

Abstract
Gene amplification, which involves the two major topographical structures double minutes (DMs) and homegeneously stained region (HSR), is a common mechanism of treatment resistance in cancer and is initiated by DNA double‐strand breaks. NHEJ, one of DSB repair pathways, is involved in gene amplification as we demonstrated previously. However, the involvement of homologous recombination, another DSB repair pathway, in gene amplification remains to be explored. To better understand the association between HR and gene amplification, we detected HR activity in DM‐ and HSR‐containing MTX‐resistant HT‐29 colon cancer cells. In DM‐containing MTX‐resistant cells, we found increased homologous recombination activity compared with that in MTX‐sensitive cells. Therefore, we suppressed HR activity by silencing BRCA1, the key player in the HR pathway. The attenuation of HR activity decreased the numbers of DMs and DM‐form amplified gene copies and increased the exclusion of micronuclei and nuclear buds that contained DM‐form amplification; these changes were accompanied by cell cycle acceleration and increased MTX sensitivity. In contrast, BRCA1 silencing did not influence the number of amplified genes and MTX sensitivity in HSR‐containing MTX‐resistant cells. In conclusion, our results suggest that the HR pathway plays different roles in extrachromosomal and intrachromosomal gene amplification and may be a new target to improve chemotherapeutic outcome by decreasing extrachromosomal amplification in cancer.
Keywords: gene amplification, DMs, HSR, HR, MTX
Short abstract
What's new?
Double‐strand DNA breaks (DSBs) initiate gene amplification, a phenomenon associated with therapeutic resistance in cancer that involves two topographical structures, double minutes (DMs) and homogeneously staining regions (HSRs). Whether DSB repair pathways, particularly homologous recombination (HR), also influence gene amplification is unknown. Here, in methotrexate‐resistant colon cancer cells, HR inhibition effectively reduced gene amplification, specifically the DM‐form, by blocking DM formation and promoting DM exclusion via micronuclei. HR inhibition had no influence on the HSR‐form of gene amplification. Loss of gene amplification by HR inhibition, through partial reversal of methotrexate resistance, may contribute to improved chemotherapeutic outcome.
Introduction
Gene amplification in the cancer genome frequently carries amplified oncogenes or drug‐resistant genes1 and is tightly associated with the development of cancer and treatment resistance.2, 3, 4 Cytogenetic studies have classified gene amplification into two major topographical structures: extrachromosomal double minutes (DMs) and the intrachromosomal homogeneously staining region (HSR).5 Various models of the mechanism of DM and HSR formation have been proposed, but the molecular mechanisms remain unclear.
Gene amplification is initiated by double‐strand DNA breaks (DSBs),6, 7 lethal damage that can be induced by radiation,8 endonucleases (i.e., I‐Sce I)9 or antineoplastic agents (i.e., methotrexate, MTX).10 Previous models of the formation of gene amplification indicate that broken DNA segments have the potential to rejoin. For example, in the B‐F‐B model, relegation between two broken chromatids can result in intrachromosomal amplification.11 In the episome model, cyclizing ligation by DNA segments themselves can result in extrachromosomal DMs.12 Thus, DSB repair pathways may be involved in gene amplification. In our previous study, we demonstrated a special role for one of the DSB pathway, NHEJ, in the formation of DMs.13 Among DSB repair pathways, homologous recombination (HR) is a classical mechanism that consists of multiple interrelated pathways. Chen et al. has classified HR into four pathways: non‐allelic homologous recombination (NAHR), gene conversion, break‐induced replication (BIR) and single‐strand annealing (SSA).14 Notably, all four processes share the same core molecule, breast cancer susceptibility gene 1 (BRCA1).15, 16 BRCA1 plays a major role in several processes, including facilitating the transition from NHEJ to HR,17 promoting the 5′‐end resection of DSBs,18 indirectly recruiting RAD51 to start HR strand invasion19, 20 and controlling the cell cycle by activating the checkpoints, such as G2/M.21
HR is commonly regarded as a high‐fidelity repair pathway in maintaining genomic stability.22 However, recent evidence has indicated that HR can tolerate and induce genomic structure variation, including duplication, deletion, and inversion by sporadic repeat regions.23 Therefore, the role of HR in gene amplification remains controversial. Some studies have suggested that HR is involved in gene amplification. For example, in the primary eukaryote Leishmania, HR is a common mechanism yielding extrachromosomal amplicons between direct repeat sequences.24 Joseph‐George has also suggested that HR may result in intrachromosomal amplification, because of the highly repetitive nature of segmental duplications in families with 9q13‐q21 duplication.25 In contrast, Ruiz‐Herrera et al. have reported that HR may inhibit gene amplification in human cells, because knocking down RAD54, an important dsDNA‐dependent ATPase of HR, increases the number of clones containing gene amplifications.26 Thus, the association between HR and gene amplification in cancer remains to be investigated.
Gene dosage is determined by the balance between the formation and elimination of gene amplification. Micronuclei (MNs) and nuclear buds (NBUDs) are the main pathways for export of nuclear material, including amplified DNA, DNA repair complexes and excess chromosomes.27, 28 In previous studies, the application of DNA synthesis or repair inhibitors, such as hydroxyurea, gemcitabine and NU7026, has been found to eliminate gene amplification by forming MN/NBUDs.13, 29, 30 The formation of MN/NBUDs tightly depends on the cell cycle,27 and the HR core protein, BRCA1, is a regulator of cell cycle checkpoints.31 However, the influence of HR on the formation of MNs/NBUDs via cell cycle progression has not yet been elucidated.
In our study, we used MTX‐resistant HT‐29 colon cancer cell lines containing the DM‐ or HSR‐form of gene amplification to directly understand the relationship between HR and gene amplification at the molecular level. Our results demonstrated that HR plays different roles in the formation of gene amplification and in MTX resistance in cancer cells.
Materials and Methods
Cell culture and medium
The HT‐29 cell line was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and was denoted MTX‐sensitive cells. HT‐29 MTX‐resistant cells were generated by continuously culturing the HT‐29 cell line with different concentrations of MTX (CalbiochemBiochemicals, Darmstadt, Germany). Cells that were resistant to 10−5 mol/L MTX and contained HSR‐form gene amplification were denoted HSR‐containing MTX‐resistant cells. Cells that were resistant to 10−4 mol/L MTX and contained DM‐form gene amplification were denoted DM‐containing MTX‐resistant cells. MTX‐sensitive and HSR‐ and DM‐containing MTX‐resistant cells were cultured in high‐glucose Dulbecco's modified Eagle's medium (DMEM) (GibcoBRL, Gaithersburg, MD) containing 15% fetal calf serum (GibcoBRL, Gaithersburg, MD) and supplemented with MTX at the indicated concentrations.
Antibodies
Antibodies used in our study are as follows: mouse monoclonal anti‐DHFR (Abnova, Taipei, Taiwan), anti‐Cyclin B (Santa Cruz Biotechnology Inc., TX), anti‐MRE11 (GeneTex, Hsinchu, Taiwan), anti‐γH2AX (Millipore, MA), anti‐CDK1 (Cell Signaling, Boston) and anti‐GAPDH (Kang Chen Bio‐tech, Shanghai, China); rabbit monoclonal anti‐BRCA1, anti‐RAD51 (Santa Cruz Biotechnology Inc., TX) and anti‐Histone H3 (Abcam, Cambridge, UK); CF488 goat anti‐mouse IgG and CF488 goat anti‐rabbit IgG (Biotium, CA); IRDye 800 conjugated affinity purified anti‐mouse IgG and IRDye 700 conjugated affinity purified anti‐rabbit IgG (Rockland, Philadelphia).
shRNA transfection
Lentivirus of two shRNA targeting sequences (5'‐ cagcggatacaacctcaaa'3‐ and 5'‐ ggataccatgcaacataac‐3') and one nontarget control sequence (GeneChem, Shanghai, China) were transfected into DM‐ and HSR‐containing MTX‐resistant HT‐29 cells at MOI = 50. The transfected DM‐containing cells were named as sh‐BRCA1–1 (DMs), sh‐BRCA1–2 (DMs) and sh‐control (DMs) and the transfected HSR‐containing cells were named as sh‐BRCA1–1 (HSR), sh‐BRCA1–2 (HSR) and sh‐control (HSR).
BRCA1 rescue assay
A lentivirus particles for negative control (LPP‐NEG‐Lv105–100) and BRCA1 (LPP‐H0047‐Lv105–400) (GeneCopoeia, Guangzhou, China) were transfected into sh‐BRCA1–1 (DMs) cells respectively by MOI = 10 and were named as sh‐BRCA1‐ov‐NC (DMs) and sh‐BRCA1‐ov‐BR (DMs). All other steps were carried out as instruction of lentiviral transfection. Verification was done by Western Blotting.
HR‐GFP repair assay
HR‐GFP reporter plasmid (#26476, AddGene, Cambridge, Britain) was transfected into DM‐containing MTX‐resistant cells with Lipo2000 (Invitrogen, CA). Puromycin was added to select stable clones at a final concentration of 0.3 μg/mL. After the establishment of stable clone, siRNA of BRCA1/control (Ribobio, Guangzhou, China) and I‐SceI plasmid (#26477, AddGene, Cambridge, Britain) were transfected into the cells with Lipo2000 (Invitrogen, CA). In BRCA1 rescue assay, BRCA1‐expressing lentivirus was used 8 hour after siBRCA1 had been transfected. Three days after the transfection, half of these cells were tested to verify the expression of BRCA1 by Western blotting, and half of the cells were detected to verify the percentage of cells with GFP by flow cytometry assay (BD Bioscience, WA).
Real‐time PCR
Genomic DNA was extracted using a QIAmp DNA Mini Kit (Qiagen, Dusseldorf, Germany). Real‐time PCR was performed using a Light Cycler 480 SYBRGreen Kit (Roche Applied Science, Mannheim, Germany) according to the manufacturer's instructions. The DNA primers were as follows: DHFR‐F: ATTTTGTTCAGTGCCTACCACA and DHFR‐R: GCCTGAATGATATCTACAAGCTG, RAD1‐F: TGTCAGTTGCGTGTCTTCAT and RAD1‐R: AGACAGTAAAACTCCCATCA, PLK2‐F: ACCCTATGGGACTCCTCTTT and PLK2‐R: GTATGCCTTAGCCTGTTCTG, ZFYVE16‐F: AGGAAGCAACCACCACAAC and ZFYVE16‐R: CAGCACCACCAACAGATACA, MSH3‐F: TGTCTGGTGTTTCGCCTGAT and MSH3‐R: TTAGCCAATAACCGCTCTAC, CCNH‐F: GTATTGCAGCACTGATTATGTCC and CCNH‐R: TCATGAAAATAGCCATAGGTGA, GLRX‐F: CCCACATTGTAGGGAATCAT and GLRX‐R: CCCACAGTCTATTCGTAGCA, CAST‐F: TTGACTCCATAGCCAACCTT and CAST‐R: GTCACTTTTCCCAGAATCCG, ACTN‐F: CTTCTACAATGAGCTGCGTG and ACTN‐R: AAGCAAATAGAACCTGCAGAG.
Western blot assay
Briefly, total protein was extracted from cells by using RIPA buffer, and nuclear protein was extracted by Nuclear‐Cytosol Extraction kit (Applygen, Beijing, China). The protein concentration was determined with a BCA Protein Assay Kit (Beyotime, Shanghai, China). Proteins were separated with 8% SDS–PAGE and then transferred to PVDF membranes (Millipore, MA). The membranes were blocked with 5% nonfat milk (BD Biosciences, WA) and 0.1% Tween‐20 in Tris‐buffered saline and immunoblotted overnight with primary antibodies at 4 °C with gentle shaking. Subsequently, the membranes were stained with fluorochrome‐labeled secondary antibody. Immunoreactivity was detected with an Odyssey fluorescence scanning system (LI‐COR, NE) at wavelengths of 800 nm and 700 nm.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 10 min at room temperature and washed with PBS before permeabilization with 0.5% Triton X‐100 for 10 min. Nonspecific binding sites were blocked with 5% BSA containing 0.5% Trion X for 1 h before incubation with primary antibodies overnight at 4 °C. The cells were washed three times with PBS and incubated with secondary antibodies for 1 h at room temperature, and the nuclei were visualized by DAPI counterstaining for 5 min. Immunofluorescence images were observed using a fluorescence microscope equipped with the MetaMorph Imaging System (Universal Imaging Corporation).
Fluorescence in situ hybridization (FISH)
In brief, metaphase spreads of cells were prepared according to standard cytogenetic methods. BAC clones, RP11‐90A9, RP11‐91I22 and RP11‐957J15 (BAC PAC Resources Center, Oakland, CA), were extracted using the Genopure Plasmid Midi kit (Roche, Basel, Switzerland) according to the manufacturer's protocol and were labeled with Cy3‐dUTP, Cy5‐dUTP or Green‐dUTP with a BioPrime DNA Labeling System kit (Invitrogen, CA). The slides with interphase or metaphase spreads were treated with RNase for 40 min at 37 °C before being washed with 2× saline sodium citrate (SSC). Subsequently, the slides were dehydrated with a graded ethanol series, then incubated with pepsin for 15 min at 37 °C and washed again with PBS. The cells were fixed in 1% paraformaldehyde for 10 min and then washed with 1× PBS. After dehydration, the slides were treated in 70% formamide for 3 min at 75 °C and subsequently washed twice in pre‐chilled 2× SSC for 3 min. After dehydration, the cells were hybridized with labeled probes, mounted with rubber cement and incubated for 48 h at 37 °C. The slides were then immersed for 15 min in 50% formamide pre‐warmed to 44 °C and subsequently washed twice with 2× SSC. After dehydration, the slides were counterstained with DAPI (Thermo, MA), and the cells were counted. The images were obtained with a fluorescence microscope equipped with the MetaMorph Imaging System (Universal Imaging Corporation).
Cell cytotoxicity assay
Cells in the logarithmic growth phase were seeded into 96‐well plates at a density of 4500 cells/well and incubated for 72–96 h in the presence of MTX. Cell viability was measured using a CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, WI), and the OD value was read on a microplate reader (Tecan Austria GmbH, Grödig Austria) at 490 nm.
Flow cytometry assay
Cells were synchronized by culturing in serum‐free medium through a conventional method,32 and the block was released for another 24 h. In brief, the cells were fixed, washed and then analyzed with a CycleTEST PLUS DNA kit (BD Bioscience, WA). Finally, the cells were promptly subjected to flow cytometry (BD Bioscience, WA).
Statistical analysis
All results were based on at least three independent experiments. Western blot and real‐time PCR results were compared with Student's t‐test followed by Bonferroni's adjustment. The rank sum test followed by Bonferroni's adjustment was used to assess differences in the number of DMs and RAD51 or γH2AX foci. A Chi‐squared analysis followed by Bonferroni's adjustment was used to assess differences in the total MN/NBUDs, the formation rates of DHFR‐containing MN/NBUDs, and the proportion of cells in the G2 phase of the cell cycle. When comparing two groups among three groups, P < 0.025 was considered significant.
Results
Increased HR is associated with gene amplification in MTX‐resistant HT‐29 cells
To assess the involvement of HR in gene amplification, we first measured the expression level of BRCA1 in HT‐29 MTX‐sensitive cells as well as HSR‐ and DM‐containing MTX‐resistant cells and found that the BRCA1 protein level was higher in both HSR‐ and DM‐containing MTX‐resistant cells than in MTX‐sensitive cells (Fig. 1 a). We then measured the accumulation of RAD51 in nuclei to evaluate homologous strand invasion.33 As presented in the left panel of Figure 1 b, the number of RAD51 foci was significantly increased in HSR‐ and DM‐containing cells compared with MTX‐sensitive cells. Moreover, the RAD51 nuclear protein level was also markedly increased in HSR‐ and DM‐containing cells (Fig. 1 c). These results suggested that the HR pathway may have a correlation with gene amplification.
Figure 1.
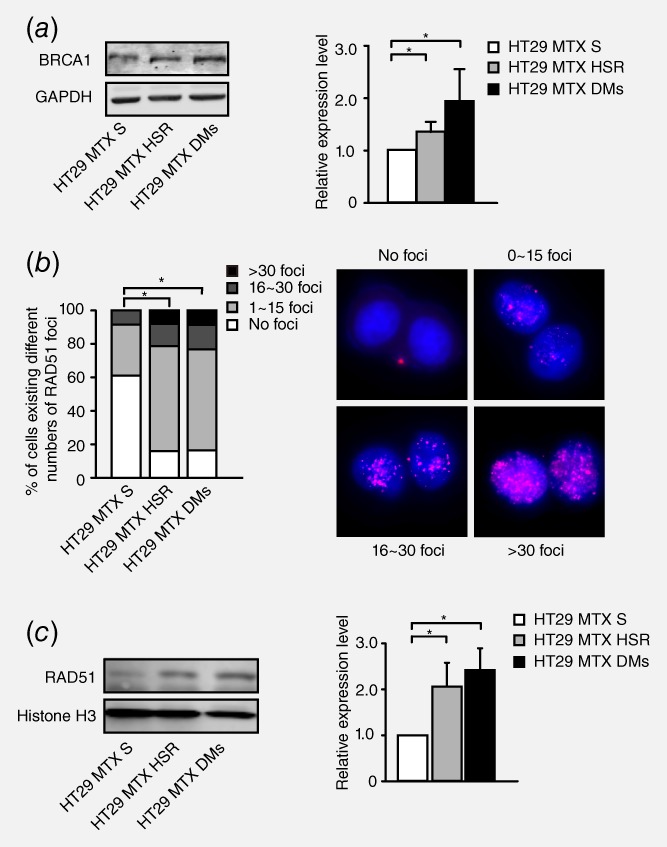
Correlation between the level of HR and gene amplification in HT29 MTX‐sensitive and MTX‐resistant cells. a. Western blot analysis of the BRCA1 protein level in HT29 MTX‐sensitive cells and HSR‐ and DM‐containing MTX‐resistant cells. The right panel shows densitometry values. Protein densities are normalized to the GAPDH expression level, and the BRCA1 expression levels of HSR‐ or DM‐containing cells are compared with that of MTX‐sensitive cells. Values are mean ±SD (n = 3, *p<0.025). b. Immunofluorescence assay for RAD51 foci in HT29 MTX‐sensitive, HSR‐ and DM‐containing cells. Cells are grouped into four categories according to the number of RAD51 foci: no foci, 1–15 foci, 16–30 foci and >30 foci. Group standards are shown in the right panel. The left panel shows the proportion of these cells in each category (n>75, *p<0.025). c. Western blot analysis of RAD51 nuclear protein levels in HT29 MTX‐sensitive, HSR‐containing and DM‐containing MTX‐resistant cells. The right panel shows densitometry values (n = 3, *p<0.025).
HR inhibition significantly attenuates extrachromosomal amplification in MTX‐resistant cells
To investigate the association between HR and extrachromosomal amplification in DM‐containing MTX‐resistant cells, we stably silenced BRCA1 in these cells by using shRNA transfection (Fig. 2 a). DSBs were more extensive in BRCA1‐depleted cells, as evidenced by γH2AX foci (Fig. 2 b) and increased levels of γH2AX (Fig. 2 c). To further confirm the influence of BRCA1 silencing on HR activity, we measured the expression level of MRE11 and RAD51. In HR repair pathway, MRE11 programs DNA end resection and recruits RPA to single‐stranded DNA.34 As expected, in accordance with BRCA1 depletion, MRE11 was significantly downregulated (Fig. 2 d) and the accumulation of RAD51 in nuclei was also markedly decreased after BRCA1 depletion (Fig. 2 e and 2 f). We also rescued the protein level of BRCA1 in BRCA1‐depleted clones (Fig. 2 g) and found that both γH2AX foci and γH2AX protein level reverted (Fig. S1, Supporting Information). As expected, RAD51 foci and protein level (Fig. 2 h and 2 i) were also reverted. MRE11 protein level was reverted as well after BRCA1 rescued (Fig. S2, Supporting Information). HR‐GFP reporter assay also demonstrated a decrease of HR by BRCA1 depletion and a revert of HR by BRCA1 overexpression (Fig. 2 j). The results above in DM‐containing MTX‐resistant cells are consistent with previous existing results that the depletion of BRCA1 promoted DSBs and inhibited HR.
Figure 2.
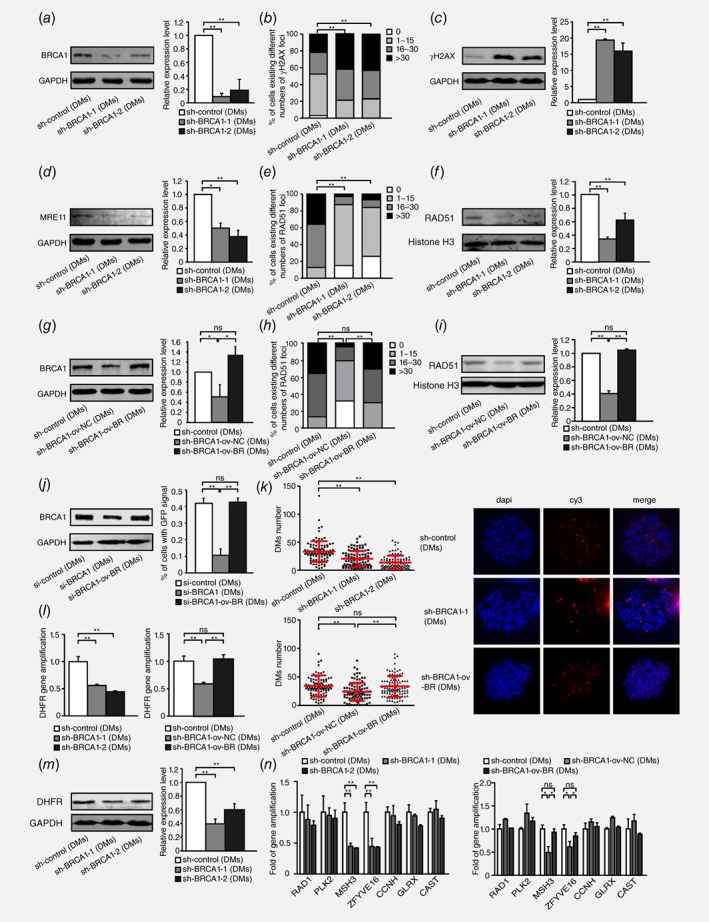
Inhibition of BRCA1 exacerbates double‐strand DNA breaks, suppresses HR and decreases DM‐form amplification in DM‐containing MTX‐resistant cells. a. Western blot analysis of BRCA1 protein levels and densitometry values in DM‐containing cells: control and two BRCA1‐depleted clones (n = 3, **p<0.005). b. IF assay for γH2AX protein in DM‐containing control and two BRCA1‐depleted clones (n ≥ 100, **p<0.005). c. Western blot bands for the γH2AX protein level, densitometry values in DM‐containing control and two BRCA1‐depleted clones (n = 3, **p<0.005). d. Western blot analysis of MRE11 protein level and densitometry values in DM‐containing control and two BRCA1‐depleted clones (n = 3, *p<0.025, **p<0.005). e. IF assay for RAD51 foci in DM‐containing control and two BRCA1‐depleted clones (n ≥ 100, **p<0.005). f. Western blot analysis of RAD51 nuclear protein levels and densitometry values in DM‐containing control and two BRCA1‐depleted clones (n = 3, **p<0.005). g. Western blot analysis of BRCA1 protein level and densitometry values in DM‐containing control, BRCA1‐depleted control and BRCA1‐depleted rescued cells (n = 3, *p<0.025). h. IF assay for RAD51 foci in DM‐containing control, BRCA1‐depleted control and BRCA1‐depleted rescued cells (n ≥ 100, **p<0.005). i. Western blot analysis of RAD51 nuclear protein levels and densitometry values in DM‐containing control, BRCA1‐depleted control and BRCA1‐depleted rescued cells (n = 3, **p<0.005). j. Western blot analysis of BRCA1 protein level (left panel) and HR‐GFP repair assay of the percentage of GFP+ cells (right panel) in DM‐containing si‐control, si‐BRCA1 and si‐BRCA1 rescued cells (n = 3, **p<0.005). k. Quantification of DMs with DHFR signal in DM‐containing control and two BRCA1‐depleted clones(left upper panel), and control, BRCA1‐depleted control and BRCA1‐depleted rescued clones (left lower panel), on the basis of FISH analysis of metaphase spreads. Values are mean ±SD. BAC‐containing DHFR was used as a probe and is marked in red; nuclei were stained with DAPI and are marked in blue (right panel) (n ≥ 100, **p<0.005). l. Real‐time PCR analysis of DHFR amplification in DM‐containing control and two BRCA1‐depleted clones (left panel), and control, BRCA1‐depleted control and BRCA1‐depleted rescued clones (right panel) (n = 3, **p<0.005). m. Western blot analysis of DHFR protein level and densitometry values in DM‐containing control and two BRCA1‐depleted clones (n = 3, **p<0.005). n. Real‐time PCR analysis of other genes that co‐localized with DHFR in chromosome 5, including RAD1, PLK2, MSH3, ZFYVE16, CCNH, GLRX and CAST, in DM‐containing control and two BRCA1‐depleted clones (left panel), and control, BRCA1‐depleted control and BRCA1‐depleted rescued clones (right panel) (n = 3, *p<0.025, **p<0.005).
To assess the effect of HR inhibition on the formation of DMs, we next detected the copy numbers of genes amplified in the form of DMs after BRCA1 depletion in DM‐containing MTX‐resistant cells. Because we have found the classical MTX‐resistant gene, DHFR, to be highly amplified on DMs in our previous study, we counted the number of DMs by locating DHFR. We found that the number of DHFR (red signal)‐carried DMs sharply decreased after BRCA1 silencing (Fig. 2 k). The decreases in the DHFR copy number and DHFR protein level were also confirmed after BRCA1 silencing, as shown in Figure 2 l and 2 m. Beyond DHFR, our previous aCGH results have identified a panel of genes on chromosome 5. For example, ZFYVE16, MSH3, CCNH, GLRX and CAST were co‐localized with DHFR within the same amplicon in HSR‐containing cells, whereas only ZFYVE16 and MSH3 showed similar co‐localization in DM‐containing cells. RAD1 and PLK2 were not amplified on chromosome 5 during the development of MTX resistance and were consequently used as negative controls. To further elucidate whether the inhibition of HR decreased incidence of cytogenetically manifested gene amplification in MTX‐resistant cells, we evaluated the copy number of the genes in the above panel at the DNA level and found that both ZFYVE16 and MSH3 amplification dramatically decreased in BRCA1‐depleted cells, as observed for DHFR, whereas the levels of RAD1, PLK2, CCNH, GLRX, and CAST were not affected (Fig. 2 n). To further confirm the role of HR in DMs, we also detected the copy numbers of genes amplified in the form of DMs by BRCA1 rescue assays. We found that the number of DHFR‐carried DMs (Fig. 2 k), the copy number and protein level of DHFR (Fig. 2 l and Fig. S3, Supporting Information) and genes (MSH3 and ZFYVE16) localized in the same amplicon of DMs (Fig. 2 n) reverted after BRCA1 was rescued. DM‐containing MTX‐resistant cells have already contained DMs in which the change of DMs after BRCA1 depletion may be a balance of formation and exclusion. To further verify the role of HR in DMs formation, we chose HT‐29 8 × 10−5 M MTX‐resistant cells, of which MTX concentration was approaching 10−4 M but did not have DMs, to deplete BRCA1 expression and induced the cells to form DMs by adding 10−4 M. A month later, cells transfected with control formed DMs, but the ones transfected with BRCA1 shRNA were dead. Overall, the inhibition of HR decreased extrachromosomal amplification in MTX‐resistant cells.
HR inhibition does not affect intrachromosomal amplification in MTX‐resistant cells
To examine the effect of HR inhibition on intrachromosomal amplification, we also silenced BRCA1 (Fig. 3 a), which promoted DSBs, as evidenced by the level of γH2AX (Fig. 3 b and 3c) in HSR‐containing MTX‐resistant cells. The HR activity assay indicated that MRE11 protein levels decreased after BRCA1 silencing (Fig. 3 d). BRCA1 depletion also significantly changed the number of RAD51 foci in nuclei or the RAD51 nuclear protein levels (Fig. 3 e and 3 f). The results above suggested that the inhibition of BRCA1 also promoted DSB and inhibited HR in HSR‐containing MTX‐resistant cells.
Figure 3.
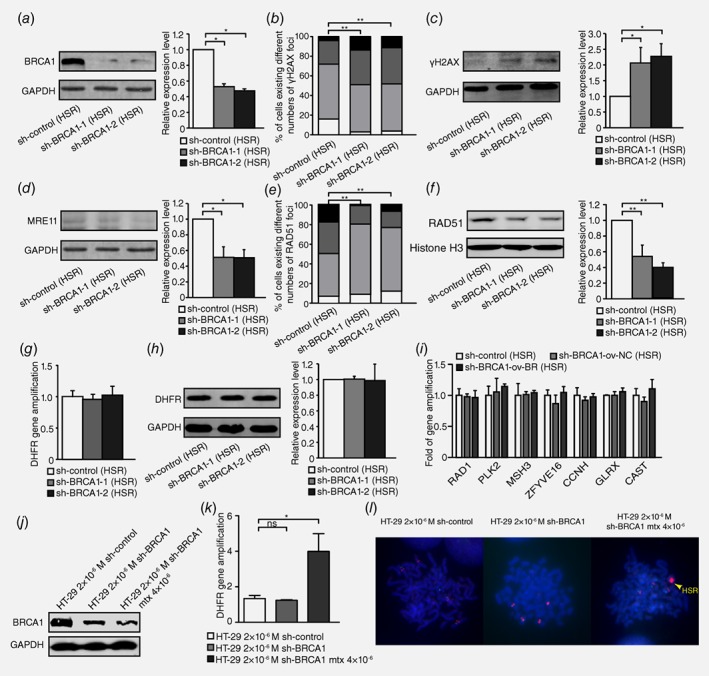
Inhibition of BRCA1 exacerbates double‐strand DNA breaks and suppresses HR but does not affect gene amplification in HSR‐containing MTX‐resistant cells. a. Western blot analysis of BRCA1 protein level and densitometry values in HSR‐containing cells: control and two BRCA1‐depleted clones (n = 3, *p<0.025). b. IF assay for γH2AX protein in HSR‐containing control and two BRCA1‐depleted clones (n ≥ 100, **p<0.005). c. Western blot for γH2AX protein level and densitometry values in HSR‐containing control and two BRCA1‐depleted clones (n = 3, *p<0.025). d. Western blot for MRE11 protein level and densitometry values in HSR‐containing control and two BRCA1‐depleted clones (n = 3, *p<0.025). e. IF assay for RAD51 in HSR‐containing control and two BRCA1‐depleted clones (n ≥ 100, **p<0.005). f. Western blot for RAD51 nuclear protein level and densitometry values in HSR‐containing control and two BRCA1‐depleted clones (n = 3, **p<0.005). g. Real‐time PCR analysis of DHFR amplification in HSR‐containing control and two BRCA1‐depleted clones (n = 3, p>0.025). h. Western blot analysis of DHFR protein level and densitometry values in HSR‐containing control and two BRCA1‐depleted clones (n = 3, p>0.025). i. Real‐time PCR analysis of other genes that co‐localized with DHFR in chromosome 5, including RAD1, PLK2, MSH3, ZFYVE16, CCNH, GLRX and CAST, in HSR‐containing control and two BRCA1‐depleted clones (n = 3, p>0.025). j. Western blot analysis of BRCA1 protein level in 2 × 10−6 M MTX‐resistant control, BRCA1‐depleted clone and sh‐BRCA1 clone adding 4 × 10−6 M MTX cells. k. Real‐time PCR analysis of DHFR amplification in 2 × 10−6 M MTX‐resistant control, BRCA1‐depleted clone and sh‐BRCA1 clone adding 4 × 10−6 M MTX cells (n = 3, *p<0.025). l. FISH analysis of HSR formation in 2 × 10−6 M MTX‐resistant control, BRCA1‐depleted clone and sh‐BRCA1 clone adding 4 × 10−6 M MTX cells. BAC‐containing DHFR was used as a probe and is marked in red; nuclei were stained with DAPI and are marked in blue. Yellow arrow points HSR.
To assess the effect of HR inhibition on the formation of HSR, we measured the genomic copy number of DHFR. However, the gene copies of DHFR did not change, and its expression did not differ between BRCA1‐depleted cells and control cells (Fig. 3 g and 3 h). We also evaluated the effect of HR inhibition on the incidence of cytogenetically manifested gene amplification in HSR‐containing cells. However, the dosage of HSR‐form amplified genes remained unchanged (Figure 3 i) in HSR‐containing MTX‐resistant cells. Because HSR was also observed in some DM‐containing cells, we counted cells with or without HSR amplification in both control and BRCA1‐depleted clones of DM‐containing MTX‐resistant cells for at least 50 karyotypes, and differences between each group were analyzed with the Chi‐squared test. As shown in Table S1, no significant differences were detected between groups. To further verify the role of HR in the formation of HSR, we chose 2 × 10−6 M MTX‐resistant HT‐29 cells which did not have HSR and depleted BRCA1 followed by adding 4 × 10−6 M MTX to induce the formation of HSR. Because among all MTX‐resistant cell lines we established, cells resistant to 4 × 10−6 M MTX had an apparent HSR. A month later, BRCA1 depletion still continued after adding 4 × 10−6 M MTX for a month (Fig. 3 j); however, the copy number of DHFR increased significantly in 4 × 10−6 M MTX‐added BRCA1‐depleted cells (Fig. 3 k) and as Figure 3 l demonstrated, an obviously small HSR had already formed. These results suggested that HR inhibition did not affect intrachromosomal amplification in MTX‐resistant cells.
HR inhibition eliminates extrachromosomal amplification via MN/NBUDs in association with cell cycle acceleration in MTX‐resistant cells
The formation of MN/NBUDs can eliminate amplified genes from the nucleus.28 To determine whether the inhibition of HR promotes the exclusion of DMs in this manner, we detected the formation of MN/NBUDs that contain amplified DHFR after BRCA1 depletion. Figure 4 a showed the MN/NBUDs with or without a DHFR signal. As presented in Figure 4 b, BRCA1‐depleted cells showed a significant increase in the formation of MN/NBUDs. In addition, the formation of MN/NBUDs with DHFR signal also markedly increased. After BRCA1 rescued, both the formation of MN/NBUDs and the formation of MN/NBUDs with DHFR signal reverted (Fig. 4 b). Therefore, we suggested that the inhibition of HR promoted the elimination of DMs with DHFR amplification via MN/NBUDs.
Figure 4.
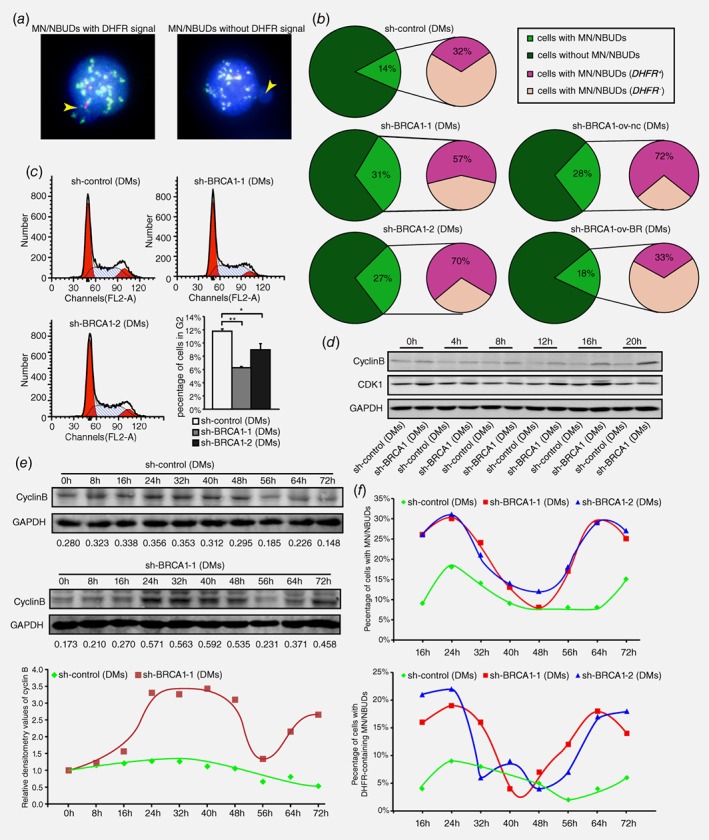
HR inhibition results in G2/M abrogation and cell cycle acceleration accompanied by promoting the exclusion of DMs via MN/NBUDs. a. FISH analyses of MN/NBUDs in DM‐containing control and BRCA1‐depleted cells probed with BAC‐containing DHFR and the centromere of chromosome 5. The yellow arrow indicated the MN/NBUDs of nuclei. The MN/NBUDs were grouped into two categories: with DHFR signal (left panel) and without DHFR signal (right panel; DHFR in green; centromere of chromosome 5 in red; DAPI in blue). b. analyses of MN/NBUDs formation and exclusion of DHFR via MN/NBUDs in DM‐containing control, two BRCA1‐depleted clones, BRCA1‐depleted control and BRCA1‐depleted rescued clone. (**p < 0.005 for MN/NBUDs formation between control and two BRCA1‐depleted clones, n ≥ 100; **p < 0.005 for MN/NBUDs formation between BRCA1‐depleted control and BRCA1‐depleted rescued clone, n ≥ 100; *p < 0.025 for exclusion of DHFR via MN/NBUDs between control and two BRCA1‐depleted clones; *p < 0.025 for exclusion of DHFR via MN/NBUDs between BRCA1‐depleted control and BRCA1‐depleted rescued clone.) c. Flow assay analyses of cell cycle distribution in DM‐containing control and two BRCA1‐depleted clones. The left panel shows distributions of G1, S and G2 phases. The right panel shows both the G2 phase percentage and cell number of G2 phase for 3 repetitions. (*p < 0.025, **p < 0.005, by Chi‐squared test and Bonferroni adjustment) d. Western blot of cyclin B and CDK1 in DM‐containing control and BRCA1‐depleted cells harvested at different time points of releasing in complete culture (0 to 20 h) and then recorded once every 4 h. E. Western blot analyses of cyclin B in DM‐containing control and BRCA1‐depleted cells released at different time points of releasing in complete culture (0 to 72 h) and recorded once every 8 h. The numbers under the bands represent the relative densitometry values. The lower panel showed the trends of cyclin B expression in DM‐containing control and BRCA1‐depleted cells. f. Analyses of MN/NBUDs formation and exclusion of DHFR via MN/NBUDs in DM‐containing control and two BRCA1‐depleted clones harvested at different time points of releasing in complete culture (16 to 72 hr) and recorded once every 8 h.
The formation of MN/NBUDs tightly depends on cell cycle progression and mainly occurs during the S and M phases.27, 35 Notably, the HR core protein BRCA1 is a regulator that activates the checkpoint regulation of the S and G2/M phases.31 Thus, we explored the influence of BRCA1 on the cell cycle in DM‐containing MTX‐resistant cells. As shown in Figure 4 c, cells in the G2 phase decreased significantly in BRCA1‐depleted cells compared with the control. This finding suggested that the G2/M checkpoint might be inhibited after BRCA1 depletion. We then released starved cells at 0, 4, 8, 12, 16 and 20 h and detected the protein levels of cyclin B and CDK1, which regulate the G2/M checkpoint, in both control and BRCA1‐depleted cells. The results showed that the protein levels of CDK1 and cyclin B were much higher in BRCA1‐depleted cells than in control cells (Fig. 4 d), thus indicating that cells prematurely entered the M phase after BRCA1 silencing in DM‐containing MTX‐resistant cells. We hypothesized that the entire cell cycle was shortened after BRCA1 depletion. To test this hypothesis, starved cells were released at 0, 8, 16, 24, 32, 48, 56, 64 and 72 h before the protein levels of cyclin B were measured (Fig. 4 e). As expected, BRCA1‐depleted cells reached one and a half peaks of cyclin B expression, whereas control cells reached the peak only once (lowest panel of Fig. 4 e). To further explore whether the formation of MN/NBUDs were tightly connected with cell cycle, we starved cells and released at 16, 24, 32, 48, 56, 64 and 72 h before the number of MN/NBUDs and MN/NBUDs with DHFR signal were counted. As presented in the upper panel of Figure 4 f, in BRCA1‐depleted cells, the formation of MN/NBUDs and MN/NBUDs with DHFR signal reached two peaks but in control cells they only reached one and a half from 16 h to 72 h. The trends of MN/NBUDs formation in both BRCA1‐depleted and control cells were similar to the protein level of cyclin B (Fig. 4 e). It demonstrated that the formation of MN/NBUDs and MN/NBUDs with DHFR signal was also accelerated as was cell cycle in BRCA1‐depleted cells compared with control cells. Hence, inhibition of HR promoted the entire cell cycle and might simultaneously promote the elimination of extrachromosomal gene amplification by MN/NBUDs.
Inhibition of HR increases MTX sensitivity in DM‐containing MTX‐resistant cells
DHFR amplification is the main machinery that underlies MTX resistance in cancer cells. Previous results have demonstrated that the inhibition of HR effectively decreases DM‐form amplification. Thus, to assess the effect of HR inhibition on the MTX sensitivity of cells, we examined the IC50 value of both HSR‐ and DM‐containing MTX‐resistant cells. Two BRCA1‐depleted clones showed a 2.58‐fold and 2.05‐fold decrease in the IC50 value, respectively compared with control DM‐containing cells. When BRCA1 expression was rescued, the value of IC50 reverted to the level of DM‐containing control. However, this difference was not identified in HSR‐containing cells (Table 1), thus indicating that the inhibition of HR partially reverted the sensitivity to MTX in DM‐containing cells but not in HSR‐containing cells.
Table 1.
IC50 values of DM‐ or HSR‐containing control and BRCA1‐depleted cells
| HT‐29 cell line | IC50(mol/L) | Fold Change |
|---|---|---|
| sh‐control (DMs) | 0.003941 ± 0.000467 | 1 |
| sh‐BRCA1–1 (DMs) | 0.001526 ± 0.000282 | 2.58** |
| sh‐BRCA1‐2 (DMs) | 0.001922 ± 0.000247 | 2.05** |
| sh‐BRCA1‐ov‐nc (DMs) | 0.001307 ± 0.000362 | 3.05** |
| sh‐BRCA1‐ov‐br (DMs) | 0.003400 ± 0.000506 | 1.15 |
| sh‐control (HSR) | 0.001507 ± 0.000401 | 1 |
| sh‐BRCA1–1 (HSR) | 0.000928 ± 0.000151 | 1.62 |
| sh‐BRCA1–2 (HSR) | 0.001644 ± 0.000687 | 0.92 |
p<0.0025
Discussion
The amplification of drug‐resistant genes appears to be a common mechanism of acquired resistance that frequently causes cancer therapy failure. Therefore, the molecular mechanisms that underlie the formation of gene amplification need to be explored. In our study, we found a significant increase in the expression of BRCA1 and RAD51 in both HSR‐ and DM‐containing MTX‐resistant cells. Therefore, we investigated the association between HR and the development of gene amplification.
To examine the involvement of HR in the formation of DMs, we first inhibited HR by interfering with BRCA1 in DM‐containing MTX‐resistant cells. We found a dramatic decrease in HR proteins, including MRE11 and RAD51. Then HR‐GFP assay demonstrated a decrease of GFP‐cells after BRCA1 inhibition, thus indicating that HR function was successfully inhibited. To investigate the effect of HR inhibition on DMs, we detected the number of DMs and level of amplified DM‐form genes. We found a sharp decrease in the copy number of DMs and the level of DM‐form amplified genes and this situation reverted after BRCA1 was resued. In addition, cell death was caused after interacting with 10−4 M MTX in BRCA1‐depleted 8 × 10−5 M MTX‐resistant HT‐29 cells. This may suggest that the reason for cell death was that cells were not able to form DMs to defend higher concentration of MTX without HR. Thus this finding indicates that HR is involved in the formation of DMs in HT‐29 MTX‐resistant cells. In general, HR is regarded as an error‐free DSB repair pathway that faithfully maintains genome stability. However, our findings indicated that even HR can tolerate mistakes that lead to genome instability. This finding is consistent with evidence showing that HR contributes to the formation of extrachromosomal amplicons in many species, such as S. cerevisiae, Leishmania, Arabidopsis thaliana and Drosophila. Korbel et al.36 and Chen et al.37 have also found long homologous sequences near amplicon breakpoints in humans, and they have suggested that HR may reconnect broken amplicons and consequently induce DNA duplications. In accordance with this conclusion, Colnaghi et al. have suggested that NAHR can induce an intrachromosomal deletion of a gene, and the deleted section might loop out of the chromosome, thus forming an extrachromosomal circular DNA.38 Nonetheless, the above findings are all indirect and have primarily been inferred from specific sequence characteristics, such as direct tandem repeats or homologous recombination elements, and direct molecular evidence showing the participation of HR in the formation of extrachromosomal DMs in humans is lacking. Because HR may be able to loop out extrachromosomal amplicons if repetitive elements or transposable elements exist,39, 40 we hypothesize that DMs may originate from the amplicon‐abundant region, HSR, which would allow for HR to loop out DMs (Fig. 5). However, our results do not corroborate those of Ruiz‐Herrera's study, which has demonstrated an increase in the number of clones containing DMs after inhibition of the HR protein RAD54. This contradiction may be due to the different genetic backgrounds of HeLa cells, which lack HSR and sufficient repetitive segments in the genome for the HR pathway to loop out DMs. Thus, the formation of DMs after RAD54 inhibition may rely on other mechanisms, such as NHEJ or A‐NHEJ, two additional DSB pathways. In addition, some clones (13/24, 54%) continued to exhibit DMs formation after RAD54 inhibition, thus suggesting that HR may be the trigger.
Figure 5.
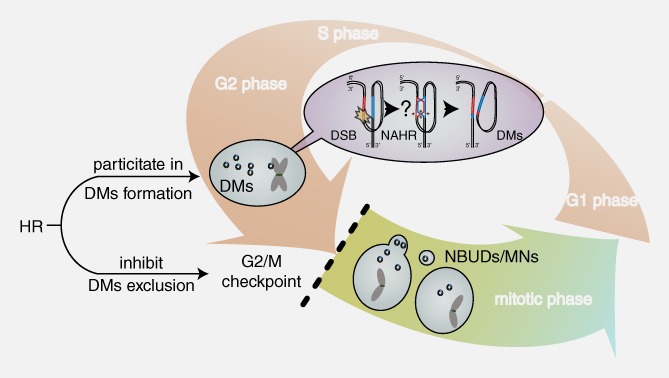
The association between HR and DMs. HR may be involved in the formation of DMs and inhibit the exclusion of DMs by MN/NBUDs by activating the G2/M checkpoint and arresting the cell cycle. The pink‐white and yellow‐green gradient arrows represent the interphase and mitotic phase of the cell cycle, respectively. The purple gradient oval represents the potential mechanism by which HR is involved in DM formation. The black lines and small cycles represent DNA double strands and DMs, respectively. The question mark represents that the repair progression of NAHR is a speculation that the broken DNA may find a homologous region on its own chromosome as a template and form double minutes simultaneously. The red and blue regions on DNA strands represent amplified homologous regions. The brown arrows represent the resecting and reconnecting sites of HR Holliday junctions. The black dashed line represents the activity of the G2/M checkpoint and cell cycle arrest.
In contrast, HSR was not affected by HR inhibition, because the number of cells containing HSR‐form gene amplification and the copy number of HSR‐form amplified genes did not significantly change. Moreover, BRCA1 depletion had no influence on the formation of HSR after adding higher concentration of MTX in our study. Therefore, HR may not participate in the formation of intrachromosomal amplicons. This finding is consistent with those reported by Gibaud A's, in which whole‐genome analysis detected no breakpoint junction sequences that would support reconnection of broken amplicon ends by HR.41 Although the classical model of HSR formation based on the fusion of two sister chromatids, B‐F‐B, most probably correlates with HR, our results do not demonstrate a relationship between these two pathways. Thus, the mechanism of HSR formation remains to be clarified.
Previous studies have demonstrated that different mechanisms are responsible for the formation of DMs and HSR. Classical models of DM formation include episome resection,12 looping out during G1/G242 and chromothripsis,43 whereas B/F/B cycles have been implicated as a model of HSR formation.11 Thus, we hypothesize that the formation of HSR and DMs may depend on different molecular mechanisms. For example, NHEJ is mainly involved in DM formation but not HSR.13 In our study, we observed different roles of HR in the formation of DM and HSR. Specifically, our findings indicates that HR is involved at a later stage (i.e., the formation of DMs) and not in early stages (i.e., the formation of HSR) of gene amplification in MTX‐resistant cells. These results may guide clinical targeted therapy on the basis of different amplification forms.
The accumulation of DSBs induced by external pressures, such as hydroxyurea or low‐dose radiation, often promotes the formation of MN/NBUDs that entrap DNA fragments.29, 44 Notably, a dysfunction in DNA repair pathways usually increases DNA breaks, thereby promoting the capture of DMs by MN/NBUDs formation.45 Moreover, we simultaneously observed increases in DSBs after HR inhibition, the formation of MN/NBUDs and the exclusion of DMs by MN/NBUDs after the inhibition of HR. As Tanaka et al. have claimed, the formation of MN/NBUDs strongly depends on cell cycle progression46, 47, 48 and the inhibition of the HR core protein BRCA1 may promote premature entry into cell.47, 48 In our study, we found that HR inhibition abrogated the G2/M checkpoint, and cells consequently prematurely progressed to mitosis, thereby accelerating the entire cell cycle. By testing the formation of MN/NBUDs and DHFR‐containing MN/NBUDs at different time points, we found highly similar formation trends in cell cycle progression. The formation of MN/NBUDs and DHFR‐containing MN/NBUDs was also accelerated in BRCA1‐depleted DM‐containing MTX‐resistant cells. Thus, we propose that the increase in the exclusion of DMs by MN/NBUDs after HR inhibition may be due to cell cycle acceleration caused by the weakening of the G2/M checkpoint (Fig. 5).
Defining the factors that drive the formation of gene amplification would aid in identifying molecular targets to reverse drug resistance. Because BRCA1 is the core molecule of HR, its depletion or mutation can induce malignant transformation and reverse drug resistance.15, 49 For example, Wiedemeyer et al. have reported that targeting BRCA1 and HR reverses platinum resistance in high‐grade serous ovarian carcinoma.50 In our study, we found that BRCA1‐depletion increased MTX sensitivity in DM‐containing cells and rescuing BRCA1 expression in BRCA1‐depleted cells decreased MTX sensitivity to the level of sh‐control. Because both the copy number and expression of the MTX‐resistant gene DHFR were downregulated after BRCA1 depletion, we suggest that the inhibition of HR may partially reverse MTX resistance by decreasing DHFR amplification in cells. Moreover, because HR inhibition did not influence MTX sensitivity in HSR‐containing MTX‐resistant cells, we propose that HR inhibition enhances chemosensitivity solely by decreasing the effect of DMs but not HSR.
In conclusion, our findings illuminated the important role of HR inhibition in effectively decreasing extrachromosomal amplification and reversing drug resistance in MTX‐resistant cells. HR may be involved in the formation of DMs and the inhibition of DM exclusion by facilitating cell cycle arrest at the G2/M checkpoint (Fig. 5). Therefore, we propose that decreasing gene amplification via HR is an effective strategy to reverse drug resistance and enhance the efficacy of chemotherapy.
Supporting information
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Figure 4
Supplemental Figure 5
Supplemental Figure 6
Supplemental Figure 7
Supplemental Figure 8
Table S1
Disclosure: The authors declare that there are no conflicts of interest.
Contributor Information
Xiangning Meng, Email: mengxiangn@ems.hrbmu.edu.cn.
Songbin Fu, Email: fusb@ems.hrbmu.edu.cn.
REFERENCES
- 1. Turner KM, Deshpande V, Beyter D, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017;543:122–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mahon FX, Deininger MW, Schultheis B, et al. Selection and characterization of BCR‐ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood 2000;96:1070–9. [PubMed] [Google Scholar]
- 3. Matheson EC, Hogarth LA, Case MC, et al. DHFR and MSH3 co‐amplification in childhood acute lymphoblastic leukaemia, in vitro and in vivo. Carcinogenesis 2007;28:1341–6. [DOI] [PubMed] [Google Scholar]
- 4. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole‐genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489–94. [DOI] [PubMed] [Google Scholar]
- 5. Storlazzi CT, Lonoce A, Guastadisegni MC, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res 2010;20:1198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reams AB, Roth JR. Mechanisms of gene duplication and amplification. Cold Spring Harbor Perspect Biol 2015;7:a016592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singer MJ, Mesner LD, Friedman CL, et al. Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proc Natl Acad Sci USA 2000;97:7921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double‐strand break repair pathway choice. Cell Res 2008;18:134–47. [DOI] [PubMed] [Google Scholar]
- 9. Rouet P, Smih F, Jasin M. Introduction of double‐strand breaks into the genome of mouse cells by expression of a rare‐cutting endonuclease. Molecular and cellular biology 1994;14:8096–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li JC, Kaminskas E. Accumulation of DNA strand breaks and methotrexate cytotoxicity. Proc Natl Acad Sci USA 1984;81:5694–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marotta M, Chen X, Watanabe T, et al. Homology‐mediated end‐capping as a primary step of sister chromatid fusion in the breakage‐fusion‐bridge cycles. Nucleic Acids Res 2013;41:9732–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith CA, Vinograd J. Small polydisperse circular DNA of HeLa cells. J Mol Biol 1972;69:163–78. [DOI] [PubMed] [Google Scholar]
- 13. Meng X, Qi X, Guo H, et al. Novel role for non‐homologous end joining in the formation of double minutes in methotrexate‐resistant colon cancer cells. J Med Genetics 2015;52:135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen JM, Cooper DN, Ferec C, et al. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol 2010;20:222–33. [DOI] [PubMed] [Google Scholar]
- 15. Moynahan ME, Chiu JW, Koller BH, et al. Brca1 controls homology‐directed DNA repair. Mol Cell 1999;4:511–8. [DOI] [PubMed] [Google Scholar]
- 16. Towler WI, Zhang J, Ransburgh DJ, et al. Analysis of BRCA1 variants in double‐strand break repair by homologous recombination and single‐strand annealing. Human Mutation 2013;34:439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Isono M, Niimi A, Oike T, et al. BRCA1 Directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep 2017;18:520–32. [DOI] [PubMed] [Google Scholar]
- 18. Yun MH, Hiom K. CtIP‐BRCA1 modulates the choice of DNA double‐strand‐break repair pathway throughout the cell cycle. Nature 2009;459:460–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carreira A, Hilario J, Amitani I, et al. The BRC repeats of BRCA2 modulate the DNA‐binding selectivity of RAD51. Cell 2009;136:1032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang F, Fan Q, Ren K, et al. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res : MCR 2009;7:1110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shabbeer S, Omer D, Berneman D, et al. BRCA1 targets G2/M cell cycle proteins for ubiquitination and proteasomal degradation. Oncogene 2013;32:5005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 2006;7:739–50. [DOI] [PubMed] [Google Scholar]
- 23. Startek M, Szafranski P, Gambin T, et al. Genome‐wide analyses of LINE‐LINE‐mediated nonallelic homologous recombination. Nucleic Acids Res 2015;43:2188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grondin K, Papadopoulou B, Ouellette M. Homologous recombination between direct repeat sequences yields P‐glycoprotein containing amplicons in arsenite resistant Leishmania. Nucleic Acids Res 1993;21:1895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Joseph‐George AM, He Y, Marshall CR, et al. Euchromatic 9q13‐q21 duplication variants are tandem segmental amplifications of sequence reciprocal to 9q13‐q21 deletions. Journal of medical genetics 2011;48:317–22. [DOI] [PubMed] [Google Scholar]
- 26. Ruiz‐Herrera A, Smirnova A, Khouriauli L, et al. Gene amplification in human cells knocked down for RAD54. Genome Integrity 2011;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fenech M, Kirsch‐Volders M, Natarajan AT, et al. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011;26:125–32. [DOI] [PubMed] [Google Scholar]
- 28. Fenech M, Crott JW. Micronuclei, nucleoplasmic bridges and nuclear buds induced in folic acid deficient human lymphocytes‐evidence for breakage‐fusion‐bridge cycles in the cytokinesis‐block micronucleus assay. Mutat Res 2002;504:131–6. [DOI] [PubMed] [Google Scholar]
- 29. Yu L, Zhao Y, Quan C, et al. Gemcitabine eliminates double minute chromosomes from human ovarian cancer cells. PloS one 2013;8:e71988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valent A, Benard J, Clausse B, et al. In vivo elimination of acentric double minutes containing amplified MYCN from neuroblastoma tumor cells through the formation of micronuclei. Am J Pathol 2001;158:1579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 2006;34:1416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cai D, Byth KF, Shapiro GI. AZ703, an imidazo[1,2‐a]pyridine inhibitor of cyclin‐dependent kinases 1 and 2, induces E2F‐1‐dependent apoptosis enhanced by depletion of cyclin‐dependent kinase 9. Cancer Res 2006;66:435–44. [DOI] [PubMed] [Google Scholar]
- 33. Sung P. Catalysis of ATP‐dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 1994;265:1241–3. [DOI] [PubMed] [Google Scholar]
- 34. Hopfner KP, Karcher A, Craig L, et al. Structural biochemistry and interaction architecture of the DNA double‐strand break repair Mre11 nuclease and Rad50‐ATPase. Cell 2001;105:473–85. [DOI] [PubMed] [Google Scholar]
- 35. Shimizu N, Itoh N, Utiyama H, et al. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. The J Cell Biol 1998;140:1307–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korbel JO, Urban AE, Affourtit JP, et al. Paired‐end mapping reveals extensive structural variation in the human genome. Science 2007;318:420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen CP, Su YN, Chen YT, Chen WL, Hsu LJ, Wang W. Prenatal diagnosis of directly transmitted benign 4q12‐q13.1 quadruplication associated with tandem segmental amplifications of the LPHN3 gene. Taiwanese journal of obstetrics & gynecology 2011;50:401–4. [DOI] [PubMed] [Google Scholar]
- 38. Colnaghi R, Carpenter G, Volker M, et al. The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin Cell Dev Biol 2011;22:875–85. [DOI] [PubMed] [Google Scholar]
- 39. Ade C, Roy‐Engel AM, Deininger PL. Alu elements: an intrinsic source of human genome instability. Curr Opin Virol 2013;3:639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sen SK, Han K, Wang J, et al. Human genomic deletions mediated by recombination between Alu elements. Am J Human Genetics 2006;79:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gibaud A, Vogt N, Brison O, et al. Characterization at nucleotide resolution of the homogeneously staining region sites of insertion in two cancer cell lines. Nucleic Acids Res 2013;41:8210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roelofs H, Tasseron‐de Jong JG, van der Wal‐Aker J, et al. Gene amplification in a human osteosarcoma cell line results in the persistence of the original chromosome and the formation of translocation chromosomes. Mutation Res 1992;276:241–60. [DOI] [PubMed] [Google Scholar]
- 43. Kloosterman WP, Koster J, Molenaar JJ. Prevalence and clinical implications of chromothripsis in cancer genomes. Curr Opin Oncol 2014;26:64–72. [DOI] [PubMed] [Google Scholar]
- 44. Schoenlein PV, Barrett JT, Kulharya A, et al. Radiation therapy depletes extrachromosomally amplified drug resistance genes and oncogenes from tumor cells via micronuclear capture of episomes and double minute chromosomes. Int J Radiat Oncol Biol Phys 2003;55:1051–65. [DOI] [PubMed] [Google Scholar]
- 45. Haaf T, Raderschall E, Reddy G, et al. Sequestration of mammalian Rad51‐recombination protein into micronuclei. J Cell Biol 1999;144:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanaka T, Shimizu N. Induced detachment of acentric chromatin from mitotic chromosomes leads to their cytoplasmic localization at G(1) and the micronucleation by lamin reorganization at S phase. J Cell Sci 2000;113(Pt 4):697–707. [DOI] [PubMed] [Google Scholar]
- 47. Kim SS, Cao L, Li C, et al. Uterus hyperplasia and increased carcinogen‐induced tumorigenesis in mice carrying a targeted mutation of the Chk2 phosphorylation site in Brca1. Mol Cellul Biol 2004;24:9498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu X, Weaver Z, Linke SP, et al. Centrosome amplification and a defective G2‐M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform‐deficient cells. Mol Cell 1999;3:389–95. [DOI] [PubMed] [Google Scholar]
- 49. Bai F, Smith MD, Chan HL, et al. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal‐to‐basal mammary tumor transformation. Oncogene 2013;32:2715–25. [DOI] [PubMed] [Google Scholar]
- 50. Wiedemeyer WR, Beach JA, Karlan BY. Reversing Platinum Resistance in High‐Grade Serous Ovarian Carcinoma: Targeting BRCA and the Homologous Recombination System. Front Oncol 2014;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Figure 4
Supplemental Figure 5
Supplemental Figure 6
Supplemental Figure 7
Supplemental Figure 8
Table S1