

Abstract
Cartilage‐hair hypoplasia syndrome (CHH) is a rare autosomal recessive condition characterized by metaphyseal chondrodysplasia and characteristic hair, together with a myriad of other symptoms, being most common immunodeficiency and gastrointestinal complications. A 15‐year‐old Mexican male initially diagnosed with Hirschsprung disease and posterior immunodeficiency, presents to our department for genetic and complementary evaluation for suspected CHH. Physical, biochemical, and genetic studies confirmed CHH together with IGF‐1 deficiency. For this reason, we propose IGF‐1 replacement therapy for its well‐known actions on hematopoiesis, immune function and maturation, and metabolism. © 2016 Wiley Periodicals, Inc.
Keywords: cartilage‐hair hypoplasia, IGF‐1, RMRP gene, GHR
INTRODUCTION
Cartilage‐hair hypoplasia syndrome (CHH) is a rare autosomal recessive condition that was originally described by McKusik in 1964 in Amish children. In this population, it accounts for one in 1,300 newborns [McKusick et al., 1965], while in the Finnish population it accounts for one in 23,000 newborns [Mäkitie and Kaitila, 1993]. Outside those populations it is extremely rare, with no incidence data available [Mäkitie and Kaitila, 1993]. The genetic locus for this disease falls within the RNA Component Of Mitochondrial RNA Processing Endoribonuclease (RMRP) gene, localized in chromosome 9p12, which codes for the RMRP, believed to be required for cell cycle, mtDNA replication and rRNA processing [Ridanpää et al., 2001; Hermanns et al., 2005; Thiel et al., 2005; Martin and Li, 2007]. The RMRP RNA product associates with at least seven proteins whose interactions and roles remain to be discovered [Welting et al., 2004].
The hallmark of this condition is a metaphyseal chondrodysplasia and characteristic hair (fine, sparse with scalp, blond eyelashes—93%), with other several signs and symptoms including short stature and skeletal dysplasia (100%) with varum deformation of lower extremities, hypoplastic anemia during childhood (79%), immunodeficiency (during childhood they may present different recurrent infections secondary to an immunodeficiency that may be humoral, B‐cell or T‐cell, or combined—56%) [Pierce and Polmar, 1982; Mäkitie et al., 1998, 2000b; Baradaran‐Heravi et al., 2008] with higher risk of malignancies, skin and nails abnormalities, gastrointestinal diseases (such as Hirschsprung disease [HD], malabsorption, anal stenosis and esophageal atresia—18%) [Mäkitie et al., 2002, 2000a], and failure to thrive [Mäkitie et al., 1992; Glass and Tifft, 1999; Riley et al., 2015]. The growth failure is progressive, owing, partly, to a weak or absent pubertal growth spurt; pubertal maturation is normal; testicular size is abnormal in some patients with normal serum concentrations of testosterone, inhibin B and gonadotropins [Mäkitie et al., 2001].
These patients are symptomatic since birth, showing short‐limb dwarfism and short puffy hands, with normal head circumference [Giedion, 1998]. The final adult heights range from 100.7 to 149 cm in males and 103.7 to 137.9 cm in females, most of them are overweight eventually becoming obese [Riley et al., 2015]. Even though life expectancy reaches adulthood, an early diagnosis is important in order to better detect and control possible hematologic and immunologic abnormalities, as well as malignancy development.
CLINICAL REPORT
A 15‐year‐old Mexican male with a probable clinical diagnosis of CHH presents to our department for genetic and complementary evaluation. He was born at 40 weeks with 3200 g of body weight, 45 cm and a cephalic perimeter of 36 cm, from non‐consanguineous healthy parents of the same race. At birth, he was diagnosed with HD and treated surgically without complications. During his early childhood, he suffered multiple episodes of otitis media, two pneumonias, several gastroenteric infections, and failure to thrive. At the age of 4, he was diagnosed humoral immunodeficiency (IgA and IgG deficiency with normal cellular response) and he begun treatment with intravenous gamma globulin until present. During the following years, he developed varus curvature of both lower extremities and intermittent claudication with shortened limbs and femurs along varus hands with short fingers. He was evaluated by a pediatric orthopedist without surgical outcome.
At age 7, and taking into account the abovementioned progression, a plausible diagnosis of CHH was coined, with no genetic confirmation; immunological treatment was continued.
Upon admission to our department he was re‐assessed: new clinical, physical, biochemical, and radiological findings are shown in Table SI. Clinically, he has a normal cognitive development. He displays, at present, short stature and several skeletal complications, with no cardiac, respiratory, digestive, or urinary specific symptoms. Moreover, since the onset with gamma globulin treatment he refers an improvement in the frequency of recurrent infections. His last infection was on September 2015: nail abscess, without further complications.
Physical Examination
A 15‐year‐old male with 103.5 cm height (<9 SD below 3rd centile for age) and a weight of 19.5 kg (− 4 SD below 3rd centile for age). Short‐limbs, limited elbow extension, short and pudgy hands, lumbar lordosis, and genu varum were present. He has thin, sparse, and brittle hair, as well as very fine and sparse eyelashes; without any dental, skin, or nails abnormalities. Genital examination revealed small testicular size for his age (6.91 cc, Tanner stage II), while the penis size was normal for his age (6.5 cm, Tanner stage V). Radiological studies showed disproportionate short‐limbs, as well as coxa vara and femoral bowing. Lumbar lordosis was also observed; no subluxations or rotations were found (Fig. 1B–D).
Figure 1.
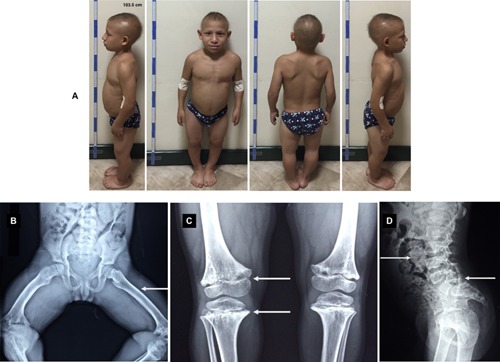
A) Patient profiles at age 15. Short stature, scarce hair, genu varum and lordosis can be appreciated. B) Radiographic features of CHH patient at 9 years of age: genu varum (arrow) with metaphyseal sclerotic irregularities (arrow). C) Radiographic features of CHH patient at 10 years of age: Metaphyseal ends are flared, scalloped, and irregularly sclerotic with cystic areas (arrow). D) Radiographic features of CHH patient at 8 years of age: Increased lumbar lordosis (arrow) and a marked bowel dilatation and fecal impaction can observe (arrow). [Color figure can be viewed at wileyonlinelibrary.com].
Biochemical Findings
Normal hemoglobin (14.5 g/dl) and total cell count (leucocytes 7.200 per µl), normal levels of inhibin A (1.2 pg/ml) and testosterone (28.4 ng/dl), with high levels of sex steroid‐binding globulin (84.8 nmol/L). Regarding the immunological study, he showed decreased levels of IgA, IgM, and IgG. He showed decreased circulating IGF‐1 levels (79 ng/ml, normal range for his age:187–599 ng/ml) and IGFBP‐3 (2.3 mg/L, normal range for his age: 3.3–10 ng/ml), together with high basal GH levels (3.98 ng/ml).
Genetic Diagnosis
Sequencing of the patient's DNA (Illumina TruSight One, CA) revealed an upstream single nucleotide variant and a nucleotide insertion for the RMRP gene (NM_174923.2c.‐542C > C/T; NM_174923.2c.‐380_‐379insT, respectively). There are no single nucleotide polymorphisms (SNP) reported for these positions in the NCBI SNP database.
In addition, a heterozygous missense variant of Growth Hormone Receptor (GHR) gene was identified (NM_001242399.2:c.1651A > C), already reported in the NCBI SNP database as rs6180. Such mutation affects a cytoplasmic domain of the receptor and hence no growth hormone binding protein (GHBP) was found in blood. Aditionally, a heterozygous missense variant of JAK2 gene was identified in this patient (NM_004972.3:c.3170C > T), with possible deleterious effects as determined by sift (0) and PolyPhen (1) algorithms, which has not been previously reported in the NCBI SNP database.
DISCUSSION
As previously mentioned, CHH is a rare autosomal recessive disease with several distinctive features with our patient presenting most of them. At present, this patient does not have any gastrointestinal problems. Despite at birth he was diagnosed with HD, it was treated surgically with any further symptoms. However, some studies have shown that those with CHH and HD have a higher predisposition to a more severely immunocompromized outcome that patients without HD [Mäkitie et al., 2002]. In this aspect, cellular immunity, humoral immunity, or both can be impaired, leading to several recurrent infections, without apparent correlation between laboratory findings and infection susceptibility [Mäkitie et al., 2000b]. Even though the most frequent finding is cellular immune deficiency, this patient presents a partial humoral immunodeficiency, well controlled with gamma globulin treatment.
Another common finding during childhood is anemia, which is secondary to defective erythropoiesis and found related to the IGF‐1 system [Mäkitie et al., 2000a]. At present, this patient has normal hemoglobin without any past history of anemia.
A normal pubertal maturation has been described; even though a lack of pubertal growth spurt is present. Only one report of spermatogenesis has been published so far, demonstrating abnormal semen analysis with low sperm count, decreased motility, and morphological changes [Mäkitie et al., 2001]. Some patients showed small testicular size, but normal hormone levels, so neither infertility nor erectile dysfunction were present. This patient presents small testicular size, being classified as Tanner II, but with normal penis size, as well as normal inhibin and testosterone levels.
Clinical features, except short stature, vary among these patients and over the time, it is thus recommended a genetic characterization to achieve a robust diagnosis. It is well described the mutation in the RMRP gene, that functions as a structural RNA subunit of the mitochondrial RNA processing ribonuclease (RMRP), believed to be required for cell cycle, mtDNA replication and rRNA processing [Ridanpää et al., 2001; Hermanns et al., 2005; Thiel et al., 2005; Martin and Li, 2007]. Such gene was found mutated in this patient, hence confirming the pathology. Together with this, mutations in the IGF‐1 system where found (GHR and JAK2) which could explain such low circulating levels of IGF‐1 and high GH, as GH is responsible for liver stimulation and secretion of ≃ 75% circulating IGF‐1 and, contrarily, IGF‐1 acts to negatively regulate GH secretion in the brain [Buul‐offers and Kooijman, 1998]. JAK2 is an adaptor protein with SH2 homology domains used by GHR to phosphorylate and activate several STATs, which in turn are responsible for IGF‐1 mRNA transcription in the liver and several immune functions [Clark, 1997; Donaghy et al., 2002] (Fig. S1). These mutations could not only help explain short stature, but also immune dysfunction.
We, thereby, suggest that IGF‐1 replacement therapy would be very beneficial to this patient, accounting to the multiple positive effects related to IGF‐1: longitudinal bone growth, immune function, erythropoiesis, metabolic, antioxidant (aiming for cancer prevalence found in some CHH patients), antiinflammatory, hypogonadism, hepato‐, neuro‐, and mitochondrial protection [Claustres et al., 1987; Kurtz et al., 1988; Pascual et al., 2000; García‐Fernández et al., 2003; Conchillo et al., 2005; Puche et al., 2008; Smith, 2010; Castilla‐Cortázar et al., 2011, 2015; Puche and Castilla‐Cortázar, 2012; Aguirre et al., 2016].
To sum up, the patient carries a mutation related to CHH, confirmed by clinical findings and literature reports. Due to the positive effects attributed to IGF‐1, only when levels are below physiological ones, and because our patient harbors mutations in the signaling pathway of its synthesis, we recommend its replacement therapy with rhIGF‐1 (Mecasermin or Increlex). It happens that much of the IGF‐1 positive effects match with most of the symptoms of these patients, hence, potentially being advantageous for his condition.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. The GHR signaling pathway.
Table S1. Somatometric parameters.
ACKNOWLEDGMENTS
First of all, we would like to express our sincere gratitude for Zvi Laron's admirable labor; alike Mexican physicians that aim to improve current standard treatment for primary growth hormone insensitivity and severe evolution in never treated patients.
We thank the medical teams of the Departments of Pediatrics, Endocrinology, and Internal Medicine of Hospital San Jose and Hospital Zambrano Hellion (Monterrey, Nuevo Leon, Mexico) for their generous an efficient help and availability.
This work was supported by “Fundacion de Investigacion HM Hospitales” and “Tecnologico de Monterrey”.
Castilla‐Cortázar I, Rodríguez De Ita J, Martín‐Estal I, Castorena F, Aguirre GA, García de la Garza R, Elizondo MI. 2017. Clinical and molecular diagnosis of a cartilage‐hair hypoplasia with IGF‐1 deficiency. Am J Med Genet Part A 173A: 537–540.
Conflict of interest: The authors have no conflicts of interest relevant to this article to disclose.
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
REFERENCES
- Aguirre GA, De Ita JR, de la Garza RG, Castilla‐Cortazar I. 2016. Insulin‐like growth factor‐1 deficiency and metabolic syndrome. J Transl Med 14:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran‐Heravi A, Thiel C, Rauch A, Zenker M, Boerkoel CF, Kaitila I. 2008. Clinical and genetic distinction of Schimke immuno‐osseous dysplasia and cartilage‐hair hypoplasia. Am J Med Genet Part A 146A:2013–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buul‐offers SC, Kooijman R. 1998. The role of growth hormone and insulin‐like growth factors. Cell Mol Life Sci 54:1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla‐Cortázar I, García‐Fernández M, Delgado G, Puche JE, Sierra I, Barhoum R, González‐Barón S. 2011. Hepatoprotection and neuroprotection induced by low doses of IGF‐II in aging rats. J Transl Med 9:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla‐Cortázar I, Gago A, Muñoz Ú, Ávila‐Gallego E, Guerra‐Menéndez L, Sádaba MC, García‐Magariño M, Olleros Santos‐Ruiz M, Aguirre GA, Puche JE. 2015. Mechanisms underlying testicular damage and dysfunction in mice with partial IGF‐1 deficiency and the effectiveness of IGF‐1 replacement therapy. Urology 86:e1–e9. [DOI] [PubMed] [Google Scholar]
- Clark R. 1997. The somatogenic hormones and insulin‐like growth factor‐1: Stimulators of lymphopoiesis and immune function. Endocr Rev 18:157–179. [DOI] [PubMed] [Google Scholar]
- Claustres M, Chatelain P, Sultan C. 1987. Insulin‐like growth factor I stimulates human erythroid colony formation in vitro. J Clin Endocrinol Metab 65:78–82. [DOI] [PubMed] [Google Scholar]
- Conchillo M, De Knegt RJ, Payeras M, Quiroga J, Sangro B, Herrero JI, Castilla‐Cortazar I, Frystyk J, Flyvbjerg A, Yoshizawa C, Jansen PLM, Scharschmidt B, Prieto J. 2005. Insulin‐like growth factor I (IGF‐I) replacement therapy increases albumin concentration in liver cirrhosis: Results of a pilot randomized controlled clinical trial. J Hepatol 43:630–636. [DOI] [PubMed] [Google Scholar]
- Donaghy AJ, Delhanty PJD, Ho KK, Williams R, Baxter RC. 2002. Regulation of the growth hormone receptor/binding protein, insulin‐like growth factor ternary complex system in human cirrhosis. J Hepatol 36:751–758. [DOI] [PubMed] [Google Scholar]
- García‐Fernández M, Castilla‐Cortázar I, Díaz‐Sánchez M, Díez Caballero F, Castilla A, Díaz Casares A, Varela‐Nieto I, González‐Barón S. 2003. Effect of IGF‐I on total serum antioxidant status in cirrhotic rats. J Physiol Biochem 59:145–146. [DOI] [PubMed] [Google Scholar]
- Giedion A. 1998. Phalangeal cone‐shaped epiphyses of the hand: Their natural history, diagnostic sensitivity, and specificity in cartilage hair hypoplasia and the trichorhinophalangeal syndromes I and II. Pediatr Radiol 28:751–758. [DOI] [PubMed] [Google Scholar]
- Glass RB, Tifft CJ. 1999. Radiologic changes in infancy in McKusick cartilage hair hypoplasia. Am J Med Genet 86:312–315. [DOI] [PubMed] [Google Scholar]
- Hermanns P, Bertuch AA, Bertin TK, Dawson B, Schmitt ME, Shaw C, Zabel B, Lee B. 2005. Consequences of mutations in the non‐coding RMRP RNA in cartilage‐hair hypoplasia. Hum Mol Genet 14:3723–3740. [DOI] [PubMed] [Google Scholar]
- Kurtz A, Zapf J, Eckardt KU, Clemons G, Froesch ER, Bauer C. 1988. Insulin‐like growth factor I stimulates erythropoiesis in hypophysectomized rats. Proc Natl Acad Sci USA 85:7825–7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkitie O, Marttinen E, Kaitila I. 1992. Skeletal growth in cartilage‐hair hypoplasia. A radiological study of 82 patients. Pediatr Radiol 22:434–439. [DOI] [PubMed] [Google Scholar]
- Mäkitie O, Kaitila I. 1993. Cartilage‐hair hypoplasia–clinical manifestations in 108 Finnish patients. Eur J Pediatr 152:211– 217 [DOI] [PubMed] [Google Scholar]
- Mäkitie O, Kaitila I, Savilahti E. 1998. Susceptibility to infections and in vitro immune functions in cartilage‐hair hypoplasia. Eur J Pediatr 157:816–820. [DOI] [PubMed] [Google Scholar]
- Mäkitie O, Juvonen E, Dunkel L, Kaitila I, Siimes MA. 2000a. Anemia in children with cartilage‐hair hypoplasia is related to body growth and to the insulin‐like growth factor system. J Clin Endocrinol Metab 85:563–568. [DOI] [PubMed] [Google Scholar]
- Mäkitie O, Kaitila I, Savilahti E. 2000b. Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr 137:487–492. [DOI] [PubMed] [Google Scholar]
- Mäkitie OM, Tapanainen PJ, Dunkel L, Siimes MA. 2001. Impaired spermatogenesis: An unrecognized feature of cartilage‐hair hypoplasia. Ann Med 33:201–205. [DOI] [PubMed] [Google Scholar]
- Mäkitie O, Heikkinen M, Kaitila I, Rintala R. 2002. Hirschsprung's disease in cartilage‐hair hypoplasia has poor prognosis. J Pediatr Surg 37:1585–1588. [DOI] [PubMed] [Google Scholar]
- Martin AN, Li Y. 2007. RNase MRP RNA and human genetic diseases. Cell Res 17:219–226. [DOI] [PubMed] [Google Scholar]
- McKusick VA, Eldridge R, Hostetler JA, Ruangwit U, Egeland JA. 1965. Dwarfism in the amish II: Cartilage‐hair hypoplasia. Bull Johns Hopkins Hosp 116:285–326. [PubMed] [Google Scholar]
- Pascual M, Castilla‐Cortazar I, Urdaneta E, Quiroga J, Garcia M, Picardi A, Prieto J. 2000. Altered intestinal transport of amino acids in cirrhotic rats: The effect of insulin‐like growth factor‐I. Am J Physiol Gastrointest Liver Physiol 279:G319–G324. [DOI] [PubMed] [Google Scholar]
- Pierce GF, Polmar SH. 1982. Lymphocyte dysfunction in cartilage‐hair hypoplasia: Evidence for an intrinsic defect in cellular proliferation. J Immunol 129:570–575. [PubMed] [Google Scholar]
- Puche JE, Castilla‐Cortázar I. 2012. Human conditions of insulin‐like growth factor‐I (IGF‐I) deficiency. J Transl Med 10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puche JE, García‐Fernández M, Muntané J, Rioja J, González‐Barón S, Cortazar IC. 2008. Low doses of insulin‐like growth factor‐I induce mitochondrial protection in aging rats. Endocrinology 149:2620–2627. [DOI] [PubMed] [Google Scholar]
- Ridanpää M, Van Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, VanVenrooij W, Pruijn G, Salmela R, Rockas S, Mäkitie O, Kaitila I, de la Chapelle A. 2001. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage‐hair hypoplasia. Cell 104:195–203. [DOI] [PubMed] [Google Scholar]
- Riley P, Weiner DS, Leighley B, Jonah D, Morton DH, Strauss KA, Bober MB, Dicintio MS. 2015. Cartilage hair hypoplasia: Characteristics and orthopaedic manifestations. J Child Orthop 9:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ. 2010. Insulin‐like growth factor‐I regulation of immune function: A potential therapeutic target in autoimmune diseases? Pharmacol Rev 62:199–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel CT, Horn D, Zabel B, Ekici AB, Salinas K, Gebhart E, Rüschendorf F, Sticht H, Spranger J, Müller D, Zweier C, Schmitt ME, Reis A, Rauch A. 2005. Severely incapacitating mutations in patients with extreme short stature identify RNA‐processing endoribonuclease RMRP as an essential cell growth regulator. Am J Hum Genet 77:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welting TJM, Van Venrooij WJ, Pruijn GJM. 2004. Mutual interactions between subunits of the human RNase MRP ribonucleoprotein complex. Nucleic Acids Res 32:2138–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. The GHR signaling pathway.
Table S1. Somatometric parameters.