

Abstract
Autism spectrum disorder (ASD) involves thousands of alleles in over 850 genes, but the current functional inference tools are not sufficient to predict phenotypic changes. As a result, the causal relationship of most of these genetic variants in the pathogenesis of ASD has not yet been demonstrated and an experimental method prioritizing missense alleles for further intensive analysis is crucial. For this purpose, we have designed a pipeline that uses Caenorhabditis elegans as a genetic model to screen for phenotype-changing missense alleles inferred from human ASD studies. We identified highly conserved human ASD-associated missense variants in their C. elegans orthologs, used a CRISPR/Cas9-mediated homology-directed knock-in strategy to generate missense mutants and analyzed their impact on behaviors and development via several broad-spectrum assays. All tested missense alleles were predicted to perturb protein function, but we found only 70% of them showed detectable phenotypic changes in morphology, locomotion or fecundity. Our findings indicate that certain missense variants in the C. elegans orthologs of human CACNA1D, CHD7, CHD8, CUL3, DLG4, GLRA2, NAA15, PTEN, SYNGAP1 and TPH2 impact neurodevelopment and movement functions, elevating these genes as candidates for future study into ASD. Our approach will help prioritize functionally important missense variants for detailed studies in vertebrate models and human cells.
Introduction
Many psychiatric disorders such as autism spectrum disorder (ASD, OMIM: 209850) have been linked to genetic variants that disrupt but do not necessarily eliminate protein functions. Missense variants in particular account for approximately half of the genetic changes known to cause disease (1), but most studies focus on identifying likely gene-disruptive mutations (e.g. nonsense, frameshift or splice-site) instead of missense variants. The severity of ASD is thought to be correlated with the average contribution of familial influences and de novo mutations (2); individuals with ASD are more likely to carry a de novo missense mutation (3). Missense mutations account for a large number of variants of uncertain significance, which are genomic variants that have an unclear effect on protein function and clinical significance due to inadequate or conflicting information (4,5). Given that some missense alleles have been validated, one challenge is to identify the subset of ASD-associated mutations that are deleterious.
Because missense variants are numerous, functional inference tools are widely used to predict the damaging effects of specific missense variants. Most current software relies heavily on sequence conservation to predict the potency of missense variants as conserved regions are considered more likely to be affected by purifying selection (6) but only 27% of missense mutations predicted by sequence conservation showed disrupted protein function in a recent rodent study (7). Given that any gene carries a certain chance of containing a missense mutation and every individual will have a different subset of missense mutations in their genome, computational analyses are insufficient for predicting the functional importance of such mutations (7). Additionally, interpretation of these data is inadequate due to variable penetrance, dosage sensitivity and functional redundancy of mutated proteins and can result in a high false-positive rate of prediction (1,8). On the other hand, variants that scored as neutral/benign may impact other physiological functions that were not expected (9). Therefore, the functional inference tools used to predict damaging effects are not accurate enough to be used as the sole basis for a conclusion, and a test of broad biological phenotypes is necessary to understand the nature of missense variants with uncertain significance.
Evaluation of missense variants in vivo is essential to accurately interpret available data as only 13% of identified de novo missense variants are suspected to contribute to the risk of ASD (3,10). Due to the large number of missense variants, an efficient pipeline is needed to evaluate the functional consequence of all residues in vivo. There have been only few studies conducted to validate the functional consequence of missense variants in vivo. Chen et al. (11) evaluated the disruptiveness of a mutation exclusively on its capacity to disrupt protein interactions using the yeast two-hybrid method. Another study by Miosge et al. (7) compared the deleterious effects predicted computationally to the actual N-ethyl-N-nitrosourea (ENU) induced mutant rodent models. Despite such exciting findings, a comprehensively targeted screen to test whether ASD-associated missense mutations are function-disrupting in a multi-cellular model organism has not yet been done. The short life cycle and easily accessible genome in Caenorhabditis elegans make it a useful tool to rapidly evaluate whether a particular disease-associated missense variant results in phenotypic consequences (12–14).
In this study, we established a pipeline for identifying ASD-associated protein-disrupting missense residues in the orthologous C. elegans proteins (Fig. 1A). First, the C. elegans residues corresponding to human missense variants were identified based on sequence conservation. The C. elegans equivalents of human missense mutants were generated using clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 and homology-directed genome editing (`knock-in’). We then analyzed the effects of these autism-associated missense alleles by comparing observable phenotypes from these missense mutants to the wild-type and known loss-of-function mutant controls. Missense mutants with phenotypic changes reflect alteration in protein function, indicating the importance of these alleles. We found that 19% of the ASD-associated missense variants are conserved in C. elegans. We evaluated the effects of 20 missense alleles that were predicted to be phenotype altering and found that only 70% of them displayed phenotypic changes in morphology, locomotion and fecundity. Our method demonstrates our ability to screen for subtle phenotypic changes and, in doing so, illustrates the functional importance of the effect of missense mutations on human disease.
Figure 1.
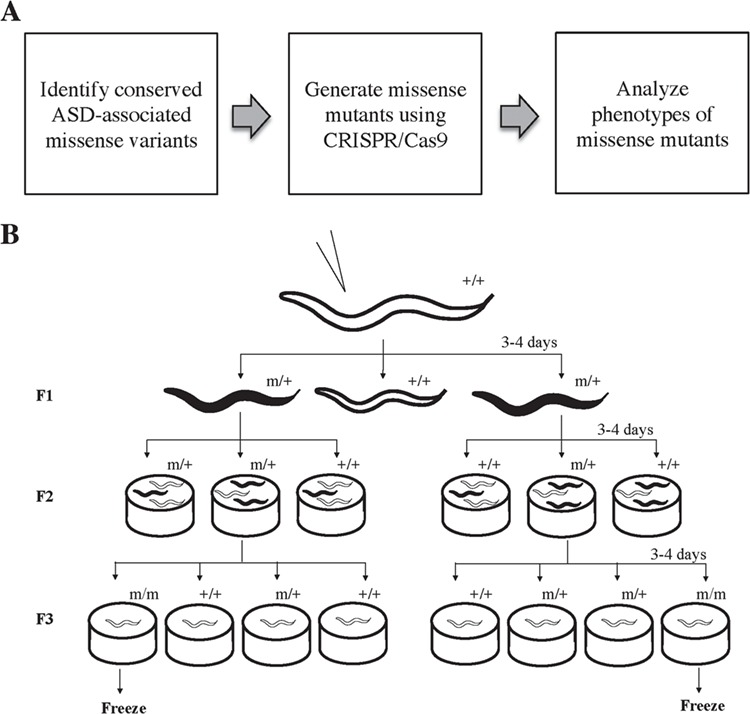
Generation of ASD-associated missense variants in C. elegans. (A) Experimental pipeline. First, we used bioinformatics to identify human ASD-associated missense variants conserved in C. elegans protein sequences. We generated the C. elegans missense mutants using CRISPR/Cas9 and homology-directed knock-in technique. We then analyzed the effects of these autism-associated alleles by comparing observable phenotypes from these alleles to both wild-type and known loss-of-function mutant controls. (B) Mutant strain screening process. Three days after injection (P0), F1 offspring expressing co-conversion marker, such as dumpy (black), were selected into individual plates. We genotyped the F2 offspring to identify the heterozygous plates. For each heterozygous plate containing the successful knock-in target missense residue (m), we randomly selected 16–20 F2 wild type-looking offspring (white) and separated them into individual plates (F3). A second round of genotyping process was then conducted to identify homozygous plates, which were cryo-preserved for future studies.
Results
Identifying C. elegans analogs of ASD-associated missense mutations
In order to identify the functionally important missense variants implicated in complex human diseases, we established a pipeline to screen for functional changes in orthologous proteins in C. elegans (Fig. 1A). Of the 1811 human ASD-associated missense variants from 423 human genes, 778 alleles (43%) from 221 human genes were identified in C. elegans orthologs. Most of the human genes were aligned to one C. elegans ortholog, but ~20% of the genes (47 of 221) had more than one orthologous protein in C. elegans (Fig. 2A). In some cases, human genes from the same family share the same C. elegans orthologous protein (e.g. both human CHD7 and CHD8 genes share the same C. elegans orthologs chd-7). Our goal was to identify each orthologous protein and corresponding equivalent residue based on sequence conservation. To achieve this, our software utilized comparative genomics and multiple alignments to ensure that the detected residue reflects conservation across the evolutionary tree and gene family (Fig. 2B). We found that 345 (19%) of missense loci from 157 human genes not only have orthologs in C. elegans but also had at least one conserved amino acid residue between human and C. elegans (Fig. 2A). Sometimes, one human residue could be matched to multiple orthologs in C. elegans (37 of the 345 conserved residues). For example, GLRA2 has multiple orthologous proteins in C. elegans, (glc-1, glc-2, glc-3, glc-4, avr-14 and avr-15). In these cases, we picked the worm residue candidate that had the sgRNA sequence most likely to produce an efficient CRISPR-Cas9 double strand break based on an online sgRNA prediction tool (15). For each allele, we identified the corresponding C. elegans ortholog, assessed the residues affected by missense mutations for evolutionary conservation and selected genes with a known phenotype for their loss-of-function mutation in C. elegans (from existing mutants or RNAi). To prioritize genes for functional screening, we focused on those genes with multiple missense variants as the chance of one causing a phenotypic defect increases when multiple missense mutations are observed in a single gene (16). We also prioritized genes involved in multiple biological pathways (17) or genes with other mutations resulting in a stop codon.
Figure 2.
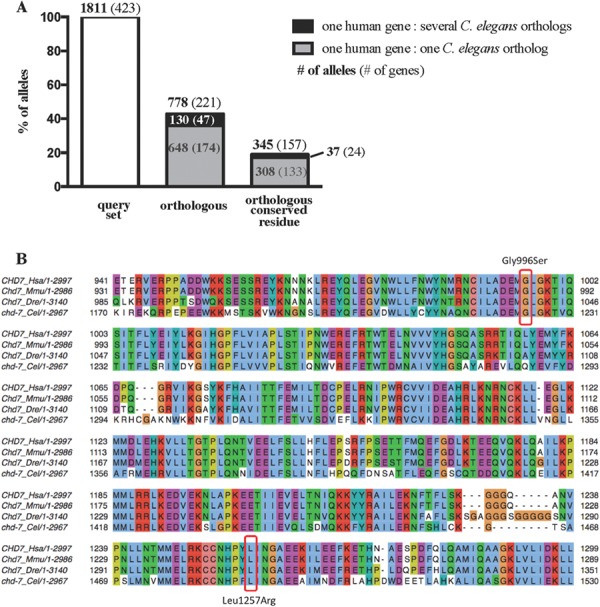
Detection of conserved ASD-associated missense residues in C. elegans. (A) For each missense allele, the C. elegans ortholog of the corresponding human gene was identified using the Ensembl Compara method. Each Ensembl ortholog pair was underpinned by a protein multiple-sequence alignment, which can be used to identify the putative orthologous C. elegans amino acid for a given human amino acid. Forty-three percent of the human missense variants had at least one C. elegans ortholog. A total of 130 of the 778 orthologous missense variants had more than one C. elegans ortholog (black bar). Overall, only 19% of the missense residues were orthologous and conserved in C. elegans. (B) Section of the protein multiple alignment for human (Hsa) CHD7 and its orthologs in mouse (Mmu), zebrafish (Dre) and C. elegans (Cel). Residues in the alignment have been colored by JalView (PMID: 19151095) using the Clustal X coloring scheme. Circled are two ASD-linked missense variants in CHD7 that occur in a highly conserved region across all the species.
To capture the impacts of missense mutations in diverse physiological functions, we sampled 20 ASD-associated missense changes in residues conserved in the C. elegans orthologs of 11 human genes (Table 1; Supplementary Material, Fig. S1). These ASD-associated missense mutations were identified in genes that were known to have a role in synaptic function (i.e. DLG4, SYNGAP1, CACNA1D and GLRA2), gene expression regulation (i.e. CHD7, CHD8 and CUL3) or neuronal signaling and cytoskeleton functions (i.e. PTEN, MAPK3, TPH2 and NAA15). Multiple aspects of physiological functions were examined, including morphology, locomotion and fecundity. These well-established quantitative assays enabled us to detect subtle changes in morphology, movement and coordination, as well as reproduction and completion of embryonic development (14,18).
Table 1.
Strain information
| Human gene | Human cDNA changea | Human protein change | Inheritance pattern | C. elegans gene (allele) | C. elegans protein change | Strain name |
|---|---|---|---|---|---|---|
| CACNA1D | c.1105G>A | V369M | Unknown | egl-19(sy849) | V331M | PS7085 |
| CACNA1D | c.1112A>C | Y371S | Unknown | egl-19(sy850) | Y333S | PS7156 |
| CHD7 | c.2986G>A | G996S | De novo | chd-7(sy861) | G1225S | PS7293 |
| CHD7 | c.3770T>G | L1257R | De novo | chd-7(sy855) | L1487R | PS7317 |
| CHD8 | c.2501T>C | L834P | De novo | chd-7(sy859) | L1220P | PS7318 |
| CHD8 | c.494C>T | P165L | Unknown | chd-7(sy1049) | P253L | PS7267 |
| CUL3 | c.2156A>G | H719R | De novo | cul-3(sy874) | H728R | PS7387 |
| DLG4 | c.2281G>A | V761I | Unknown | dlg-1(sy872) | V964I | PS7343 |
| GLRA2 | c.407A>G | N136S | De novo | avr-15(sy873) | N347S | PS7384 |
| GLRA2 | c.458G>A | R153Q | De novo | avr-15(sy851) | R364Q | PS7257 |
| MAPK3 | c.833G>A | R278Q | De novo | mpk-1(sy870) | R332Q | PS7382 |
| NAA15 | c.1319T>C | L440S | Familial | hpo-29(sy877) | L575S | PS7394 |
| PTEN | c.66C>G | D22E | Familial | daf-18(sy879) | D66E | PS7439 |
| PTEN | c.208C>G | L70V | Unknown | daf-18(sy887) | L115V | PS7432 |
| PTEN | c.278A>G | H93R | De novo | daf-18(sy881) | H138R | PS7436 |
| PTEN | c.369C>G | H123Q | Unknown | daf-18(sy885) | H168Q | PS7430 |
| PTEN | c.392C>T | T131I | De novo | daf-18(sy882) | T176I | PS7434 |
| SYNGAP1 | c.698G>A | C233Y | De novo | gap-2 (sy889) | C417Y | PS7433 |
| SYNGAP1 | c.1288C>T | L430F | Familial | gap-2(sy886) | L660F | PS7457 |
| TPH2 | c.674G>A | R225Q | Familial | tph-1(sy878) | R259Q | PS7395 |
aThe virtual cDNA was provided by the SFARI database.
Morphology of missense mutants
To examine changes in morphology, we utilized a quantitative tracking system to measure the length, width and body area of these missense mutants under freely moving condition. Alterations in size were detected in avr-15/GLR2, chd-7/CHD7 or CHD8; cul-3/CUL3; daf-18/PTEN; gap-2/SYNGAP1; egl-19/CACNA1D; hpo-29/NAA15; and tph-1/TPH2 (Table 2). Every chd-7 mutant tested showed a significant decrease in body width and area. A null mutant, chd-7(sy956), displayed the most severe defects. Other missense alleles, chd-7(L1220P), chd-7(L1487R), chd-7(G1225S) and chd-7(P253L), showed milder degree of defects. One of the egl-19 missense mutants, egl-19(Y333S), displayed a smaller decrease in body length, width and areas compared to the semidominant allele, egl-19(n2368) (19). Another egl-19 mutant, egl-19(V331M), showed a similar body size as the N2 wild-type strain. Similarly, the tph-1(R259Q) mutant showed a decrease in body length and area, and the change was milder in the missense mutant as compared to the null mutant tph-1(mg280) (20). One missense mutation in avr-15, avr-15(R364Q), caused a decrease in body length, width and area, similar to its null mutant, avr-15(ad1051) (21). Another avr-15 missense mutant, avr-15(N347S), showed no morphological changes. Missense mutant hpo-29(L575S) exhibited shorter body length. Missense mutants cul-3(H728R), daf-18(H168Q) and gap-2(C417Y) displayed increased body width and area. Missense mutants of dlg-1/DLG4 and mpk-1/MAPK3 did not show morphological changes.
Table 2.
Morphological phenotypes of missense alleles and their controls
| Gene | Length (μm) | Width (μm) | Area (μm2) |
|---|---|---|---|
| N2 | 1105 ± 5 | 87.8 ± 0.7 | 98 798 ± 1147 |
| avr-15(N347S) | 1101 ± 7 | 87.9 ± 1.5 | 98 214 ± 1273 |
| avr-15(R364Q) | 989 ± 7a | 77.3 ± 0.9a | 77 860 ± 1457a |
| avr-15(ad1051) | 1033 ± 10a | 78.5 ± 0.8a | 82 468 ± 1410a |
| chd-7(P253L) | 1110 ± 9 | 78.9 ± 0.9a | 86 572 ± 1815a |
| chd-7(L1220P) | 978 ± 10a | 76.2 ± 1.0a | 75 909 ± 1432a |
| chd-7(G1225S) | 1038 ± 11a | 80.8 ± 1.3a | 85 400 ± 1977a |
| chd-7(L1487R) | 1033 ± 13a | 81.7 ± 1.3a | 86 060 ± 2277a |
| chd-7(sy956) | 954 ± 6a | 75.4 ± 0.9a | 73 201 ± 1254a |
| cul-3(H728R) | 1111 ± 9 | 96.1 ± 2.8a | 108 771 ± 3865a |
| daf-18(D66E) | 1125 ± 14 | 91.5 ± 1.1 | 104 804 ± 2496 |
| daf-18(L115V) | 1128 ± 9 | 94.2 ± 1.1 | 108 208 ± 1902 |
| daf-18(H138R) | 1130 ± 6 | 94.6 ± 1.2 | 108 706 ± 1366 |
| daf-18(H168Q) | 1148 ± 6 | 103.3 ± 2.7a | 120 936 ± 3535a |
| daf-18(T176I) | 1135 ± 9 | 93.1 ± 1.4 | 107 395 ± 1711 |
| dlg-1(V964I) | 1129 ± 4 | 83.2 ± 0.9 | 95 502 ± 1210 |
| egl-19(V331M) | 1082 ± 4 | 83.1 ± 1.1 | 91 504 ± 1448 |
| egl-19(Y333S) | 1052 ± 8a | 81.2 ± 0.7a | 86 896 ± 1310a |
| egl-19(n2368sd) | 639 ± 10a | 68.1 ± 0.8a | 44 402 ± 1104a |
| gap-2(C417Y) | 1128 ± 14 | 98.7 ± 2.6a | 113 351 ± 3713a |
| gap-2(L660F) | 1120 ± 13 | 89.0 ± 1.7 | 101 634 ± 3020 |
| hpo-29(L575S) | 1044 ± 12a | 94.0 ± 2.4 | 99 849 ± 3111 |
| mpk-1(R332Q) | 1145 ± 7 | 88.0 ± 1.7 | 10 4188 ± 1889 |
| tph-1(R259Q) | 1026 ± 9a | 84.5 ± 1.1 | 88 238 ± 1151a |
| tph-1(mg280) | 993 ± 20a | 80.9 ± 1.3a | 81 918 ± 2765a |
a P < 0.01 via one-way analysis of variance and multiple comparison. All values are presented as mean ± SEM.
Movement and coordination of missense mutants
To examine movement and coordination in these missense mutants, a quantitative tracking system was used to measure moving speed, reversal rate and sinusoidal wavelength and amplitude. Locomotion defects were found in missense mutants of chd-7/CHD7 or CHD8, daf-18/PTEN, gap-2/GLRA2 and hpo-29/NAA15 (Fig. 3; Supplementary Material, Table S2). Less severe than null mutant, all missense mutants in chd-7, except chd-7(P253L), exhibited decreased speed. Missense mutant hpo-29(L575S) also showed a significant decrease in speed. In terms of reversal rate, most chd-7 mutants, except chd-7(P253L), showed a significant reduction in turns per minute. Missense mutants daf-18(H138R) and gap-2(C417Y) displayed an increased reversal rate. Missense mutations in avr-15/GLRA2, cul-3/CUL3, dlg-1/DLG4, egl-19/CACNA1D, mpk-1/MAPK3 and tph-1/TPH2 did not result in differences in speed and reversal rate.
Figure 3.
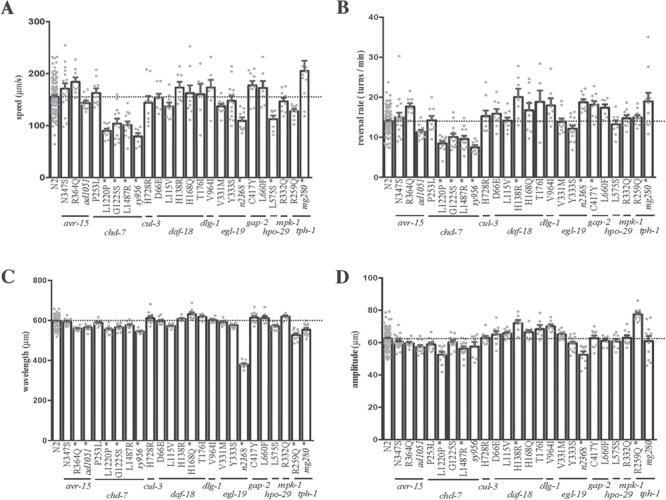
Locomotion phenotypes of missense alleles and their controls. (A) Speed and (B) reversal rate are measurement of locomotion while (C) wavelength and (D) amplitude represent sinusoidal shape of movement. avr-15(R364Q) mutant showed decreased wavelength. Most chd-7 missense mutants displayed a weaker version of the null mutant phenotypes in all measurements. daf-18(H138R) and daf-18(H168Q) exhibited larger sinusoidal wavelength and amplitude. gap-2(C417Y) showed higher reversal rate. hpo-29(L575S) displayed slower speed. tph-1(R259Q) exhibited changes in sinusoidal movement. Bars were presented as the mean ± SEM. Each dot represented the average of one plate containing 8–10 worms. *P < 0.01 via one-way analysis of variance and multiple comparison to wild type. Horizontal line indicated the mean of wild type.
Locomotion in C. elegans is typically expressed as the wavelength and amplitude of a sinusoidal wave (22). Motor coordination defects have been associated with ASD (23) and were found in missense mutants of avr-15/GLRA2, chd-7/CHD7 or CHD8, daf-18/PTEN and tph-1/TPH2 (Fig. 3; Supplementary Material, Table S2). The tph-1(R259Q) mutant exhibited significantly lower wavelength and higher amplitude, indicating a curvier sinusoidal wave similar to but less severely than its null mutant (Supplementary Material, Fig. S2). One of the avr-15 missense mutants, avr-15(R364Q), showed a decrease in wavelength, slightly milder than the null mutant, avr-15(ad1051) (21). Another avr-15 mutant, avr-15(N347S), showed normal sinusoidal shape. All missense mutants in chd-7, except chd-7(P253L), displayed a decreased wavelength and/or amplitude. The mutation in chd-7(L1220P) resulted in a decrease in both wavelength and amplitude. Mutations in chd-7(G1225S) and chd-7(L1487R) led to a decrease in wavelength and amplitude, respectively. A null mutant of chd-7 also displayed a decrease wavelength. Two of the daf-18 missense mutants, daf-18(H138R) and daf-18(H168Q), exhibited an increase in amplitude and wavelength, respectively. Missense mutations in cul-3/CUL3, dlg-1/DLG4, egl-19/CACNA1D, gap-2/SYNGAP1, hpo-29/NAA15 and mpk-1/MAPK3 did not lead to differences in the sinusoidal wave.
Fecundity of missense mutants
We used the fecundity assay to examine larvae viability in genes with reported sterile or lethal phenotypes in null mutants. Fecundity defects were found in missense mutants of chd-7/CHD7 or CHD8, cul-3/CUL3 and dlg-1/DLG4 (Fig. 4). Three of four missense mutations in the chromatin modifier gene chd-7 displayed a reduced fecundity phenotype compared to the wild-type control strain N2. Specifically, the chd-7(L1220P) allele had a median fecundity of 119 (P < 10−6); chd-7(G1225S) had a median fecundity of 176 (P = 1.2 × 10−5); chd-7(L1487R) had a median fecundity of 168 (P = 4.4 × 10−5); and chd-7(P253L) had a median fecundity of 254.5 compared to a median fecundity of 228 for N2 control. These missense alleles showed weaker fecundity defects compared to its deletion (chd-7(tm6139)) or frameshift (chd-7(sy956)) controls, which had median fecundity of 38 and 47, respectively (P < 10−6). Missense variants in the DNA replication gene, cul-3(H728R), also displayed a decreased fecundity of 168.5 (P = 10−6). In addition, the missense variant dlg-1(V964I) showed a reduced median fecundity of 154.5 (P < 10−6), which is slightly less severe than the 67% reduction in a previous RNAi study (24). We did not observe changes in fecundity in missense mutants in other genes, namely avr-15/GLRA2, daf-18/PTEN, egl-19/CACNA1D, gap-2/SYNGAP1, hpo-29/NAA15, mpk-1/MAPK3 and tph-1/TPH2.
Figure 4.
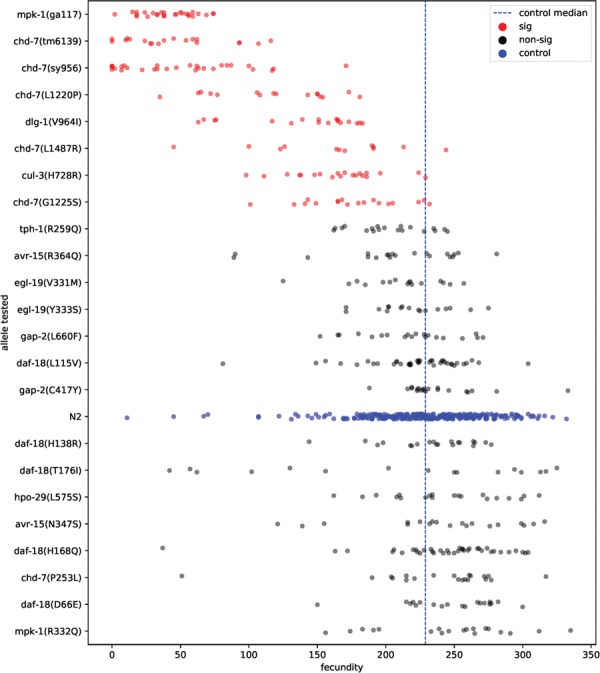
Fecundity phenotype of missense alleles and their controls. Most chd-7 mutants displayed decreased fecundity. The defects were more subtle than its deletion (tm6139) or frameshift (sy956) null controls. cul-3(H728R) and dlg-1(V964I) also showed decrease in fecundity. Each dot represented total number of living larvae from one animal. Approximately 20 animals were tested in each strain. Wild type (N2) and its median values (dotted vertical line) were shown in blue. Mutants significantly different (sig) from wild type were shown in red. P < 0.01/(total test number) via non-parametric bootstrap analysis.
Comparison with phenotype-predicting software
To examine the accuracy of our biological platform, we compared our results to the existing prediction software Sorting Intolerant From Tolerant (SIFT) and Polymorphism Phenotyping v.2 (PolyPhen-2) (Table 3). SIFT emphasizes sequence conservation and the physical properties of amino acids (25) whereas PolyPhen-2 considers both the analysis of multiple sequence alignments and protein 3D structures (26). Both software programs are commonly used to predict the effects of non-synonymous amino acid changes. For SIFT, all the alleles tested were predicted to be damaging due to having a similar approach of analyzing sequence conservation as our software. Our phenotypic assays identified six residues (among the 20 predictions) that did not align with the prediction. As compared to PolyPhen-2’s prediction, 35% (7/20) of the phenotypic results do not agree with the predictions. Among the seven strains that did not match, five were predicted to have damaging effects but had no phenotypic change in our functional assays (false positive), and two were predicted to be benign but displayed phenotypic changes (false negative). Overall, our results demonstrated that 70% (14 of 20) missense alleles predicted to be damaging by at least one functional inference tool actually showed detectable phenotypic changes in morphology, locomotion, and fecundity.
Table 3.
Comparison of behavioral results to software prediction
| Human gene | C. elegans gene | PolyPhen-2a | (1-SIFT)a | Phenotype |
|---|---|---|---|---|
| CACNA1D(V369M) | egl-19(V331M) | 0.995 | 0.99 | No |
| CACNA1D(Y371S) | egl-19(Y333S) | 1 | 1 | Morphology changes |
| CHD7(G996S) | chd-7(G1225S) | 0.998 | 1 | Morphology changes, locomotion variants and reduced fecundity |
| CHD7(L1257R) | chd-7(L1487R) | 1 | 1 | Morphology changes, locomotion variants and reduced fecundity |
| CHD8(L834P) | chd-7(L1220P) | 1 | 1 | Morphology changes, locomotion variants and reduced fecundity |
| CHD8(P165L) | chd-7(P253L) | 0.996 | 0.86 | Morphology changes |
| CUL3(H719R) | cul-3(H728R) | 1 | 0.96 | Morphology changes and reduced fecundity |
| DLG4(V761I) | dlg-1(V964I) | 0.001 | 0.84 | Reduced fecundity |
| GLRA2(N136S) | avr-15(N347S) | 0.979 | 1 | Locomotion variants |
| GLRA2(R153Q) | avr-15(R364Q) | 0.997 | 1 | Locomotion variants |
| MAPK3(R278Q) | mpk-1(R332Q) | 0.997 | 1 | No |
| NAA15(L440S) | hpo-29(L575S) | 0.999 | 0.96 | Morphology changes and locomotion variants |
| PTEN(D22E) | daf-18(D66E) | 0.297 | 0.93 | No |
| PTEN(L70V) | daf-18(L115 V) | 0.999 | 1 | No |
| PTEN(H93R) | daf-18(H138R) | 1 | 0.97 | Locomotion variants |
| PTEN(H123Q) | daf-18(H168Q) | 1 | 1 | Morphology changes and locomotion variants |
| PTEN(T131I) | daf-18(T176I) | 1 | 0.82 | No |
| SYNGAP1(C233Y) | gap-2(C417Y) | 0.940 | 1 | No |
| SYNGAP1(L430F) | gap-2(L660F) | 1 | 1 | Morphology changes and locomotion variants |
| TPH2(R225Q) | tph-1(R259Q) | 0.162 | 0.92 | Morphology changes and locomotion variants |
aPolyPhen-2 and SIFT prediction scores were based on human sequence (1 = probably damaging).
Discussion
In this study, we have developed a fast and tractable pipeline to comprehensively screen for ASD-associated missense mutations. Our analysis finds that 43% of the human disease-associated alleles have an ortholog in the genome of C. elegans, which is consistent with previous estimates (27). Among the 19% conserved loci, we evaluated 20 missense alleles that were predicted to be damaging and found 70% of them actually cause detectable phenotypic changes. We have successfully prioritized 14 missense variants that are functionally significant in C. elegans orthologs of human genes. These are the first animal models with deliberately engineered missense mutations in these loci. Our approach is useful for characterizing novel missense alleles that are potentially relevant to human disease and be used as a tool to identify functionally consequential alleles.
Compared to null mutants, most of the phenotypically altered missense alleles displayed milder phenotypes, indicating that our assays can detect relatively subtle changes in protein functions. For example, the chd-7 missense mutants and tph-1(R259Q) displayed hypomorphic phenotypes less severe than their null mutants (28). The cul-3(H728R) and dlg-1(V964I) missense mutants displayed a smaller reduction in fecundity compared to previous RNAi studies (24,29,30). avr-15(R364Q) showed defects in morphology and locomotion similar to its null mutant, avr-15(ad1051), even though it did not recapitulate the spontaneous reversal rate defect documented in an RNAi study (31). The functional consequences of missense alleles can vary in different assays. For instance, the egl-19(Y333S) showed milder morphological changes similar to its null mutant (28) but displayed normal functions in locomotion and fecundity. Missense mutants hpo-29(L575S) and gap-2(C417Y) displayed defects in morphology and locomotion, but they did not show the fecundity defects reported in RNAi studies (30,32). The daf-18(H138R) and daf-18(H168Q) missense mutants showed defects in morphology and locomotion, which were not documented before, suggesting a role for our biological screening platforms to detect subtle phenotypic changes in different physiological functions.
As pointed out in the previous literature, computational inference tends to have a higher false-positive rate of identifying protein function-disrupting missense alleles (1). This study demonstrated that predictions based solely on sequence conservation did not effectively distinguish missense mutations that cause phenotypic changes from ones that exhibit no observable phenotype. Only 70% of our behavioral results agreed with the predictions from two commonly used computational programs, PolyPhen-2 and SIFT. Most of the discrepancies are false positive predictions. Absence of a phenotype in vivo may occur due to genetic redundancy and robust gene networks compensating for the inhibition of a single component, especially in tightly regulated cellular networks involving in signaling, metabolic and transcriptional pathways (33), or we simply did not observe every possible phenotype. More pointedly, our study showed that two missense alleles, predicted by PolyPhen-2 as benign, presented phenotypes. The fecundity defect found in dlg-1(V964I) can be recapitulated by RNAi whereas the tph-1(R259Q) displayed hypomorphic phenotypes similar to its null mutant (24). The false negatives predicted by the software indicate a void in current prediction algorithms, suggesting a need for a screening platform in a multicellular model organism such as our own. Our in vivo screening platform not only selects for genes that display sequence conservation across evolution but also reflects the complex nature in biological system, such as redundancy and compensation. Our phenotypic results can also provide feedback to improve the accuracy of prediction algorithm.
In contrast with previous studies on the phenotypic consequences of missense mutations, our platform examines gene functions in its endogenous multicellular context. Compared to a previous study using yeast two-hybrid to verify the effects of missense mutations in protein interaction experimentally and computationally (11), our strategy captures the overall readout of mutation effects and intercellular interaction. Furthermore, our use of endogenous proteins allows all other molecular interactions to remain intact and thus avoids potential confounding factors, such as intron disruption and isoform imbalance (34,35). As a result, using CRISPR to knock-in a DNA missense template is more efficient and may more accurately reflect the consequence of a variant than does a `humanized' model organism (36–38). Our high-throughput screening strategy occupies an unusual niche in primary screening for the consequence of missense mutations in vivo.
Using C. elegans as a model for psychiatric disorders has some limitations, including a lack of highly complex behaviors and some neurotransmitter systems (e.g. norepinephrine). However, C. elegans and humans share essential physiological pathways (e.g. insulin signaling, Ras/Notch signaling, p53 and many miRNAs), neurotransmitter systems and receptor pharmacology (14,27). The transparency and easy access genetic tools make C. elegans a powerful model for dissecting the mechanisms of pathological conditions and drug target identification. The short generation time of C. elegans enable high-throughput screening for numerous targets (such as missense variants) before embarking on less efficient and more costly animal models (27). In addition, with the tissue-specific promoters and conditional knockout techniques available in C. elegans, it is possible to decipher the effects of these disease-associated missense mutations spatially and temporally (39–41). For genetic candidates that show correlated expression, our platform also can be used to investigate the interaction between missense variants by generating double/multiple missense mutations model.
The discovery of novel genetic variants associated with human diseases has accelerated due to technical improvements and decreasing costs of next-generation sequencing. However, it is difficult to assess the impact of single missense mutations due to the complexity of human genetic backgrounds. One solution is to test variants in a model organism with an isogenic background to quickly identify variants producing changes in protein function. Here, we developed an experimental pipeline to investigate the functional consequences of ASD-associated missense variants in C. elegans. Our approach will help prioritize consequential missense variants for detailed studies in vertebrate models or human cells. This pipeline will serve as a stepping stone for defining molecular mechanisms in complex human diseases such as ASD.
Materials and Methods
Mapping locations of human residues to the C. elegans genome
ASD-associated missense variants were obtained from the SFARI Gene–Human Gene Module (42) (Supplementary Material, Table S1). We used the comparative genomics resources provided by Ensembl (release 90), which integrates in-house annotation for nearly 100 vertebrate genomes (e.g. human, mouse and zebrafish) with reference annotation for selected invertebrate model organisms (e.g. C. elegans, with genome and annotation provided by WormBase). Ensembl provides a protein multiple alignment and evolutionary trees for each gene family and asserts orthology and paralogy relationships between pairs of genes (43). These data were organized with a custom automated pipeline (44): for a given human genome coordinate, (a) identify which human protein-coding gene (if any) coincided with the provided coordinate; (b) obtain the amino acid coordinates in that protein; (c) check if the human gene has a C. elegans ortholog; (d) if so, use the multiple alignment associated with the orthology assertion to identify the orthologous amino acid in the C. elegans protein; and (e) from the protein coordinates, obtain the corresponding position in the C. elegans reference genome.
Strains
The Bristol N2 C. elegans strain was used as the wild-type control and background for all CRISPR experiments (13). The control strains for functional assays were obtained from laboratory stock, the Caenorhabditis Genetics Center (CGC) and the National BioResource Project—C. elegans (NBRP). Loss-of-function mutant controls were JD105 avr-15(ad1051) (21), FX17094 chd-7(tm6139), PS3071 egl-19(n2368sd) (19), SD464 mpk-1(ga117) (45) and PS3156 tph-1(mg280) (20). All strains were maintained on nematode growth medium (NGM) agar plates seeded with Escherichia coli OP50 at room temperature (20°C–22°C).
Generation of missense mutant strains
The Cas9 protein-based CRISPR knock-in protocol was adapted from Paix et al. (46). The sgRNA sequences were selected using the C. elegans CRISPR guide RNA tool (15). Single-stranded donor oligonucleotides contained 35 bp of flanking homology on both sides of the mutated region. An online tool for restriction analysis, WatCut, was used to assist in designing restriction sites that did not affect protein sequence. The crRNA, tracrRNA and donor oligonucleotides were commercially synthesized and dissolved in Nuclease Free Duplex Buffer (Integrated DNA Technologies Inc., Coralville, IA). Purified Cas9 protein was a kind gift from Dr Tsui-Fen Chou (LA BioMed). gRNA duplexes were generated by mixing crRNA and tracrRNA at 1:1 ratio and incubating at 94°C for 2 min. The Cas9 protein (25 μm final concentration) and gRNA duplex (27 μm final concentration) were mixed and incubated at room temperature for 5 min before adding donor oligonucleotides (0.6 μm final concentration). To facilitate screening, dpy-10(cn64) or unc-58(e665) was used as a co-conversion marker and made up part of the crRNA and donor oligo used (47) (Fig. 1B). A crRNA ratio [marker: target gene] of 1:4 and 2:3 were used for dpy-10 and unc-58, respectively. A donor ratio [marker: target gene] of 1:2 was used for both dpy-10 and unc-58.
The F1 offspring displaying the co-conversion phenotype were genotyped as follows: about 5 worms were picked into 10 μl lysis buffer (10 mm Tris, 50 mm KCl, 2 mm MgCl2, pH 8.0) with proteinase K (500 ng/ml; Invitrogen, Carlsbad, CA) and incubated at 65°C for an hour to extract genomic DNA. The genomic prep was amplified in a PCR reaction and then treated with a restriction enzyme (NEB, Ipswich, MA) to check the presence of targeted missense mutation. Mutants with the correct length were confirmed by sequencing (Laragen, Culver City, CA). When available, we saved two independent missense mutant lines. While there are little to no off-targets effects of Cas9 (48), C. elegans N2 suffers approximately one mutation per generation so it was useful to have more than one strain for each locus.
Fecundity assay
Well-fed C. elegans were synchronized at the L4 stage. Individual L4 hermaphrodites were placed on separate NGM plates seeded with OP50 and these animals were subsequently transferred to a new plate every day. The number of newly hatched larvae progeny was counted for every plate 1 day after the adult was transferred. The total fecundity consisted of the sum of progeny produced for 3 days per animal.
Locomotion tracking
Well-fed L4 hermaphrodites were picked at ~16 h before the experiment to provide synchronized young adults. On the day of the experiment, eight young adults were picked onto NGM plates freshly seeded with a 50 μl drop of a saturation-phase culture of OP50. The worms were given 30 min for habituation and then tracked for 4 min. Strains were tracked between 1 p.m. and 6 p.m. across several days. WormLab (MBF Bioscience, Williston, VT) equipment and software were used for tracking and analyses. The camera was a Nikon AF Micro 60/2.8D with zoom magnification. A 2456 × 2052 resolution, 7.5 fps camera with a magnification that results in 8.2 μm per pixel and an FOV of roughly 2 × 2 cm2 were used. Approximately 8–10 plates were tracked per experimental strain. The mean of each plate was first calculated and then the total mean of all plates of the same genotype was computed.
Statistical analysis
The fecundity assay was analyzed using a non-parametric bootstrap analysis (D. Angeles-Albores & P.W. Sternberg, unpublished). Initially, the two datasets were mixed, samples were selected at random with replacement from the mixed population into two new datasets and then the difference in the averages of these new datasets were calculated; this process was iterated 106 times. We reported the P-value as the probability when the difference in the average of simulated datasets was greater than the difference in the average of the original datasets. If P < 0.01/(total testing number), we rejected the null hypothesis that the average values of the two datasets were not equal to each other. Morphology and locomotion were analyzed by one-way analysis of variance using GraphPad Prism version 6 (GraphPad, La Jolla, CA). Dunnett multiple comparisons were performed between wild-type and mutant strains. The significant level was defined as P < 0.01.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank WormBase for genome information. The authors thank CGC (funded by National Institute of Health (NIH) Office of Research Infrastructure Programs, P40 OD010440) and NBRP for providing strains. They also thank Tsui-Fen Chou for providing the Cas9 protein. The authors thank Shahla Gharib for the assistance in strain generation. The authors thank David Angeles-Albores for sharing his data analysis software. They also thank Hillel Schwartz and Han Wang for comments on manuscript.
Conflict of Interest statement. None declared.
Web resources
SFARI Gene–Human Gene Module, https://gene.sfari.org/database/human-gene/
Ensembl, www.ensembl.org
WormBase, www.wormbase.org
C. elegans CRISPR guide RNA tool, http://genome.sfu.ca/crispr/
WatCut, http://watcut.uwaterloo.ca
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Funding
This work was supported by Simons Foundation (SFARI award # 367560 to P.W.S.). K.B. was supported by NIH pre-doctoral training grant T32GM007616. P.W.S. was an investigator with the Howard Hughes Medical Institute during part of this study.
References
- 1. Andrews T.D., Sjollema G. and Goodnow C.C. (2013) Understanding the immunological impact of the human mutation explosion. Trends Immunol., 34, 99–106. [DOI] [PubMed] [Google Scholar]
- 2. Robinson E.B., Samocha K.E., Kosmicki J.A., McGrath L., Neale B.M., Perlis R.H. and Daly M.J. (2014) Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl. Acad. Sci. U. S. A., 111, 15161–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E. et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Han P.K.J. (2013) Conceptual, methodological, and ethical problems in communicating uncertainty in clinical evidence. Med. Care Res. Rev., 70, 14S–36S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petrucelli N., Lazebnik N., Huelsman K.M. and Lazebnik R.S. (2002) Clinical interpretation and recommendations for patients with a variant of uncertain significance in BRCA1 or BRCA2: a survey of genetic counseling practice. Genet. Test., 6, 107–113. [DOI] [PubMed] [Google Scholar]
- 6. Alfoldi J. and Lindblad-toh K. (2013) Comparative genomics as a tool to understand evolution and disease. Genome Res., 23, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miosge L.A., Field M.A., Sontani Y., Cho V., Johnson S., Palkova A., Balakishnan B., Liang R., Zhang Y., Lyon S. et al. (2015) Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. U. S. A., 112, E5189–E5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G. et al. (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science, 337, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Billack B. and Monteiro A.N.A. (2004) Methods to classify BRCA1 variants of uncertain clinical significance: the more the merrier. Cancer Biol. Ther., 3, 458–459. [DOI] [PubMed] [Google Scholar]
- 10. Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron, 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen S., Fragoza R., Klei L., Liu Y., Wang J., Roeder K., Devlin B. and Yu H. (2018) An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat. Genet., 50, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim S., Twigg S.R.F., Scanlon V.A., Chandra A., Hansen T.J., Alsubait A., Fenwick A.L., McGowan S.J., Lord H., Lester T. et al. (2017) Localized TWIST1 and TWIST2 basic domain substitutions cause four distinct human diseases that can be modeled in Caenorhabditis elegans. Hum. Mol. Genet., 26, 2118–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Engleman E.A., Katner S.N. and Neal-beliveau B.S. (2016) Caenorhabditis elegans as a model to study the molecular and genetic mechanisms of drug addiction. Prog. Mol. Biol. Transl. Sci., 137, 229–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Au V., Li-Leger E., Raymant G., Flibotte S., Chen G., Martin K., Fernando L., Doell C., Rosell F.I., Wang S. et al. (2019) CRISPR/Cas9 Methodology for the Generation of Knockout Deletions in Caenorhabditis elegans. G3, 9, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geisheker M.R., Heymann G., Wang T., Coe B.P., Turner T.N., Stessman H.A.F., Hoekzema K., Kvarnung M., Shaw M., Friend K. et al. (2017) Hotspots of missense mutation identify novel neurodevelopmental disorder genes and functional domains. Nat. Neurosci., 20, 1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krumm N., O’Roak B.J., Shendure J. and Eichler E.E. (2014) A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci., 37, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Bono M. and Villu Maricq A. (2005) Neuronal substrates of complex behaviors in C. elegans. Annu. Rev. Neurosci., 28, 451–501. [DOI] [PubMed] [Google Scholar]
- 19. Lee R.Y., Lobel L., Hengartner M., Horvitz H.R. and Avery L. (1997) Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J., 16, 6066–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sze J.Y., Victor M., Loer C., Shi Y. and Ruvkun G. (2000) Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature, 403, 560–564. [DOI] [PubMed] [Google Scholar]
- 21. Dent J.A., Davis M.W. and Avery L. (1997) avr-15 encodes a chloride channel subunit that mediates inhibitory glutamatergic neurotransmission and ivermectin sensitivity in Caenorhabditis elegans. EMBO J., 16, 5867–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cronin C.J., Mendel J.E., Mukhtar S., Kim Y.M., Stirbl R.C., Bruck J. and Sternberg P.W. (2005) An automated system for measuring parameters of nematode sinusoidal movement. BMC Genet., 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fournier K.A., Hass C.J., Naik S.K., Lodha N. and Cauraugh J.H. (2010) Motor coordination in autism spectrum disorders: a synthesis and meta-analysis. J. Autism Dev. Disord., 40, 1227–1240. [DOI] [PubMed] [Google Scholar]
- 24. Pilipiuk J., Lefebvre C., Wiesenfahrt T., Legouis R. and Bossinger O. (2009) Increased IP3/Ca2+ signaling compensates depletion of LET-413/DLG-1 in C. elegans epithelial junction assembly. Dev. Biol., 327, 34–47. [DOI] [PubMed] [Google Scholar]
- 25. Ng P.C. and Henikoff S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S. and Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Markaki M. and Tavernarakis N. (2010) Modeling human diseases in Caenorhabditis elegans. Biotechnol. J., 5, 1261–1276. [DOI] [PubMed] [Google Scholar]
- 28. Yemini E., Jucikas T., Grundy L.J., Brown A.E.X. and Schafer W.R. (2013) A database of Caenorhabditis elegans behavioral phenotypes. Nat. Methods, 10, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sonnichsen B., Koski L.B., Walsh A., Marschall P., Neumann B., Brehm M., Alleaume A.-M.M., Artelt J., Bettencourt P., Cassin E. et al. (2005) Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature, 434, 462–469. [DOI] [PubMed] [Google Scholar]
- 30. Maeda I., Kohara Y., Yamamoto M. and Sugimoto A. (2001) Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol., 11, 171–176. [DOI] [PubMed] [Google Scholar]
- 31. Cook A., Aptel N., Portillo V., Siney E., Sihota R., Holden-Dye L. and Wolstenholme A. (2006) Caenorhabditis elegans ivermectin receptors regulate locomotor behaviour and are functional orthologues of Haemonchus contortus receptors. Mol. Biochem. Parasitol., 147, 118–125. [DOI] [PubMed] [Google Scholar]
- 32. Rual J.-F., Ceron J., Koreth J., Hao T., Nicot A., Hirozane-kishikawa T., Vandenhaute J., Orkin S.H., Hill D.E. and Vidal M. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res., 14, 2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. El-Brolosy M.A. and Stainier D.Y.R. (2017) Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet., 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reble E., Dineen A. and Barr C.L. (2018) The contribution of alternative splicing to genetic risk for psychiatric disorders. Genes Brain Behav., 17, 1–12. [DOI] [PubMed] [Google Scholar]
- 35. Robison A.J. (2014) Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci., 37, 653–662. [DOI] [PubMed] [Google Scholar]
- 36. McDiarmid T.A., Au V., Loewen A.D., Liang J., Mizumoto K., Moerman D.G. and Rankin C.H. (2018) CRISPR-Cas9 human gene replacement and phenomic characterization in Caenorhabditis elegans to understand the functional conservation of human genes and decipher variants of uncertain significance. Dis. Model. Mech., 10.1242/dmm.036517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baruah P.S., Beauchemin M., Parker J.A. and Bertrand R. (2017) Expression of human Bcl-xL (Ser49) and (Ser62) mutants in Caenorhabditis elegans causes germline defects and aneuploidy. PLoS One, 12, e0177413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walsh N., Kenney L., Jangalwe S., Aryee K., Greiner D.L., Brehm M.A., Shultz L.D. and Harbor B. (2017) Humanized mouse models of clinical disease. Annu. Rev. Pathol., 24, 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shen Z., Zhang X., Chai Y., Zhu Z., Yi P., Feng G., Li W. and Ou G. (2014) Conditional knockouts generated by engineered CRISPR-Cas9 endonuclease reveal the roles of coronin in C. elegans neural development. Dev. Cell, 30, 625–636. [DOI] [PubMed] [Google Scholar]
- 40. Hubbard E.J.A. (2014) FLP/FRT and Cre/lox recombination technology in C. elegans. Methods, 68, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Voutev R. and Hubbard E.J.A. (2008) A ‘FLP-out’ system for controlled gene expression in Caenorhabditis elegans. Genetics, 180, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fischbach G.D. and Lord C. (2010) The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron, 68, 192–195. [DOI] [PubMed] [Google Scholar]
- 43. Herrero J., Muffato M., Beal K., Fitzgerald S., Gordon L., Pignatelli M., Vilella A.J., Searle S.M.J., Amode R., Brent S. et al. (2016) Ensembl comparative genomics resources. Database (Oxford), 2016, bav096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yates A., Beal K., Keenan S., McLaren W., Pignatelli M., Ritchie G.R.S., Ruffier M., Taylor K., Vullo A. and Flicek P. (2015) The Ensembl REST API: Ensembl data for any language. Bioinformatics, 31, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lackner M.R., Kornfeld K., Miller L.M., Horvitz H.R. and Kim S.K. (1994) A MAP kinase homolog, mpk-1, is involved in ras-mediated induction of vulval cell fates in Caenorhabditis elegans. Genes Dev., 8, 160–173. [DOI] [PubMed] [Google Scholar]
- 46. Paix A., Folkmann A., Rasoloson D. and Seydoux G. (2015) High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR/Cas9 ribonucleoprotein complexes. Genetics, 201, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arribere J., Bell R., Fu B., Artiles K., Hartman P. and Fire A. (2014) Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics, 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chiu H., Schwartz H.T., Antoshechkin I. and Sternberg P.W. (2013) Transgene-free genome editing in Caenorhabditis elegans using CRISPR-Cas. Genetics, 195, 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.