

Abstract
Though kinase inhibitors have been heavily investigated in the clinic to combat advanced hepatocellular carcinoma (HCC), clinical outcomes have been disappointing overall, which may be due to the absence of kinase‐addicted subsets in HCC patients. Recently, strategies that simultaneously inhibit multiple kinases are increasingly appreciated in HCC treatment, yet they are challenged by the dynamic nature of the kinase networks. This study aims to identify clustered kinases that may cooperate to drive the malignant growth of HCC. We show that anaplastic lymphoma kinase, fibroblast growth factor receptor 2, and ephrin type‐A receptor 5 are the essential kinases that assemble into a functional cluster to sustain the viability of HCC cells through downstream protein kinase B–dependent, extracellular signal–regulated kinase–dependent, and p38‐dependent signaling pathways. Their coactivation is associated with poor prognosis for overall survival in about 13% of HCC patients. Moreover, their activities are tightly regulated by heat shock protein 90 (Hsp90). Thereby Combined kinase inhibition or targeting of heat shock protein 90 led to significant therapeutic responses both in vitro and in vivo. Conclusion: Our findings established a paradigm that highlights the cooperation of anaplastic lymphoma kinase, fibroblast growth factor receptor 2, and ephrin type‐A receptor 5 kinases in governing the growth advantage of HCC cells, which might offer a conceptual “combined therapeutic target” for diagnosis and subsequent intervention in a subgroup of HCC patients.
Abbreviations
- AKT
protein kinase B
- ALK
anaplastic lymphoma kinase
- EphA5
ephrin type‐A receptor 5
- ERK
extracellular signal–regulated kinase
- FGFR2
fibroblast growth factor receptor 2
- HCC
hepatocellular carcinoma
- Hsp90
heat shock protein 90
- LTK
leukocyte receptor tyrosine kinase
- p‐
phosphorylated
- RTV
relative tumor volume
- SEM
standard error of mean
- siRNA
small interfering RNA
Hepatocellular carcinoma (HCC) represents the major histological subtype of primary liver cancer and is associated with multiple etiological factors such as viral infection and alcohol consumption.1, 2, 3 In the clinic, most individuals are diagnosed at a late stage, when effective curative therapies are not feasible, rendering advanced HCC one of the most lethal cancer types worldwide.4, 5, 6 Recently, encouraged by the success of kinase inhibition in several “oncogene addiction”–defined tumor types, especially non‐small cell lung cancer, kinase inhibitors have become the mainstay in combating this systemic disease.7, 8, 9, 10 However, their overall clinical results are rather disappointing. For instance, treatment with the two Food and Drug Administration–approved drugs, sorafenib and regorafenib, only improved the overall survival of patients by about 2‐3 months.11, 12 Meanwhile, many subsequent clinical trials targeting diverse aberrantly activated kinases that are responsible for tumor growth or angiogenesis in HCC, such as c‐Met, epidermal growth factor receptor, and platelet‐derived growth factor receptor, all failed to achieve positive endpoints due to a lack of efficiency or intolerance.13, 14 One major reason for these failures is lack of consensus of addiction to kinases as revealed by comprehensive genomic studies.15, 16 Unlike other solid tumors, many well‐recognized or targetable driving alterations in kinase genes, such as epidermal growth factor receptor mutation and echinoderm microtubule–associated protein kinase–like 4/anaplastic lymphoma kinase (ALK) rearrangement, are rarely detected in HCC patient samples.17, 18, 19 These observations suggested that stratifying patients according to their genetic kinase alterations seems unfeasible in the setting of HCC treatment.
Recently, concurrent inhibition of several overactivated kinases has been increasingly recognized for its potential to gain therapeutic advantages.18, 20, 21, 22, 23 Nevertheless, recent clinical investigations that randomly cotargeted some kinases have been quite disappointing. These failures may arise from the intrinsic dynamic nature of the kinase network.24, 25, 26 In this study, we hypothesized that precisely cotargeting a limited cluster of critical kinases that stringently cooperated to sustain the viability of HCC cells may result in optimal therapeutic outcome. We tested this possibility by profiling and stratifying a group of pivotal kinases, accounting for the growth advantage of HCC cells and the prognosis of patients. We further depicted several rational therapeutic approaches for the clinical management of kinase coactivation in a defined subcohort of HCC patients.
Materials and Methods
CELL CULTURE AND REAGENTS
SMMC‐7721, ZIP177, QGY‐7703, BEL‐7402, SK‐Hep‐1, and QSG‐7701 cells were obtained from the Cell Bank, Chinese Academy of Sciences (Shanghai, China). HepG2, Hep3B, and Huh‐7 cells were obtained from the American Type Culture Collection (Manassas, VA). All cell lines from the American Type Culture Collection were authenticated by short tandem repeat testing (Genesky Biopharma Technology, Shanghai, China). All cell lines were maintained in appropriate medium as the suppliers suggested.
The inhibitors used for in vitro studies were obtained from Selleck Chemicals (Shanghai, China) and dissolved to 10 mmol/L with dimethyl sulfoxide as stock solution. Ceritinib, AZD4547, and dasatinib for in vivo studies were obtained from Melonepharma (Dalian, China).
HUMAN RECEPTOR TYROSINE KINASE PHOSPHORYLATION ARRAY
Cells were seeded in 100‐mm dishes and incubated at 37°C for 24 hours. Cells were washed twice with cold phosphate‐buffered saline, followed by solubilization at 2 × 107 cells/mL in 1 × lysis buffer provided by Raybiotech (Norcross, GA). After centrifugation at 14,000g for 20 minutes for removal of cell debris, the whole lysates of HCC cell lines were subjected to Human RTK Phosphorylation Arrays C1; the experimental operation was conducted by Raybiotech (Guangzhou, China).
PATIENT SAMPLES AND TISSUE MICROARRAYS
The HCC tissue microarrays and paired frozen tumor samples were obtained from Zuo Cheng Bio (Shanghai, China). Immunohistochemical analysis was performed following routine protocols. The primary antibodies used were as follows: phosphorylated ALK (p‐ALK; GTX16377; Genetex, Irvine, CA), 1:50; phosphorylated fibroblast growth factor receptor 2 (p‐FGFR2; ab111124; Abcam, Cambridge, MA), 1:50; phosphorylated ephrin type‐A receptor 5 (p‐EphA5; GTX17348; Genetex), 1:50; cluster of differentiation 34 antibody (CD34 ab8158; Abcam), 1:100; and Ki‐67 antibody (ab16667; Abcam), 1:100. Antirabbit or antimouse peroxidase‐conjugated antibodies (Santa Cruz, Paso Robles, CA) were used as secondary antibodies. The array staining and data assessment were conducted by Zuo Cheng Bio. Kaplan‐Meier plotter analysis was conducted to assess the relative influence of prognostic factors on overall survival. For immunoblotting analysis, tumor samples and paired normal tissues were homogenized in cold radio immunoprecipitation assay lysis buffer (Beyotime, Nantong, China) supplemented with protease and phosphatase inhibitors (Merck, Darmstadt, Germany), quantitated using a bicinchoninic acid assay (Beyotime), and boiled with sodium dodecyl sulfate lysis buffer.
HCC CELL LINE XENOGRAFTS
Six‐week‐old female BALB/c nude mice were obtained from Vital River (Beijing, China). All studies were done in compliance with the Institutional Animal Care and Use Committee guidelines of the Shanghai Institute of Materia Medica. Tumors were generated by transplanting 5 × 106 SMMC‐7721 cells resuspended in phosphate‐buffered saline (200 μL/mouse) into the right flank. Mice were maintained under specific pathogen‐free conditions. Prior to initiation of treatment, mice were randomized among control and treated groups (n = 6 per group). Ceritinib (0.5% methylcellulose and 0.5% Tween‐80 in a ratio of 1:1), AZD4547 (1% Tween‐80), or dasatinib (0.05% sodium carboxymethyl cellulose) was orally administered once a day. Ganetespib (10% dimethyl sulfoxide, 18% Cremophor RH40, 3.6% dextrose in water) was intraperitoneally injected three times a week. For combination treatment, the drugs were given concurrently. Tumor volume (V) was measured three times a week and calculated by caliper measurements of the width (W) and length (L) of each tumor using the formula V = (L × W2)/2. The individual relative tumor volume (RTV) was calculated as follows: RTV = Vt/V0, where Vt is the volume on each day and V0 represents the volume at the beginning of the treatment. RTV was shown on indicated days as means ± standard error of the mean (SEM) indicated for groups of mice. At designated times, mice were sacrificed and tumor tissues were resected, homogenized in cold radio immunoprecipitation assay lysis buffer (Beyotime) supplemented with protease and phosphatase inhibitors (Merck), and then processed for immunoblotting.
PATIENT‐DERIVED TUMOR XENOGRAFTS
Tumor xenografts were established directly from patient tumors and routinely passaged by subcutaneous engraftment of female BALB/c nude mice (Wuxi Apptech). All experimental procedures were approved by the Wuxi Apptech Laboratory Animal Care and Use Committee. When the tumor volume reached around 100‐150 mm3 the animals were treated following randomization (n = 6 per group). Ceritinib (0.5% methylcellulose and 0.5% Tween‐80 in a ratio of 1:1), AZD4547 (1% Tween‐80), or dasatinib (0.05% sodium carboxymethyl cellulose) was orally administered once a day. Ganetespib (10% dimethyl sulfoxide, 18% Cremophor RH40, 3.6% dextrose in water) was intraperitoneally injected three times a week. For combination treatment, the drugs were given concurrently. Tumor volume (V) was measured twice a week and calculated by caliper measurements of the width (W) and length (L) of each tumor using the formula V = (L × W2)/2. The individual RTV was calculated as follows: RTV = Vt/V0, where Vt is the volume on each day and V0 represents the volume at the beginning of the treatment. RTV was shown on indicated days as means ± SEM indicated for groups of mice. At designated times, mice were sacrificed and tumor tissues were resected, homogenized in cold radio immunoprecipitation assay lysis buffer (Beyotime) supplemented with protease and phosphatase inhibitors (Merck), and then processed for immunoblotting.
STATISTICAL ANALYSIS
The difference between experimental groups was compared using an unpaired two‐tailed Student t test or two‐way analysis of variance. P < 0.05 was considered statistically significant, * P < 0.05, ** P < 0.01, *** P < 0.001. All analyses were conducted with GraphPad Prism 6 software (GraphPad Software, Inc., San Diego, CA).
Results
COACTIVATION OF ALK, FGFR2, AND EphA5 REGULATES THE VIABILITY OF HCC CELLS
We initiated this study to evaluate the basal activation status of tyrosine kinases in HCC cells. To this end, the phosphorylation of 71 tyrosine kinases was profiled across eight HCC cell lines using human receptor tyrosine kinase phosphorylation arrays. As expected, pairs of kinases were aberrantly activated in HCC cells, but the detailed activation pattern varied among these cell lines (Supporting Table S1). Given this intrinsic heterogeneity, we further selected the 20 top‐ranked kinases in each cell line for enriched cluster analysis to seek commonly coactivated kinases in most tested cell lines. Out of the total 41 enrolled kinases, including ALK and c‐Met, 17 were found to be consistently overactivated in more than five cell lines (Fig. 1A; Supporting Table S2). In accordance with the clinical observations, no obvious functional genomic alterations of these 17 kinases were detected (Supporting Table S3). Then, we applied genetic manipulation (RNA interference; Supporting Table S4) and pharmacologic perturbation (kinase inhibitor; Supporting Table S2) to determine their oncogenic roles. Intriguingly, inhibition of these kinases individually in ZIP177 and SMMC‐7721 cells barely showed significant growth inhibition (Supporting Fig. S1A‐C). These findings indicated the lack of dependence of HCC cells on a single kinase, which in part explained the failure of the “kinase addiction”–based practice in the clinic.
Figure 1.
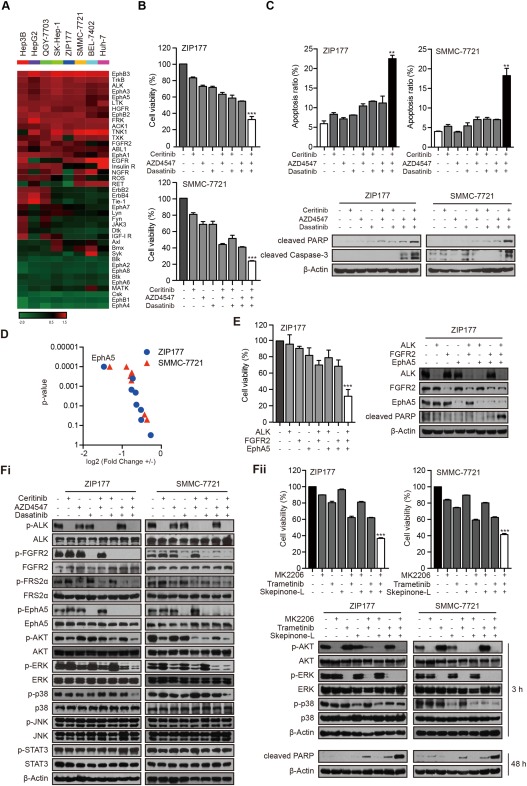
ALK, FGFR2, and EphA5 collaborate to regulate the viability of HCC cells through AKT‐dependent, ERK‐dependent, and p38‐dependent pathways. (A) The basal phosphorylation of the 20 top‐ranked kinases in HCC cell lines was selected for cluster analysis. (B) ZIP177 and SMMC‐7721 cells were treated with ceritinib (1 μM), AZD4547 (1 μM), and dasatinib (1 μM) respectively or in combination, for 72 hours. Cell viability was assessed by sulforhodamine B assay. Bars represent means ± standard deviation. (C) Cells were treated with indicated inhibitors at 1 μM for 48 hours. Apoptosis was analyzed by flow cytometry and immunoblotting. Bars represent means ± standard deviation. (D) ZIP177 and SMMC‐7721 cells were transfected with siRNAs for each indicated dasatinib target with or without ceritinib and AZD4547 for 72 hours. Mean values were compared by two‐way analysis of variance. +, ceritinib+ AZD4547+; –, ceritinib– AZD4547–. (E) ZIP177 cells were transfected with indicated siRNAs for 72 hours and analyzed by immunoblotting. (Fi) ZIP177 and SMMC‐7721 cells were treated with inhibitors at 1 μM for 3 hours, followed by immunoblotting analysis. (Fii) Cells were treated with MK2206 (1 μM), trametinib (1 μM) and skepinone‐L (10 μM). Cell viability was assessed by sulforhodamine B assay after 72h, and cell lysates were subjected to immunoblotting after 3h or 48h. Abbreviation: PARP, poly(adenosine diphosphate ribose) polymerase.
Then, we asked whether several key kinases could assemble into a stringent functional network to sustain the growth advantage of HCC cells. To determine this possibility, we used kinase inhibitors as probes and applied combinatory screenings to seek an effective synergistic toolbox, in which the clinically available drugs were preferred. The results of a two‐step screening demonstrated that, in contrast to their marginal effects individually, the coadministration of ceritinib, AZD4547, and dasatinib profoundly suppressed the growth of ZIP177 and SMMC‐7721 cells (Fig. 1B; Supporting Fig. S1D,E). Similar effects were also observed in several other HCC cell lines (Supporting Fig. S1F). Accordingly, a significant induction of apoptosis was also observed after cotreatment with ceritinib, AZD4547, and dasatinib, as determined by the annexin V/propidium iodide staining and evidenced by the promoted cleavage of poly(adenosine diphosphate ribose) polymerase and caspase‐3 (Fig. 1C). Moreover, this combination barely affected the viability of non‐malignant hepatocyte cells (Supporting Fig. S1G).
Thereafter, we explored the possibly targeted kinases by these three inhibitors. Of note, FGFR2 is the only kinase we selected for cluster analysis that targeted by AZD4547, a pan‐FGFR inhibitor (Supporting Table S2).27 As multiple kinases were modulated by dasatinib (Supporting Table S2),28 we made efforts to dissect its predominant targets in HCC cells. To this end, kinases known to be targeted by dasatinib were individually knocked down and their abilities to suppress cell growth assessed, with or without cotreatment with ceritinib and AZD4547. Among the tested kinases, only disruption of EphA5 led to the most significant effect on growth inhibition in both cell lines, suggesting that EphA5 represents the main target of dasatinib in HCC cells (Fig. 1D). Ceritinib has been approved for targeting ALK.29 Intriguingly, we also identified its strong potency toward leukocyte receptor tyrosine kinase (LTK) activity (data not shown), suggesting ceritinib as a dual ALK/LTK inhibitor. We further narrowed our focus to ALK as the disruption of ALK, but not LTK, combined with the cosilencing of FGFR2 and EphA5, recapitulated the notable growth repression observed using three kinase inhibitors (Fig. 1E; Supporting Fig. S1H,J). Meanwhile, substantial induction of apoptosis was observed after the concurrent interruption of ALK, FGFR2, and EphA5 (Fig. 1E; Supporting Fig. S1I). All of these data indicated that ALK, FGFR2, and EphA5 kinases, targeted by ceritinib, AZD4547, and dasatinib, respectively, might synergize to govern the survival of HCC cells. We also probed their downstream signaling cascades. Combined inhibition of ALK, FGFR2, and EphA5 remarkably blocked several key signaling pathways, namely phosphorylated protein kinase B (p‐AKT), phosphorylated extracellular signal–regulated kinase (p‐ERK) and p‐p38 (Fig. 1Fi). In turn, simultaneous blockade of AKT, ERK, and p38 signaling caused a similar effect, inducing growth inhibition and apoptosis (Fig. 1Fii).
In conclusion, these findings implied that ALK, FGFR2, and EphA5 are essential to sustain the survival of HCC cells through downstream AKT‐dependent, ERK‐dependent, and p38‐dependent signaling cascades. Their coinhibition will lead to a remarkable therapeutic response.
ABERRANTLY COACTIVATED ALK, FGFR2, AND EphA5 CORRELATE WITH POOR PROGNOSIS IN A SUBCOHORT OF HCC PATIENTS
The essential role of ALK, FGFR2, and EphA5 in HCC cells prompted us to evaluate whether their coactivation is suitable for patient stratification for both treatment and prognosis of HCC. We found that p‐ALK, p‐FGFR2, and p‐EphA5 were highly activated in most HCC cell lines compared to QSG‐7701, an immortalized hepatocyte cell line (Fig. 2A). We further profiled the phosphorylation of these kinases in 24 paired frozen tumor tissues of HCC patients. In agreement with the observations obtained in HCC cells, the levels of p‐ALK, p‐FGFR2, and p‐EphA5 were abundant in tumor tissues compared with their normal counterparts (Fig. 2B,C), indicating the aberrant activation of ALK, FGFR2, and EphA5 in HCC patients. Then, we applied tissue microarray analysis to gain insight into their potential clinical relevance. A total of 250 eligible HCC patient samples from three independent cohorts were enrolled and assessed by immunochemical staining. As expected, p‐ALK, p‐FGFR2, and p‐EphA5 showed variable abundance in different patients (Fig. 2D). Among them, 32 patient samples were stained for simultaneous high abundance of p‐ALK, p‐FGFR2, and p‐EphA5 (Fig. 2E), suggesting that about 13% of the patients may be selected for further treatment. We also applied Kaplan‐Meier plotter analysis to ascertain the effect of p‐ALK, p‐FGFR2, and p‐EphA5, alone or in combination, on the clinical outcome of HCC patients. Intriguingly, activation of these kinases individually showed no significant correlation to overall survival (Fig. 2F). Further subgroup analysis revealed that a subcohort of patients harboring coactivation of these three kinases showed worse prognosis compared to other subtypes (Fig. 2F). Collectively, these data suggested that concurrent activation of ALK, FGFR2, and EphA5 could help stratify a subcohort of HCC patients with poor prognosis as suitable for further therapeutic intervention.
Figure 2.
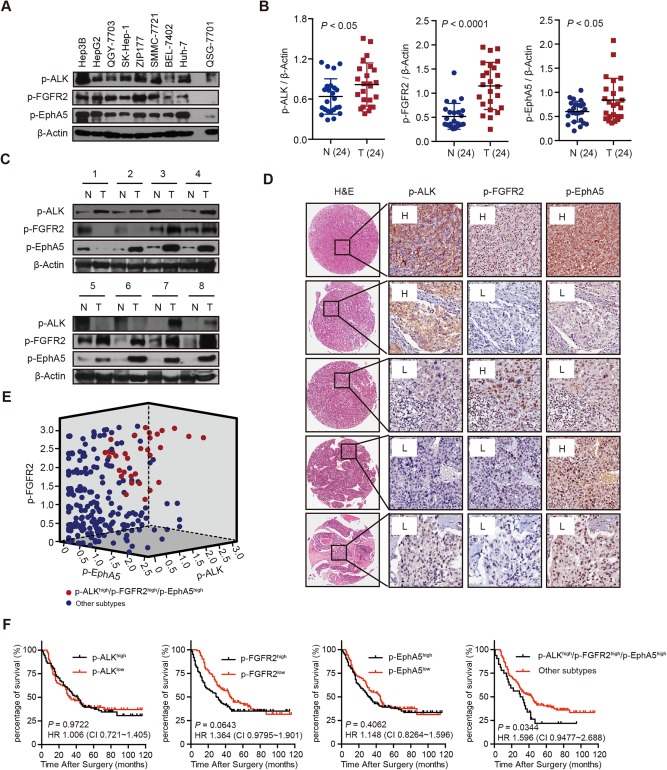
Coactivation of ALK, FGFR2, and EphA5 is highly correlated with poor prognosis in a subpopulation of HCC patients. (A) The expression of p‐ALK, p‐FGFR2, and p‐EphA5 in HCC cell lines and QSG‐7701 was analyzed by immunoblotting. (B,C) Expression of p‐ALK, p‐FGFR2, and p‐EphA5 in 24 paired tumor tissues was detected with immunoblotting. Dot plots represent the protein levels normalized to β‐actin (B), and representative immunoblotting results are shown (C). (D) Representative immunochemical staining results for p‐ALK, p‐FGFR2, and p‐EphA5 with different abundance are shown (original magnification, ×200). (E) Scatter plot of the p‐ALK, p‐FGFR2, and p‐EphA5 scores generated from the image analysis intensity algorithm. Red plots depict the subtype of patients with triple strong staining. (F) Retrospective analyses for overall survival of HCC patients were conducted using the Kaplan‐Meier method. Abbreviations: CI, confidence interval; H, relative abundance higher than the median; H&E, hematoxylin and eosin; HR, hazard ratio; L, relative abundance lower than the median; N, nontumor; T, tumor.
CONCURRENT BLOCKADE OF ALK, FGFR2, AND EphA5 SUPPRESSES TUMOR GROWTH IN VIVO
We further investigated whether coinhibition of ALK, FGFR2, and EphA5 could show therapeutic benefit in the subset of HCC. Mice bearing SMMC‐7721 xenografts were treated with ceritinib at 25 mg/kg, AZD4547 at 12.5 mg/kg, and dasatinib at 25 mg/kg, alone or in combination, for a consecutive 2 weeks; and their abilities to suppress tumor growth were assessed. Tumor volume was examined three times a week, and the intratumoral expression of the signaling axis was detected after mice were sacrificed on the last day. As expected, ceritinib or dasatinib treatment alone had no effect on tumor growth. AZD4547 treatment could result in partial tumor growth inhibition. Importantly, the concurrent treatment of ceritinib, AZD4547, and dasatinib dramatically suppressed tumor growth compared to the vehicle group (treatment/control = 19.5%; Fig. 3A). Accordingly, the intratumoral expression of p‐ALK, p‐FGFR2, and p‐EphA5 as well as of their downstream p‐AKT, p‐ERK, and p‐p38 was profoundly modulated after the cotreatment (Fig. 3B). The results of immunochemical staining further confirmed these findings (Fig. 3C). We also noticed that the expression of cluster of differentiation 34 was decreased by treatment with AZD4547, probably resulting from the antiangiogenesis effect due to KDR inhibition by AZD4547 in vivo (Fig. 3C).27 Together, these observations revealed the ability of combined inhibition of ALK, FGFR2, and EphA5 kinases and their downstream signaling network for HCC treatment.
Figure 3.
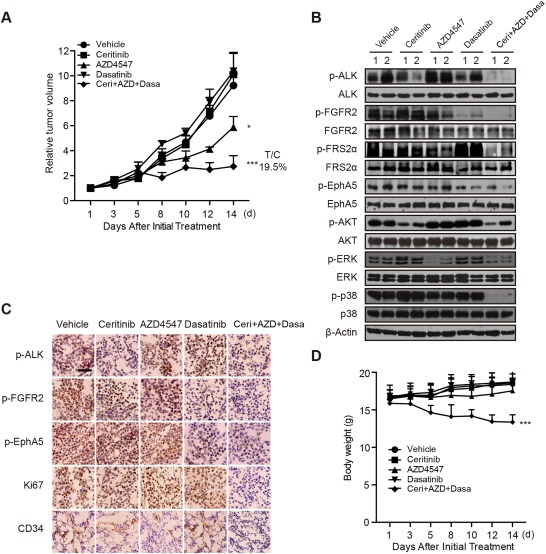
Concurrent blockade of ALK, FGFR2, and EphA5 suppresses tumor growth in vivo. (A) Mice bearing SMMC‐7721 xenografts were treated with vehicle, ceritinib (25 mg/kg, once a day), AZD4547 (12.5 mg/kg, once a day), and dasatinib (25 mg/kg, once a day), alone or in combination, for a consecutive 2 weeks; tumor volume was measured three times a week. Bars represent means ± SEM. (B) Tumor lysates were prepared and subjected to immunoblotting analysis with indicated antibodies. (C) Representative immunochemical staining results for p‐ALK, p‐FGFR2, p‐EphA5, cluster of differentiation 34, and Ki67 are shown. Scale bar, 50 μm. (D) Body weights of mice in each group were measured three times a week. Abbreviations: C+A+D, Ceri+AZD+Dasa; CD34, cluster of differentiation 34; T/C, treatment/control.
ALK, FGFR2, AND EphA5 ARE CLIENT PROTEINS OF HEAT SHOCK PROTEIN 90
All of the data above have confirmed the existence of a limited cluster of key kinases in HCC cells and provided therapeutic indications for cotargeting ALK, FGFR2, and EphA5 in a subset of HCC patients. However, to translate this combinatory strategy into clinical practice remains challenging and unfeasible due to the difficulty of concomitant application of multiple kinase inhibitors. Meanwhile, it is noteworthy that the body weights in the cotreatment group declined significantly (Fig. 3D), suggesting uncertain severe toxicities in vivo. In addition, the rational design of inhibitors that directly target ALK, FGFR2, and EphA5 seems reasonable but remains unachievable at present. We thus asked whether there exists an alternative therapeutic option. Of note, kinases are the most prevalent class of client proteins of Hsp90, which can strictly regulate kinases activity.30, 31, 32 Therefore, we explored whether inhibition of Hsp90 can effectively modulate the activity of ALK, FGFR2, and EphA5 in HCC cells. To test this possibility, we conducted coimmunoprecipitation experiments to test whether ALK, FGFR2, and EphA5 are client proteins of Hsp90 in HCC cells. As expected, the endogenous mutual binding of Hsp90 to ALK, FGFR2, and EphA5, respectively, and vice versa, was detected in ZIP177 and SMMC‐7721 cells (Fig. 4A,B). As a small molecule Hsp90 inhibitor, PU‐H71‐conjugated beads selectively bind dysregulated oncoproteins in tumor.33, 34 For further confirmation, we synthesized and prepared PU‐H71‐conjugated beads to pull down Hsp90 binding proteins, and found that ALK, FGFR2, and EphA5 were effectively precipitated by PU‐H71 beads (Fig. 4C). In addition, all of these bindings were significantly impaired after pretreatment with ganetespib, a putative Hsp90 inhibitor, at 1 μM for 4 hours in ZIP177 and SMMC‐7721 cells (Fig. 4D,E). Finally, we treated SMMC‐7721 cells with 0.1 μM ganetespib for different times and found that the protein levels of ALK, FGFR2, and EphA5 were notably decreased at 12 hours and markedly diminished at 24 hours, whereas their mRNA expression remained constant (Supporting Fig. S2A,B). Importantly, the down‐regulation of these kinases by ganetespib was largely rescued by pretreatment with proteasome inhibitor MG132 (Fig. 4F), indicating protein stability change upon Hsp90 inhibition. Together, these results implied that ALK, FGFR2, and EphA5 are recognized client proteins of Hsp90 in HCC cells.
Figure 4.
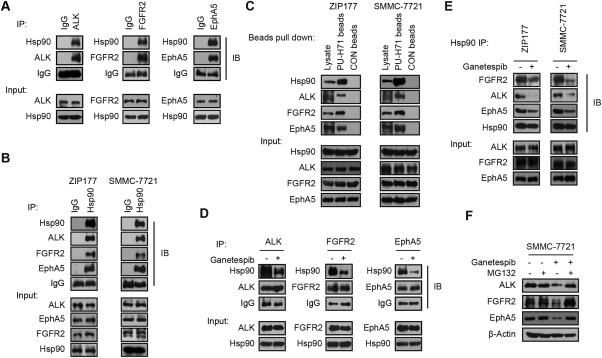
ALK, FGFR2, and EphA5 are stringent client proteins of Hsp90. (A) Lysates of SMMC‐7721 cells were immunoprecipitated with ALK, FGFR2, or EphA5 antibodies. Their bindings to endogenous Hsp90 were detected by immunoblotting. Antibody against immunoglobulin G was used for internal control. (B) Lysates of ZIP177 and SMMC‐7721 cells were immunoprecipitated with Hsp90 or immunoglobulin G antibodies and subjected to immunoblotting analysis with indicated antibodies. (C) Lysates of ZIP177 and SMMC‐7721 cells were immunoprecipitated with PU‐H71 beads or CON beads and analyzed by immunoblotting. (D) SMMC‐7721 cells were treated with ganetespib at 1 μM for 4 hours in serum‐free medium, then the cell lysates were immunoprecipitated with ALK, FGFR2, or EphA5 antibodies and analyzed by immunoblotting. (E) ZIP177 and SMMC‐7721 cells were treated with ganetespib at 1 μM for 4 hours, followed by the immunoprecipitation with Hsp90 antibody and analysis with immunoblotting. (F) SMMC‐7721 cells were preincubated with MG‐132 at 10 μM for 6 hours, washed, and then treated with ganetespib at 0.1 μM for 24 hours. Cell lysates were subjected to immunoblotting. Abbreviations: IB, immunoblot; IgG, immunoglobulin G; IP, immunoprecipitate.
Hsp90 INHIBITION IMPACTS THE GROWTH OF HCC CELLS THROUGH ABROGATING ALK, FGFR2, AND EphA5 ACTIVITY IN VITRO AND IN VIVO
Then, we thought to determine whether Hsp90 modulation could be considered as an alternative approach for targeting ALK, FGFR2 and EphA5 in HCC cells. To this end, we first tested the expression pattern of Hsp90 in HCC patients using the same paired frozen tumor tissues and tumor tissue microarrays. Clearly, Hsp90 was highly expressed in tumor tissues compared to their normal counterparts (Fig. 5A,B). In addition, the degree of highly expressed Hsp90 was tightly correlated with ALK, FGFR2, and EphA5 activation pattern, much higher in the coactivated subgroup (Fig. 5C). All these data suggested the possibility of perturbing Hsp90 in ALK, FGRF2 and EphA5 coactivated subpopulation. Indeed, several Hsp90 inhibitors, ganetespib in particular, have demonstrated promising effects in clinic for HCC patients.35, 36, 37 However, their most sensitive subpopulations and precise mechanisms of action remain obscure. Thus we intended to ascertain whether inhibition of Hsp90 impacted the viability of HCC cells through perturbation of the activity of ALK, FGFR2, and EphA5. Obviously, compared to sorafenib, HCC cells showed exquisite sensitivity to Hsp90 inhibitors, such as ganetespib, NVP‐AUY922, and 17‐dimethylaminoethylamino‐17‐demethoxygeldanamycin, confirming the effectiveness of Hsp90 inhibition in HCC with an acceptable safety profile (Fig. 5D; Supporting Fig. S3A). Thereafter, ZIP177 and SMMC‐7721 cells were selected to reveal the role of Hsp90 in regulating cell survival. As expected, disruption of Hsp90 using small interfering RNAs (siRNAs) resulted in significant impaired proliferation and induced apoptosis of HCC cells (Fig. 5E,F). The immunoblotting results verified that knockdown of Hsp90 remarkably decreased the protein levels of ALK, FGFR2, and EphA5 and the downstream signaling pathways to promote the cleavage of poly(adenosine diphosphate ribose) polymerase and caspase‐3 (Fig. 5G). The consistent conclusion was observed by treatment with ganetespib in a dose‐dependent manner (Supporting Fig. S3B‐D), suggesting the involvement of a converged network of ALK, FGFR2, and EphA5 in Hsp90 inhibition–attributed growth arrest in HCC cells. Interestingly, ganetespib treatment had no effect on most of the tested kinases, only down‐regulated limited kinases including epidermal growth factor receptor, hepatocyte growth factor receptor, insulin receptor, ABL1, and TNK1 (Supporting Fig. S3E). Nevertheless, inhibition of these kinases did not affect the growth of HCC cells (Supporting Fig. S1C‐E). All together, these findings clearly clarified that the coinhibition of ALK, FGFR2 and EphA5 also accounts for the mechanistic role of Hsp90 inhibition in HCC cells, providing the rational for their cotargeting using Hsp90 inhibitors.
Figure 5.
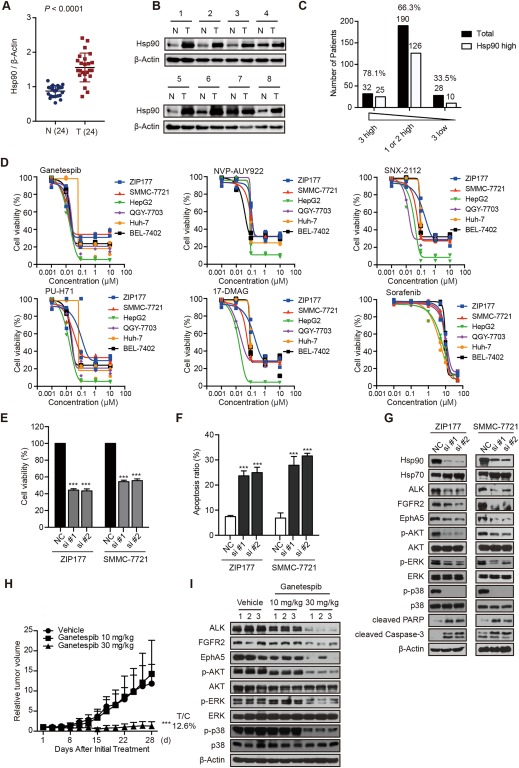
Hsp90 inhibition impacts cell growth through abrogating ALK, FGFR2, and EphA5 activity in vitro and in vivo. (A,B) Expression of Hsp90 in 24 paired tumor tissues was detected with immunoblotting. Dot plots represent the protein levels normalized to β‐actin (A), and representative immunoblotting results are shown (B). (C) Hsp90 high expression ratio was analyzed in HCC patients with 3 high (n = 32) vs 1or 2 high (n=190) vs 3 low (n = 28) kinases activity. (D) HCC cells were treated with Hsp90 inhibitors (ganetespib, NVP‐AUY922, SNX‐2112, PU‐H71, and 17‐DMAG) or sorafenib for 72 hours. Cell viability was assessed by sulforhodamine B assay. (E‐G) ZIP177 and SMMC‐7721 cells were transfected with negative control or Hsp90 siRNAs for 72 hours. Cell viability was assessed by sulforhodamine B assay (E), cell apoptosis was analyzed by flow cytometry (F), and cell lysates were subjected to immunoblotting analysis (G). Bars represent means ± standard deviation. (H,I) Mice bearing SMMC‐7721 xenografts were treated with vehicle or ganetespib (10 mg/kg or 30 mg/kg, three times per week) for a consecutive 4 weeks; tumor volume was measured three times a week (H). Bars represent means ± SEM. Tumor lysates were subjected to immunoblotting analysis (I). Abbreviations: 17‐DMAG, 17‐dimethylaminoethylamino‐17‐demethoxygeldanamycin; N, nontumor; NC, negative control; PARP, poly(adenosine diphosphate ribose) polymerase; T, tumor.
We then proceeded to in vivo studies to further confirm the therapeutic benefit of Hsp90 inhibition. Mice bearing SMMC‐7721 xenografts were treated with ganetespib at 10 mg/kg or 30 mg/kg for a consecutive 4 weeks, and their abilities to suppress tumor growth as well as intratumoral protein expression were assessed. Obviously, treatment with ganetespib at 30 mg/kg significantly decreased tumor growth, while no body weight loss was observed (treatment/control = 12.6%; Fig. 5H; Supporting Fig. S3F). Meanwhile, as expected, the expression of ALK, FGFR2, EphA5, and their downstream signaling cascades profoundly declined after ganetespib treatment at 30 mg/kg (Fig. 5I). These data indicated the involvement of ALK, FGFR2, and EphA5 kinases in Hsp90 inhibition–caused growth suppression of HCC cells.
ABROGATING ALK, FGFR2 AND EphA5 ACTIVITY LEADS TO HCC PATIENT–DERIVED XENOGRAFT GROWTH SUPPRESSION
Finally, we further investigate employed patient derived xenograft models to mimic clinical settings and evaluate the therapeutic potential possibility of the concurrent inhibition translating our strategies of cotargeting ALK, FGFR2 and EphA5 into clinic, we screened for stratified HCC patients. To this end, the activation of the three kinases in liver cancer patient derived xenograft array containing 32 models was detected using immunostaining. Among the 32 models, ALK, FGFR2, and EphA5 were coactivated in eight models including LI0752 (Supporting Table S5). We then tested the anticancer activity of the ceritinib, AZD4547, and dasatinib combination or ganetespib in the LI0752 patient–derived xenograft model. Mice bearing the LI0752 model were treated for a consecutive 3 weeks, and tumor growth as well as intratumoral protein expression were assessed. Ceritinib, AZD4547, and dasatinib individually did not affect tumor growth, while the drug combination significantly decreased tumor volume without affecting body weight (treatment/control = 35.9%; Fig. 6A; Supporting Fig. S4A). Intratumoral signaling evaluation showed that the inhibitors combination remarkably blocked the activity of their targets and further downstream AKT, ERK and p38 pathways (Fig. 6B). LI0752 treated with ganetespib at 30 mg/kg and 50 mg/kg showed tumor growth without body weight loss (30 mg/kg treatment/control = 54.6%, 50 mg/kg treatment/control= 40.6%; Fig. 6C) Supporting Fig. S4B, and the high‐dose treatment led to ALK, FGFR2, and EphA5 degradation and downstream signaling reduction (Fig. 6D), while slightly affecting other kinases (Supporting Fig. S4C). All these observations suggested the potential application of co‐targeting of ALK, FGFR2 and EphA5 in clinic in the future. In conclusion, Our data uncovered that the coactivation of ALK, FGFR2 and EphA5 kinases, together with downstream pathways, is essential for the survival of HCC cells and subpopulation of HCC patients. Therapeutic intervention of these kinases with drug combination or Hsp90 inhibition alone will potentially result in remarkable clinical benefits. (Fig. 6E).
Figure 6.
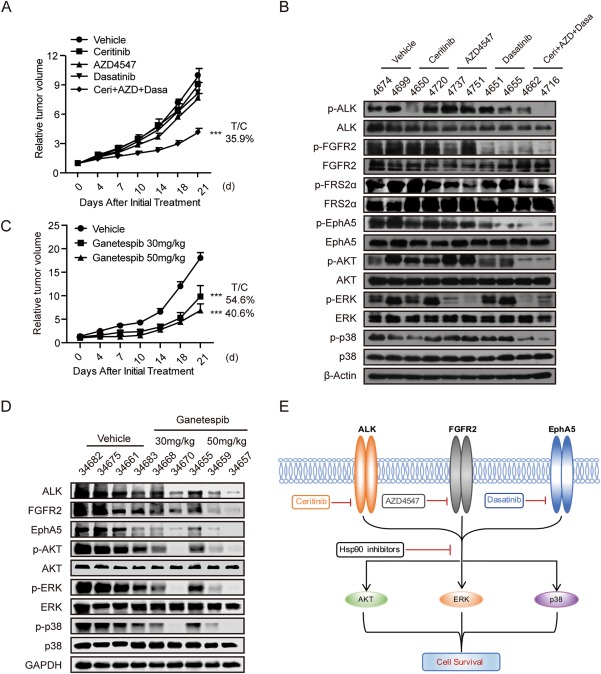
Abrogating ALK, FGFR2, and EphA5 activity leads to HCC patient–derived xenograft growth suppression. (A) Mice bearing HCC patient–derived xenograft model LI0752 were treated with vehicle or ceritinib (25 mg/kg), AZD4547 (12.5 mg/kg), or dasatinib (25 mg/kg) individually or in combination once a day for a consecutive 3 weeks. Tumor volume was measured twice a week. Bars represent means ± SEM, n = 6. (B) Tumor lysates were prepared and subjected to immunoblotting analysis with indicated antibodies. (C) Mice bearing HCC patient–derived xenograft model LI0752 were treated with vehicle or ganetespib (30 mg/kg or 50 mg/kg, three times per week). Tumor volume was measured twice a week. Bars represent means ± SEM, n = 6. (D) Tumor lysates were prepared and subjected to immunoblotting analysis with indicated antibodies. (E) A proposed overview of ALK, FGFR2 and EphA5 co‐activation in HCC cells and therapeutic strategies to block their activities were illustrated. Abbreviations: FRS2, FGFR substrate 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; T/C, treatment/control.
Discussion
In recent years, the application of inhibitors to target mutated driving kinases, termed “oncogene addiction,” has become a mainstay for clinical management of cancer patients. However, the practice in HCC has been largely challenged owing to an absence of explicit kinase addictions in the clinic. Our data also revealed that none of the 17 kinases that are aberrantly activated in HCC cells demonstrated notable functional genomic alterations. In line with these observations, HCC cells are insensitive to specific kinase inhibitors, leading to their clinical failure. Therefore, unlike other well‐defined oncogene‐addicted solid tumors, the discovery of a single candidate driving kinase in HCC seems unlikely.
Alternatively, a common recognition that several key kinases should be targeted concurrently to improve therapeutic response is increasing. Due to the intrinsic dynamic feature of the kinase network, however, it is difficult to dissect the limited cluster of essential kinases that cooperate in HCC cells.38 In this study, we provided evidence to demonstrate that, although many kinases were aberrantly activated, only ALK, FGFR2, and EphA5 served as core kinases in HCC cells. Their coactivation is required for cell growth and is clinically highly correlated with poor prognoses for overall survival. Importantly, their coactivation stratified about 13% of the HCC patients. These findings may have remarkable significance for the clinical management of HCC patients. Guided by the direction of stratification medicine, the triple‐positive status of p‐ALK/p‐FGFR2/p‐EphA5 can be considered as a conceptual “combined therapeutic target” for treatment. Meanwhile, they can be selected as a diagnostic or “predictive” biomarker for patient enrollment in eligible subpopulations.
Despite the observed therapeutic promise, the direct translation of combined targeting of ALK, FGFR2, and EphA5 with multiple kinase inhibitors into clinical practice remains challenging. Meanwhile, the in vivo observations suggested that the side effects of this strategy will be an inevitable obstacle for application. Theoretically, an ideal approach is to identify compounds that directly target ALK, FGFR2, and EphA5 with favorable tolerance. Although unachievable at the moment, our findings could pave the way for drug development for the prospective treatment of a subclass of HCC patients in the future.
As a functional kinome buffer, Hsp90 has been recognized as an important therapeutic target in a range of tumors, especially kinase‐controlled subgroups.39, 40, 41 Our data clearly clarified that ALK, FGFR2, and EphA5 are the essential client proteins of Hsp90 in HCC cells. Correspondingly, inhibition of Hsp90 significantly modulated their activity and resulted in profound growth arrest. This suggested that targeting Hsp90 might be taken as an alternative method to abrogate these kinases for treatment of HCC patients. On the other hand, the relationship between Hsp90 and ALK, FGFR2, and EphA5 illustrated the precise mechanism of action for Hsp90 inhibitors tested in HCC patients. Of note, the triple‐positive p‐ALK/p‐FGFR2/p‐EphA5 subcohort may be given priority as the most sensitive population of HCC patients for treatment with Hsp90 inhibitors.
In conclusion, our study suggests that coactivation of ALK, FGFR2, and EphA5 might be an effective marker for clinical diagnosis in a subcohort HCC patients for therapeutic interventions with kinase inhibitors combination or Hsp90 inhibitors.
Author names in bold designate shared co‐first authorship.
Supporting information
Acknowledgment
We are grateful to Dr. Boyun Tang (Gene Company Ltd., Shanghai, China) who helped us with the bioinformatics analysis, and we thank Dr. Sheng Guo and Dr. Wubin Qian (Crownbio, Taicang, China) for helping with the genomic interpretation.
Potential conflict of interest: Nothing to report.
Supported by the National Natural Science Foundation of China (81673472, 81402966, to A.S.; 91229205, to J.D.); the National Program on Key Basic Research Project of China (2012CB910704, to M.G.); the Natural Science Foundation of China for Innovation Research Group (81321092, to J.D.); Personalized Medicines‐Molecular Signature‐based Drug Discovery and Development, Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12020101, to J.D.; XDA12020105, to A.S.).
Contributor Information
Aijun Shen, Email: shenaj@simm.ac.cn.
Jian Ding, Email: jding@simm.ac.cn.
Meiyu Geng, Email: mygeng@simm.ac.cn.
References
- 1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012;379:1245‐1255. [DOI] [PubMed] [Google Scholar]
- 2. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV‐ and HCV‐associated hepatocellular carcinoma. Nat Rev Cancer 2013;13:123‐135. [DOI] [PubMed] [Google Scholar]
- 3. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118‐1127. [DOI] [PubMed] [Google Scholar]
- 4. El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557‐2576. [DOI] [PubMed] [Google Scholar]
- 5. Villanueva A, Hernandez‐Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol 2013;10:34‐42. [DOI] [PubMed] [Google Scholar]
- 6. Bertino G, Di Carlo I, Ardiri A, Calvagno GS, Demma S, Malaguarnera G, et al. Systemic therapies in hepatocellular carcinoma: present and future. Future Oncol 2013;9:1533‐1548. [DOI] [PubMed] [Google Scholar]
- 7. McCormick F. Cancer therapy based on oncogene addiction. J Surg Oncol 2011;103:464‐467. [DOI] [PubMed] [Google Scholar]
- 8. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015;12:408‐424. [DOI] [PubMed] [Google Scholar]
- 9. Weinstein IB, Joe A. Oncogene addiction. Cancer Res 2008;68:3077‐3080. [DOI] [PubMed] [Google Scholar]
- 10. Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction—a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006;3:448‐457. [DOI] [PubMed] [Google Scholar]
- 11. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 12. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;389:56‐66. [DOI] [PubMed] [Google Scholar]
- 13. Abou‐Alfa GK, Venook AP. The antiangiogenic ceiling in hepatocellular carcinoma: does it exist and has it been reached? Lancet Oncol 2013;14:e283‐e288. [DOI] [PubMed] [Google Scholar]
- 14. Worns MA, Galle PR. HCC therapies—lessons learned. Nat Rev Gastroenterol Hepatol 2014;11:447‐452. [DOI] [PubMed] [Google Scholar]
- 15. Llovet JM, Hernandez‐Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res 2014;20:2072‐2079. [DOI] [PubMed] [Google Scholar]
- 16. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010;29:4989‐5005. [DOI] [PubMed] [Google Scholar]
- 17. Han ZG. Functional genomic studies: insights into the pathogenesis of liver cancer. Annu Rev Genomics Hum Genet 2012;13:171‐205. [DOI] [PubMed] [Google Scholar]
- 18. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008;48:1312‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nault JC, Villanueva A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clin Cancer Res 2015;21:1786‐1788. [DOI] [PubMed] [Google Scholar]
- 20. Galuppo R, Ramaiah D, Ponte OM, Gedaly R. Molecular therapies in hepatocellular carcinoma: what can we target? Dig Dis Sci 2014;59:1688‐1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Worns MA. Systemic therapy and synergies by combination. Dig Dis 2013;31:104‐111. [DOI] [PubMed] [Google Scholar]
- 22. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non‐oncogene addiction. Cell 2009;136:823‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu AM, Huang PH. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res 2010;70:3857‐3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu AX, Kang YK, Rosmorduc O, Evans TR, Ross PJ, Santoro A, Carrilho FJ, et al. SEARCH: a phase III, randomized, double‐blind, placebo‐controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2015;33:559‐566. [DOI] [PubMed] [Google Scholar]
- 25. Finn RS, Poon RT, Yau T, Klumpen HJ, Chen LT, Kang YK, et al. Phase I study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J Hepatol 2013;59:1271‐1277. [DOI] [PubMed] [Google Scholar]
- 26. Graves LM, Duncan JS, Whittle MC, Johnson GL. The dynamic nature of the kinome. Biochem J 2013;450:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gavine PR, Mooney L, Kilgour E, Thomas AP, Al‐Kadhimi K, Beck S, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 2012;72:2045‐2056. [DOI] [PubMed] [Google Scholar]
- 28. Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res 2011;17:5546‐5552. [DOI] [PubMed] [Google Scholar]
- 29. Shaw AT, Engelman JA. Ceritinib in ALK‐rearranged non‐small‐cell lung cancer. N Engl J Med 2014;370:2537‐2539. [DOI] [PubMed] [Google Scholar]
- 30. Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, et al. Quantitative analysis of HSP90–client interactions reveals principles of substrate recognition. Cell 2012;150:987‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 2010;11:515‐528. [DOI] [PubMed] [Google Scholar]
- 32. Lachowiec J, Lemus T, Borenstein E, Queitsch C. Hsp90 promotes kinase evolution. Mol Biol Evol 2015;32:91‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, et al. Affinity‐based proteomics reveal cancer‐specific networks coordinated by Hsp90. Nat Chem Biol 2011;7:818‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cerchietti LC, Lopes EC, Yang SN, Hatzi K, Bunting KL, Tsikitas LA, et al. A purine scaffold Hsp90 inhibitor destabilizes BCL‐6 and has specific antitumor activity in BCL‐6‐dependent B cell lymphomas. Nat Med 2009;15:1369‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goyal L, Wadlow RC, Blaszkowsky LS, Wolpin BM, Abrams TA, McCleary NJ, et al. A phase I and pharmacokinetic study of ganetespib (STA‐9090) in advanced hepatocellular carcinoma. Invest New Drugs 2015;33:128‐137. [DOI] [PubMed] [Google Scholar]
- 36. Cheng W, Ainiwaer A, Xiao L, Cao Q, Wu G, Yang Y, et al. Role of the novel HSP90 inhibitor AUY922 in hepatocellular carcinoma: potential for therapy. Mol Med Rep 2015;12:2451‐2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Proia DA, Bates RC. Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer Res 2014;74:1294‐1300. [DOI] [PubMed] [Google Scholar]
- 38. Al‐Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post‐genomic era. Nat Biotechnol 2012;30:679‐692. [DOI] [PubMed] [Google Scholar]
- 39. Garcia‐Carbonero R, Carnero A, Paz‐Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 2013;14:e358‐e369. [DOI] [PubMed] [Google Scholar]
- 40. Ramalingam S, Goss G, Rosell R, Schmid‐Bindert G, Zaric B, Andric Z, et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second‐line therapy of advanced non‐small cell lung cancer (GALAXY‐1). Ann Oncol 2015;26:1741‐1748. [DOI] [PubMed] [Google Scholar]
- 41. Butler LM, Ferraldeschi R, Armstrong HK, Centenera MM, Workman P. Maximizing the therapeutic potential of Hsp90 inhibitors. Mol Cancer Res 2015;13:1445‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials