

Abstract
Ertugliflozin, a sodium‐glucose cotransporter 2 inhibitor for the treatment of adults with type 2 diabetes mellitus, is expected to be coadministered with sitagliptin, metformin, glimepiride, and/or simvastatin. Four separate open‐label, randomized, single‐dose, crossover studies were conducted in healthy adults to assess the potential pharmacokinetic interactions between ertugliflozin 15 mg and sitagliptin 100 mg (n = 12), metformin 1000 mg (n = 18), glimepiride 1 mg (n = 18), or simvastatin 40 mg (n = 18). Noncompartmental pharmacokinetic parameters derived from plasma concentration–time data were analyzed using mixed‐effects models to assess interactions. Coadministration of sitagliptin, metformin, glimepiride, or simvastatin with ertugliflozin had no effect on area under the plasma concentration–time profile from time 0 to infinity (AUCinf) or maximum observed plasma concentration (Cmax) of ertugliflozin (per standard bioequivalence boundaries, 80% to 125%). Similarly, ertugliflozin did not have any impact on AUCinf or Cmax of sitagliptin, metformin, or glimepiride. AUCinf for simvastatin (24%) and simvastatin acid (30%) increased slightly after coadministration with ertugliflozin and was not considered clinically relevant. All treatments were well tolerated. The lack of clinically meaningful pharmacokinetic interactions demonstrates that ertugliflozin can be coadministered safely with sitagliptin, metformin, glimepiride, or simvastatin without any need for dose adjustment.
Keywords: diabetes, drug‐drug interaction, ertugliflozin, glimepiride, metformin, pharmacokinetics, SGLT2i, simvastatin, sitagliptin
Ertugliflozin is a selective sodium‐glucose cotransporter 2 (SGLT2) inhibitor1, 2 that reduces plasma glucose and glycated hemoglobin by increasing urinary glucose excretion.3, 4 Ertugliflozin, at once‐daily doses of 5 and 15 mg, has been recently approved in the United States (US) and European Union (EU) for the treatment of adults with type 2 diabetes mellitus (T2DM). Phase 3 studies have shown that ertugliflozin significantly improves glycemic control, decreases body weight and blood pressure, and is well tolerated.5, 6, 7 The pharmacokinetics (PK) of ertugliflozin are similar in healthy subjects and patients with T2DM. It is absorbed rapidly after oral administration (time to maximum plasma concentration [Tmax] 1 hour postdose), and terminal phase half‐life (t1/2) ranges from 11 to 17 hours.1, 2 Additionally, the absolute bioavailability of ertugliflozin is approximately 100%,8 its exposure increases proportionally with increasing dose over the range of 0.5 to 300 mg, and its administration with food does not have a clinically meaningful effect on PK.9 The PK of ertugliflozin are time independent and, consistent with the half‐life, steady‐state concentrations are achieved by 4 to 6 days after initiating once‐daily dosing. Ertugliflozin is highly bound to plasma proteins (93.6%). The primary clearance mechanism of ertugliflozin is metabolism. Glucuronidation is the major metabolic pathway (86%), with minor contributions from oxidative metabolism (12%). Renal excretion of unchanged ertugliflozin accounts for 1.5% of the administered dose.
Ertugliflozin is likely to be used in combination with a number of other antihyperglycemic agents including sitagliptin, metformin, and glimepiride. Many T2DM patients also have concomitant dyslipidemia and are likely to be treated with statins, such as simvastatin, in combination with ertugliflozin. Ertugliflozin‐sitagliptin and ertugliflozin‐metformin fixed‐dose combination tablets have been approved for use in the US and EU for the treatment of adult patients with T2DM.
Sitagliptin is an oral antihyperglycemic agent from the dipeptidyl peptidase‐4 (DPP‐4) inhibitor class, which acts by inhibiting the degradation of incretins, including glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypeptide.10 Urinary excretion of unchanged drug via glomerular filtration and renal secretion by organic anion transporter‐3 (OAT‐3), organic anion transporting polypeptide 4C1 (OATP4C1), and multidrug‐resistance P‐glycoprotein are the major routes of elimination of sitagliptin, with metabolism playing only a minor role.11 Sitagliptin undergoes limited oxidative metabolism, predominantly by cytochrome P450 (CYP) 3A4 with minor contribution from CYP2C8.12
Metformin is a biguanide antihyperglycemic agent that improves glycemic control by decreasing hepatic glucose production, decreasing glucose absorption, and increasing insulin‐mediated glucose uptake.13, 14, 15 Metformin is transported by the organic cation transporters OCT1 and OCT2. The uptake of metformin in the liver, which is the primary target of metformin, is mediated primarily by OCT1. Studies in healthy volunteers suggest that human multidrug and toxin extrusion 1 (MATE1) and MATE2‐K also contribute to the renal excretion of metformin.13
Glimepiride is an orally active blood glucose–lowering agent from the sulfonylurea class, the action of which is dependent on stimulating the release of insulin from pancreatic beta cells by blocking adenosine triphosphate–dependent potassium channels on the beta cell membrane. Glimepiride is extensively metabolized in the liver, mainly by CYP2C9.16
Simvastatin is a 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitor that is hydrolyzed to the active metabolite simvastatin acid. Both simvastatin and simvastatin acid are metabolized by CYP3A4.17
Ertugliflozin is metabolized mainly by uridine diphosphate glucuronosyltransferase (UGT) isoenzyme 1A9 and UGT2B7 to 2 pharmacologically inactive glucuronide metabolites.2 CYP3A4 is the predominant CYP enzyme involved in the minor oxidative metabolism of ertugliflozin with some contributions by CYP2C8 and CYP3A5. At clinically relevant concentrations, ertugliflozin is a substrate for the P‐glycoprotein and breast cancer resistance protein efflux transporters; however, no clinically relevant interaction is expected with inhibitors of these transporters based on the oral bioavailability of ertugliflozin of approximately 100%, and dose‐proportional increases in exposure over the dose range of 0.5 mg to 300 mg. Ertugliflozin is not a substrate for OATP1B1, OATP1B3, OATP2B1, OCT1, OAT1, OAT3, and OCT2 uptake transporters.
Based on in vitro studies, ertugliflozin and its glucuronide metabolites are not inhibitors or inducers of CYP isozymes or transporters (including OATP1B1); therefore, it is not anticipated to alter the PK of coadministered drugs, sitagliptin, metformin, glimepiride, or simvastatin. Ertugliflozin is a weak inhibitor of OAT3 and OCT2 transporters, but the unbound clinical maximum observed plasma concentration (Cmax)/half maximal inhibitory concentration is < 0.1; thus, ertugliflozin is not anticipated to alter the PK of coadministered sitagliptin (substrate of OAT3) or metformin (substrate of OCT2).
There is no evidence and/or data in the literature that sitagliptin, metformin, glimepiride, or simvastatin (or simvastatin acid) either induce or inhibit UGT1A9 or UGT2B7 at clinically relevant plasma levels.18 Therefore, these coadministered drugs are not anticipated to alter the PK of ertugliflozin.
Four drug‐drug interaction (DDI) studies were conducted in healthy volunteers to quantify the effect of the coadministered drugs on the PK of ertugliflozin, and vice versa, following oral administration of a single dose of ertugliflozin 15 mg coadministered with a single dose of either sitagliptin 100 mg, metformin 1000 mg, glimepiride 1 mg, or simvastatin 40 mg. The secondary objective of all 4 studies was to evaluate the safety and tolerability of single doses of ertugliflozin 15 mg and the coadministered drugs.
Methods
All 4 phase I, open‐label, randomized, 3‐period, 6‐sequence, single oral dose, crossover DDI studies were conducted at the Pfizer Clinical Research Unit in Brussels, Belgium, in compliance with the ethical principles of the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice guidelines. The final protocols and informed consent documentation of all 4 DDI studies were reviewed and approved by the independent ethics committee (Comité d'Ethique Hospitalo‐Facultaire Erasme‐ULB, Brussels, Belgium) at the investigational center participating in each study.
Key Eligibility Criteria
Healthy male and/or female subjects aged 18 to 55 years inclusive, with a body mass index of 17.5 to 30.5 kg/m2 and total body weight >50 kg, who had provided a signed, dated informed consent form and were willing and able to comply with the study plan, were eligible for inclusion in this study. Subjects with the following were excluded: positive urine screen for drugs, either due to abuse or recreational use; history of alcohol abuse or binge drinking and/or any other illicit drug use or dependence within 6 months of screening; known hypersensitivity or intolerance to any SGLT2 inhibitor, sitagliptin (or any other DPP‐4 inhibitor), metformin, glimepiride (or any other sulfonylurea or sulfonamide), or simvastatin (or any other 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitor) in respective trials. For the sitagliptin study, subjects with a calculated creatinine clearance of <80 mL/min based on the Cockcroft‐Gault formula19 were excluded. Similarly, for the metformin, glimepiride, and simvastatin studies, subjects with an estimated glomerular filtration rate of <90 mL/min/1.73 m2 based on the 4‐variable Modification of Diet in Renal Disease equation20 were excluded. Subjects were not permitted to eat or drink grapefruit or grapefruit‐related citrus fruits (eg, Seville oranges, pomelos) from 7 days prior to the first dose of study medication until collection of the final PK blood sample in period 3.
Study Design and Treatment
Each study consisted of a screening visit and 3 study‐treatment periods: (1) ertugliflozin 15 mg; (2) sitagliptin 100 mg/metformin 1000 mg/glimepiride 1 mg/simvastatin 40 mg; (3) ertugliflozin 15 mg + sitagliptin 100 mg/metformin 1000 mg/glimepiride 1 mg/simvastatin 40 mg. Screening occurred within 28 days of the first dose of study medication in period 1. In each study, eligible subjects admitted to the clinical research unit on day 0 were randomized to 1 of 6 treatment sequences and received their assigned treatment in the morning of day 1 of each treatment period after an overnight fast of at least 8 hours. In the coadministered period, subjects received the single‐dose oral treatments within 5 minutes of each other, with ertugliflozin administered first. Dosing in consecutive crossover periods was separated by a washout period of at least 5 days. To minimize the risk of hypoglycemia with glimepiride, all study treatments in the ertugliflozin‐glimepiride study were administered with approximately 240 mL of a 20% glucose solution, followed by approximately 60 mL of 20% glucose solution administered approximately every 15 minutes until approximately 4 hours after the dose in each period. Subjects were discharged from the clinical research unit after collection of the 24‐hour PK sample in each period and returned to the clinical research unit in an outpatient setting for collection of the 48‐ and 72‐hour PK samples. Sample sizes of 12 subjects for the sitagliptin study and 18 subjects for each of the metformin, glimepiride, and simvastatin studies provided 90% confidence intervals (CIs) for the difference between treatments of ±0.1832 and ±0.2222 at most, on the natural logarithm scale for area under the plasma concentration–time profile from time 0 extrapolated to infinite time (AUCinf) and Cmax, respectively, with 80% coverage probability. Within‐subject variability estimates were obtained from the weighted average of previous crossover studies.
Assessments
Serial blood samples for PK analyses were collected from each subject at the following times in each treatment period:
Ertugliflozin‐sitagliptin study: predose (0 hour) and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours postdose
Ertugliflozin‐metformin study: predose (0 hour) and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 8, 12, 24, 48, and 72 hours postdose
Ertugliflozin‐glimepiride and ertugliflozin‐simvastatin studies: predose (0 hour) and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48, and 72 hours postdose
Plasma samples were analyzed for ertugliflozin, metformin, glimepiride, simvastatin, and simvastatin acid concentrations at WuXi AppTec (Shanghai, China), and for sitagliptin concentrations by inVentiv Health Clinique (Quebec, Canada) using validated, sensitive, and specific high‐performance liquid chromatography–tandem mass spectrometry methods. The method for bioanalysis of ertugliflozin has been described previously.21
Sitagliptin was extracted from 100 μL of human plasma by protein precipitation with acetonitrile and sitagliptin‐d4 as the internal standard. The extracted sample was injected into a Waters Atlantis HILIC Silica column 3 μm (2.1 mm × 50 mm) (Waters Corporation, Milford, Massachusetts) using a mobile phase of acetonitrile/water (80/20, v/v) containing 10 mM NH4Ac (pH 4.7). Detection was performed by Sciex API 4000 (SCIEX, Framingham, Massachusetts) in the positive ion mode. The multiple reaction monitoring ion transition was m/z 408 → 235 for sitagliptin and m/z 412 → 239 for the internal standard. The calibration curve was linear over the range of 1.0 to 1000 ng/mL.
Metformin was extracted from 50 μL of human plasma by protein precipitation with acetonitrile and metformin‐d6 as the internal standard. The supernatant was diluted with 10 mM NH4Ac in acetonitrile containing 0.5% formic acid. The extracted sample was injected into a high‐performance liquid chromatography column (Waters Atlantis HILIC Silica 5 μm [2.1 mm × 50 mm]), with gradient mobile phase containing 0.5% formic acid and 10 mM NH4Ac in water, and 0.5% formic acid in acetonitrile. Detection was performed by Sciex API 4000 in the positive ion mode. The multiple reaction monitoring ion transition was m/z 130 → 71 for metformin and m/z 136 → 77 for the internal standard. The calibration curve was linear over the range of 2.0 to 1000 ng/mL.
Glimepiride was extracted from 50 μL of human plasma using a solid‐phase extraction 96‐well plate with methanol and water, and glimepiride‐d5 as the internal standard. The extracted sample was injected into a Luna C18(2), 5 μm (2.0 mm × 50 mm) high‐performance liquid chromatography column (Phenomenex, Inc., Torrance, California) with gradient mobile phase containing 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Detection was performed by Sciex API 4000 in the positive ion mode. The multiple reaction monitoring ion transition was m/z 491 → 352 for glimepiride and m/z 496 → 357 for the internal standard. The calibration curve was linear over the range of 0.1 to 200 ng/mL.
Simvastatin and simvastatin acid were extracted from 200 μL of human plasma using a solid‐phase extraction C18 96‐well plate with NH4Ac and acetonitrile, and simvastatin‐d6 and simvastatin acid‐d6 as the internal standards, respectively. The extracted sample was injected into a Waters Atlantis dc18, 3 μm (2.1 mm × 100 mm) column with gradient mobile phase containing ammonium hydroxide and acetonitrile. Detection was performed by Sciex API 5000 (SCIEX) in the positive ion mode for simvastatin and negative mode for simvastatin acid. The multiple reaction monitoring ion transition was m/z 420 → 199 for simvastatin, m/z 425 → 199 for simvastatin‐d6, m/z 435 → 319 for simvastatin acid, and m/z 441 → 319 for simvastatin acid‐d6. The calibration curve was linear over the range of 0.05 to 50.0 ng/mL for simvastatin and 0.05 to 10.0 ng/mL for simvastatin acid.
Safety assessments were conducted from screening and throughout the duration of study participation. Subjects also received a follow‐up phone call 14 ± 3 days after administration of the last dose of study medication in period 3 to assess for adverse events (AEs). In the glimepiride study, additional fingerstick blood glucose measurements were conducted to monitor for hypoglycemia at predose (0 hour), and at 2, 4, 7, 12 and 24 hours postdose.
Data Analysis
PK parameters were calculated for each subject for each treatment using noncompartmental analysis of plasma concentration–time data via an in‐house validated software system. Plasma concentrations that were below the lower limit of quantification were set to 0 for analysis. Natural log‐transformed AUCinf, area under the plasma concentration–time profile from time 0 to time of the last quantifiable concentration (AUClast), and Cmax were analyzed using separate mixed‐effects models with sequence, period, and treatment as fixed effects, and subject within sequence as a random effect. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (Test/Reference) and 90% CI for the ratios.
Results
Subjects
A total of 12 subjects were enrolled in the sitagliptin study, and 18 subjects were enrolled in each of the metformin, glimepiride, and simvastatin studies (each subject participated in 1 study only). The majority of subjects enrolled were male, white, and non‐Hispanic. The age and body mass index of the subjects ranged from 19 to 55 years and 19.3 to 30.4 kg/m2, respectively (Table 1). All subjects completed their respective study and were included in the analyses.
Table 1.
Subject Demographic Characteristics
| Characteristic | Sitagliptin Study (N = 12) | Metformin Study (N = 18) | Glimepiride Study (N = 18) | Simvastatin Study (N = 18) |
|---|---|---|---|---|
| Gender, n | ||||
| Male | 5 | 10 | 10 | 10 |
| Female | 7 | 8 | 8 | 8 |
| Age, n | ||||
| 18–44 y | 9 | 14 | 15 | 16 |
| 45–64 y | 3 | 4 | 3 | 2 |
| Mean (SD), y | 34.7 (10.5) | 37.6 (9.8) | 32.6 (10.1) | 31.3 (9) |
| Range, y | 25–53 | 24–55 | 19–50 | 19–51 |
| Race, n | ||||
| White | 12 | 18 | 16 | 15 |
| Black | 0 | 0 | 2 | 2 |
| Other | 0 | 0 | 0 | 1 |
| Ethnicity, n | ||||
| Hispanic/Latino | 1 | 0 | 1 | 0 |
| Non‐Hispanic/Latino | 11 | 18 | 17 | 18 |
| Weight, kg | ||||
| Mean (SD) | 70.6 (13.1) | 76.1 (11.3) | 72.2 (14.4) | 73.2 (12.8) |
| Range | 55.1–104.3 | 54.1–99.5 | 50.6–107.7 | 57.1–95.3 |
| BMI, kg/m2 | ||||
| Mean (SD) | 23.4 (2.5) | 24.6 (3.0) | 24.4 (3.1) | 24.4 (3.0) |
| Range | 19.3–28.0 | 20.9–30.3 | 19.3–30.4 | 19.6–28.9 |
BMI, body mass index; n, number of subjects; SD, standard deviation.
PK Results
Effect of Coadministered Drugs on the PK of Ertugliflozin
PK parameters of ertugliflozin with and without the coadministered drugs are summarized in Table 2. Following oral administration of a single dose of ertugliflozin 15 mg alone, or in combination with either a single dose of oral sitagliptin 100 mg, metformin 1000 mg, glimepiride 1 mg, or simvastatin 40 mg, mean plasma ertugliflozin concentration–time profiles were nearly superimposable (Figure 1). While not prospectively specified, the 90% CIs for the geometric mean ratios of ertugliflozin AUCinf and Cmax were within the equivalence bounds of 80% to 125%, indicating that coadministration of ertugliflozin with sitagliptin, metformin, glimepiride, or simvastatin had no clinically meaningful effect on the PK of ertugliflozin (Table 3 and Figure 2).
Table 2.
Descriptive Summary of Plasma Ertugliflozin, Sitagliptin, Metformin, Glimepiride, and Simvastatin (and Simvastatin Acid) Pharmacokinetic Parameter Values
| Ertugliflozin‐Sitagliptin Study | |||||
|---|---|---|---|---|---|
| Ertugliflozin Pharmacokinetic Summarya by Treatment | Sitagliptin Pharmacokinetic Summarya by Treatment | ||||
| Ertugliflozin 15 mg (N = 12) | Ertugliflozin 15 mg + Sitagliptin 100 mg (N = 12) | Sitagliptin 100 mg (N = 12) | Ertugliflozin 15 mg + Sitagliptin 100 mg (N = 12) | ||
| n | 12 | 12 | n | 12 | 12 |
| AUCinf, ng • h/mL | 1456 (380.55) | 1485 (364.31) | AUCinf, μM • hb | 7.018 (1.484) | 7.130 (1.493) |
| AUClast, ng • h/mL | 1428 (379.51) | 1450 (353.16) | AUClast, μM • hb | 6.953 (1.501) | 7.049 (1.511) |
| Cmax, ng/mL | 270.3 (66.67) | 266.0 (67.23) | Cmax, nMb | 814.3 (213.64) | 825.9 (195.29) |
| Tmax, h | 1.00 (1.00–3.00) | 1.00 (0.50–2.10) | Tmax, h | 2.00 (1.00–4.00) | 3.00 (1.00–6.00) |
| t1/2, h | 12.63 ± 5.15 | 14.17 ± 4.55 | t1/2, h | 11.00 ± 2.89 | 11.79 ± 2.98 |
| Ertugliflozin‐Metformin Study | |||||
|---|---|---|---|---|---|
| Ertugliflozin Pharmacokinetic Summarya by Treatment | Metformin Pharmacokinetic Summarya by Treatment | ||||
| Ertugliflozin 15 mg (N = 18) | Ertugliflozin 15 mg + Metformin 1000 mg (N = 18) | Metformin 1000 mg (N = 18) | Ertugliflozin 15 mg + Metformin 1000 mg (N = 18) | ||
| n | 17 | 17 | 13 | 13 | |
| AUCinf, ng • h/mL | 1401 (356.05) | 1424 (340.33) | 13 170 (3172.9) | 12 640 (3167.2) | |
| AUClast, ng • h/mL | 1382 (339.78) | 1399 (323.71) | 12 940 (3161.5) | 12 560 (2739.0) | |
| Cmax, ng/mL | 279.1 (60.68) | 269.3 (50.71) | 2041 (473.72) | 1890 (468.89) | |
| Tmax, h | 1.02 (1.00–2.00) | 1.29 (1.00–3.00) | 2.00 (0.50–4.00) | 2.00 (1.00–3.00) | |
| t1/2, h | 11.79 ± 2.34 | 13.48 ± 4.65 | 10.23 ± 2.39 | 14.47 ± 6.94 | |
| Ertugliflozin‐Glimepiride Study | |||||
|---|---|---|---|---|---|
| Ertugliflozin Pharmacokinetic Summarya by Treatment | Glimepiride Pharmacokinetic Summarya by Treatment | ||||
| Ertugliflozin 15 mg (N = 18) | Ertugliflozin 15 mg + Glimepiride 1 mg (N = 18) | Glimepiride 1 mg (N = 18) | Ertugliflozin 15 mg + Glimepiride 1 mg (N = 18) | ||
| N, n | 17c, 17 | 16d, 16 | 18, 13 | 16c, 11 | |
| AUCinf, ng • h/mL | 1247 (257.88) | 1294 (249.90) | 249.3 (213.55) | 296.7 (306.9) | |
| AUClast, ng • h/mL | 1232 (256.73) | 1278 (247.81) | 217.5 (185.36) | 283.9 (245.9) | |
| Cmax, ng/mL | 145.8 (24.41) | 146.7 (27.01) | 34.35 (19.90) | 33.47 (15.79) | |
| Tmax, h | 2.0 (1.5–3.0) | 2.0 (1.5–3.0) | 3.00 (1.00–12.0) | 4.00 (1.50–12.0) | |
| t1/2, h | 10.63 ± 2.44 | 11.27 ± 3.28 | 5.89 ± 2.79 | 6.68 ± 4.02 | |
| Ertugliflozin–Simvastatin Study | ||||||
|---|---|---|---|---|---|---|
| Ertugliflozin Pharmacokinetic Summarya by Treatment | Simvastatin Pharmacokinetic Summarya by Treatment | Simvastatin Acid Pharmacokinetic Summarya by Treatment | ||||
| Ertugliflozin 15 mg (N = 18) | Ertugliflozin 15 mg + Simvastatin 40 mg (N = 18) | Simvastatin 40 mg (N = 18) | Ertugliflozin 15 mg + Simvastatin 40 mg (N = 18) | Simvastatin 40 mg (N = 18) | Ertugliflozin 15 mg + Simvastatin 40 mg (N = 18) | |
| n | 18 | 18 | 12 | 18 | 16 | 14 |
| AUCinf, ng • h/mL | 1407 (313.51) | 1446 (337.63) | 43.78 (19.23) | 59.49 (42.03) | 31.44 (23.13) | 46.97 (29.13) |
| AUClast, ng • h/mL | 1384 (311.06) | 1419 (329.16) | 43.85 (29.11) | 57.52 (41.32) | 30.94 (22.46) | 40.63 (28.31) |
| Cmax, ng/mL | 273.2 (58.49) | 289.8 (69.03) | 9.283 (5.67) | 12.01 (9.26) | 2.605 (2.62) | 3.079 (2.843) |
| Tmax, h | 1.5 (1.0–2.5) | 1.0 (1.0–2.0) | 1.00 (0.50–12.0) | 1.25 (0.50–12.00) | 4.00 (1.50–12.0) | 4.00 (2.50–8.00) |
| t1/2, h | 12.34 ± 3.07 | 12.58 ± 3.98 | 5.88 ± 1.96 | 7.44 ± 2.72 | 8.44 ± 6.00 | 8.60 ± 2.91 |
AUCinf, area under the plasma concentration–time profile from time 0 extrapolated to infinite time; AUClast, area under the plasma concentration–time profile from time 0 to time of the last quantifiable concentration; Cmax, maximum observed plasma concentration; N, number of subjects contributing to summary statistics; n, number of subjects with reportable t½ and AUCinf; t1/2, terminal half‐life; Tmax, time for maximum observed plasma concentration.
Arithmetic mean (standard deviation) for all except median (range) for Tmax.
Sitagliptin concentration of 1.00 ng/mL is equivalent to 2.46 nM.
One subject from ertugliflozin 15 mg treatment vomited within 2 times the median ertugliflozin Tmax; therefore, the pharmacokinetic data from this subject was excluded.
Two subjects from ertugliflozin 15 mg + glimepiride 1 mg treatment vomited within/close to 2 times the median ertugliflozin Tmax and within 2 times the median glimepiride Tmax; therefore, the pharmacokinetic data from these 2 subjects were excluded.
Figure 1.
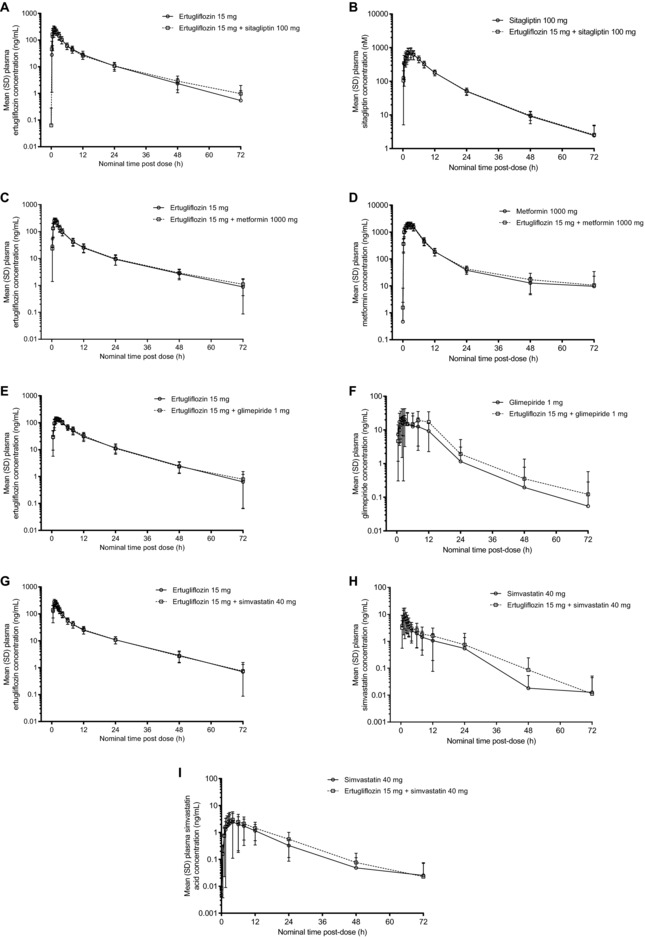
Mean (± standard deviation) plasma concentration–time profiles for (A) ertugliflozin alone and in combination with sitagliptin; (B) sitagliptin alone and in combination with ertugliflozin; (C) ertugliflozin alone and in combination with metformin; (D) metformin alone and in combination with ertugliflozin; (E) ertugliflozin alone and in combination with glimepiride; (F) glimepiride alone and in combination with ertugliflozin; (G) ertugliflozin alone and in combination with simvastatin; and (H) simvastatin and (I) simvastatin acid after administration of simvastatin alone and in combination with ertugliflozin. Summary statistics were calculated by setting concentration values below the lower limit of quantification to zero. SD, standard deviation.
Table 3.
Statistical Summary of Treatment Comparisons for Ertugliflozin, Sitagliptin, Metformin, Glimepiride, and Simvastatin
| GMR (90% CI) | ||||
|---|---|---|---|---|
| Analyte | Test | Reference | AUCinf | Cmax |
| Ertugliflozin‐Sitagliptin Study | ||||
| Ertugliflozin | Ertugliflozin + Sitagliptin | Ertugliflozin | 102.3 (99.7, 104.9) | 98.2 (91.2, 105.7) |
| Sitagliptin | Ertugliflozin + Sitagliptin | Sitagliptin | 101.7 (98.4, 105.0) | 101.7 (91.7, 112.8) |
| Ertugliflozin‐Metformin Study | ||||
| Ertugliflozin | Ertugliflozin + Metformin | Ertugliflozin | 100.3 (97.4, 103.3) | 97.1 (88.8, 106.3) |
| Metformin | Ertugliflozin + Metformin | Metformin | 100.9 (90.6, 112.4) | 94.0 (82.9, 106.6) |
| Ertugliflozin‐Glimepiride Study | ||||
| Ertugliflozin | Ertugliflozin + Glimepiride | Ertugliflozin | 102.1 (97.2, 107.3) | 98.2 (92.2, 104.6) |
| Glimepiride | Ertugliflozin + Glimepiride | Glimepiride | 109.8 (98.1, 122.9) | 97.4 (71.1, 133.5) |
| Ertugliflozin–Simvastatin Study | ||||
| Ertugliflozin | Ertugliflozin + Simvastatin | Ertugliflozin | 102.4 (99.6, 105.3) | 105.2 (98.3, 112.5) |
| Simvastatin | Ertugliflozin + Simvastatin | Simvastatin | 123.8 (90.9, 168.7) | 119.1 (97.2, 145.8) |
| Simvastatin Acid | Ertugliflozin + Simvastatin | Simvastatin | 130.5 (108.3, 157.1) | 115.7 (95.7, 139.7) |
AUCinf, area under the plasma concentration–time profile from time 0 extrapolated to infinite time; CI, confidence interval; Cmax, maximum observed plasma concentration; GMR, geometric mean ratio.
Figure 2.
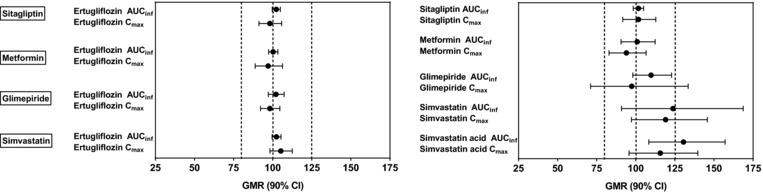
Forest plot of statistical summary of treatment comparisons for ertugliflozin, sitagliptin, metformin, glimepiride, and simvastatin. AUCinf, area under the plasma concentration–time profile from time 0 extrapolated to infinite time; CI, confidence interval; Cmax, maximum observed plasma concentration; GMR, geometric mean ratio; the vertical dashed lines are 80%, 100%, and 125%.
Tmax and t1/2 values of ertugliflozin when administered alone or with coadministered drugs were also similar. Plasma concentrations of ertugliflozin increased rapidly with median Tmax ranging from 1.0 to 3.0 hours when administered alone versus 0.5 to 3.0 hours when coadministered with sitagliptin, metformin, glimepiride, or simvastatin. The mean t1/2 of ertugliflozin administered alone ranged from 10.63 to 12.63 hours compared with 11.27 to 14.17 hours when coadministered with other drugs (Table 2).
Effect of Ertugliflozin on the PK of Coadministered Drugs
Following oral administration of sitagliptin 100 mg alone or metformin 1000 mg alone or either drug in combination with ertugliflozin 15 mg, mean plasma sitagliptin and metformin concentration–time profiles were nearly superimposable between treatments (Figure 1), and AUCinf, Cmax, and AUClast values were equivalent between treatments in both ertugliflozin‐sitagliptin and ertugliflozin‐metformin studies. While not prospectively specified, the 90% CIs for the geometric ratios of sitagliptin and metformin AUCinf and Cmax were within the equivalence bounds of 80% to 125%, indicating that coadministration of ertugliflozin with sitagliptin and metformin did not have any meaningful impact on sitagliptin or metformin PK (Table 3).
Following oral administration of glimepiride 1 mg alone or with ertugliflozin 15 mg, mean plasma glimepiride concentration–time profiles were similar (Figure 1). One subject in the study, who received ertugliflozin 15 mg, vomited within 2 times the median ertugliflozin Tmax after administration; 2 subjects, who received ertugliflozin 15 mg and glimepiride 1 mg, vomited within 2 times the median ertugliflozin or glimepiride Tmax after administration; therefore, as per the statistical analysis plan, data from these 3 subjects for the affected treatments were excluded from the PK analysis. The 90% CI for the geometric mean ratios of glimepiride AUCinf fell within the equivalence bounds of 80% to 125%. The 90% CI for the geometric mean ratio of Cmax for glimepiride was wider (71.1%, 133.5%; Table 3). For most subjects, 2 peaks were observed in the plasma glimepiride concentration–time profile, which resulted in a wide range of Tmax (1.0‐12.0 h) and an intrasubject variability of 53.5% for Cmax.
Following oral administration of simvastatin 40 mg alone or with ertugliflozin 15 mg, mean plasma simvastatin and simvastatin acid concentration–time profiles were similar (Figure 1). Geometric mean AUCinf and Cmax ratios for simvastatin were increased slightly (24% and 19%, respectively) when simvastatin was coadministered with ertugliflozin than when administered alone. Geometric mean ratios of AUCinf and Cmax for simvastatin acid also increased in the presence of ertugliflozin by 30% and 16%, respectively (Table 3). The Tmax and t1/2 of simvastatin and simvastatin acid were comparable between the 2 treatments (Table 2).
Safety Results
A single dose of ertugliflozin 15 mg was well tolerated when administered alone or in combination with sitagliptin, metformin, glimepiride, or simvastatin in healthy subjects. Treatment with coadministered drugs was also well tolerated in all studies. No deaths, serious/severe AEs, or discontinuations due to AEs were reported. The most commonly reported AEs were gastrointestinal disorders in the metformin and glimepiride studies, and headache in the simvastatin study. Treatment‐emergent AEs occurring in ≥2 subjects in any treatment group for each study are summarized in Table S1.
Discussion
Ertugliflozin has been recently approved in the US and EU for the treatment of adults with T2DM, and has been shown to improve glycemic control and reduce body weight and systolic blood pressure in patients with inadequate glycemic control.5, 7 It is likely to be used in combination with a number of other antihyperglycemic and lipid‐lowering agents (eg, statins) for comorbidities associated with diabetes. The efficacy of ertugliflozin in combination with sitagliptin and metformin was assessed in 3 and 4 phase III studies, respectively.5, 7, 22, 23 Coadministration of ertugliflozin with sitagliptin 100 mg provided greater glycemic control compared with each individual dose of ertugliflozin and sitagliptin with inadequate control on either metformin or diet and exercise.5 Similar to these results, on a background of metformin, ertugliflozin at 5‐ and 15‐mg doses provided sustained glycemic control over 52 weeks.23 Ertugliflozin as an add‐on to metformin, and metformin and sitagliptin, also provided clinically meaningful body weight and systolic blood pressure reductions.5, 7, 22
Due to the linear and time‐independent PK of ertugliflozin, single‐dose PK are predictive of multiple‐dose PK of ertugliflozin. There is minimal accumulation observed after repeated ertugliflozin dosing and, based on in vitro data, drug interaction between ertugliflozin and concomitant medications is not expected. Therefore, the single‐dose study design used was considered to be adequate for determining the drug interaction with co‐administered drugs. Ertugliflozin 15 mg was used in these studies as it is the highest dose evaluated in the phase 3 development program. Across all 4 DDI studies presented here, the plasma concentrations of ertugliflozin observed were as expected and similar to other studies using a single ertugliflozin 15‐mg dose.8, 24, 25
Glucuronidation (UGT1A9 and UGT2B7) is the major elimination pathway of ertugliflozin, with only minor contributions from oxidative metabolism (CYP3A4/5, CYP2C8) and renal excretion.2 Although ertugliflozin is a substrate of P‐glycoprotein and breast cancer resistance protein, no clinically relevant interaction is expected with inhibitors of these transporters based on ertugliflozin's oral bioavailability of approximately 100%.8 As expected, no differences were observed in ertugliflozin PK when coadministered with a single dose of sitagliptin (100 mg), metformin (1000 mg), glimepiride (1 mg), or simvastatin (40 mg), compared with a single dose of ertugliflozin (15 mg) alone. The geometric mean ratios of AUCinf and Cmax for ertugliflozin fell within the accepted equivalence limits (80% to 125% in all 4 studies), suggesting that there was no meaningful impact of these commonly coadministered drugs on ertugliflozin PK.
No differences were observed in sitagliptin or metformin PK when coadministered with ertugliflozin, compared with administration of single doses alone, as the 90% CIs for the geometric mean ratios for AUCinf and Cmax for sitagliptin and metformin were within the accepted equivalence limits of 80% to 125%.
In the ertugliflozin‐glimepiride DDI study, ertugliflozin had no meaningful impact on the PK of glimepiride as median plasma glimepiride concentration–time profiles with or without ertugliflozin were similar. The 90% CI for the geometric mean ratio for glimepiride AUCinf, with or without ertugliflozin, also fell within the equivalence limit of 80% to 125%. The majority of subjects had 2 peaks in the plasma glimepiride concentration–time profile, resulting in high variability in Tmax. Similarly, Cmax values for glimepiride in both treatment arms were also variable; this high variability and occurrence of 2 peaks was likely due to the administration of the 20% glucose solution until approximately 4 hours after the dose in each period. Due to the high variability, the 90% CI for the geometric mean ratio for Cmax fell outside the equivalence limits (90% CI, 71.1, 133.5). However, the geometric mean ratio for glimepiride Cmax was 97.4%, indicating no clinically meaningful differences in glimepiride PK when coadministered with ertugliflozin compared with administration of glimepiride alone. A similar PK profile for glimepiride with 2 peaks was observed in a study conducted to assess the interaction between dapagliflozin (another SGLT2 inhibitor) and glimepiride, in which a glucose solution was also administered.26
Coadministration of ertugliflozin 15 mg and simvastatin 40 mg slightly increased simvastatin AUCinf and Cmax by 24% and 19%, respectively, and increased simvastatin acid AUCinf and Cmax by 30% and 16%, respectively; the upper limits of the 90% CIs for the geometric mean ratios were outside the equivalence limits. Other SGLT2 inhibitors such as canagliflozin, dapagliflozin, and empagliflozin have also been evaluated for their interaction with simvastatin. Like ertugliflozin, both canagliflozin and dapagliflozin slightly increased the AUC of simvastatin and simvastatin acid by 12% to 19% and 18% to 30%, respectively.27 Empagliflozin had no effect on the AUCinf and Cmax of simvastatin and simvastatin acid.28 Ertugliflozin and its 2 major glucuronide metabolites have been evaluated extensively in vitro for their potential to inhibit or induce the drug‐metabolizing enzyme (CYP3A4) or transporters (OATP) involved in the disposition of simvastatin. These in vitro studies have shown that neither ertugliflozin nor the glucuronide metabolites are inhibitors or inducers of CYP3A4 or OATP. As such, the mechanism for the small increases in simvastatin and its active metabolite's exposure by ertugliflozin are not known. However, these changes in simvastatin and simvastatin acid exposure are not considered clinically relevant, as these modest increases are not expected to alter the active 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitory activity of simvastatin.
Overall, in vitro data indicate that clinically meaningful drug interactions via ertugliflozin‐mediated inhibition or induction of CYP/UGT isozymes and transporters with concomitant medications that are substrates of OAT3 (sitagliptin), OCT2 (metformin), CYP isozymes (glimepiride, statins), and OATP1B1 (statins) are not anticipated. The observed lack of interaction between ertugliflozin and sitagliptin, metformin, glimepiride, and simvastatin was consistent with that predicted by the known metabolism of each drug as well as in vitro inhibition data.
The results of the ertugliflozin DDI studies with concomitant medications presented here are consistent with those reported for other SGLT2 inhibitors. Canagliflozin is metabolized by UGT1A9 and UGT2B4 and did not exhibit clinically relevant DDIs when administered with metformin or simvastatin.29 Dapagliflozin is also metabolized mainly in the liver and kidneys by UGT1A9,28 and a lack of clinically meaningful DDI was reported when dapagliflozin was coadministered with either sitagliptin, metformin, glimepiride, or simvastatin.26, 27 Similarly, no relevant DDIs were observed when empagliflozin was coadministered with simvastatin,30 metformin,31 or sitagliptin.32
In summary, the lack of PK interactions demonstrates that ertugliflozin can be administered with sitagliptin, metformin, simvastatin, or glimepiride without any need for dose adjustments. However, when ertugliflozin is coadministered with sulfonylureas, the sulfonylurea dose may need to be reduced to decrease the risk of hypoglycemia.
Supporting information
Table S1. All‐Causality (Treatment‐Related) AEs Occurring in ≥2 Subjects in Any Treatment Group
Acknowledgments
We thank Kyle Matschke, MAS, of Pfizer Inc. and Lata Maganti, PhD, of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, for reviewing the design and statistical analysis results.
Declaration of Conflicting Interests
H.S., Y.L., A.H., D.S., S.G.T., and V.S. are employees of Pfizer Inc. V.K.D. and C.A. were employees of Pfizer Inc. at the time of study conduct. S.Z. and R.K. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey. D.L.C. was an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, at the time of study conduct.
Funding
These studies (2014‐002400‐24; 2014‐002816‐17; 2014‐005208‐11; 2014‐004275‐24) were sponsored by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, in collaboration with Pfizer Inc., New York, New York. Editorial support was provided by Helen Jones, PhD, and Radhika Bhatia, PhD, of Engage Scientific Solutions and was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, in collaboration with Pfizer Inc., New York, New York.
References
- 1. Kalgutkar AS, Tugnait M, Zhu T, et al. Preclinical species and human disposition of PF‐04971729, a selective inhibitor of the sodium‐dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus. Drug Metab Dispos. 2011;39(9):1609–1619. [DOI] [PubMed] [Google Scholar]
- 2. Miao Z, Nucci G, Amin N, et al. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF‐04971729) in healthy male subjects. Drug Metab Dispos. 2013;41(2):445–456. [DOI] [PubMed] [Google Scholar]
- 3. Amin NB, Wang X, Jain SM, Lee DS, Nucci G, Rusnak JM. Dose‐ranging efficacy and safety study of ertugliflozin, a sodium‐glucose co‐transporter 2 inhibitor, in patients with type 2 diabetes on a background of metformin. Diabetes Obes Metab. 2015;17(6):591–598. [DOI] [PubMed] [Google Scholar]
- 4. Amin NB, Wang X, Mitchell JR, Lee DS, Nucci G, Rusnak JM. Blood pressure‐lowering effect of the sodium glucose co‐transporter‐2 inhibitor ertugliflozin, assessed via ambulatory blood pressure monitoring in patients with type 2 diabetes and hypertension. Diabetes Obes Metab. 2015;17(8):805–808. [DOI] [PubMed] [Google Scholar]
- 5. Pratley RE, Eldor R, Raji A, et al. Ertugliflozin plus sitagliptin versus either individual agent over 52 weeks in patients with type 2 diabetes mellitus inadequately controlled with metformin: The VERTIS FACTORIAL randomized trial. Diabetes Obes Metab. 2018;20(5):1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Terra SG, Focht K, Davies M, et al. Phase III, efficacy and safety study of ertugliflozin monotherapy in people with type 2 diabetes mellitus inadequately controlled with diet and exercise alone. Diabetes Obes Metab. 2017;19(5):721–728. [DOI] [PubMed] [Google Scholar]
- 7. Dagogo‐Jack S, Liu J, Eldor R, et al. Efficacy and safety of the addition of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sitagliptin: The VERTIS SITA2 placebo‐controlled randomized study. Diabetes Obes Metab. 2018;20(3):530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raje S, Callegari E, Sahasrabudhe V, et al. Determination of absolute oral bioavailability and fraction absorbed of ertugliflozin using a novel microtracer approach [published online ahead of print March 25, 2018]. Clin Transl Sci. 10.1111/cts.12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sahasrabudhe V, Fediuk DJ, Matschke K, et al. Effect of food on the pharmacokinetics of ertugliflozin and its fixed dose combinations: ertugliflozin/sitagliptin and ertugliflozin/metformin Clin Pharmacol Ther. 2017;101(S1):S86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase‐4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91(11):4612–4619. [DOI] [PubMed] [Google Scholar]
- 11. Chu XY, Bleasby K, Yabut J, et al. Transport of the dipeptidyl peptidase‐4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P‐glycoprotein. J Pharmacol Exp Ther. 2007;321(2):673–683. [DOI] [PubMed] [Google Scholar]
- 12. Vincent SH, Reed JR, Bergman AJ, et al. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab Dispos. 2007;35(4):533–538. [DOI] [PubMed] [Google Scholar]
- 13. Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(11):820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goodarzi MO, Bryer‐Ash M. Metformin revisited: re‐evaluation of its properties and role in the pharmacopoeia of modern antidiabetic agents. Diabetes Obes Metab. 2005;7(6):654–665. [DOI] [PubMed] [Google Scholar]
- 15. Hundal RS, Inzucchi SE. Metformin: new understandings, new uses. Drugs. 2003;63(18):1879–1894. [DOI] [PubMed] [Google Scholar]
- 16. Niemi M, Cascorbi I, Timm R, Kroemer HK, Neuvonen PJ, Kivisto KT. Glyburide and glimepiride pharmacokinetics in subjects with different CYP2C9 genotypes. Clin Pharmacol Ther. 2002;72(3):326–332. [DOI] [PubMed] [Google Scholar]
- 17. Igel M, Sudhop T, von Bergmann K. Metabolism and drug interactions of 3‐hydroxy‐3‐methylglutaryl coenzyme A‐reductase inhibitors (statins). Eur J Clin Pharmacol. 2001;57(5):357–364. [DOI] [PubMed] [Google Scholar]
- 18. DIDB®: the metabolism & transport drug interaction database . University of Washington; 2017. https://www.druginteractioninfo.org/. Accessed November 11, 2017.
- 19. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. [DOI] [PubMed] [Google Scholar]
- 20. Levey AS, Coresh J, Greene T, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006;145(4):247–254. [DOI] [PubMed] [Google Scholar]
- 21. Sahasrabudhe V, Terra SG, Hickman A, et al. The effect of renal impairment on the pharmacokinetics and pharmacodynamics of ertugliflozin in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2017;57(11):1432–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller SS, Krumins T, Zhou HJ, et al. Ertugliflozin and sitagliptin co‐initiation in patients with type 2 diabetes: the VERTIS SITA randomized study. Diabetes Ther. 2018;9(1):253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenstock J, Frias J, Pall D, et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET). Diabetes Obes Metab. 2018;20(3):520–529. [DOI] [PubMed] [Google Scholar]
- 24. Kumar V, Sahasrabudhe V, Liang Y, et al. Effect of rifampin on the pharmacokinetics of ertugliflozin in healthy subjects. Clin Pharmacol Ther. 2016;99(S1):S47. [DOI] [PubMed] [Google Scholar]
- 25. Kumar V, Liang Y, Matschke K, et al. A PK/PD study comparing twice‐daily to once‐daily dosing of ertugliflozin in healthy subjects. Clin Pharmacol Ther. 2017;101(S1):S71. [Google Scholar]
- 26. Kasichayanula S, Liu X, Shyu WC, et al. Lack of pharmacokinetic interaction between dapagliflozin, a novel sodium‐glucose transporter 2 inhibitor, and metformin, pioglitazone, glimepiride or sitagliptin in healthy subjects. Diabetes Obes Metab. 2011;13(1):47–54. [DOI] [PubMed] [Google Scholar]
- 27. Kasichayanula S, Chang M, Liu X, et al. Lack of pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, or digoxin. Ad Ther. 2012;29(2):163–177. [DOI] [PubMed] [Google Scholar]
- 28. Kasichayanula S, Liu X, Lacreta F, Griffen SC, Boulton DW. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium‐glucose co‐transporter type 2. Clin Pharmacokinet. 2014;53(1):17–27. [DOI] [PubMed] [Google Scholar]
- 29. Devineni D, Manitpisitkul P, Murphy J, et al. Effect of canagliflozin on the pharmacokinetics of glyburide, metformin, and simvastatin in healthy participants. Clin Pharmacol Drug Dev. 2015;4(3):226–236. [DOI] [PubMed] [Google Scholar]
- 30. Macha S, Lang B, Pinnetti S, Broedl UC. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter 2 inhibitor, and simvastatin following co‐administration in healthy volunteers. Int J Clin Pharmacol Ther. 2014;52(11):973–980. [DOI] [PubMed] [Google Scholar]
- 31. Macha S, Dieterich S, Mattheus M, Seman LJ, Broedl UC, Woerle HJ. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter‐2 (SGLT2) inhibitor, and metformin following co‐administration in healthy volunteers. Int J Clin Pharmacol Ther. 2013;51(2):132–140. [DOI] [PubMed] [Google Scholar]
- 32. Brand T, Macha S, Mattheus M, Pinnetti S, Woerle HJ. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter‐2 (SGLT‐2) inhibitor, coadministered with sitagliptin in healthy volunteers. Adv Ther. 2012;29(10):889–899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. All‐Causality (Treatment‐Related) AEs Occurring in ≥2 Subjects in Any Treatment Group