

Abstract
The understanding of the natural history of Alzheimer’s disease (AD) and temporal trajectories of in vivo molecular mechanisms requires longitudinal approaches. A behavioral and multimodal imaging study was performed at 4/8/12 and 16 months of age in a triple transgenic mouse model of AD (3xTg-AD). Behavioral assessment included the open field and novel object recognition tests. Molecular characterization evaluated hippocampal levels of amyloid β (Aβ) and hyperphosphorylated tau. Magnetic resonance imaging (MRI) included assessment of hippocampal structural integrity, blood–brain barrier (BBB) permeability and neurospectroscopy to determine levels of the endogenous neuroprotector taurine. Longitudinal brain amyloid accumulation was assessed using 11C Pittsburgh compound B positron emission tomography (PET), and neuroinflammation/microglia activation was investigated using 11C-PK1195. We found altered locomotor activity at months 4/8 and 16 months and recognition memory impairment at all time points. Substantial early reduction of hippocampal volume started at month 4 and progressed over 8/12 and 16 months. Hippocampal taurine levels were significantly decreased in the hippocampus at months 4/8 and 16. No differences were found for amyloid and neuroinflammation with PET, and BBB was disrupted only at month 16. In summary, 3xTg-AD mice showed exploratory and recognition memory impairments, early hippocampal structural loss, increased Aβ and hyperphosphorylated tau and decreased levels of taurine. In sum, the 3xTg-AD animal model mimics pathological and neurobehavioral features of AD, with early-onset recognition memory loss and MRI-documented hippocampal damage. The early-onset profile suggests temporal windows and opportunities for therapeutic intervention, targeting endogenous neuroprotectors such as taurine.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by memory deficits associated with progressive deterioration of cognitive and executive functions. Episodic memory impairment is one of the most important deficits in AD. The hippocampus, which is involved in episodic memory, is particularly affected and structural alterations have been observed in AD patients (1,2). Moreover, behavioral assessment of cognitive function is pivotal to determine the impact of AD progression.
The need to identify mechanisms of disease and new diagnostic and therapeutic tools for AD has led to the development of several transgenic mouse models to mimic AD pathophysiology (3–6). Since most genetically engineered mouse models rely on genes for early-onset familial AD, these models only partially mimic the features of human AD. However, one expects that these animal models share biological characteristics of human AD, such as brain amyloid plaques and neurofibrillary tangles, as well as the pattern of behavioral deficits observed in the human disease (7). In this study we used the triple transgenic mouse model of AD (3xTg-AD), a model of early-onset AD, which has mutant genes for amyloid precursor protein (APPSWE), APP23 presenilin 1 (PS1M146V) and tau.
Concerning the molecular characteristics of this model it has been reported that the extracellular amyloid β (Aβ) deposits become apparent in 6 months old mice in the cerebral cortex (8). These authors also described that Aβ oligomers begin to accumulate between 2 and 6 months of age, with continued age-dependent increase observed between 12 and 20 months. Concerning the human disease, it is also known that amyloid pathology starts very early on, ~22 years before clinical symptoms become apparent (9).
In order to understand disease mechanisms and test therapeutic interventions it is very important to track the natural history of the disease in a longitudinal way in the same animals. This requires the use of non-invasive techniques that allow studying molecular mechanisms in vivo. In this multimodal molecular imaging longitudinal study we have used 4 months old 3xTg-AD mice as the starting point because at this age they do not present Aβ plaques despite the presence of increased intraneuronal soluble Aβ levels that correlate with the earliest cognitive impairment (8,10). We decided to uniquely combine positron emission tomography (PET) and magnetic resonance imaging (MRI) methods longitudinally to best evaluate the natural history of the pathology, from the points of view of amyloid load, microglia activation, presence of endogenous neuroprotectors (specifically taurine) and structural integrity in key brain structures.
Concerning amyloid binding, we chose 11C Pittsburgh compound B (11C-PIB) because its binding in PET correlates with Aβ load and can be related to impaired metabolism, as measured by 18F-Fluordesoxiglucose imaging (PET-FDG), as our own human data corroborate (11). We also aimed to test the face value of 11C-PIB binding to Aβ deposits in this transgenic mouse model as compared to MR imaging. This is because it is believed that binding of that radiopharmaceutical in the brain is highly dependent on the AD model and the structure of Aβ plaques. For example, binding of 11C-PIB to Aβ-rich cortical regions was shown to occur in APP23 mice but in contrast no increases in binding were observed in aged Tg2576 or APPSWE-PS1dE9 mice in vivo although extensive Aβ deposition as assessed by immunohistochemistry was shown in APPSWE-PS1dE9 mice (12). Voxel based analysis of in vivo Aβ PET imaging studies in mouse models of AD is feasible and allows studying the PIB retention patterns in whole brain maps as further shown in a recent study of the APP/PS1 double transgenic mouse model of AD (13).
The combined use of imaging techniques is very scarce in this model, although one can identify studies using isolated modalities. A notable exception is the combined PET/MRI study focusing on amyloid load and perfusion of Maier and colleagues (14) in two amyloid precursor protein transgenic mouse models (APP23 and APP/PS1). This study showed that in the presence of cerebral amyloid angiopathy, Aβ deposition is accompanied by a decline of regional cerebral blood flow. PET-FDG does not assess amyloid load and has been used to probe the effects of therapeutic interventions in 3xTg-AD (15–17).
The demonstration that PET imaging can quantitatively map amyloid accumulation in living amyloid precursor protein transgenic mice was performed by Maeda and colleagues (18). They showed that in vivo imaging of Aβ plaque burden is feasible in mouse models of AD as a valuable translational research tool and even longitudinally to monitor treatment effects. They also showed repeated measures in relatively old APP23 animals. A study with the APP/PS1 model allowed for multi-method cross-validations for the PET results using ex vivo and in vitro methodologies, such as regional brain biodistribution, multi-label digital autoradiography, protein quantification with Enzyme-Linked Immunosorbent Assay (ELISA), fluorescence microscopy, semi-automated histological quantification and radioligand binding assays (19).
Concerning MRI studies coupled with behavior, a recent study (20) suggested that early neuroanatomic changes seem to precede major memory deficits, which further justifies imaging studies in a preclinical stage. Several behavioral tests performed with 3xTg-AD mice have previously shown that this model has both cognitive and non-cognitive deficits (10,21–26). Memory deficits are a hallmark of AD, as well as underlying hippocampal damage, and behavioral tasks in combination with methods to assess regional neural loss are therefore critical (27,28). In order to understand the natural history of core manifestations of human AD in a 3xTg-AD mouse model we performed a behavioral, molecular and multimodal imaging study. Importantly, hippocampal damage and potential underlying mechanisms were evaluated with voxel based morphometry (VBM), neurospectroscopy, molecular imaging of amyloid deposition (11C-PIB PET) and neuroinflammation(11C-PK1195) as well as molecular analysis and behavioral testing. Concerning MR spectroscopy we focused on taurine, given its role in preventing β-amyloid neurotoxicity (29).
Moreover, a recent in vitro study (30) testing whether fingolimod modulates neuroinflammatory markers in the 5xFAD mouse model of AD used high-resolution magic angle spinning magnetic resonance spectroscopy. The authors found that fingolimod decreased soluble plus insoluble Aβ measured by ELISA and that taurine levels, measured using that in vitro Magnetic resonance spectroscopy (MRS) technique, showed a very strong inverse correlation with Glial fibrillary acidic protein (GFAP) levels and ELISA measurements of Aβ, but not with activated microglia levels. The notion that taurine inversely relates with amyloid load further motivated our MRS study of taurine as a potential biomarker of impaired neuroprotection and disease progression.
Ultimately, we aimed to understand temporal patterns of disease progression, their molecular underpinnings and the amount of biological homology with the human disease. Finally, we aimed to identify the best structural and/or molecular imaging biomarkers that best predict the course of the disease.
Results
Behavioral impairment patterns in 3xTg-AD mice
The open field test evaluates the spontaneous activity of mice when exploring an environment. Several parameters can be measured as a way to determine locomotor function, namely distance and speed (Fig. 1). Wild-type (WT) mice had a better exploratory activity performance than 3xTg-AD mice at 4, 8 and 16 months, as can be seen by evaluating the overall distance traveled or the speed in the open field arena (Fig. 1). Motor deficits were detected at 4 months, and these deficits persisted until older ages. However, at 12 months of age no significant differences were detected both in distance traveled and speed between WT and 3xTg-AD mice (Distance WT at 4 months = 7.02 ± 0.39 m; Distance 3xTg-AD at 4 months = 5.09 ± 0.49 m; Distance WT at 16 months = 3.86 ±0.30 m; Distance 3xTg-AD at 16 months = 2.54 ± 0.24 m). Novel object recognition memory measures show that there was an impairment in object recognition memory in 3xTg-AD mice at all time points (Fig. 2A). WT animals spent more time exploring the novel object than the familiar object at all time points, and this difference was statistically significant at 4, 8 and 16 months of age (Fig. 2B). Moreover, at 4 and 16 months there was a statistically significant difference in the number of explorations of the novel object between groups (Fig. 3A). At 12 and 16 months WT mice significantly explored more times the novel object than the familiar one. At 4 and 8 months of age WT mice explored more times the novel object than the familiar object, but this difference was not statistically significant (Fig. 3B). In addition, concerning within group comparisons, 3xTg-AD animals did not show a statistically significant difference in exploration time (Fig. 2C) and number of explorations (Fig. 3C) between familiar and novel objects at all time points. Regarding the total exploration time, WT mice spent more time exploring both objects than transgenic mice at 4, 12 and 16 months of age. At 8 months WT mice spent more time exploring both object than 3xTg-AD mice, but this difference was not significant (Fig. 4A). Also, WT mice spent more time exploring the novel object than 3xTg-AD mice at all time points (Fig. 4B).
Figure 1.

(A) Representative trace images of WT and 3xTg-AD mice in the open field. Locomotor activity of WT and 3xTg-AD animals was evaluated by measuring the (B) distance traveled (m) and (C speed (m/s) at different time points (4, 8, 12 and 16 months of age). Results are presented as mean ± SEM and were analyzed with the Student’s t-test; *P < 0.05 and **P < 0.01 (n WT at 4 months = 21, n WT at 8 months = 20, n WT at 12 months = 17, n WT at 16 months = 17, n 3xTg-AD at 4 months = 20, n 3xTg-AD at 8 months = 23, n 3xTg-AD at 12 months = 22, n 3xTg-AD at 16 months = 22).
Figure 2.

Exploration time of WT and 3xTg-AD mice in the novel object recognition test at different time points (4, 8, 12 and 16 months of age). (A) Exploration time of both groups is compared; (B) exploration time of WT in familiar and novel object; (C) exploration time of 3xTg-AD in familiar and novel object. Results are presented as mean ± SEM and were analyzed with the Student’s t-test; *P < 0.05, **P < 0.01 and ***P < 0.001 (n WT at 4 months = 21, n WT at 8 months = 20, n WT at 12 months = 17, n WT at 16 months = 17, n 3xTg-AD at 4 months = 20, n 3xTg-AD at 8 months = 23, n 3xTg-AD at 12 months = 22, n 3xTg-AD at 16 months = 22).
Figure 3.

Number of explorations of WT and 3xTg-AD mice in the novel object recognition test at different time points (4, 8, 12 and 16 months of age). (A) Number of explorations of both groups is compared; (B) number of explorations of WT in familiar and novel object; (C) number of explorations of 3xTg-AD in familiar and novel object. Results are presented as mean ± SEM and were analyzed with the Student’s t-test; *P < 0.05, **P < 0.01 and ***P < 0.001 (n WT at 4 months = 21, n WT at 8 months = 20, n WT at 12 months = 17, n WT at 16 months = 17, n 3xTg-AD at 4 months = 20, n 3xTg-AD at 8 months = 23, n 3xTg-AD at 12 months = 22, n 3xTg-AD at 16 months = 22).
Figure 4.

Total exploration time (A) and novel object exploration time (B) of WT and 3xTg-AD mice at different time points (4, 8, 12 and 16 months of age). Results are presented as mean ± SEM and were analyzed with the Student’s t-test; *P < 0.05, **P < 0.01 and ***P < 0.001 (n WT at 4 months = 21, n WT at 8 months = 20, n WT at 12 months = 17, n WT at 16 months = 17, n 3xTg-AD at 4 months = 20, n 3xTg-AD at 8 months = 23, n 3xTg-AD at 12 months = 22, n 3xTg-AD at 16 months = 22).
Increased Aβ and p-tau protein levels in the hippocampus of 3xTg-AD mice
The two major hallmarks of AD are the diffuse and neuritic plaques that are predominantly composed by Aβ, and the neurofibrillary tangles, formed by aggregates of hyperphosphorylated tau protein (p-tau) (31). The protein levels of Aβ monomers in the hippocampus of 3xTg-AD mice increased, comparing with age-matched WT, at 4 months (373.4 ± 63.3% of WT, P < 0.01), 8 months (717.1 ± 174.7% of WT, P < 0.01), 12 months (1434.9 ± 196.1% of WT, P < 0.05) and 16 months (9447.5 ± 1338.9% of WT, P < 0.05), reaching a maximum peak at 16 months of age (Fig. 5A). We also detected an increase of p-tau levels in the hippocampus of 3xTg-AD at 4 months (434.8 ± 138.2% of WT, P < 0.01), 8 months (360.7 ± 69.4% of WT, P < 0.01), 12 months (147.4 ± 7.0% of WT, P < 0.01) and 16 months (230.2 ± 27.3% of WT, P < 0.01) (Fig. 5B). Contrarily to what was observed for Aβ, p-tau levels were higher at early time points and decreased with age.
Figure 5.

Increase of Aβ and p-tau protein levels in the hippocampus of 3xTg-AD mice. The protein levels of Aβ (A) and p-tau (B) in total hippocampal extracts from 3xTg-AD and age-matched WT mice were analyzed by western blotting. Representative images for Aβ, p-Tau and β-actin (loading control) for each time point (4, 8, 12 and 16 months of age) are presented. The results are expressed as percentage of age-matched WT animals and are presented as mean ± SEM of four to six animals. *P < 0.05 and **P < 0.01, different from WT, Mann–Whitney test.
Hippocampal structural impairment in 3xTg-AD mice
When evaluating the structural integrity of the hippocampus, volumetric analysis showed that the 3xTg-AD mice had reduced hippocampal volumes starting early on at 4 months, which was accentuated during disease progression toward 16 months. We found statistically significant between group effects (F(1,8) = 268.230, P < 0.001), and the post hoc analyses revealed that the volume of the transgenic mice hippocampi was significantly reduced at all time points compared to the WT group (P < 0.001) as shown in Figure 6. Regression curves suggested a more pronounced volumetric reduction occurring in the 3xTg-AD animals (β13xTg-AD = -0.091 mm3/month; β1WT = −0.012 mm3/month). Comparison of relative decay showed a trend for significance when comparing the slopes [F(1,36) = 3.704, P = 0.062; Fig. 6, solid lines].
Figure 6.

Hippocampal volumes of WT and 3xTg-AD mice at different time points (4, 8, 12 and 16 months). Solid lines represent the regression curves showing the temporal evolution of the hippocampal volumes. Regression curves: WT,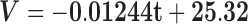 ; 3xTg-AD,
; 3xTg-AD, 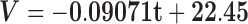 . V, volume; t, time. Results are presented as mean ± SEM and were analyzed with an ANOVA repeated measures (mixed-effect) followed by a Bonferroni post hoc test and a linear regression analysis (n WT at 4, 8, 12 and 16 months = 4, n 3xTg-AD at 4, 8, 12 and 16 months = 6).
. V, volume; t, time. Results are presented as mean ± SEM and were analyzed with an ANOVA repeated measures (mixed-effect) followed by a Bonferroni post hoc test and a linear regression analysis (n WT at 4, 8, 12 and 16 months = 4, n 3xTg-AD at 4, 8, 12 and 16 months = 6).
Morphometric analyses, which assess differences in gray matter (GM) volumes in a voxel-by-voxel basis, further corroborate these observations showing that clusters of significantly reduced GM volume are already present at 4 months of age in the 3xTg-AD group in the right and left hippocampus (Fig. 7A). At later stages, clusters are more prevalent and spread across the hippocampus with emerging clusters of GM volume reductions in the anterior regions englobing CA1, CA2, CA3 and dentate gyrus subfields, as illustrated in Fig. 7B.
Figure 7.

VBM results in a coronal view at 4 (A) and 16 months. (B) Color bar denotes the T-score magnitude, at the voxel level, with a threshold level of P < 0.05 FWE corrected (n WT at 4, 8, 12 and 16 months = 6, n 3xTg-AD at 4, 8, 12 and 16 months = 7).
Regarding perfusion imaging, which is useful to evaluate the blood–brain barrier (BBB) permeability, results revealed a time-dependent increased permeability index and decreased vascular volume (as measured by the peak amplitude) in transgenic animals at later stages of the disease, i.e. 16 months (Fig. 8). We found a significant main effect detected with Analysis of variance (ANOVA) for peak amplitude (F = 3.41, P = 0.034) and differences between groups were visible at 16 months (F(1,8) = 23.06, P = 0.001; Fig. 8A). Regarding blood–brain barrier perfusion index (BBBi), which represents the leakage/permeability of the brain barrier, the longitudinal analysis did not detect a time*group interaction. Post hoc analysis showed that 3xTg-AD animals had increased BBBi in comparison to WT at 16 months (F(1,8) = 25.47, P = 0.001; Fig. 8B).
Figure 8.

Perfusion peak amplitude and BBB index at 16 months of age. Right panel, BBB permeability index. Left panel, perfusion peak amplitude. Results are presented as mean ± SEM and were analyzed with an ANOVA repeated measures (mixed-effect) followed by a pairwise comparison employing a Bonferroni post hoc test; **P < 0.01 (n WT at 4, 8, 12 and 16 months = 4, n 3xTg-AD at 4, 8, 12 and 16 months = 6).
Decreased levels of taurine in the hippocampus of 3xTg-AD mice
The spectra enabled a reliable quantification of taurine, and its levels were found significantly reduced in the 3xTg-AD animals compared to the WT (F(1,8) = 45.804, P < 0.001) at 4 months (F(1,8) = 18.470, P < 0.003), 8 months (F(1,8) = 8.573, P < 0.019) and 16 months (F(1,8) = 21.635, P < 0.002), as summarized in Figure 9.
Figure 9.

Taurine levels of 3xTg-AD and WT mice at different time points (4, 8, 12 and 16 months of age). Results are presented as mean ± SEM and were analyzed with an ANOVA repeated measures (mixed-effect) followed by a pairwise comparison employing a Bonferroni post hoc test; *P < 0.05 and **P < 0.01(n WT at 4, 8, 12 and 16 months = 4 and n 3xTg-AD at 4, 8, 12 and 16 months = 6).
No changes in amyloid load and inflammation evaluated by PET
No significant differences were found neither for the uptake values of 11C-PIB (a marker of amyloid) or Pk11195 (a marker of inflammatory processes) between groups.
Discussion
In this multimodal molecular imaging study of 3xTg-AD, in the presence of major hallmarks of AD, increased Aβ and p-tau, we measured early abnormal hippocampal volumetry that progressed along all time points. We found that neurobehavioral deficits can also be identified using multimodal molecular imaging techniques in animals, along with the novel finding of reduced levels of taurine over several time points. In particular, we found evidence for early hippocampal structural damage, recognition memory deficits that paralleled increased hippocampal Aβ and p-tau levels.
Interestingly, in vivo MRI measures of hippocampal parameters proved to be much more sensitive in this model than PET imaging. Moreover, we found no in vivo PET evidence for microglia activation, and parameters of BBB integrity were only affected in late stages (16 months). Since motor activity can influence the outcome measurements of subsequent tests, it needs to be evaluated in order to determine its influence in tasks designed for assessing cognitive deficits that depend on motor function. Locomotor activity deficits have already been detected not only in 3xTg-AD mice, but also in APP-deficient mice and APP23 mice (32–36). The 3xTg-AD animal model used in this study shows similar deficits, as shown by measuring the velocity and distance traveled in the open field test. Similar impairments have also been observed in AD patients, and this functional impairment progresses as cognition declines (37).
The novel object recognition test describes the ability of mice to preferentially explore a novel object than a familiar one. This memory test requires that vision is used to guide behavior toward objects that are discriminated based on their novelty and specific visual features and so it can be used to evaluate cognitive deficits related to recognition memory in several preclinical models of neurodegenerative disorders, including AD. Similarly to humans several brain regions are involved in rodent recognition memory including the hippocampus, prefrontal, perirhinal and entorhinal cortex. Therefore, the study of recognition memory impairments in mouse models of AD may provide important insights regarding AD pathology in humans (38–42). Recognition memory test deficits suggest that the hippocampus or other structures of the medial temporal lobe are impaired, a behavioral pattern that is a characteristic of AD. Consequently, the independent pattern of impairment observed in the object recognition memory test in 3xTg-AD mice suggests that the episodic memory system typically impaired in AD was also impaired in this animal model.
The hippocampus plays a pivotal role in the process of memory encoding and retrieval. Besides the hippocampus, other brain structures may also be affected such as the striatum and frontal cortex. In particular, the interaction between these brain regions is important in recognition memory (43–45). Oddo and colleagues (8) detected synaptic dysfunction in the hippocampus of 3xTg-AD mice at 6 months of age. This structure is well known to have an impact in memory and cognition in AD patients. A possible explanation for this event is the intraneuronal accumulation of Aβ species. Intracellular immunoreactive Aβ species have been observed in 3xTg-AD mice in the neocortex between 3 and 4 months of age (8). Therefore, it is not surprising that we were able to observe object recognition memory impairment already at 4 months. Furthermore, structural differences between WT and 3xTg-AD groups were detected with volumetry and VBM in the hippocampus as early as 4 months of age that corroborates the behavioral results. This is equivalent to a human age of ~26 years (46), which shows that this measure is really an early sensitive marker.
Importantly, MR volumetry of the hippocampus in humans is an important diagnostic tool. Therefore, these results can be useful for further characterization of animal models of neurodegenerative diseases and are in line with evidence reporting hippocampal atrophy in AD patients (47). This is also in line with previous works in humans that reported alterations in the hippocampus volume of the same magnitude as we found here (48,49).
Despite the observation that several mouse models of AD have normal BBB permeability (50) we found differences in BBB permeability at 16 months, probably being a late consequence of the disease progression. Moreover, the decreased peak amplitude in the transgenic animals may be a sign of hypoperfusion.
We also found that the hippocampus has increased levels of Aβ and p-tau. Contrarily to what has been reported in AD patients (51–53), using PET imaging we did not detect significant differences in amyloid deposition and neuroinflammatory status in the hippocampus. The reasons why amyloid load is difficult to detect using 11C-PIB in this particular model, unlike other models, remain to be elucidated, given that we found molecular evidence for such accumulation. Also, we did not find evidence for microglia activation using a neuroinflammation marker (11C-PK1195), also suggesting that this is not a prominent feature underlying neurodegeneration in this model.
Regarding magnetic resonance spectroscopy we found that taurine levels in 3xTg-AD mice were reduced in the hippocampus, across multiple time points. Since taurine prevents the neurotoxicity of Aβ reduced levels of taurine imply lower neuroprotection, which is relevant when amyloid plaques load is high in transgenic animals. Indeed, Louzada and colleagues (29) demonstrated that taurine prevents the neurotoxicity of β-amyloid and glutamate receptor agonists, and its low levels early on in the disease process suggests failure of endogenous neuroprotection. The temporal pattern of change in taurine is quite distinct from the rate of change in hippocampus volume. Moreover, we also found reduced taurine levels in regions such as the striatum at time points while volume was still preserved. This renders unlikely that changes in taurine levels are because of volume changes.
Finally, and concerning other metabolites, we would have expected decreased levels of glutamate in the hippocampus as was observed in the Ts2 model of Down syndrome mice (54). However, we did not find glutamatergic abnormalities in our mouse model. Future studies should address these differences across models.
In summary, our findings provide interesting homologies between human and animal models, suggest further investigation of this taurine neuroprotection in humans and pave the way for studies involving early neurodegeneration, including therapeutic ones.
In AD brain atrophy is enhanced in comparison with normal aging. It has been shown that 11-month-old 3xTg-AD animals have a decrease in the brain weight and in the brain to body weight ratio (33,55). This brain shrinkage can be because of the loss of synaptic connections, reduction of neuronal arborization and neuronal loss. Consequently, the observation of a reduction of the volume of the hippocampus and recognition memory impairment in 3xTg-AD mice represents a valuable indicator in line with its use in AD diagnosis and management. However, most of the current mouse models of AD fail to show visible brain neuronal loss, a hallmark present in AD patients (56–58). Thus, the 3xTg-AD mouse model seems to be valuable in this context.
Conclusion
In this multimodal molecular imaging study of the 3xTg-AD mouse model, in the presence of the major hallmarks of AD, increased Aβ and p-tau, we measured early abnormal hippocampal volumetry that progressed along all time points. MR neurospectroscopy also showed loss of taurine, which is an important endogenous neuroprotector. PET imaging of amyloid load was less sensitive than MR, and the use of the PET marker 11C-PK1195 did not reveal microglia activation. The BBB was preserved unless until late stages. This shows that MR signatures of hippocampal volumetry and neurospectroscopy represent early valuable biomarkers that parallel neurobehavioral deficits, thus providing an in vivo multimodal validation of the 3xTg-AD mouse as an early disease model that may be applied in preclinical research using these biomarkers.
Methodology
Animals
Experiments were performed in 3xTg-AD mice harboring three human mutant genes: PS1M146V, APPSWE and tau (TauP301L) and in age-matched WT animals (WT: C57BL6/129S background). Male 3xTg-AD mice and WT mice were used to evaluate in vivo structural, molecular imaging, neurospectroscopic and behavioral changes at 4, 8, 12 and 16 months of age. WT mice used in this study have the same genetic background of the presenilin knockin embryos used to generate the 3xTg-AD mouse model, but instead of expressing mutant PS1M146V gene they express the endogenous WT mouse PS1M146V gene (8). The animals were maintained at 22 ± 1 °C, 68% relative humidity, on a 12 h light–dark cycle, with access to water and food ad libitum. In order to assess age-related changes the animals were tagged. All procedures involving animals were approved by the Animal Welfare Committee of the Coimbra Institute for Clinical and Biomedical Research, Faculty of Medicine, University of Coimbra. The animal experimentation was conducted in accordance with the European Community directive guidelines for the use of animals in laboratory (2010/63/EU), transposed into the Portuguese law in 2013 (Decreto-Lei 113/2013) and was in agreement with the Association for Research in Vision and Ophthalmology statement for animal use.
Behavioral experiments
Mice behavior (WT, n = 21; 3xTg-AD, n = 23) was evaluated using the open field and novel object recognition tests to assess changes in locomotor activity and memory performance. The behavioral tests were performed by the same researcher, who remained outside the view of each mouse while performing the test, only interfering in the test to replace the animals and clean the arenas between consecutive tests. The tests were video recorded and analyzed with Observador (http://en.pharmacology.med.uoa.gr/) and ANY-Maze software (version 4.99 m; http://www.anymaze.co.uk/). Tests were performed at 4, 8, 12 and 16 months of age in 3xTg-AD and age-matched WT animals. On the first day, the animals were placed in an open field maze during 10 min to allow their adaptation to the new environment, and locomotor activity was analyzed during this period. The parameters evaluated were the distance traveled and the speed. The performance of the animals was analyzed using the behavioral scoring program ANY-maze. Habituation to the open field arena was performed to reduce possible anxiety like behavior due to placing in a novel environment. On the second day, the animals were placed in an open field apparatus with two identical objects, and after a time interval one of the objects was kept in the arena (familiar object), while the other was replaced by a new one, approximately with the same size and texture, but different in shape (novel object). The novel object recognition test was performed in order to assess mice recognition memory. This test consisted of two 10 min sessions: the first with two identical objects (sample session) and the second (test session, 2 h after sample session) with two dissimilar objects (a familiar and a novel one). Exploration of objects was considered when animals were facing the objects with the nose up to 2 cm away from the object. The time exploring each object and the number of explorations were recorded. The performance of the animals was analyzed using the behavioral scoring program Observador.
Western blot
Hippocampus from 3xTg-AD (n = 5) and WT (n = 6) mice were dissected in ice-cold Hank’s balanced salt solution (137 mM NaCl, 5.4 mM KCl, 0.45 mM KH2PO4, 0.34 mM NaHPO, 4 mM, NaHCO3, 5 mM glucose, pH 7.4), frozen in liquid nitrogen and stored at −80°C. All samples were lysed in cell lysis buffer (Cell Signaling Technology, Danvers, Massachusetts, USA) supplemented with complete mini ethylenediaminetetraacetic acid-free protease inhibitor cocktail tablets (Roche, Basel, Switzerland), PhosSTOP phosphatase inhibitor tables (Roche), 0.1 mM phenylmethylsulfonyl fluoride (Roche) and 2 mM of dithiothreitol (DTT; Fisher, Hampton, New Hampshire, USA). After homogenization, samples were frozen and thawed three times. Then, the samples were centrifuged at 16 100 xg during 10 min at 4°C, and the supernatant was collected. After protein quantification by The bicinchoninic acid (BCA) method (Pierce Biotechnology, Waltham, Massachusetts, USA), samples were denatured by adding 6× concentrated sample buffer (0.5 M Tris, 30% glycerol, 10% sodium dodecyl sulfate, 0.6 M DTT, 0.012% bromophenol blue) and heated for 5 min at 95°C. Samples were separated in SDS-polyacrylamide gel electrophoresis and transferred onto Polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, Massachusetts, USA). The membranes were blocked with 5% BSA and were then incubated with the antibodies indicated in Table 1. Immunoreactive bands were visualized using Enhanced Chemi-Fluorescence system (GE Healthcare, Chicago, Illinois, USA) on an imaging system (Thyphoon FLA 9000, GE Healthcare) or with Enhanced Chemiluminescence system (Bio-Rad, Hercules, California, USA) on Image Quant LAS 500 (GE Healthcare). Digital quantification of band intensity was performed using Quantity One software (Bio-Rad). The membranes were then reprobed and tested for β-actin immunoreactivity to prove that similar amounts of protein were applied in the gels. Data were obtained by calculating the ratio between each target protein and β-actin and expressed as percentage of WT (control).
Table 1.
List of primary and secondary antibodies used in western blotting
| Primary antibody | Host | Dilution | Protein (μg) | Supplier |
|---|---|---|---|---|
| Anti-Amyloid-β (D54D2) | Rabbit | 1:1000 | 20 | Cell Signaling Technology |
| Anti-p-tau (ser396) | Rabbit | 1:1000 | 10 | Santa Cruz Biotechnology |
| Anti-β-Actin | Mouse | 1:5000 | 20/10 | Sigma-Aldrich |
| Secondary antibody | ||||
| HRP* Anti-rabbit | 1:10 000 | - | Bio-Rad | |
| AP** Anti-mouse | 1:10 000 | - | Sigma-Aldrich |
*HRP, Horseradish peroxidase
**AP, alkaline phosphatase
MRI acquisitions
MRI experiments were performed in a BioSpec 9.4 T scanner with a standard cross coil set-up using a volume coil for excitation (with 86/112 mm of inner/outer diameter, respectively) and quadrature mouse surface coil for signal detection (Bruker Biospin, Ettlingen, Germany). Mice (W, n = 6; 3xTg-AD, n = 7) were kept anesthetized by isoflurane (1.5%) with 100% O2 and body temperature and respiration monitoring (SA Instruments SA, Stony Brook, NY, USA).
For structural analysis, T2-weighted images were acquired in coronal planes using a rapid acquisition with relaxation enhancement sequence with the following parameters: TR = 3800 ms; TE = 33 ms; 10 averages; pixel size of 0.078 mm ×0.078 mm and slice thickness of 0.5 mm without spacing between slices (total head volume, 256 pixels × 256 pixels × 34 slices). Brain permeability was assessed after injection of Gadovist® (0.1 mmol/kg) using the dynamic contrast-enhanced (DCE) MRI technique. The following parameters were used in 2D DCE-FLASH: TE/TR = 2.5/251.446 ms, flip angle = 70°, FOV = 20 × 20 mm, matrix = 156 × 85, slice thickness of 0.5 mm, number of slices = 18, number of repetitions = 40. For localized 1H magnetic resonance spectroscopy (1H-MRS), data were collected in a volume of interest placed on the hippocampus. B0 map was acquired before spectroscopy, and shims were optimized through a mapshim voxel. A point resolved spectroscopy sequence was used in combination with outer volume suppression and variable power and optimized relaxation delays water suppression with parameters as follows: TR = 2500 ms, TE = 16.225 ms, number of averages = 720, three flip angles = 90°, 142° and 142°, bandwidth = 5000 Hz, number of acquired points = 2048 yielding a spectral resolution of 1.22 Hz/point and voxel size of 2.1 × 1.3 × 1.3 mm. For each animal, an unsuppressed water signal (TE = 16.225 ms, TR = 2500 ms, 16 averages and none water suppression) was acquired immediately before acquiring the water suppressed spectrum.
VBM
VBM was performed using the statistical parametric mapping software (SPM, Welcome Department of Cognitive Neurology, London, UK), the SPMMouse toolbox (59) and a homemade script involving the following steps: (a) T2-weighted images were corrected for the magnetic field inhomogeneity generated by the surface coil; this was done using intensity curves from T2-weighted images obtained for an homogeneous phantom and acquired with the same coil and system configuration; (b) the rigid body was aligned by registering (affine transformation) the images to the template space; (c) tissue segmentation was carried out by means of the GM, white matter and cerebrospinal fluid tissue probability maps as provided in the toolbox; (d) GM images were non-linearly normalized to the template space and were modulated to correct for volume changes that may have occurred during normalization; and (e) the obtained images were smoothed by applying a 0.5 mm full width at half maximum (FWHM) Gaussian kernel. All steps were done using the default settings of the toolbox.
Volumetry
For volumetry, after correction of magnetic field inhomogeneity (as described for VBM), images were segmented using the ITK-SNAP software based on manual delineation of the hippocampus (−1.06 to −4.16 Bregma) performed by a user blind for the two groups. The atlas from Franklyn and Paxinos was used as reference (60). The hippocampal volume was then obtained by multiplying the number of voxels by the voxel size.
DCE imaging
DCE imaging depicts the wash-in, plateau and wash-out contrast kinetics of the tissue. DCE data were processed as follows: (a) images were rescaled, filtered (excluding voxels outside the brain) and corrected for movement; (b) regions of interest (ROI) were drawn for each animal in a semi-automatic procedure, using MRICron (2015) and custom made Matlab functions. Previously reported in house Matlab functions were used for further analysis and extraction of perfusion measurements (61); (c) the perfusion curve (accumulation of contrast agent) was extracted per ROI and animal and normalized to the baseline (time window before injection); and (d) from these curves, the time to peak (time delay between injection and perfusion peak), the peak amplitude (maximum perfusion value, representing the vascular volume) and the area under the curve (AUC) were calculated. Note that the AUC parameter is the overall accumulation of contrast agent in the ROI. Additionally, the BBBi was calculated as follows: the residual enhancement at the last 10 dynamic acquisition time points/frames included the contribution from the contrast agent that had leaked into the interstitial space, and this parameter, normalized to the vascular volume measured as the peak amplitude of the perfusion curve, was used as a BBBi (62).
1H-MRS
1H-MRS spectra were saved as free induction decay signals (FIDs), corrected for the frequency drift and for residual eddy current effects using the reference water signal. Data were analyzed using the LCModel package (63), which calculates the best fit to the acquired spectrum as a linear combination of a model based on a set of brain metabolites. The Cramér–Rao lower bounds criterion was used as a measure of the reliability of the fitting and set to 21%. Representative spectra are given in Figure 10. Metabolite concentration was calculated as we have described previously (64,65). For statistical analysis, we were particularly interested in taurine, and therefore its concentration in relation to the water signal was determined.
Figure 10.

Representative 1H-MRS spectra of WT (A) and 3xTg-AD (B) mice acquired at 16 months.
PET acquisitions
For in vivo assessment of cerebral Aβ plaques deposition by means of 11C-PIB and microglia activation (11C-PK11195), mice were submitted to PET imaging in a high-resolution small animal PET-RPC scanner, prototype developed by LIP-Coimbra, in collaboration with the Institute of Nuclear Sciences Applied to Health, University of Coimbra (66). Animals were kept anesthetized and monitored, as for MRI acquisitions, and scanned after the injection of the radiopharmaceutical. Both 11C-PIB and PK11195 scans lasted for 60 min and were performed in different days. 11C-PiB had a mean activity of 12.44 ± 2.11 μCi/g. This radiopharmaceutical binds with high affinity to the Aβ plaques in the brain. PK11195, which is a marker of the translocator protein, indexing microglia activation, had a mean activity of 12.66 ± 2.26 μCi/g.
To facilitate the co-registration between PET and MRI images, and thus allowing the segmentation of regions of interest, three fiducial markers visible in both PET and MRI images were positioned on the animal platform.
PET data processing
PET images were reconstructed using the ordered subset expectation-maximization (OSEM) algorithm with a voxel volume of 0.125 mm3 (0.5 × 0.5 × 0.5 mm) using the data acquired between 20 and 60 min after injection. After reconstruction, a correction to the field of view was carried out, and images were saved as Digital Imaging and Communications in Medicine files (DICOM). 3D Slicer software was then used to manually co-register PET and MRI images using the fiducial markers as reference. Manual segmentation of the hippocampus was then performed, using ITK-SNAP, and the mean counts per mm3, normalized to the activity injected per gram and corrected for the system sensitivity were computed. For statistical analyses the ratio between the uptake in the hippocampus and cerebellum was used.
Statistical analysis
The normality of the data was assessed with Shapiro–Wilk and D’Agostino and Pearson omnibus normality tests. Statistical analysis of molecular and behavioral data was performed with GraphPad Prism Version 6 (GraphPad Software, San Diego, CA, USA) using parametric Student’s t-test or nonparametric Mann–Whitney test, as applicable. Longitudinal and between groups analyses of the hippocampal volumes, taurine concentrations, BBB permeability parameters and the uptakes of 11C-PIB and PK11195 were carried out using SPSS 22.0 (SPSS Inc., Chicago, IL, USA) with an ANOVA repeated measures (mixed-effect) followed by pairwise comparisons employing Bonferroni corrections. A regression analysis was also performed to fit the volumetric data to a linear model and assess the effect of aging and rate of volumetric reduction. Intercepts (β0) and slopes (β1) were compared using GraphPad Prism Version 6. During VBM, to analyze differences in GM volumes between groups, at each time point a Student’s t-test was performed at the voxel level. Voxels with GM values <0.2 were not included. Results were, firstly, obtained as whole brain statistical maps. After that, the analysis was repeated on the ROI and corrected for multiple comparisons using family-wise error rate (FWE) (P < 0.05). Differences were localized using the atlas from Franklyn and Paxinos. Data were considered to be significant at *P < 0.05, **P < 0.01 and ***P < 0.001. All data are reported as mean ± standard error of mean (SEM).
Conflict of Interest statement. None declared.
Funding
Santa Casa Mantero Belard Award 2015 (MB-1049-2015); Foundation for Science and Technology (SFRH/BD/52045/2012, PEst UID/NEU/04539/2013 and MEDPERSYST SAICTPAC/0010/2015); COMPETE-FEDER (POCI-01-0145-FEDER-007440 and POCI-01-0145-FEDER-016428); Centro 2020 Regional Operational Programme (CENTRO-01-0145-FEDER-000008: BrainHealth 2020 and CENTRO-01-0145-FEDER-000016: BIGDATIMAGE).
References
- 1. Ballard C., Gauthier S., Corbett A., Brayne C., Aarsland D. and Jones E. (2011) Alzheimer’s disease. Lancet, 377, 1019–1031. [DOI] [PubMed] [Google Scholar]
- 2. Jahn H. (2013) Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci., 15, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Christopher Janusa D.W. (2001) Transgenic mouse models of Alzheimer’ s disease. Physiol. Behav., 73, 873–886. [DOI] [PubMed] [Google Scholar]
- 4. Wong P.C., Cai H., David R. and Borchelt D.L.P. (2002) Genetically engineered mouse models of neurodegenerative diseases. Nat. Neurosci., 5, 633–639. [DOI] [PubMed] [Google Scholar]
- 5. Harper A. (2010) Mouse models of neurological disorders––a comparison of heritable and acquired traits. Biochim. Biophys. Acta––Mol. Basis Dis., 1802, 785–795. [DOI] [PubMed] [Google Scholar]
- 6. Hall A.M. and Roberson E.D. (2012) Mouse models of Alzheimer’s disease. Brain Res. Bull., 88, 259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bryan K.J., Lee H., Perry G., Smith M.A. and Casadesus G. (2009) Transgenic mouse models of Alzheimer’s disease: behavioral testing and considerations In Methods of Behavior Analysis in Neuroscience. Taylor & Francis Group, Abingdon OX, United Kingdom. [Google Scholar]
- 8. Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y. and LaFerla F.M. (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles. Neuron, 39, 409–421. [DOI] [PubMed] [Google Scholar]
- 9. Gordon B.A., Blazey T.M., Su Y., Hari-Raj A., Dincer A., Flores S., Christensen J., McDade E., Wang G., Xiong C. et al. (2018) Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol., 17, 211–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Billings L.M., Oddo S., Green K.N., McGaugh J.L. and LaFerla F.M. (2005) Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron, 45, 675–688. [DOI] [PubMed] [Google Scholar]
- 11. Oliveira F.P.M., Moreira A.P., De Mendonça A., Verdelho A., Xavier C., Barroca D., Rio J., Cardoso E., Cruz Â., Abrunhosa A. et al. (2018) Can 11 C-PiB-PET relative delivery R 1 or 11 C-PiB-PET perfusion replace 18 F-FDG-PET in the assessment of brain neurodegeneration? J. Alzheimers Dis., 65, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Snellman A., Lopez-Picon F.R., Rokka J., Salmona M., Forloni G., Scheinin M., Solin O., Rinne J.O. and Haaparanta-Solin M. (2013) Longitudinal amyloid imaging in mouse brain with 11C-PIB: comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of Alzheimer disease. J. Nucl. Med., 54, 1434–1441. [DOI] [PubMed] [Google Scholar]
- 13. Von Reutern B., Grünecker B., Yousefi B.H., Henriksen G., Czisch M. and Drzezga A. (2013) Voxel-based analysis of amyloid-burden measured with [11c]pib pet in a double transgenic mouse model of Alzheimer’s disease. Mol. Imaging Biol., 15, 576–584. [DOI] [PubMed] [Google Scholar]
- 14. Maier F.C., Wehrl H.F., Schmid A.M., Mannheim J.G., Wiehr S., Lerdkrai C., Calaminus C., Stahlschmidt A., Ye L., Burnet M. et al. (2014) Longitudinal PET-MRI reveals β 2-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med., 20, 1485–1492. [DOI] [PubMed] [Google Scholar]
- 15. Baek H., Ye M., Kang G.-H., Lee C., Lee G., Choi D.B., Jung J., Kim H., Lee S., Kim J.S. et al. (2016) Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget, 7, 69347–69357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye M., Chung H.S., An Y.H., Lim S.J., Choi W., Yu A.R., Kim J.S., Kang M., Cho S., Shim I. et al. (2016) Standardized herbal formula PM012 decreases cognitive impairment and promotes neurogenesis in the 3xTg AD mouse model of Alzheimer’s disease. Mol. Neurobiol., 53, 5401–5412. [DOI] [PubMed] [Google Scholar]
- 17. Ye M., Chung H.S., Lee C., Yoon M.S., Yu A.R., Kim J.S., Hwang D.S., Shim I. and Bae H. (2016) Neuroprotective effects of bee venom phospholipase A2 in the 3xTg AD mouse model of Alzheimer’s disease. J. Neuroinflammation, 13, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maeda J., Ji B., Irie T., Tomiyama T., Maruyama M., Okauchi T., Staufenbiel M., Iwata N., Ono M., Saido T.C. et al. (2007) Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J. Neurosci., 27, 10957–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manook A., Yousefi B.H., Willuweit A., Platzer S., Reder S., Voss A., Huisman M., Settles M., Neff F., Velden J. et al. (2012) Small-animal PET imaging of amyloid-beta plaques with [11c]PiB and its multi-modal validation in an APP/PS1 mouse model of Alzheimer’s disease. PLoS One, 7, e31310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kong V., Devenyi G.A., Gallino D., Ayranci G., Germann J., Rollins C. and Chakravarty M.M. (2018) Early-in-life neuroanatomical and behavioural trajectories in a triple transgenic model of Alzheimer’s disease. Brain Struct. Funct., 223, 3365–3382. [DOI] [PubMed] [Google Scholar]
- 21. Pietropaolo S., Feldon J. and Yee B.K. (2008) Age-dependent phenotypic characteristics of a triple transgenic mouse model of Alzheimer disease. Behav. Neurosci., 122, 733–747. [DOI] [PubMed] [Google Scholar]
- 22. Romberg C., Mattson M.P., Mughal M.R., Bussey T.J. and Saksida L.M. (2011) Impaired attention in the 3xTgAD model of Alzheimer’s disease assessed using a translational touchscreen method for mice: rescue by donepezil (Aricept). J. Neurosci., 31, 3500–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Orta-Salazar E., Feria-Velasco A., Medina-Aguirre G.I. and Díaz-Cintra S. (2013) Morphological analysis of the hippocampal region associated with an innate behaviour task in the transgenic mouse model (3xTg-AD) for Alzheimer disease. Neurologia, 28, 497–502. [DOI] [PubMed] [Google Scholar]
- 24. Davis K.E., Easton A., Eacott M.J. and Gigg J. (2013) Episodic-like memory for what–where–which occasion is selectively impaired in the 3xTgAD mouse model of Alzheimer’s disease. J. of Alzheimer's Dis., 33, 681–698. [DOI] [PubMed] [Google Scholar]
- 25. Stevens L.M. and Brown R.E. (2015) Reference and working memory deficits in the 3xTg-AD mouse between 2 and 15-months of age: a cross-sectional study. Behav. Brain Res., 278, 496–505. [DOI] [PubMed] [Google Scholar]
- 26. Stover K.R., Campbell M.A., Van Winssen C.M. and Brown R.E. (2015) Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res., 289, 29–38. [DOI] [PubMed] [Google Scholar]
- 27. Snigdha S., Milgram N.W., Willis S.L., Albert M., Weintraub S., Fortin N.J. and Cotman C.W. (2013) A preclinical cognitive test battery to parallel the National Institute of Health toolbox in humans: bridging the translational gap. Neurobiol. Aging, 34, 1891–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Puzzo D., Lee L., Palmeri A., Calabrese G. and Arancio O. (2014) Behavioral assays with mouse models of Alzheimer’s disease: practical considerations and guidelines. Biochem. Pharmacol., 88, 450–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Louzada P.R. (2004) Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J., 18, 511–518. [DOI] [PubMed] [Google Scholar]
- 30. Aytan N., Choi J.-K., Carreras I., Brinkmann V., Kowall N.W., Jenkins B.G. and Dedeoglu A. (2016) Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep., 6, 24939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selkoe D.J. (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev., 81, 741–767. [DOI] [PubMed] [Google Scholar]
- 32. Van Dam D., D’Hooge R., Staufenbiel M., Van Ginneken C., Van Meir F. and De Deyn P.P. (2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci., 17, 388–396. [DOI] [PubMed] [Google Scholar]
- 33. Carvalho C., Machado N., Mota P.C., Correia S.C., Cardoso S., Santos R.X., Santos M.S., Oliveira C.R. and Moreira P.I. (2013) Type 2 diabetic and Alzheimer’s disease mice present similar behavioral, cognitive, and vascular anomalies. J. Alzheimers Dis., 35, 623–635. [DOI] [PubMed] [Google Scholar]
- 34. Zheng H., Jiang M., Trumbauer M.E., Sirinathsinghji D.J.S., Hopkins R., Smith D.W., Heavens R.P., Dawson G.R., Boyce S., Conner M.W. et al. (1995) β-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell, 81, 525–531. [DOI] [PubMed] [Google Scholar]
- 35. Halagappa V.K.M., Guo Z., Pearson M., Matsuoka Y., Cutler R.G., LaFerla F.M. and Mattson M.P. (2007) Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis., 26, 212–220. [DOI] [PubMed] [Google Scholar]
- 36. Filali M., Lalonde R., Theriault P., Julien C., Calon F. and Planel E. (2012) Cognitive and non-cognitive behaviors in the triple transgenic mouse model of Alzheimer’s disease expressing mutated APP, PS1, and Mapt (3xTg-AD). Behav. Brain Res., 234, 334–342. [DOI] [PubMed] [Google Scholar]
- 37. Bridenbaugh S., Monsch A.U. and Kressig R.W. (2012) How does gait change as cognitive decline progresses in the elderly? Alzheimers Dement., 8, P131–P132. [Google Scholar]
- 38. Eichenbaum H., Yonelinas A.P. and Ranganath C. (2007) The medial temporal lobe and recognition memory. Annu. Rev. Neurosci., 30, 123–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arsenault D., Julien C., Tremblay C. and Calon F. (2011) DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS One, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cohen S.J. and Stackman R.W. (2015) Assessing rodent hippocampal involvement in the novel object recognition task. A review. Behav. Brain Res., 285, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morici J.F., Bekinschtein P. and Weisstaub N.V. (2015) Medial prefrontal cortex role in recognition memory in rodents. Behav. Brain Res., 292, 241–251. [DOI] [PubMed] [Google Scholar]
- 42. Warburton E.C. and Brown M.W. (2015) Neural circuitry for rat recognition memory. Behav. Brain Res., 285, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seger C.A. and Cincotta C.M. (2006) Dynamics of frontal, striatal, and hippocampal systems during rule learning. Cereb. Cortex, 16, 1546–1555. [DOI] [PubMed] [Google Scholar]
- 44. Herweg N.A., Apitz T., Leicht G., Mulert C., Fuentemilla L. and Bunzeck N. (2016) Theta-alpha oscillations bind the hippocampus, prefrontal cortex, and striatum during recollection: evidence from simultaneous EEG-fMRI. J. Neurosci., 36, 3579–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Headley D.B. and Paré D. (2017) Common oscillatory mechanisms across multiple memory systems. npj Sci. Learn., 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Flurkey K., MCurrer J. and Harrison D. (2007) Mouse models in aging research In The Mouse in Biomedical Research. Elsevier, Amsterdam, North-Holland, Netherlands, pp. 637–672. [Google Scholar]
- 47. Hsu P.J., Shou H., Benzinger T., Marcus D., Durbin T., Morris J.C. and Sheline Y.I. (2015) Amyloid burden in cognitively normal elderly is associated with preferential hippocampal subfield volume loss. J. Alzheimers Dis., 45, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schuff N., Woerner N., Boreta L., Kornfield T., Shaw L.M., Trojanowski J.Q., Thompson P.M., Jack C.R. and Weiner M.W. (2009) MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain, 132, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Henneman W.J.P., Sluimer J.D., Barnes J., Van Der Flier W.M., Sluimer I.C., Fox N.C., Scheltens P., Vrenken H. and Barkhof F. (2009) Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology, 72, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bien-Ly N., Boswell C.A., Jeet S., Beach T.G., Hoyte K., Luk W., Shihadeh V., Ulufatu S., Foreman O., Lu Y. et al. (2015) Lack of widespread BBB disruption in Alzheimer’s disease models: focus on therapeutic antibodies. Neuron, 88, 289–297. [DOI] [PubMed] [Google Scholar]
- 51. Edison P., Archer H.A., Gerhard A., Hinz R., Pavese N., Turkheimer F.E., Hammers A., Tai Y.F., Fox N., Kennedy A. et al. (2008) Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis., 32, 412–419. [DOI] [PubMed] [Google Scholar]
- 52. Okello A., Edison P., Archer H.A., Turkheimer F.E., Kennedy J., Bullock R., Walker Z., Kennedy A., Fox N., Rossor M. et al. (2009) Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology, 72, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schuitemaker A., Kropholler M.A., Boellaard R., Flier W.M., Kloet R.W., Doef T.F., Knol D.L., Windhorst A.D., Luurtsema G., Barkhof F. et al. (2013) Microglial activation in Alzheimer’s disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol. Aging, 34, 128–136. [DOI] [PubMed] [Google Scholar]
- 54. Kaur G., Sharma A., Xu W., Gerum S., Alldred M.J., Subbanna S., Basavarajappa B.S., Pawlik M., Ohno M., Ginsberg S.D. et al. (2014) Glutamatergic transmission aberration: a major cause of behavioral deficits in a murine model of Down’s syndrome. J. Neurosci., 34, 5099–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carvalho C., Cardoso S., Correia S.C., Santos R.X., Santos M.S., Baldeiras I., Oliveira C.R. and Moreira P.I. (2012) Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes, 61, 1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karran E., Mercken M. and De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov., 10, 698–712. [DOI] [PubMed] [Google Scholar]
- 57. Ashe K.H. and Zahs K.R. (2010) Probing the biology of Alzheimer’s disease in mice. Neuron, 66, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Onos K.D., Sukoff Rizzo S.J., Howell G.R. and Sasner M. (2016) Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res. Bull., 122, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sawiak S.J., Wood N.I., Williams G.B., Morton A.J. and Carpenter T.A. (2013) Voxel-based morphometry with templates and validation in a mouse model of Huntington’s disease. Magn. Reson. Imaging, 31, 1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Franklin K.B. and Paxinos G. (1997) The Mouse Brain in Stereotaxic Coordinates. Academic Press, Cambridge, Massachusetts, USA. [Google Scholar]
- 61. Rodrigues T., Matafome P., Sereno J., Almeida J., Castelhano J., Gamas L., Neves C., Gonçalves S., Carvalho C., Arslanagic A. et al. (2017) Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci. Rep., 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang H., Golob E.J. and Su M.Y. (2006) Vascular volume and blood–brain barrier permeability measured by dynamic contrast enhanced MRI in hippocampus and cerebellum of patients with MCI and normal controls. J. Magn. Reson. Imaging, 24, 695–700. [DOI] [PubMed] [Google Scholar]
- 63. Provencher S.W. (1993) Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn. Reson. Med., 30, 672–679. [DOI] [PubMed] [Google Scholar]
- 64. Petrella L.I., Castelhano J.M., Ribeiro M., Sereno J.V., Gonçalves S.I., Laço M.N., Hayden M.R., Rego A.C. and Castelo-Branco M. (2018) A whole brain longitudinal study in the YAC128 mouse model of Huntington’s disease shows distinct trajectories of neurochemical structural connectivity and volumetric changes. Hum. Mol. Genet., 27, 1–13. [DOI] [PubMed] [Google Scholar]
- 65. Ribeiro M., Castelhano J., Petrella L.I., Sereno J., Rodrigues T., Neves C., Letra L., Baptista F.I., Seiça R., Matafome P. et al. (2018) High-fat diet induces a neurometabolic state characterized by changes in glutamate and N-acetylaspartate pools associated with early glucose intolerance: an in vivo multimodal MRI study. J. Magn. Reson. Imaging. 48, 757–766. doi: 10.1002/jmri.25942. [DOI] [PubMed] [Google Scholar]
- 66. Martins P., Blanco A., Crespo P., Marques M.F.F., Marques R.F., Gordo P.M., Kajetanowicz M., Korcyl G., Lopes L., Michel J. et al. (2014) Towards very high resolution RPC-PET for small animals. J. Instrum., 9. [Google Scholar]