information section at the end of the article.
3. RESULTS
3.1. Direct activation of CB1R impairs mitochondrial architecture of RPTCs
To evaluate the direct role of CB1R in regulating mitochondrial dynamics in RPTCs, we tested the acute effect of AEA (10 mg/kg, ip) or vehicle in RPTC‐CB1R‐/‐ mice (lacking CB1R only in the RPTCs)9 and in their WT littermate controls. Electron micrographs of kidney sections demonstrated extensive alterations in mitochondrial architecture, mainly in mitochondrial fragmentation, following AEA administration (Figure 1A). Whereas WT mice treated with vehicle showed many elongated mitochondria, the mice treated with AEA contained round and shorter mitochondria (quantified in Figure 1B,C). The AEA‐induced impairment of mitochondrial morphology was CB1R dependent, as RPTC‐CB1R‐/‐ mice were protected from these morphological alterations (Figure 1A–C). As AEA is known to be rapidly degraded in vivo, leading to the production of arachidonic acid (AA), which, in turn, may cause mitochondrial fission on its own,24 we assessed the eCB “tone” in kidneys collected from mice 6 hours following an intraperitoneal injection of AEA. Renal AEA levels remained significantly high in comparison with Veh‐treated controls, whereas no changes in the 2‐AG and AA levels were found, highlighting the specificity of the effect documented by AEA (Figure S2). Moreover, we repeated the above‐mentioned experiment with the highly selective and stable CB1R agonist, ACEA, and found extensive mitochondrial fragmentation following its administration in vivo (10 mg/kg, ip) (Figure S3), thus supporting the notion that the effect is CB1R mediated. As AEA was given systemically, we assumed that the CB1R‐induced changes in mitochondrial architecture were not only restricted to the kidney, but could greatly affect the structure and activity of mitochondria elsewhere; indeed, an indirect assessment of mitochondrial activity in vivo revealed that acute AEA treatment resulted in decreased total oxygen consumption only in the WT animals (Figure 1D).
Figure 1.
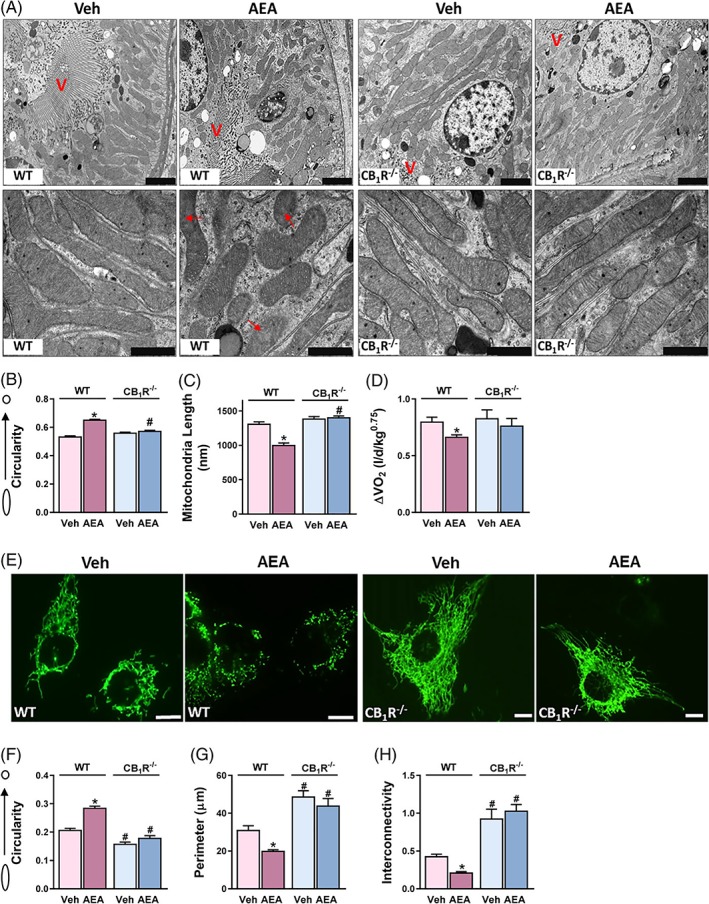
Direct activation of CB1R impairs mitochondrial architecture of RPTCs. Male, RPTC‐CB1R‐/‐ mice and their WT littermate controls received an intraperitoneal injection of either vehicle (Veh) or 10 mg/kg AEA. A, Mice were euthanized 6 hours after drug administration and their kidneys were processed for electron microscopy. Note: A disrupted mitochondrial morphology was induced by AEA treatment in WT mice only. Scale bar, 2 μm (in upper x3.9 k images) and 1 μm (in lower x23k images). Legend: V, brush border villi; red arrows, obstructed cristae. B,C, mitochondrial circularity and length were measured with adobe Photoshop C3S software. Data represent mean ± SEM of 400–700 mitochondria from 8–10 RPTCs from 2–3 mice per treated group. D, Mice were monitored by the Promethion High‐Definition Behavioral Phenotyping System (Sable Instruments, Inc.) over 6 hours. VO2, normalized to an effective body mass, was calculated. Data represent the mean ± SEM from 7–9 animals per group. E, WT‐HK‐2 or CB1R‐/‐‐HK‐2 cells were stably transfected with MitoGFP and treated with either Veh or 5 μM AEA for 6 hours. Live cells were examined under a fluorescent microscope to visualize mitochondrial morphology. F–H, mitochondrial circularity, perimeter and interconnectivity were measured using a publicly available ImageJ macro for mitochondrial morphology.23 Data represent the mean ± SEM from 30–65 cells in each group. Scale bar, 10 μm. *P < 0.05 relative to Veh‐treated animals or HK‐2 of the same cell line. # P < 0.05 relative to the same treated group in WT mice or HK‐2 cells
To further investigate the physiological role of the eCB/CB1R system in mitochondria of RPTCs, we established a cellular model in which CB1R was deleted from HK‐2 cells, stably expressing mitochondrially targeted GFP, using the CRISPR‐CAS genome editing technique (Figure S1). In accordance with our in vivo findings, we found striking differences in mitochondrial morphology associated with the presence or absence of and/or with stimulation of CB1R in vitro. AEA treatment resulted in excessive mitochondrial fragmentation in RPTCs (Figure 1E), indicated by increased mitochondrial circularity, and decreased perimeter and interconnectivity (Figure 1F–H). Interestingly, CB1R‐/‐‐HK‐2 cells displayed reduced basal circularity, increased perimeter and a hyper‐fused mitochondrial network compared to the WT cells, and AEA failed to induce mitochondrial fragmentation in the null cells. Similar findings were documented in cells treated with ACEA (5 μM), as well as with JZL195, a potent inhibitor of both fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), the primary enzymes responsible for degrading AEA and 2‐AG, respectively (Figure S4), suggesting that mitochondrial dynamics in RPTCs are regulated by CB1R.
3.2. Acute activation of CB1R in RPTCs impairs mitochondrial function and biogenesis
As mitochondrial shape is closely related to function, we next evaluated the functional consequence of CB1R activation on the mitochondria. In vitro AEA treatment resulted in a significant reduction in the basal OCR, and substantially decreased ATP levels in WT‐HK‐2 cells (Figure 2A,B). Similarly, AEA induced reduction in the NAD+/NADH ratio (Figure 2C), as well as a 15% increase in cellular lactate content (Figure 2D). All of the mitochondrial malfunctions induced by AEA were absent in CB1R‐/‐‐HK‐2 cells. Next, we assessed the effect of AEA on mitochondrial biogenesis, and found that acute activation of CB1R in WT‐HK‐2, but not in CB1R‐/‐‐HK‐2, cells resulted in a mild, yet significant, reduction in the mtDNA‐to‐nDNA ratio (Figure 2E). Likewise, the protein expression of two representative mitochondrial respiratory chain components, mtDNA‐encoded cytochrome C oxidase I (MTCO1) and nDNA‐encoded succinate dehydrogenase complex subunit A (SDHA) was downregulated following AEA exposure in WT‐HK‐2 cells (Figure 2F). As AEA has been shown to inhibit the proliferation of different cell lines,25, 26 and to induce apoptosis in others,27, 28 we wanted to exclude the possibility that the observed mitochondrial changes resulted from the deleterious effects of AEA on cell viability. Therefore, we performed CFSE staining, followed by FACS analysis, and revealed that AEA did not affect cell proliferation (Figure 2G). Moreover, no increase in the number of early or late apoptotic cells following AEA treatment was documented (Figure 2H). Taken together, our findings suggest that CB1R‐mediated mitochondrial fragmentation is followed by impaired mitochondrial function and reduced biogenesis, without compromising cell viability in the observed time frame.
Figure 2.
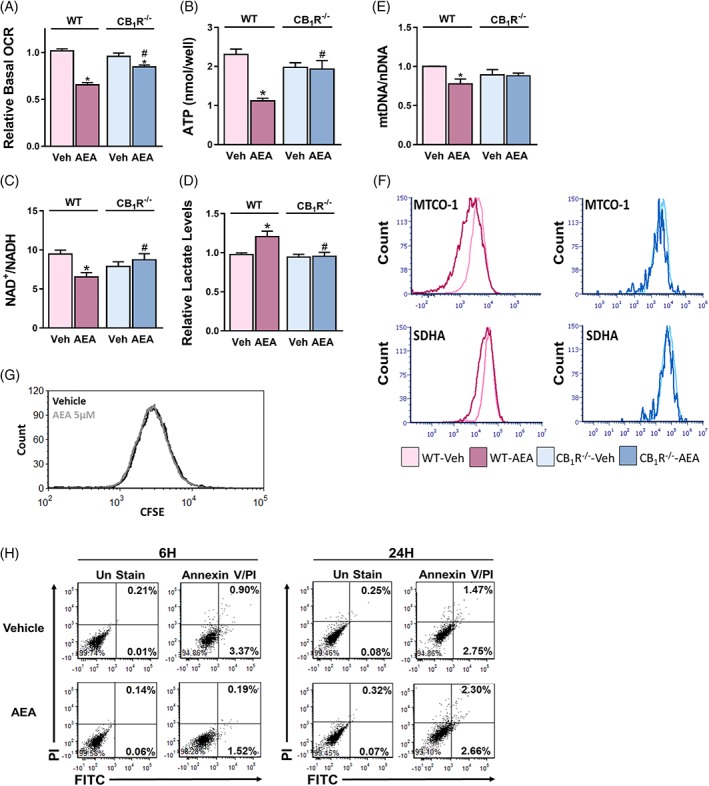
Acute activation of CB1R in HK‐2 cells impairs mitochondrial function and biogenesis. WT‐HK‐2 or CB1R‐/‐‐HK‐2 cells were treated with either vehicle (Veh) or 5 μM AEA for 6 hours and were then analysed for mitochondrial function. A, Reduced oxygen consumption rate (OCR) measured using the Seahorse XF analyzer, B, reduced ATP levels, and C, reduced NAD+/NADH ratio, as well as D, elevated cellular lactate levels, were found in WT‐HK‐2 cells treated with AEA (5 μM). Data represent the mean ± SEM of at least six replicates from three independent experiments. E, The ratio between mitochondrial and nuclear DNA. F, FACS analysis for MTCO‐1 and SDHA, demonstrating reduced biogenesis following treatment of wild‐type cells with AEA. G, H, WT‐HK‐2 cells, exposed to Veh or 5 μM AEA, were analysed by flow cytometry for proliferation (G) with CFSE staining after 24 hours of treatment or for apoptosis (H) with Annexin V and Propidium Iodide at the indicated time points. Data represent one experiment out of three performed (in triplicate). *P < 0.05 relative to Veh‐treated HK‐2 of the same cell line. # P < 0.05 relative to the same treated group in WT‐HK‐2 cells
3.3. RPTC‐CB1R induces DRP1‐mediated mitochondrial fission
To decipher the underlying molecular mechanism involved in CB1R‐induced mitochondrial fragmentation, we assessed the contribution of DRP1, a fundamental component of the fission machinery, to this phenomenon. At the mRNA level, DRP1 was slightly upregulated by AEA in WT‐HK‐2 cells (Figure 3A). On the other hand, a reduction in its phosphorylation on S637 was induced by AEA treatment in WT‐ but not in CB1R‐/‐‐HK‐2 cells (Figure 3B,C). Interestingly, the basal ratio between the S637‐phosphorylated and total DRP1 in CB1R‐/‐‐HK‐2 cells was higher than in the WT‐HK‐2 cells (Figure 3B,C), concurrent with their hyper‐fused mitochondrial network (Figure 1E–H). ACEA and JZL195 also reduced this ratio in a way similar to AEA (Figure S4D,E). When phosphorylated on S637, DRP1 is retained in the cytoplasm and, upon de‐phosphorylation, it is translocated to the mitochondria and induces fission.29, 30 Indeed, in a cellular fractionation analysis, AEA treatment led to decreased recovery of both S637‐phosphorylated DRP1 and total DRP1 from the cytoplasm of RPTCs, and increased the recovery of total DRP1 from the mitochondrial fraction compared with vehicle‐treated cells (Figure 3D). The fractionation fidelity and efficiency were controlled using anti‐GFP, which could be recovered only from the mitochondria. Furthermore, by using immune‐fluorescence, we found that DRP1 was distributed throughout the cytoplasm in vehicle‐treated RPTCs, whereas, in cells treated with AEA, the signal co‐localized with HSP60 at the peri‐nuclear region, indicating increased mitochondrial localization (Figure 3E,F). No significant changes in the DRP1 expression pattern or cellular localization were documented in CB1R‐/‐‐HK‐2 cells treated with AEA.
Figure 3.
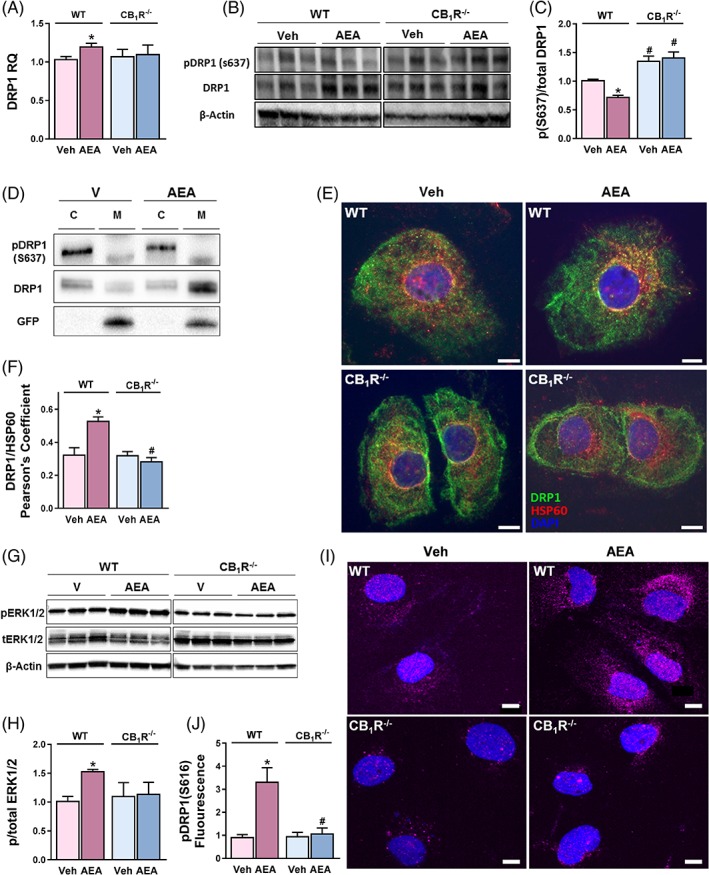
RPTC‐CB1R induces DRP1‐mediated mitochondrial fission. A, qPCR analysis of DRP1. B,C, Reduced expression of the phosphorylated (S637) form of DRP1 was measured in AEA (5 μM)‐treated WT‐HK‐2 cells. D, Cellular fractionation revealed increased localization of DRP1 in the mitochondria of RPTCs treated with AEA. E, Co‐staining of HSP60 and DRP1 in RPTCs. F, Co‐localization was quantified by calculating Pearson correlation coefficient [P(r)] of two fluorescent signals using CellSens Dimension Software. G,H, Increased phosphorylation of ERK1/2, as well as I, J, DRP1 (S616), in RPTCs treated with AEA. Data represent the mean ± SEM of at least five replicates from two–three independent experiments. *P < 0.05 relative to Veh‐treated animals or HK‐2 of the same cell line. # P < 0.05 relative to the same treated group in WT mice or HK‐2 cells. B, G Representative images from three–five independent experiments performed in triplicates
The inhibitory phosphorylation of DRP1 on S637 is dependent on protein kinase A (PKA),30, 31 whose activity is decreased when CB1R is stimulated,32, 33 providing a possible explanation for the involvement of CB1R in this process. Moreover, activating CB1R and inhibiting PKA, in turn, may lead to increased phosphorylation and the subsequent activity of ERK1/2, which was recently shown to directly phosphorylate DRP1 on S616 to promote fission.34, 35 Indeed, AEA treatment increased the phosphorylated‐to‐total ERK1/2 ratio (Figure 3G,H) as well as the levels of S616‐phosphorylated DRP1 (Figure 3I,J) in WT‐HK‐2 cells, but not in CB1R‐/‐‐HK‐2 cells, indicating that this signaling cascade is also involved in DRP1‐induced mitochondrial fragmentation in RPTCs. Taken together, our findings suggest that CB1R plays a pivotal role in inducing mitochondrial fission, via regulation of DRP1 phosphorylation or dephosphorylation and its cellular localization.
3.4. CB1R‐mediated mitochondrial fragmentation is cAMP‐dependent
To further corroborate the PKA signaling pathway in this process, we treated both cell lines with Forskolin, a potent inducer of cAMP generation. As expected, this treatment resulted in reduced mitochondrial circularity in both WT‐HK‐2 and CB1R‐/‐‐HK‐2 cells (Figure 4A,B), an effect that could be attributed to increased S637 phosphorylation of DRP1 (Figure 4C,D). The decreased circularity was reversed by AEA treatment in WT‐HK‐2 cells only, but not in HK‐2 cells lacking the CB1R (Figure 4A,B). Moreover, direct inhibition of PKA with H‐89 induced mitochondrial fragmentation (Figure 4E,F) and reduced S637 phosphorylation (Figure 4G,H) in both cell lines, suggesting that PKA is located downstream of CB1R and upstream of DRP1 in this cascade.
Figure 4.
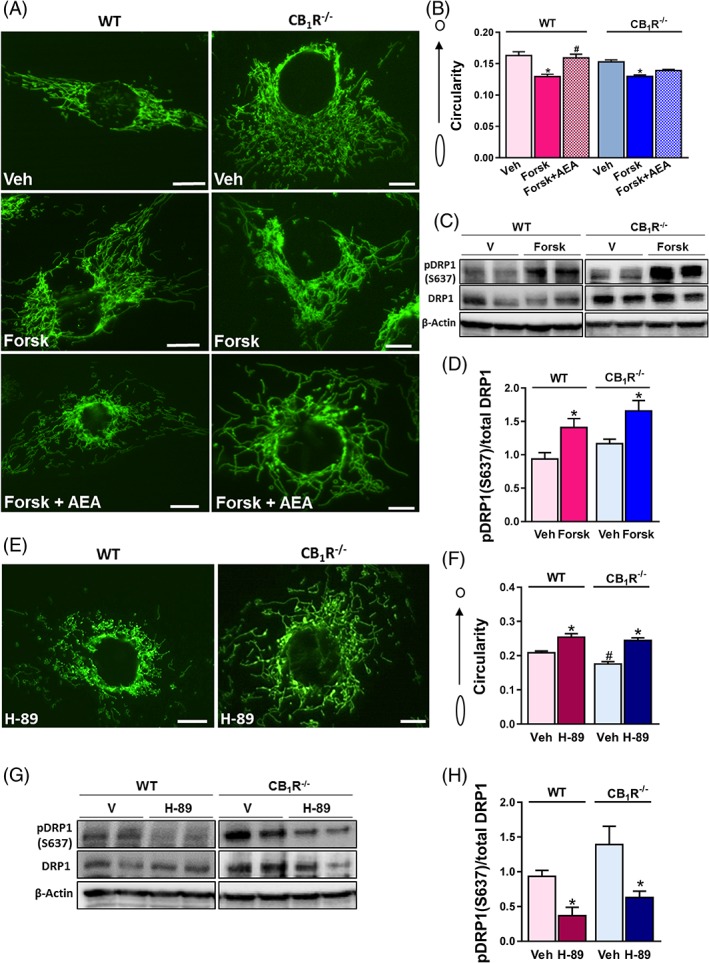
CB1R‐mediated mitochondrial fragmentation is cAMP‐dependent. A, WT‐HK‐2 or CB1R‐/‐‐HK‐2 cells, stably transfected with MitoGFP, were treated with vehicle (Veh) or 25 μM Forskolin for 1 hour to induce cAMP generation. AEA was then added to selected wells and live cells were examined under a fluorescent microscope to visualize mitochondrial morphology. B, Circularity was quantified using ImageJ macro.23 Data represent the mean ± SEM from >30 cells in each group. C, D, Western blot analysis and quantification of pDRP1 (s637). E, 10 μM of H‐89 were added to both cell lines for 3 hours, and F, circularity was quantified using ImageJ macro.23 Data represent the mean ± SEM from 30–120 cells in each group. G, H, Western blot analysis and quantification of pDRP1 (s637). Western blot data represent the mean ± SEM of four–five replicates from two independent experiments. *P < 0.05 relative to Veh‐treated animals or HK‐2 of the same cell line. # P < 0.05 relative to the same treated group in WT mice or HK‐2 cells
3.5. Deletion of CB1R ameliorates fatty acid‐induced mitochondrial fragmentation in RPTCs
High‐fat diet (HFD) feeding was shown to induce vast changes in mitochondrial architecture in the kidney, leading to proximal tubular injury and glomerulopathy.36 We recently reported that CB1R blockade protects against HFD‐induced reduction in mitochondrial fatty acid β‐oxidation and ATP production.9 This, together with the current data, encouraged us to explore the possibility that the eCB/CB1R system is also involved in HFD‐mediated changes in mitochondrial architecture. Specifically, we evaluated the effect of an acute fatty acid overload on mitochondrial dynamics in RPTCs both in vivo and in vitro. To that end, we fed RPTC‐CB1R‐/‐ mice and their WT littermate controls with either STD or HFD for a short‐term period, and followed mitochondrial shape and fat accumulation in the RPTCs. Interestingly, HFD‐fed WT animals exhibited an extensive impairment of mitochondrial morphology in RPTCs, while RPTC‐CB1R‐/‐ mice were almost completely protected from these alterations (Figure 5A). These alterations included increased mitochondrial circularity (Figure 5B) and reduced mitochondrial length (Figure 5C), as well as increased accumulation of fat droplets within RPTCs (Figure 5A, yellow arrows). As expected, both mouse strains gained a similar body weight (data not shown), as well as increased whole‐body oxygen consumption and total fat oxidation (Figure 5D,E). Likewise, mitochondrial fragmentation was documented in an in vitro model for fatty acid flux, in which wild‐type‐MitoGFP or CB1R‐/‐‐MitoGFP HK‐2 cells were incubated with a mixture of 0.1 mM O:P for 24 hours. The exposure to O:P resulted in excessive mitochondrial fragmentation (Figure 5F,G), and reduced basal OCR (Figure 5H), conferring the same functional decline as a direct activation of CB1R (Figure 1D). Additionally, reduced levels of DRP1 S637 phosphorylation in the WT‐HK2 cells was documented (Figure 5I,J). Conversely, HK‐2 cells that do not express CB1R had significantly lower circularity scores in response to O:P (Figure 5G), with no significant changes in basal OCR or DRP1‐S637 phosphorylated levels, suggesting that CB1R mediates the changes documented in mitochondrial architecture that were induced by increased fatty acid flux. Interestingly, both direct and fatty acid‐induced activation of CB1R in wild type cells resulted in intracellular lipid accumulation (Figure 5K,L) and elevated levels of reactive oxygen species (Figure 5M,N), which are often associated with mitochondrial dysfunction,37, 38 supporting a common mechanism that mediates mitochondrial damage in both experimental models.
Figure 5.
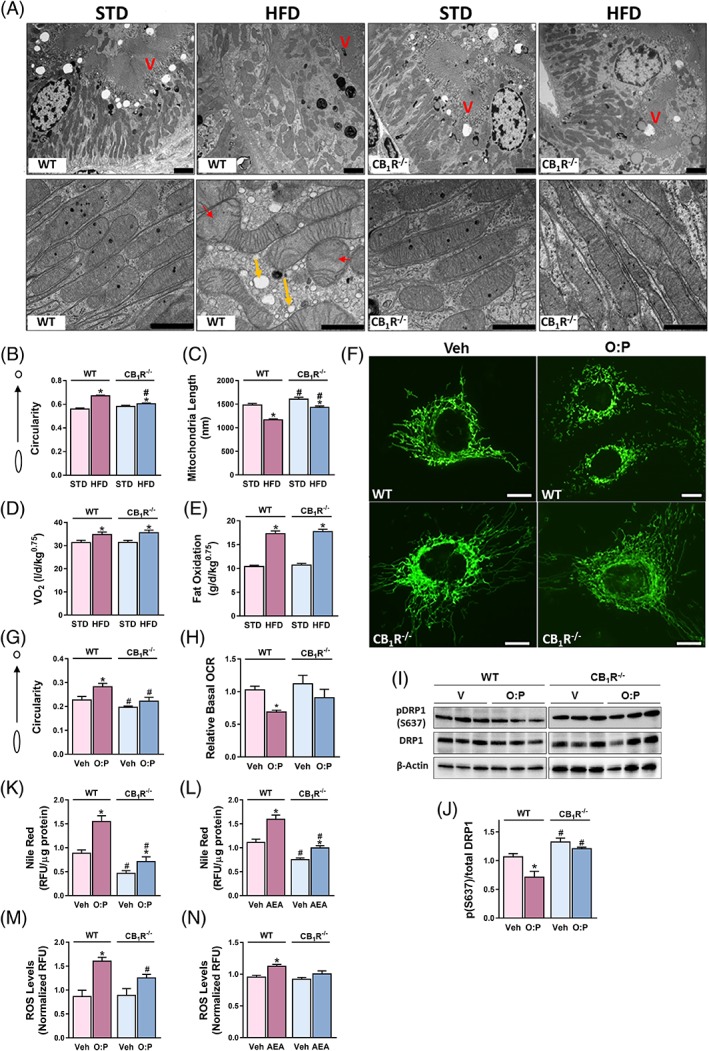
Deletion of CB1R ameliorates fatty acid‐induced mitochondrial fragmentation in RPTCs. Male RPTC‐CB1R‐/‐ mice and their WT littermate controls received STD or HFD for 7 days. A, Kidneys were processed for electron microscopy. Disrupted mitochondrial morphology induced by HFD feeding was documented only in WT mice. Scale bar, 2 μm (in upper x3.9 k images) and 1 μm (in lower in x23k images). Legend: V, Brush border villi; red arrows, obstructed cristae; yellow arrows, lipid vacuoles. B,C, Mitochondrial circularity and length were measured with Adobe Photoshop C3S software. Data represent mean ± SEM of at least 300 mitochondria from 5–7 RPTCs per group. D, E, Mice were monitored by the Promethion High‐Definition Behavioral Phenotyping System (Sable Instruments, Inc.) over the last 24 hours of the experiment. VO2 and total fat oxidation, normalized to an effective body mass, were calculated. Data represent the mean ± SEM from four–six animals per group. F, WT‐HK‐2 or CB1R‐/‐‐HK‐2 cells stably transfected with MitoGFP were examined under a fluorescent microscope to visualize mitochondrial morphology following an exposure to 0.1 mM mixture of O:P for 24 hours. Representative images from three independent experiments performed in triplicates. Scale bar, 10 μm. G, Circularity was quantified using ImageJ macro.23 Data represent the mean ± SEM from >20 cells for each group. H, Reduced oxygen consumption rate (OCR) in WT‐HK‐2 cells treated with fatty acids, as measured by the Seahorse XF analyzer. I, J, Reduced expression of the phosphorylated (S637) form of DRP1 was measured in O:P‐treated WT‐HK‐2 cells. K, L Quantification of intracellular fat accumulation following O:P and AEA treatment by using Nile‐Red staining. M, N, ROS production was measured in cell lysates using commercial kits, as described in the methods section (Supporting Information). Data represents mean ± SEM of at least 15 replicates per group, from three independent experiments. *P < 0.05 relative to Veh‐treated animals or HK‐2 of the same cell line. # P < 0.05 relative to the same treated group in WT mice or HK‐2 cells
4. DISSCUSSION
The ability of the eCB/CB1R system to regulate energy balance and metabolism is well established, including its effects on appetite39, 40 and energy expenditure.41 These effects are not only limited to central regulation, because the eCB system, specifically via the CB1R, controls cellular metabolism in peripheral organs also, including the liver, adipose tissue and kidney.42, 43, 44, 45, 46 Whereas several lines of investigation suggest that the eCB/CB1R system regulates bioenergetics by directly regulating mitochondrial function (reviewed in Reference 19), no evidence implicating mitochondrial dynamics in this phenomenon has yet been reported. Here, we documented, for the first time, the involvement of CB1R in regulating mitochondrial morphology and assessed its relevance to renal lipotoxicity.
Previous reports clearly link altered metabolic flux with mitochondrial shape and function (reviewed in Reference 47). Specifically, metabolic insults, such as elevated levels of glucose48, 49 or fatty acids,50 result in mitochondrial fragmentation and dysfunction. As CB1R activation recapitulates the conditions of excess nutrients, it was very exciting to discover that AEA, its stable analog, ACEA, or increased eCB “tone” induces excessive mitochondrial fragmentation in RPTCs. This notion is further supported by our findings that short‐term HFD feeding in vivo and acute exposure to fatty acids in vitro result in mitochondrial fragmentation, mediated also via the CB1R. Similarly, long‐term HFD feeding has been shown to induce mitochondrial fragmentation in the kidney,36 and treating obese mice with SS‐31, which protects cristae structure, preserves mitochondrial architecture and restores normal mitochondrial function,36 emphasizing the tight link between architecture and function. Likewise, our findings suggest that activating the eCB system via CB1R may link fatty acid flux with mitochondrial fragmentation. These findings are of great importance, considering the well‐documented contribution of mitochondrial dysfunction to the development of kidney injury,15, 16, 17, 51, 52 making CB1R a compelling target for treating lipotoxicity‐related kidney damage. Indeed, our recent work supports this notion as peripheral CB1R blockade, as well as its specific genetic deletion from RPTCs, ameliorates obesity‐induced kidney dysfunction, inflammation and tubulointerstitial fibrosis by preventing lipid accumulation and subsequent lipotoxicity, which is associated with abnormal mitochondrial function of RPTCs.9 The current work further highlights the contribution of CB1R‐induced mitochondrial fragmentation to such cellular damage. These findings could generally be associated with regulating energy utilization because the kidneys are second only to the heart in mitochondrial abundance and oxygen consumption,53 and because the RPTCs constitute approximately half of the kidney mass and use the majority of oxygen consumed by the organ.54
A link between mitochondrial morphology and bioenergetics was first reported decades ago,55 with a substantial amount of supporting data accumulating ever since. The relationship between the two mitochondrial features is reciprocal, as evident from studies showing that manipulation of mitochondrial dynamics led to deficiencies in mitochondrial respiration and ATP depletion.56 In contrast, a few studies reported that inhibiting complex I, or complementing it genetically, can affect mitochondrial fragmentation.1, 57 In accordance with recent reports demonstrating that AEA induces reduced OXPHOS and ATP production,58, 59 we documented here reduced OCR, as well as reduced ATP levels and the NAD+/NADH ratio in WT‐ but not in CB1R‐/‐‐HK‐2 cells treated with AEA, suggesting a functional decline in mitochondrial respiration following CB1R activation.
Previous studies have linked activated CB1R with decreased mitochondrial biogenesis in other tissues.21, 60 Similarly, we found decreased mtDNA content and mitochondrial protein expression following treatment of WT‐HK‐2 cells with AEA. However, we could not detect any appreciable changes in the mRNA expression levels of Complex I or Complex IV components (data not shown). Taken together, these findings support the key role of CB1R in modulating mitochondrial function and, possibly, biogenesis. Notably, we cannot rule out the involvement of enhanced autophagy rather than impaired biogenesis in reducing mitochondrial content.
Our findings are also in agreement with those of others (reviewed in Reference 61) who describe the role of the eCB system in regulating ROS production and the redox state. Whereas several studies reported that activating the eCB system confers protection from oxidative stress,62, 63 others demonstrated that its stimulation results in ROS generation and cell death.20, 28 Interestingly, Mukhopadhyay and colleagues found that pharmacological inhibition or genetic deletion of CB1R alleviates cisplatin‐induced kidney injury by attenuating MAPK activation and, consequently, cell death associated with oxidative stress.64 As AEA also activates CB2R and interacts with other membrane receptors, ion channels and effector proteins,65 it is possible that some of the AEA‐induced changes are mediated via other receptors. In fact, there is evidence that AEA also exerts some of its actions in CB1R null mice.65 Nevertheless, as most of the effects of AEA observed here were negated in RPTC‐CB1R‐/‐ mice and in CB1R‐/‐‐HK‐2 cells, as well as the fact that both ACEA‐ and JZL195‐induced eCB production resulted in a similar degree of mitochondrial fragmentation in RPTCs, off‐target effects are less likely to mediate the entire phenomenon described here.
The observed CB1R‐dependent mitochondrial fragmentation is mediated, at least in part, by regulating the phosphorylation levels of DRP1, one of the key post‐translational modifications of DRP1. Our findings are in agreement with studies showing that PKA‐mediated phosphorylation of S637 inhibits fission by retaining DRP1 in the cytoplasm.30, 31 On the other hand, others have demonstrated that phosphorylation of the same conserved residue by CaMKIIα or ROCK1 enhances DRP1 recruitment to mitochondria,66, 67 therefore promoting fission. This alleged contradiction most likely suggests that the functional consequence of DRP1 phosphorylation on its activity is stimulus and/or cell‐type dependent. The peri‐nuclear accumulation of both S616‐phosphorylated and total‐DRP1 following CB1R stimulation further supports enhanced fission, as such peri‐nuclear clustering of fragmented mitochondria following the induction of fission has been reported previously.68
After CB1R was identified as a classic plasma membrane receptor, the effects of cannabinoids on mitochondrial function were thought to be either the indirect result of stimulating CB1Rs present on the plasma membrane or the result of unspecific changes in the mitochondrial membranes by these lipid ligands.69 Conversely, consistent evidence shows that mitochondria contain G proteins and their downstream effectors. In fact, 30% of neuronal mitochondria carry CB1Rs (mtCB1R), and both Δ9‐THC and eCBs suppress electron transport complex I activity and respiration in isolated mitochondria, probably by reducing cAMP levels and PKA activity.70 Since this initial discovery, others have suggested the presence of CB1Rs in the mitochondria of different organs.71, 72 Whether cannabinoids also modulate mitochondrial dynamics via a direct effect on mtCB1R in RPTCs remains to be determined, and we are currently conducting studies to elucidate this phenomenon.
In conclusion, our data describe a novel role for CB1R in modulating mitochondrial physiology by regulating the phosphorylation levels of DRP1 (Figure 6). We suggest that a direct CB1R activation, or a fatty environment, indirectly, may inhibit the activity of PKA, thereby reducing the phosphorylation of DRP1 on S637. In addition, CB1R stimulation led to increased phosphorylation of ERK1/2 which, in turn, may contribute to the enhanced phosphorylation of DRP1 on S616. Consequently, increased mitochondrial fission occurs, leading to deterioration in mitochondrial function. This, in turn, may lead to RPTC dysfunction, documented in obesity and other metabolic diseases associated with kidney damage.
Figure 6.
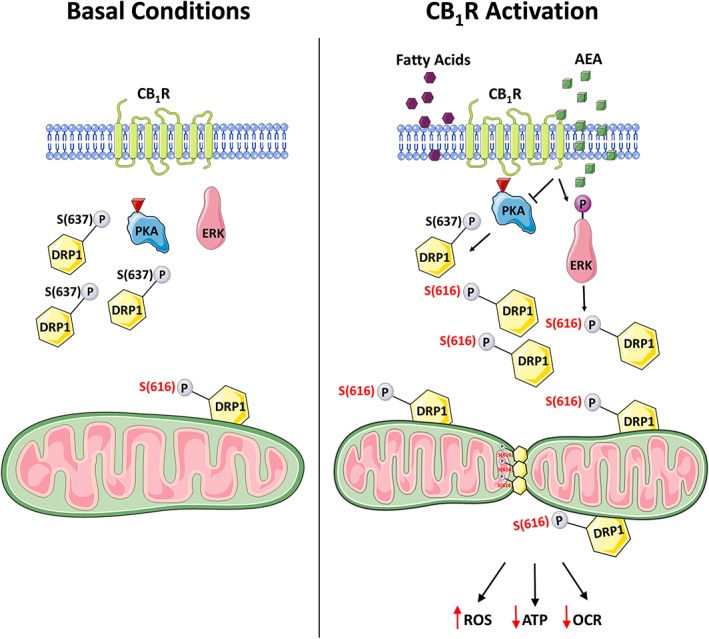
Suggested mechanism linking CB1R and mitochondrial dynamics. Left panel describes normal conditions in which PKA is active, thereby phosphorylating DRP1 on S637. This inhibitory phosphorylation retains DRP1 in the cytoplasm and fission is limited. Right panel describes either a direct activation of CB1R with AEA or a fatty acid flux‐induced indirect activation of CB1R that inhibits PKA, leading to decreased phosphorylation of S637, which allows DRP1 to target mitochondria and induce fission. PKA inhibition subsequently activates ERK1/2, which phosphorylates DRP1 on S616, further promoting fission. Excessive mitochondrial fragmentation under these conditions is followed by a functional decline of mitochondria
Supporting information
Figure S1 Generation of CB1R−/−‐HK‐2 cells. A, Cas9‐expressing vector containing gRNA sequence to target all hCNR1 isoforms was designed and provided by GeneCopoeiaTM. Wild‐type HK‐2 were transfected with the vector, and selected for Hygromycin resistance. Resistant cells were serially diluted, seeded in single‐cell density and expanded. B, DNA from different clones was isolated and sequenced. Clone no.23 had a T insertion mutation within the gRNA target sequence and low CB1R mRNA expression (C). It was designated as CB1R−/−‐HK‐2 in the manuscript
Figure S2. Renal endocannabinoid levels following AEA administration. Male, C57Bl6/J mice were administered an intraperitoneal injection of either vehicle (Veh) or 10 mg/kg AEA. Six hours following the administration, AEA (A), 2‐AG (B), and AA (C) were quantified by LC/MS‐MS. Data represent the mean ± SEM from 5 mice in each group. *P < 0.05 relative to Veh‐treated animals.
Figure S3. CB1R activation by ACEA induces in vivo mitochondrial fission in RPTCs. Male, C57Bl6/J mice were administered an intraperitoneal injection of either vehicle (Veh) or 10 mg/kg ACEA. Mice were euthanized 6 hours after drug administration and their kidneys were processed for electron microscopy. A, Note the disrupted mitochondrial morphology induced by ACEA treatment in RPTCs. A scale bar is indicated for each picture. Legend: V, brush border villi; red arrows, obstructed cristae. B, C, Mitochondrial circularity and length were measured with Adobe Photoshop C3S software. Data represent mean ± SEM of 650–850 mitochondria from 11–12 RPTCs (from 2 (Veh) or 3 (ACEA) mice per group). *P < 0.05 relative to Veh‐treated animals.
Figure S4. in vitro mitochondrial fission induced by ACEA or increased endocannabinoid ‘tone’ is CB1R mediated. WT‐HK‐2 or CB1R‐/‐‐HK‐2 cells were stably transfected with MitoGFP and treated with either vehicle (Veh), 5 μM ACEA, or 250 nM JZL195 for 6 hours. A, Live cells were examined under a fluorescent microscope to visualize the mitochondrial morphology. B, Mitochondrial circularity was measured using a publically available version of ImageJ macro for mitochondrial morphology. Data represent the mean ± SEM from 45‐02 cells in each group. Scale bar, 10 μm. *P < 0.05 relative to Veh‐treated HK‐2 of the same cell line. #P < 0.05 relative to the same treated group in WT‐HK‐2 cells. C, Increased endocannabinoid ‘tone’ after JZL195 administration in HK‐2 cells. Measurements of AEA, 2‐AG, and AA were done using LC‐MS/MS, and normalized to cellular protein content. Data represent the mean ± SEM from 12 replicates in each group. *P < 0.05 relative to Veh‐treated HK‐2 cells. D, E, Reduced ratio between the phosphorylated (S637) and total forms of DRP1 was measured in ACEA (5 μM), and JZL195 (250 nM)‐treated WT‐HK‐2 cells. Data represent the mean ± SEM of 3–6 replicates from 2 independent experiments. *P < 0.05 relative to Veh‐treated HK‐2 of the same cell line. #P < 0.05 relative to the same treated group in WT‐HK‐2 cells.
Table S1. Primer list
ACKNOWLEDGMENTS
We are grateful to Dr Yael Friedman from the Bio‐Imaging Unit, The Alexander Silberman Institute of Life Science of the Hebrew University for her technical assistance with transmission electron microscopy. We also appreciate the insights and assistance of Mr Dekel Assaf in implementing the quantification methods of mitochondrial morphology described in this work as well as the technical assistance of Dr Liad Hinden and Mr Shahar Azar.
Conflict of interest
The authors declare that no conflict of interest exists.
Author contributions
A. D. conducted the experiments and analysed the data. A. P. assisted in conducting the experiments. S. U. conducted and analyzed the in vivo metabolic assessment. R. H. provided reagents and technical assistance. A. N. conducted the LC–MS/MS analysis. A. D. and J. T. designed and supervised the experiments and wrote the manuscript.
Drori A, Permyakova A, Hadar R, Udi S, Nemirovski A, Tam J. Cannabinoid‐1 receptor regulates mitochondrial dynamics and function in renal proximal tubular cells. Diabetes Obes Metab. 2019;21:146–159. 10.1111/dom.13497
Funding information This work was supported by an ERC‐2015‐StG grant (#676841) as well as a grant from the Israel Science Foundation (ISF; 158/18) to J. T.
REFERENCES
- 1. Benard G, Bellance N, James D, et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838‐848. [DOI] [PubMed] [Google Scholar]
- 2. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105‐117. [DOI] [PubMed] [Google Scholar]
- 3. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fride E, Bregman T, Kirkham TC. Endocannabinoids and food intake: newborn suckling and appetite regulation in adulthood. Exp Biol Med. 2005;230:225‐234. [DOI] [PubMed] [Google Scholar]
- 5. Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73‐100. [DOI] [PubMed] [Google Scholar]
- 6. Engeli S, Bohnke J, Feldpausch M, et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes. 2005;54:2838‐2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Engeli S. Dysregulation of the endocannabinoid system in obesity. Neuroendocrinology. 2008;20(suppl 1):110‐115. [DOI] [PubMed] [Google Scholar]
- 8. Pagotto U, Vicennati V, Pasquali R. The endocannabinoid system and the treatment of obesity. Ann Med. 2005;37:270‐275. [DOI] [PubMed] [Google Scholar]
- 9. Udi S, Hinden L, Earley B, et al. Proximal tubular cannabinoid‐1 receptor regulates obesity‐induced CKD. J Am Soc Nephrol. 2017;28:3518‐3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiland C, Ahr HJ, Vohr HW, Ellinger‐Ziegelbauer H. Characterization of primary rat proximal tubular cells by gene expression analysis. Toxicol In Vitro. 2007;21:466‐491. [DOI] [PubMed] [Google Scholar]
- 11. Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. 2014;171:1917‐1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Casemayou A, Fournel A, Bagattin A, et al. Hepatocyte nuclear factor‐1beta controls mitochondrial respiration in renal tubular cells. J Am Soc Nephrol. 2017;28:3205‐3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210‐4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol. 2011;22:431‐436. [DOI] [PubMed] [Google Scholar]
- 16. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83:568‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hall AM, Unwin RJ. The not so 'mighty chondrion': emergence of renal diseases due to mitochondrial dysfunction. Nephron Physiol. 2007;105:1‐10. [DOI] [PubMed] [Google Scholar]
- 18. Hebert‐Chatelain E, Desprez T, Serrat R, et al. A cannabinoid link between mitochondria and memory. Nature. 2016;539:555‐559. [DOI] [PubMed] [Google Scholar]
- 19. Lipina C, Irving AJ, Hundal HS. Mitochondria: a possible nexus for the regulation of energy homeostasis by the endocannabinoid system? Am J Physiol Endocrinol Metab. 2014;307:E1‐E13. [DOI] [PubMed] [Google Scholar]
- 20. Siegmund SV, Qian T, de Minicis S, et al. The endocannabinoid 2‐arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB J. 2007;21:2798‐2806. [DOI] [PubMed] [Google Scholar]
- 21. Tedesco L, Valerio A, Dossena M, et al. Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: the role of eNOS, p38 MAPK, and AMPK pathways. Diabetes. 2010;59:2826‐2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hinden L, Udi S, Drori A, et al. Modulation of renal GLUT2 by the cannabinoid‐1 receptor: implications for the treatment of diabetic nephropathy. J Am Soc Nephrol. 2018;29:434‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dagda RK, Cherra SJ III, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843‐13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cereghetti GM, Costa V, Scorrano L. Inhibition of Drp1‐dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010;17:1785‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laezza C, Pisanti S, Crescenzi E, Bifulco M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006;580:6076‐6082. [DOI] [PubMed] [Google Scholar]
- 26. Ligresti A, Bisogno T, Matias I, et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology. 2003;125:677‐687. [DOI] [PubMed] [Google Scholar]
- 27. Patsos HA, Greenhough A, Hicks DJ, et al. The endogenous cannabinoid, anandamide, induces COX‐2‐dependent cell death in apoptosis‐resistant colon cancer cells. Int J Oncol. 2010;37:187‐193. [DOI] [PubMed] [Google Scholar]
- 28. Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Mackie K, Pacher P. Cannabinoid‐1 receptor activation induces reactive oxygen species‐dependent and ‐independent mitogen‐activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol. 2010;160:688‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin‐related protein Drp1. Ann N Y Acad Sci. 2010;1201:34‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP‐dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang CR, Blackstone C. Cyclic AMP‐dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583‐21587. [DOI] [PubMed] [Google Scholar]
- 32. Melck D, Rueda D, Galve‐Roperh I, De Petrocellis L, Guzman M, Di Marzo V. Involvement of the cAMP/protein kinase A pathway and of mitogen‐activated protein kinase in the anti‐proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463:235‐240. [DOI] [PubMed] [Google Scholar]
- 33. Mackie K. Cannabinoid receptors: where they are and what they do. Neuroendocrinology. 2008;20(suppl 1):10‐14. [DOI] [PubMed] [Google Scholar]
- 34. Kashatus JA, Nascimento A, Myers LJ, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK‐driven tumor growth. Mol Cell. 2015;57:537‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prieto J, Leon M, Ponsoda X, et al. Early ERK1/2 activation promotes DRP1‐dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. 2016;7:11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high‐fat diet‐induced glomerulopathy and proximal tubular injury. Kidney Int. 2016;90:997‐1011. [DOI] [PubMed] [Google Scholar]
- 37. Panov A, Schonfeld P, Dikalov S, Hemendinger R, Bonkovsky HL, Brooks BR. The neuromediator glutamate, through specific substrate interactions, enhances mitochondrial ATP production and reactive oxygen species generation in nonsynaptic brain mitochondria. J Biol Chem. 2009;284:14448‐14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang CH, Wu SB, Wu YT, Wei YH. Oxidative stress response elicited by mitochondrial dysfunction: implication in the pathophysiology of aging. Exp Biol Med. 2013;238:450‐460. [DOI] [PubMed] [Google Scholar]
- 39. Koch M. Cannabinoid receptor signaling in central regulation of feeding behavior: a mini‐review. Front Neurosci. 2017;11:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cota D, Marsicano G, Tschop M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112:423‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Osei‐Hyiaman D, Liu J, Zhou L, et al. Hepatic CB1 receptor is required for development of diet‐induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118:3160‐3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jourdan T, Djaouti L, Demizieux L, Gresti J, Verges B, Degrace P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet‐induced obese mice. Diabetes. 2010;59:926‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tam J, Vemuri VK, Liu J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953‐2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu J, Zhou L, Xiong K, et al. Hepatic cannabinoid receptor‐1 mediates diet‐induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tam J, Cinar R, Liu J, et al. Peripheral cannabinoid‐1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Janiak P, Poirier B, Bidouard JP, et al. Blockade of cannabinoid CB1 receptors improves renal function, metabolic profile, and increased survival of obese Zucker rats. Kidney Int. 2007;72:1345‐1357. [DOI] [PubMed] [Google Scholar]
- 47. Galloway CA, Yoon Y. Perspectives on: SGP symposium on mitochondrial physiology and medicine: what comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol. 2012;139:455‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653‐2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose‐induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Molina AJ, Wikstrom JD, Stiles L, et al. Mitochondrial networking protects beta‐cells from nutrient‐induced apoptosis. Diabetes. 2009;58:2303‐2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Decleves AE, Zolkipli Z, Satriano J, et al. Regulation of lipid accumulation by AMP‐activated kinase [corrected] in high fat diet‐induced kidney injury. Kidney Int. 2014;85:611‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mount P, Davies M, Choy SW, Cook N, Power D. Obesity‐related chronic kidney disease‐the role of lipid metabolism. Metabolites. 2015;5:720‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Forbes JM. Mitochondria‐power players in kidney function? Trends Endocrinol Metab. 2016;27:441‐442. [DOI] [PubMed] [Google Scholar]
- 55. Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 1966;30:269‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parone PA, Da Cruz S, Tondera D, et al. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One. 2008;3:e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol. 2005;289:C881‐C890. [DOI] [PubMed] [Google Scholar]
- 58. Athanasiou A, Clarke AB, Turner AE, et al. Cannabinoid receptor agonists are mitochondrial inhibitors: a unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochem Biophys Res Commun. 2007;364:131‐137. [DOI] [PubMed] [Google Scholar]
- 59. Zaccagnino P, Corcelli A, Baronio M, Lorusso M. Anandamide inhibits oxidative phosphorylation in isolated liver mitochondria. FEBS Lett. 2011;585:429‐434. [DOI] [PubMed] [Google Scholar]
- 60. Tedesco L, Valerio A, Cervino C, et al. Cannabinoid type 1 receptor blockade promotes mitochondrial biogenesis through endothelial nitric oxide synthase expression in white adipocytes. Diabetes. 2008;57:2028‐2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lipina C, Hundal HS. Modulation of cellular redox homeostasis by the endocannabinoid system. Open Biol. 2016;6:150276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Carracedo A, Geelen MJ, Diez M, Hanada K, Guzman M, Velasco G. Ceramide sensitizes astrocytes to oxidative stress: protective role of cannabinoids. Biochem J. 2004;380:435‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA. Involvement of protein kinase A in cannabinoid receptor‐mediated protection from oxidative neuronal injury. J Pharmacol Exp Ther. 2005;313:88‐94. [DOI] [PubMed] [Google Scholar]
- 64. Mukhopadhyay P, Pan H, Rajesh M, et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol. 2010;160:657‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Di Marzo V, De Petrocellis L, Fezza F, Ligresti A, Bisogno T. Anandamide receptors. Prostaglandins Leukot Essent Fatty Acids. 2002;66:377‐391. [DOI] [PubMed] [Google Scholar]
- 66. Han XJ, Lu YF, Li SA, et al. CaM kinase I alpha‐induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang W, Wang Y, Long J, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Frieden M, James D, Castelbou C, Danckaert A, Martinou JC, Demaurex N. Ca(2+) homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. J Biol Chem. 2004;279:22704‐22714. [DOI] [PubMed] [Google Scholar]
- 69. Fisar Z, Singh N, Hroudova J. Cannabinoid‐induced changes in respiration of brain mitochondria. Toxicol Lett. 2014;231:62‐71. [DOI] [PubMed] [Google Scholar]
- 70. Benard G, Massa F, Puente N, et al. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat Neurosci. 2012;15:558‐564. [DOI] [PubMed] [Google Scholar]
- 71. Koch M, Varela L, Kim JG, et al. Hypothalamic POMC neurons promote cannabinoid‐induced feeding. Nature. 2015;519:45‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mendizabal‐Zubiaga J, Melser S, Benard G, et al. Cannabinoid CB1 receptors are localized in striated muscle mitochondria and regulate mitochondrial respiration. Front Physiol. 2016;7:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Generation of CB1R−/−‐HK‐2 cells. A, Cas9‐expressing vector containing gRNA sequence to target all hCNR1 isoforms was designed and provided by GeneCopoeiaTM. Wild‐type HK‐2 were transfected with the vector, and selected for Hygromycin resistance. Resistant cells were serially diluted, seeded in single‐cell density and expanded. B, DNA from different clones was isolated and sequenced. Clone no.23 had a T insertion mutation within the gRNA target sequence and low CB1R mRNA expression (C). It was designated as CB1R−/−‐HK‐2 in the manuscript
Figure S2. Renal endocannabinoid levels following AEA administration. Male, C57Bl6/J mice were administered an intraperitoneal injection of either vehicle (Veh) or 10 mg/kg AEA. Six hours following the administration, AEA (A), 2‐AG (B), and AA (C) were quantified by LC/MS‐MS. Data represent the mean ± SEM from 5 mice in each group. *P < 0.05 relative to Veh‐treated animals.
Figure S3. CB1R activation by ACEA induces in vivo mitochondrial fission in RPTCs. Male, C57Bl6/J mice were administered an intraperitoneal injection of either vehicle (Veh) or 10 mg/kg ACEA. Mice were euthanized 6 hours after drug administration and their kidneys were processed for electron microscopy. A, Note the disrupted mitochondrial morphology induced by ACEA treatment in RPTCs. A scale bar is indicated for each picture. Legend: V, brush border villi; red arrows, obstructed cristae. B, C, Mitochondrial circularity and length were measured with Adobe Photoshop C3S software. Data represent mean ± SEM of 650–850 mitochondria from 11–12 RPTCs (from 2 (Veh) or 3 (ACEA) mice per group). *P < 0.05 relative to Veh‐treated animals.
Figure S4. in vitro mitochondrial fission induced by ACEA or increased endocannabinoid ‘tone’ is CB1R mediated. WT‐HK‐2 or CB1R‐/‐‐HK‐2 cells were stably transfected with MitoGFP and treated with either vehicle (Veh), 5 μM ACEA, or 250 nM JZL195 for 6 hours. A, Live cells were examined under a fluorescent microscope to visualize the mitochondrial morphology. B, Mitochondrial circularity was measured using a publically available version of ImageJ macro for mitochondrial morphology. Data represent the mean ± SEM from 45‐02 cells in each group. Scale bar, 10 μm. *P < 0.05 relative to Veh‐treated HK‐2 of the same cell line. #P < 0.05 relative to the same treated group in WT‐HK‐2 cells. C, Increased endocannabinoid ‘tone’ after JZL195 administration in HK‐2 cells. Measurements of AEA, 2‐AG, and AA were done using LC‐MS/MS, and normalized to cellular protein content. Data represent the mean ± SEM from 12 replicates in each group. *P < 0.05 relative to Veh‐treated HK‐2 cells. D, E, Reduced ratio between the phosphorylated (S637) and total forms of DRP1 was measured in ACEA (5 μM), and JZL195 (250 nM)‐treated WT‐HK‐2 cells. Data represent the mean ± SEM of 3–6 replicates from 2 independent experiments. *P < 0.05 relative to Veh‐treated HK‐2 of the same cell line. #P < 0.05 relative to the same treated group in WT‐HK‐2 cells.
Table S1. Primer list