

Abstract
Perinatal exposure to endocrine disrupting chemicals negatively impacts health, but the mechanism by which such toxicants damage long-term reproductive and metabolic function is unknown. Lipid metabolism plays a pivotal role in steroid hormone synthesis as well as energy utilization and storage; thus, aberrant lipid regulation may contribute to phthalate-driven health impairments. In order to test this hypothesis, we specifically examined epigenetic disruptions in lipid metabolism pathways after perinatal phthalate exposure. During gestation and lactation, pregnant Long–Evans rat dams were fed environmentally relevant doses of phthalate mixture: 0 (CON), 200 (LO), or 1000 (HI) µg/kg body weight/day. On PND90, male offspring in the LO and HI groups had higher body weights than CON rats. Gene expression of lipid metabolism pathways was altered in testis and adipose tissue of males exposed to the HI phthalate dosage. Specifically, Srebf1 was downregulated in testis and Srebf2 was upregulated in adipose tissue. In testis of HI rats, DNA methylation was increased at two loci and reduced at one other site surrounding Srebf1 transcription start site. In adipose tissue of HI rats, we observed increased DNA methylation at one region within the first intron of Srebf2. Computational analysis revealed several potential transcriptional regulator binding sites, suggesting functional relevance of the identified differentially methylated CpGs. Overall, we show that perinatal phthalate exposure affects lipid metabolism gene expression in a tissue-specific manner possibly through altering DNA methylation of Srebf1 and Srebf2.
Keywords: gene expression, lipid metabolism, transcription, hypermethylation, epigenetic mechanisms
Introduction
Phthalates are environmental toxicants that have endocrine disrupting effects. Phthalates can be found in many household plastic and cosmetic products [1, 2] and as contaminants in foods such as poultry and dairy [3]. Due to phthalate influence on hormone balance, epidemiological studies have extensively examined the relationship between phthalates and male reproductive health. Phthalate exposure in adulthood has been associated with reduced sperm quality and testosterone levels [4–6], while prenatal phthalate exposure results in shorter anogenital distance in infants [7]. Several animal models have confirmed and expanded upon these observations. Early-life exposure has been of particular interest due to heightened sensitivity during the critical period and the potential to impact long-term health outcomes. In addition to validating findings in human studies [8, 9], rodent models have shown that perinatal phthalate exposure disrupts Leydig [10, 11] and Sertoli [12, 13] cell populations as well as induces aberrant DNA methylation [14, 15] and histone modifications [10, 16] in the testis.
Additionally, studies have quantified metabolic outcomes after phthalate exposure. While some have suggested a positive association between early-life phthalate exposure and body mass index (BMI) [17, 18], others have found no correlation [19] or a negative correlation between perinatal phthalates, BMI, waist circumference, and body fat percentage [20, 21]. Similar inconsistencies have been reported in animal models. Indeed, early-life phthalate exposure in rodents has been shown to have positive [22, 23], negative [24–26], and no relationship [27, 28] with body weight. There remains no clear consensus as to whether early-life phthalate exposure produces sustained metabolic complications. Thus, understanding the possible mechanisms by which phthalates affect reproduction and metabolism is critical in clarifying the long-term health implications of environmental exposure.
Proper metabolic and reproductive function involves appropriate regulation of lipid synthesis, catabolism, and storage. Obesity is characterized by excessive fat storage and hyperlipidemia. As for reproductive health, not only does adiposity impact endocrine function, but cholesterol itself is also a precursor of androgen and estrogen. Previous experimentation in animal and in vitro models suggests that phthalates may induce lipid accumulation by upregulating expression of fatty acid synthesis and lipid transport genes [28–34]. Such metabolic processes are highly regulated and involve a number of transcription factors. Given their widespread regulatory role in lipid metabolism, sterol regulatory element binding proteins (SREBPs) have been hypothesized to underlie phthalate-induced metabolic dysregulation [35, 36]. When cellular sterol levels are low, SREBP cleavage-activating protein (SCAP) escorts SREBP from the endoplasmic reticulum to the Golgi apparatus, where it is cleaved and released by Site-1 protease and Site-2 protease [37]. SREBP then translocates to the nucleus and can bind sterol response elements to induce transcription of genes such as fatty acid synthase (Fasn), acetyl CoA carboxylase (Acc), and HMG-CoA synthase (Hmgcs) [38, 39]. SREBP1 upregulates lipogenesis and triglyceride synthesis while SREBP2 promotes cholesterol uptake and synthesis. Thus, SREBPs are critical in responding to dietary conditions and maintaining metabolic health across tissues.
We sought to uncover the impact of perinatal phthalate exposure on lipid metabolism in key metabolic and reproductive tissues. We hypothesized that aberrant DNA methylation and elevated expression of SREBP genes may underlie the metabolic and reproductive abnormalities caused by phthalates. In the current experiment, we asked whether perinatal phthalate exposure impacts lipid metabolism in adult offspring. Long–Evans rat dams consumed environmentally relevant doses of phthalates during gestation and lactation. Male offspring were sacrificed at postnatal day (PND) 90 and gene expression and DNA methylation were measured in testis, adipose, and liver. Overall we show that phthalate exposure drives tissue-specific changes in gene expression and potentially operates through an epigenetic mechanism.
Results
Body Weight and Composition
Pregnant Long–Evans rat dams were fed one of three phthalate dosages from gestational day 2 until PND10: 0 [control (CON)], 200 [low (LO)], or 1000 [high (HI)] µg/kg of body weight/day. Body weights of male offspring were measured at PND10 and 90. At PND10, the LO group had significantly lower body weights than controls (P = 0.015; Fig. 1A). However, at PND90, this trend was reversed, as both LO and HI animals had higher body weights than control (P = 0.0080 and P = 0.070, respectively; Fig 1B). Additionally, body fat and lean mass were measured on PND90. LO rats had slightly lower and HI rats had slightly higher average body fat percentage than CON, however neither of these were significant (P = 0.29 and P = 0.31, respectively; Fig. 1C).
Figure 1:
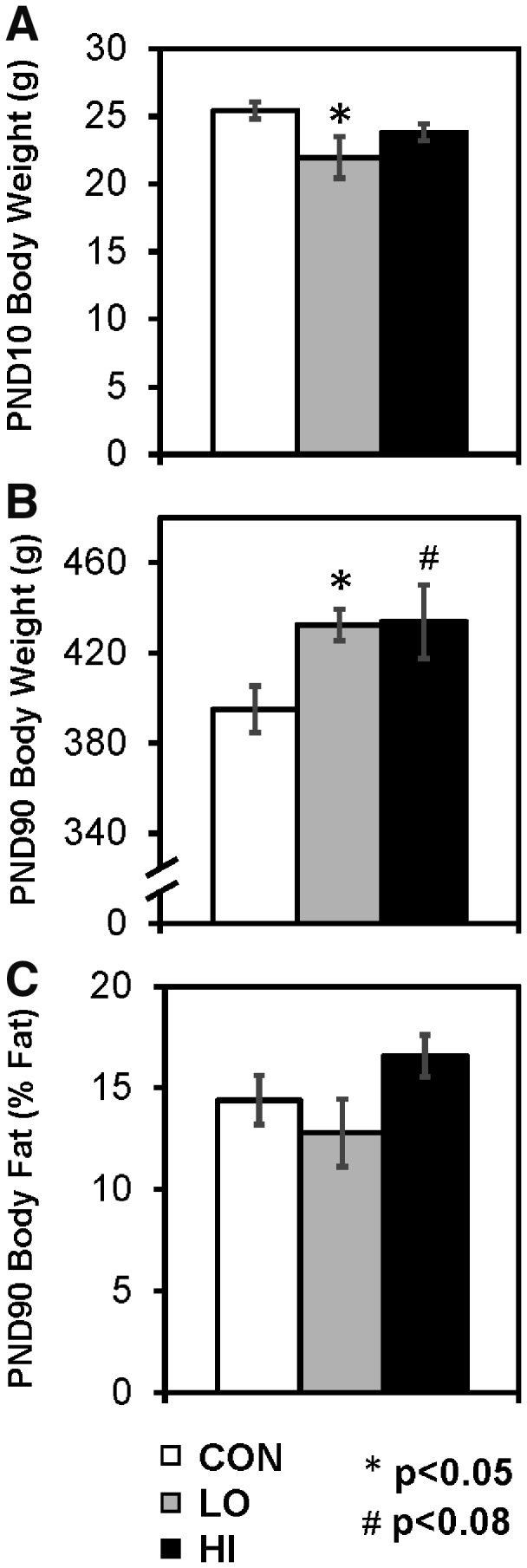
body weights for male offspring on (A) PND10 and (B) PND90. (C) Body fat percentage on PND90. Values are represented as mean ± SEM. *P < 0.05, #P < 0.08
Gene Expression
On PND90, animals were sacrificed and testis, gonadal adipose, and liver were removed for analysis. Due to the endocrine disrupting properties of phthalates and the important role of lipids in hormone synthesis, we measured genes involved in tissue-specific as well as general lipid metabolic processes. In testis, we measured cholesterol and hormone synthesis, lipogenesis, and transcriptional regulation genes. In phthalate-exposed animals, we found a decrease in Niemann–Pick intracellular cholesterol transporter 1 (Npc1; P = 0.042), fatty acid synthase (Fasn; P = 0.012), and Srebf1 (P = 0.027; Table 1). These differences were only observed in the HI group. Moreover, we saw a slight decrease in glycerol-3-phosphate acyltransferase (Gpam; P = 0.076) and stearoyl-CoA desaturase (Scd; P = 0.084). In the LO group, there was a non-significant increase in Srebf2 expression (P = 0.065).
Table 1:
gene expression in testis at PND90 (values are represented as mean ± SEM)
| Gene | CON | LO | HI |
|---|---|---|---|
| Cholesterol and hormone synthesis | |||
| Cyp11a1 | 0.56 (0.17) | 0.60 (0.13) | 0.44 (0.021) |
| Cyp19a1 | 1.29 (0.24) | 1.52 (0.22) | 1.15 (0.11) |
| Hmgcr | 1.08 (0.16) | 1.50 (0.17) | 1.2 (0.077) |
| Hsd17b3 | 0.51 (0.054) | 0.70 (0.10) | 0.54 (0.04) |
| Insig1 | 1.10 (0.041) | 1.17 (0.11) | 1.00 (0.047) |
| Npc1 | 0.99 (0.16) | 0.84 (0.097) | 0.64 (0.034)* |
| Scap | 0.87 (0.16) | 0.92 (0.14) | 0.68 (0.032) |
| Scp2 | 1.04 (0.054) | 1.06 (0.11) | 1.07 (0.051) |
| Star | 0.56 (0.099) | 0.57 (0.082) | 0.57 (0.052) |
| Tspo | 0.80 (0.16) | 0.81 (0.13) | 0.57 (0.031) |
| Fatty acids and triacylglycerols synthesis | |||
| Acacb | 1.03 (0.10) | 1.19 (0.081) | 1.01 (0.052) |
| Fasn | 0.82 (0.059) | 0.78 (0.073) | 0.66 (0.025)* |
| Gpam | 0.96 (0.18) | 0.76 (0.061) | 0.64 (0.016)** |
| Scd | 0.89 (0.17) | 0.78 (0.084) | 0.60 (0.014) |
| Transcriptional regulators | |||
| Lxra | 0.97 (0.045) | 1.05 (0.14) | 0.91 (0.042) |
| Srebf1 | 0.77 (0.088) | 0.72 (0.047) | 0.56 (0.015)* |
| Srebf2 | 0.94 (0.053) | 1.18 (0.10)** | 0.92 (0.031) |
P < 0.05.
P < 0.08.
In adipose tissue, we measured an increase in Fasn (P = 0.040) and Srebf2 (P = 0.031; Table 2). We found slight increases in expression of cholesterol-regulating gene, insulin induced gene 1 (Insig1; P = 0.061) as well as Scd (P = 0.073). In liver, only minimal gene expression differences were observed (Table 3). Scd was upregulated in HI group, but this was not statistically significant (P = 0.066).
Table 2:
gene expression in gonadal adipose at PND90 (values are represented as mean ± SEM)
| Gene | CON | LO | HI |
|---|---|---|---|
| Cholesterol | |||
| Hmgcr | 1.14 (0.12) | 1.11 (0.12) | 1.18 (0.13) |
| Insig1 | 1.51 (0.22) | 2.07 (0.48) | 2.86 (0.58)** |
| Npc1 | 1.50 (0.12) | 1.21 (0.15) | 1.49 (0.28) |
| Scap | 1.19 (0.13) | 1.21 (0.20) | 1.38 (0.20) |
| Fatty acids and triacylglycerols synthesis | |||
| Acacb | 2.13 (0.41) | 1.51 (0.29) | 1.92 (0.37) |
| Fasn | 0.57 (0.12) | 0.73 (0.19) | 1.87 (0.53)* |
| Gpam | 1.32 (0.20) | 1.13 (0.19) | 1.69 (0.26) |
| Scd | 1.30 (0.42) | 1.21 (0.33) | 4.34 (1.27)** |
| Transcriptional regulators | |||
| Srebf1 | 1.32 (0.25) | 1.46 (0.24) | 1.42 (0.20) |
| Srebf2 | 0.90 (0.12) | 1.23 (0.15) | 1.61 (0.20)* |
| Lxra | 1.93 (0.36) | 1.89 (0.37) | 1.96 (0.30) |
P < 0.05.
P < 0.08.
Table 3:
gene expression in liver at PND90 (values are represented as mean ± SEM)
| Gene | CON | LO | HI |
|---|---|---|---|
| Lipid import | |||
| Ldlr | 1.22 (0.23) | 1.25 (0.16) | 1.67 (0.33) |
| Scarb1 | 0.75 (0.070) | 0.79 (0.065) | 0.81 (0.11) |
| Lipid export | |||
| Mttp | 1.13 (0.089) | 1.08 (0.055) | 1.12 (0.14) |
| ApoB | 0.84 (0.056) | 0.76 (0.064) | 0.85 (0.13) |
| ApoE | 1.44 (0.20) | 1.07 (0.20) | 1.09 (0.16) |
| Cholesterol | |||
| Hmgcr | 1.40 (0.20) | 1.06 (0.21) | 1.50 (0.37) |
| Insig1 | 1.27 (0.35) | 1.02 (0.12) | 1.44 (0.29) |
| Npc1 | 0.86 (0.16) | 0.75 (0.083) | 0.76 (0.10) |
| Scap | 0.78 (0.049) | 0.88 (0.071) | 0.92 (0.092) |
| Fatty acids and triacylglycerols synthesis | |||
| Acacb | 1.10 (0.27) | 1.44 (0.27) | 1.42 (0.29) |
| Fasn | 0.73 (0.10) | 1.00 (0.33) | 1.28 (0.34) |
| Gpam | 0.64 (0.077) | 0.78 (0.15) | 0.73 (0.15) |
| Scd | 1.03 (0.31) | 1.61 (0.53) | 2.61 (0.70)** |
| Transcriptional regulators | |||
| Lxra | 1.44 (0.17) | 1.50 (0.13) | 1.73 (0.26) |
| Srebf1 | 0.76 (0.17) | 1.02 (0.22) | 1.06 (0.18) |
| Srebf2 | 0.82 (0.049) | 0.87 (0.075) | 0.87 (0.064) |
P < 0.05.
P < 0.08.
DNA Methylation
Next, we tested the hypothesis that differences in gene expression may be caused by phthalate-mediated alterations in DNA methylation. We identified several differentially expressed lipid metabolism genes, but due to their potential for widespread transcriptional regulation, we only measured DNA methylation associated with Srebf1 and Srebf2. MSP was performed for several sites surrounding the transcription start site (TSS) of the two genes. The HI phthalate had reduced testicular Srebf1 expression, whereas in adipose and liver, Srebf1 expression was unchanged (Fig. 2A). The sequence of Srebf1 includes one cytosine phosphate guanine dinucleotides (CpG) island straddling the TSS (Fig. 2B). We measured DNA methylation of three CpG loci located in islands and one locus in either shore. In testis, we detected increased DNA methylation in the HI animals within the upstream and downstream shores (P = 0.061 and P = 0.048, respectively; Fig. 2C). Additionally, there was a decrease in methylation in the HI group in the island CpGs located 295 bp downstream of the TSS (P = 0.080). There were not DNA methylation differences in adipose tissue (Fig. 2D). In liver, there was increased DNA methylation in the LO group in the island CpGs 295 bp downstream of the TSS (P = 0.011; Fig. 2E).
Figure 2:

(A) mRNA expression of Srebf1 across tissues. (B) CpG distribution around the Srebf1 transcription start site (TSS). Black line graph represents the guanine-cytosine content across the region (GC%). Blue line graph represents observed/expected CpG ratio (observed/expected). Red bars represent CpG sites. Light blue shaded area represents CpG island. Green arrows represent location of CpGs measured by MSP. Bottom three graphs show DNA methylation in (C) testis, (D) adipose, and (E) liver. Gene expression and DNA methylation values are represented as mean fold change compared to CON ± SEM. *P < 0.05, #P < 0.08
DNA methylation changes around Srebf2 were minimal. Gene expression in adipose tissue was significantly increased in the HI group. In testis, there was a slight increase in the LO group (Fig. 3A). Surrounding the Srebf2 TSS, there is one large CpG island (Fig. 3B). Another CpG island is located ∼500 bp downstream. In testis, there was a non-significant decrease in DNA methylation in the LO group at CpGs located 78 bp upstream of the TSS (P = 0.058; Fig. 3A). The same locus was differentially methylated in the liver, where it was hypermethylated in the HI group (P = 0.0021; Fig. 3E). In adipose, there was increased DNA methylation at an intronic site located 368 bp downstream of the TSS and within the CpG island (P = 0.048; Fig. 3D).
Figure 3:

(A) mRNA expression of Srebf2 across tissues. (B) CpG distribution around the Srebf2 transcription start site (TSS). Black line graph represents the guanine-cytosine content across the region (GC%). Blue line graph represents observed/expected CpG ratio (observed/expected). Red bars represent CpG sites. Light blue shaded area represents CpG island. Green arrows represent location of CpGs measured by MSP. Bottom three graphs show DNA methylation in (C) testis, (D) adipose, and (E) liver. Gene expression and DNA methylation values are represented as mean fold change compared to CON ± SEM. *P < 0.05, #P < 0.08
Transcription Factor Prediction
Finally, we attempted to predict whether the identified differentially methylated regions might impact transcription and expression of Srebf1 and Srebf2. We performed a bioinformatics analysis of the differentially methylated regions using PROMO. First, we looked at three regions around Srebf1 that were differentially methylated in the testis. Within the site 130–100 bp upstream of Srebf1, we found potential binding sites for SMAD3, ELK1, E2F, DP1, and STAT4 (Fig. 4A). Within the region 280–310 bp downstream of the Srebf1 TSS, there was a potential binding site for nuclear factor I/CAAT box transcription factor (NFI/CTF; Fig. 4B). Within the region 355–400 bp downstream of the Srebf1 TSS, we identified putative binding sites for VDR, MYB, and POU3F2 (Fig. 4C). In Srebf2, we examined the region 355–395 bp downstream of the TSS and found sites for ELK1, VDR, and MYB binding (Fig. 4D).
Figure 4:

computationally predicted transcription factor binding sites at differentially methylated loci around (A, B, C) Srebf1 and (D) Srebf2. Red boxes represent differentially methylated CpGs as measured by MSP in testis (Srebf1) and adipose (Srebf2)
Discussion
In this paper, we have demonstrated the long-lasting effect of endocrine disruptors on lipid metabolism. During the perinatal period, Long–Evans rats were administered a mixture of phthalates at concentrations observed in human populations. Phthalates altered early postnatal and adolescent body weight but did not alter fat deposition. Gene expression analysis at PND90 revealed minor alterations in hepatic lipid metabolism genes, but significant changes in cholesterol and fatty acid synthesis in gonadal adipose tissue. Finally, high doses of perinatal phthalates significantly altered cholesterol trafficking and fatty acid metabolism genes in testis. In particular the transcriptional regulator Srebf1 was downregulated, possibly through hypermethylation of its promoter and coding region. Early-life phthalate exposure might interfere with adipose and testicular fatty acid and cholesterol synthesis through epigenetic mechanisms, and thus alter reproductive function.
Phthalates and Lipid Metabolism
Lipid metabolism is an integral part of reproductive and general metabolic health. Through highly regulated pathways, fatty acids, triglycerides, and cholesterol are synthesized in response to environmental and homeostatic stimuli in order to meet metabolic demands (Fig. 5). Such processes are transcriptionally regulated by factors including SREBP1, which upregulates fatty acid and triglyceride synthesis; SREBP2, which promotes cholesterol uptake and synthesis; and LXR, which opposes the effects of SREBP2 and activates SREBP1 (Fig. 5). In testis, cholesterol plays a particularly critical role as a precursor of testosterone (Fig. 5).
Figure 5:

summary of the effects of perinatal phthalate exposure on lipid metabolism pathways. Gene expression changes in different tissue types are noted with stars (P < 0.08)
Previous investigation has suggested that exposure to individual phthalates impacts lipid metabolism. In Caenorhabditis elegans, life-long exposure to phthalates DEHP and DEP increased fat accumulation along with survival and reproductive function, possibly related to the upregulation of fatty acid synthesis (fasn-1, pod-2, fat-5, elo-2, acs-6, sbp-1) and lipid transport genes (vit-2, -4, -5, and -6) [29]. In zebrafish, water-exposure to diisononyl phthalate (DiNP) activated orexigenic signals in the brain and steatohepatitis in adult animals [30]. In human cells, exposure to monoethylhexyl phthalate (MEHP) affected adipocyte size, possibly via increased lipolysis, glucose uptake, and oxygen consumption [31]. In placental cells MEHP produced lipidome-wide changes with specific alterations in glycerolipids and glycerophospholipids and promoted triacylglycerols accumulation [34]. Interestingly, prenatal DEHP exposure in rats was shown to target adrenal gland peroxisome proliferator-activated receptor (PPAR) and cholesterol biosynthesis pathways [33], which rendered offspring more susceptibility to second-hit stressor challenge [32]. Similarly, in utero DEHP exposure has been shown to potentiate the effect of a postnatal high fat-diet [28]. In our study, we used a phthalate mixture to mimic human environmental exposure. However just as individual phthalates induce transcriptional changes in lipid metabolism genes in different tissues, we also observed specific adipose tissue and testicular gene expression changes in cholesterol and fatty acid synthesis pathways.
Lipid metabolism genes that are affected by phthalates vary among tissues, and depend on the window of exposure. In our paper, prenatal exposure to low or high doses of phthalates produced no significant effects in liver. This finding contradicts previous studies that have shown phthalate-mediated changes in hepatic morphology and Ppara gene expression [40, 41]. This difference might be due to the low phthalate dose used in our study (1000× lower). Furthermore, the lack of gene expression changes in liver might not be reflective of the ability of the liver to handle fatty acids and cholesterol, given that the animals were exposed to neither a high fat-diet nor a second stressor later in life.
In adipose tissue, we showed a significant increase in cholesterol, fatty acid, and lipid metabolism-related genes after phthalate treatment. A trend toward increased Insig1 expression might speak to altered cholesterol signaling, which is vital for the production of hormones within adipose tissue. Importantly, Srebf2 (Srebp2) was significantly upregulated by a high dose of phthalates, suggesting higher cholesterol synthesis as its main target genes include Hmgcr and Hmgcs, key enzymes for cholesterol biosynthesis [37, 42]. Moreover, increases in Fasn and Scd indicate a prominent effect on lipid synthesis with high phthalate doses, possibly affecting the ability of adipocytes to accumulate fat.
Lastly, a high dosage of perinatal phthalates decreased expression of genes involved with cholesterol, fatty acid, and triglyceride metabolism, namely Npc1, Fasn, Gpam, and Srebf1. Dysregulation of Srebf1 (Srepb1) is known to affect both fatty acid and cholesterol metabolism via the two known isoforms of SREBP protein 1a and 1c [43]. Indeed, Npc1, Fasn, Gpam are transcriptional targets of Srebp1 [38, 44, 45]. This suggests that the phthalate-induced decrease in expression of these genes may be a result of decreased SREBP1 binding. Previous studies have shown that exposure to DiBP (600 mg/kg BW/day) caused dysregulation of testicular fatty acid and cholesterol-metabolism genes, which impacts steroidogenesis and reproductive function [41]. Studies have shown that androgen production does not require the accumulation of SREBP2 protein [46], thus highlighting the effect of Srebf1 in regulating fatty acid and cholesterol metabolism, and in turn modulating the hormone production of testis and reproductive function.
Phthalates and DNA Methylation
In this study, we provide evidence that early-life phthalate exposure induces different DNA methylation patterns around the Srebf1 and Srebf2 promoters. Several others have highlighted the epigenetic implications of phthalates. In rat testis, maternal phthalate exposure resulted in hypermethylation of steroidogenic factor-1 (SF-1) and specific protein-1 (Sp-1) [14] and hypomethylation of the mineralocorticoid receptor [47]. Phthalates also induced decreased global DNA hydroxymethylation [48] and increased global DNA methylation [49, 50]. In adipose tissue, we have previously observed decreased methylation in the adipogenesis gene, frizzled 1 (Fzd1), and the triglyceride cleaving enzyme, lipoprotein lipase (Lpl) [51]. Phthalate administration in rodent models also resulted in hepatic hypomethylation of the oncogene, c-Myc [52, 53], while another study reported no change in global DNA methylation [54]. Our results add to the literature by demonstrating that phthalates alter DNA methylation in a tissue-specific manner.
Our findings support the idea that DNA methylation in the promoter prevents transcription factor binding and induces a closed chromatin confirmation in order to repress transcription. We found three regions around the Srebf1 TSS that were differentially methylated in the testis. Two of the three were located in CpG shores and were hypermethylated after phthalate exposure. This finding is consistent with previous literature showing that CpG shores are more vulnerable to differential methylation than CpG islands [55, 56]. Additionally, Srebf1 gene expression was downregulated in testis; thus, an increase in DNA methylation follows the canonical assumption that DNA methylation blocks transcription. Transcription factor analysis identified SMAD3, ELK1, E2F, DP1, and STAT4 binding sites within the upstream shore (130–100 bp upstream of Srebf1 TSS). Each of these factors has been shown to activate transcription [57–61]. Within the downstream shore (355–4000 bp downstream of the Srebf1 TSS), there was a potential binding site for VDR, MYB, and POU3F2, which have also been demonstrated to have activational properties [62–64].
We identified two differentially methylated CpG loci that showed a positive association between DNA methylation and gene expression. First, in testis we observed decreased DNA methylation at the CpG site 295 bp downstream of the Srebf1 TSS. This decrease was associated with decreased gene expression. Upon examination of the transcription factor binding sites, we saw that relatively few factors bind to the region. NFI/CTF may activate or repress transcription depending on recruitment of other cofactors and chromatin modifiers [65]. However, the same locus was highly methylated in the liver, despite no change in gene expression. Thus, it may be the case that DNA methylation at this site is not critical for the transcriptional regulation of Srebf1. Secondly, in adipose tissue we observed increased Srebf2 expression accompanied by increased DNA methylation at the region 355–395 bp downstream of the TSS. Binding sites for ELK1, VDR, and MYB were found. Although much of the literature points to the activating role of these transcription factors, there is evidence that ELK1 and MYB display repressive characteristics in certain contexts [66–68]. It is possible that increased methylation could increase transcription by inhibiting repressor binding.
Conclusion
Here we demonstrate the tissue-specific implications of perinatal phthalate exposure. We are among the first to investigate phthalate-induced DNA methylation across tissues; however, our experiments should be viewed in light of a few limitations. First, our sample size was relatively small (n = 8–9/group). This might be one reason that we found several genes and CpGs that were slightly but not statistically altered with phthalate treatment. Our data can be used to generate hypotheses and should be replicated in a larger cohort. Secondly, while we measured body weight and body composition as metabolic outcomes, we did not quantify reproductive outcomes beyond changes in gene expression. Future experimentation should focus on discovering associations between DNA methylation at the identified loci and physiological outcomes such as sperm count and quality as well as other fertility measures. Additionally, we only looked at a limited number of CpG sites using MSP. To gain a more comprehensive picture of phthalate-mediated DNA methylation, genome-wide methods such as bisulfite sequencing or methylated DNA immunoprecipitation sequencing (MeDIP-seq) may be utilized. Finally, future experiments can confirm the impact of DNA methylation and TF binding within the identified loci. We measured differential methylation at CpGs within the Srebf1 and Srebf2 promoters, but it is unclear whether these differences directly impact transcription or whether they are consequences of gene expression changes. Moreover, we report several computationally predicted TF binding sites around Srebf1 and Srebf2, however these findings should be validated for transcriptional relevance through appropriate in vitro assays.
Overall, we show that perinatal phthalate treatment induces distinct gene expression changes in lipid metabolism pathways in testis and adipose tissue but not in the liver. Importantly, expression and DNA methylation of Srebf1 and Srebf2 were altered by phthalates. We propose that perinatal phthalate exposure alters DNA methylation around lipid-related transcription factors, changes the expression of their gene targets, and alters lipid metabolism. This study provides insight into the mechanism by which phthalates might disrupt reproductive and general metabolic outcomes.
Methods
Animals
Three-month old Long–Evans hooded female rats (n = 8–9 per group) were obtained from Harlan Laboratories (Indianapolis, IN). Before breeding, rats were housed in same-sex pairs and were given ad libitum access to a low phytoestrogen diet (Harlan 2020X; Taklad Diets, Madison, WI). During breeding, dams were placed in suspended wire-bottom cages, and once a sperm plug was detected, dams were removed and individually housed. From gestational day 2 until PND10, dams received one of three phthalate treatments, 0 (CON), 200 (LO), or 1000 µg/kg body weight/day (HI).
The phthalate mixture was designed to reflect exposure levels found in pregnant women in Champaign—Urbana, Illinois (unpublished data) as well as exposure levels in the general US population [69]. The mixture consisted of ∼35% diethyl (DEP), 21% bis(2-ethylhexyl) (DEHP), 15% dibutyl (DBP), 15% diisononyl (DiNP), 8% diisobutyl (DiBP), and 5% benzyl butyl (BBP) phthalate. The phthalates were suspended in tocopherol-stripped corn oil at a concentration of 0, 0.6, or 3 mg phthalates/ml for the 0, 200, or 1000 µg phthalates/kg body weight/day doses, respectively. The phthalate mixture was administered orally on top of half a cookie (Newman’s Own organic alphabet cookie, vanilla flavor). Dams were acclimatized for 2 days with half a cookie topped with tocopherol-stripped corn oil, and from gestational day 2 through PND10, dams voluntarily ate the half cookie and phthalate mixture. Additionally, dams were given ad libitum access a control diet (15.8% kcal fat, 63.9% kcal carbohydrate, 20.3% kcal protein; D10012G, Research Diets Inc., New Brunswick, NJ).
On PND25, one male offspring from each dam was randomly selected and moved to its own individual cage (CON: n = 8, LO: n = 9, HI: n = 9). Offspring continued on the D10012G diet and received no phthalates for the remaining duration of the experiment. On PND90, body composition was measured using the EchoMRI-700 Body Composition Analyzer (Echo Medical Systems, Houston, TX). The rats were then sacrificed and liver, testis, and gonadal adipose tissue was dissected out. All tissues were snap-frozen in liquid nitrogen and stored at −80°C for genetic analysis. All procedures were approved by the University of Illinois Institutional Care and Use Committee and adhere to the National Institutes of Health guidelines on the ethical use of animals.
Gene Expression
Frozen tissue was ground in liquid nitrogen and placed in a 1.5 ml tube containing Trizol. Before loading the adipose sample into the column, the tube was spun and the lipid layer was removed. Total RNA was extracted using Direct-zol RNA MiniPrep Plus (Zymo Research, Irvine, CA) with in-column DNase I treatment to eliminate DNA contamination. cDNA synthesis and qRT-PCR were performed as previously described [70]. Primers were designed (VectorNTI software; Life Technologies, Grand Island, NY), validated for specificity (Basic Local Alignment Search Tool; BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi), and synthesized (IDT; Coralville, IA). All mRNA primer sequences and amplification efficiencies can be found in Supplementary Table S1.
DNA Methylation
Genomic DNA was isolated (ZR Genomic DNA Tissue MiniPrep kit; Zymo Research) and bisulfite converted (EZ DNA Methylation Gold Kit; Zymo Research). The EZ DNA Methylation Gold Kit was chosen for its high bisulfite conversion efficiency, which has repeatedly been reported to range between 99 and 100% [71–74]. Thus, we can confidently assume that methylation differences are likely not artifacts of different conversion efficiencies. To perform the conversion reaction, samples were heated to 98°C for 10 min, 64°C for 2.5 h, and held at 4°C. Samples were desulphonated and diluted to 5 ng/μl for methylation specific PCR (MSP) using Sybr Green (Quanta Biosciences, Beverly, MA). Quantitative real-time PCR was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). CT values were quantified based on a serially diluted standard curve and percent methylation was calculated by dividing quantity of methylated DNA by the sum of methylated and unmethylated DNA. Methylation was normalized to the CON group.
Methylated and unmethylated primer pairs were manually designed (Primer Express 3.0.1 software’s Primer Probe Test Tool; Life Technologies). The Tm for each primer was between 68 and 70°C and the CG content was between 30 and 80%. Primers were validated in the IDT OligoAnalyzer to minimize dimers and hairpin loops. Unmethylated DNA was measured using both unmethylated forward and reverse primers. Methylated DNA was measured by pairing one methylated primer with the corresponding unmethylated primer. All MSP primer sequences and amplification efficiencies can be found in Supplementary Table S2.
Transcription Factor and Statistical Analysis
Computational prediction of transcription factor binding was performed with PROMO version 3.0.2 [75, 76]. Identified transcription factor sequences were validated with the Jaspar Database [77]. One way ANOVA followed by pairwise comparisons between CON vs LO and CON vs HI was used to determine statistical significance for body weight, body composition, gene expression, and DNA methylation. Statistical analysis was performed in R version 3.3.2.
Supplementary Material
Acknowledgements
This work was supported, in part, by the Center grant that produced these animals for our analyses (USEPA RD 83543401—Project 3 and NIEHS P01 ES002848—Project 3). L.M., D.G.K., J.M.J., H.C., and Y.X.P. conceived the study; L.M., D.H.S., and D.G.K. conducted the experiments; L.M. analysed the data; L.M., H.C., and Y.X.P. wrote the manuscript.
Conflict of interest statement. None declared.
References
- 1. Koniecki D, Wang R, Moody RP, Zhu J.. Phthalates in cosmetic and personal care products: concentrations and possible dermal exposure. Environ Res 2011;111:329–36. [DOI] [PubMed] [Google Scholar]
- 2. Sathyanarayana S. Phthalates and children's health. Curr Probl Pediatr Adolesc Health Care 2008;38:34–49. [DOI] [PubMed] [Google Scholar]
- 3. Serrano SE, Braun J, Trasande L, Dills R, Sathyanarayana S.. Phthalates and diet: a review of the food monitoring and epidemiology data. Environ Health 2014;13:43.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hauser R, Meeker JD, Duty S, Silva MJ, Calafat AM.. Altered semen quality in relation to urinary concentrations of phthalate monoester and oxidative metabolites. Epidemiology 2006;17:682–91. [DOI] [PubMed] [Google Scholar]
- 5. Chang WH, Li SS, Wu MH, Pan HA, Lee CC.. Phthalates might interfere with testicular function by reducing testosterone and insulin-like factor 3 levels. Hum Reprod 2015;30:2658–70. [DOI] [PubMed] [Google Scholar]
- 6. Meeker JD, Ferguson KK.. Urinary phthalate metabolites are associated with decreased serum testosterone in men, women, and children from NHANES 2011–2012. J Clin Endocrinol Metab 2014;99:4346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Swan SH, Main KM, Liu F, Stewart Sl, Kruse Rl, Calafat AM, Mao CS, Redmon JB, Ternand Cl, Sullivan S. et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect 2005;113:1056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gray LE Jr, Furr J, Tatum-Gibbs KR, Lambright C, Sampson H, Hannas BR, Wilson VS, Hotchkiss A, Foster PM.. Establishing the “biological relevance” of dipentyl phthalate reductions in fetal rat testosterone production and plasma and testis testosterone levels. Toxicol Sci 2016;149:178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li N, Chen X, Zhou X, Zhang W, Yuan J, Feng J.. The mechanism underlying dibutyl phthalate induced shortened anogenital distance and hypospadias in rats. J Pediatr Surg 2015;50:2078–83. [DOI] [PubMed] [Google Scholar]
- 10. Kilcoyne KR, Smith LB, Atanassova N, Macpherson S, McKinnell C, van den Driesche S, Jobling MS, Chambers TJ, De Gendt K, Verhoeven G. et al. Fetal programming of adult Leydig cell function by androgenic effects on stem/progenitor cells. Proc Natl Acad Sci USA 2014;111:E1924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wakui S, Shirai M, Motohashi M, Mutou T, Oyama N, Wempe MF, Takahashi H, Inomata T, Ikegami M, Endou H, Asari M.. Effects of in utero exposure to di(n-butyl) phthalate for estrogen receptors alpha, beta, and androgen receptor of Leydig cell on rats. Toxicol Pathol 2014;42:877–87. [DOI] [PubMed] [Google Scholar]
- 12. Auharek SA, de Franca LR, McKinnell C, Jobling MS, Scott HM, Sharpe RM.. Prenatal plus postnatal exposure to Di(n-Butyl) phthalate and/or flutamide markedly reduces final Sertoli cell number in the rat. Endocrinology 2010;151:2868–75. [DOI] [PubMed] [Google Scholar]
- 13. Wang Y, Yang Q, Liu W, Yu M, Zhang Z, Cui X.. Di(2-ethylhexyl) phthalate exposure in utero damages Sertoli cell differentiation via disturbance of sex determination pathway in fetal and postnatal mice. Toxicol Sci 2016;152:53–61. [DOI] [PubMed] [Google Scholar]
- 14. Sekaran S, Jagadeesan A.. In utero exposure to phthalate downregulates critical genes in Leydig cells of F1 male progeny. J Cell Biochem 2015;116:1466–77. [DOI] [PubMed] [Google Scholar]
- 15. Chen J, Wu S, Wen S, Shen L, Peng J, Yan C, Cao X, Zhou Y, Long C, Lin T. et al. The mechanism of environmental endocrine disruptors (DEHP) induces epigenetic transgenerational inheritance of cryptorchidism. PLoS One 2015;10:e0126403.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu C, Qian P, Yang L, Zhang L, Chen C, He M, Lu Y, Feng W, Li M, Zhang Y. et al. Pubertal exposure to di-(2-ethylhexyl)-phthalate inhibits G9a-mediated histone methylation during spermatogenesis in mice. Arch Toxicol 2016;90:955–69. [DOI] [PubMed] [Google Scholar]
- 17. Buckley JP, Engel SM, Braun JM, Whyatt RM, Daniels Jl, Mendez MA, Richardson DB, Xu Y, Calafat AM, Wolff MS. et al. Prenatal phthalate exposures and body mass index among 4- to 7-year-old children: a pooled analysis. Epidemiology 2016;27:449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hatch EE, Nelson JW, Qureshi MM, Weinberg J, Moore Ll, Singer M, Webster TF.. Association of urinary phthalate metabolite concentrations with body mass index and waist circumference: a cross-sectional study of NHANES data, 1999–2002. Environ Health 2008;7:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shoaff J, Papandonatos GD, Calafat AM, Ye X, Chen A, Lanphear BP, Yolton K, Braun JM.. Early-life phthalate exposure and adiposity at 8 years of age. Environ Health Perspect 2017;125:097008.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buckley JP, Engel SM, Mendez MA, Richardson DB, Daniels Jl, Calafat AM, Wolff MS, Herring AH.. Prenatal phthalate exposures and childhood fat mass in a New York city cohort. Environ Health Perspect 2016;124:507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maresca MM, Hoepner LA, Hassoun A, Oberfield SE, Mooney SJ, Calafat AM, Ramirez J, Freyer G, Perera FP, Whyatt RM. et al. Prenatal exposure to phthalates and childhood body size in an urban cohort. Environ Health Perspect 2016;124:514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hao C, Cheng X, Guo J, Xia H, Ma X.. Perinatal exposure to diethyl-hexyl-phthalate induces obesity in mice. Front Biosci (Elite Ed) 2013;5:725–33. [DOI] [PubMed] [Google Scholar]
- 23. Hao C, Cheng X, Xia H, Ma X.. The endocrine disruptor mono-(2-ethylhexyl) phthalate promotes adipocyte differentiation and induces obesity in mice. Biosci Rep 2012;32:619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang JR, Sun Wl, Jing YF, Liu SB, Ma Z, Hong Y, Ma L, Qin C, Liu Q, Stratton HJ. et al. Prenatal exposure to di-n-butyl phthalate induces anorectal malformations in male rat offspring. Toxicology 2011;290:322–6. [DOI] [PubMed] [Google Scholar]
- 25. Wine RN, Li LH, Barnes LH, Gulati DK, Chapin RE.. Reproductive toxicity of di-n-butylphthalate in a continuous breeding protocol in Sprague-Dawley rats. Environ Health Perspect 1997;105:102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li M, Qiu L, Zhang Y, Hua Y, Tu S, He Y, Wen S, Wang Q, Wei G.. Dose-related effect by maternal exposure to di-(2-ethylhexyl) phthalate plasticizer on inducing hypospadiac male rats. Environ Toxicol Pharmacol 2013;35:55–60. [DOI] [PubMed] [Google Scholar]
- 27. Saillenfait AM, Gallissot F, Sabate JP, Remy A.. Prenatal developmental toxicity studies on diundecyl and ditridecyl phthalates in Sprague-Dawley rats. Reprod Toxicol 2013;37:49–55. [DOI] [PubMed] [Google Scholar]
- 28. Strakovsky RS, Lezmi S, Shkoda I, Flaws JA, Helferich WG, Pan YX.. In utero growth restriction and catch-up adipogenesis after developmental di (2-ethylhexyl) phthalate exposure cause glucose intolerance in adult male rats following a high-fat dietary challenge. J Nutr Biochem 2015;26:1208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pradhan A, Olsson PE, Jass J.. Di(2-ethylhexyl) phthalate and diethyl phthalate disrupt lipid metabolism, reduce fecundity and shortens lifespan of Caenorhabditis elegans. Chemosphere 2018;190:375–82. [DOI] [PubMed] [Google Scholar]
- 30. Forner-Piquer I, Maradonna F, Gioacchini G, Santangeli S, Allara M, Piscitelli F, Habibi HR, Di Marzo V, Carnevali O.. Dose-specific effects of di-isononyl phthalate on the endocannabinoid system and on liver of female zebrafish. Endocrinology 2017;158:3462–76. [DOI] [PubMed] [Google Scholar]
- 31. Chiang HC, Kuo YT, Shen CC, Lin YH, Wang Sl, Tsou TC.. Mono(2-ethylhexyl)phthalate accumulation disturbs energy metabolism of fat cells. Arch Toxicol 2016;90:589–601. [DOI] [PubMed] [Google Scholar]
- 32. Lee S, Martinez-Arguelles DB, Campioli E, Papadopoulos V.. Fetal exposure to low levels of the plasticizer DEHP predisposes the adult male adrenal gland to endocrine disruption. Endocrinology 2017;158:304–18. [DOI] [PubMed] [Google Scholar]
- 33. Martinez-Arguelles DB, Campioli E, Lienhart C, Fan J, Culty M, Zirkin BR, Papadopoulos V.. In utero exposure to the endocrine disruptor di-(2-ethylhexyl) phthalate induces long-term changes in gene expression in the adult male adrenal gland. Endocrinology 2014;155:1667–78. [DOI] [PubMed] [Google Scholar]
- 34. Petit J, Wakx A, Gil S, Fournier T, Auzeil N, Rat P, Laprevote O.. Lipidome-wide disturbances of human placental JEG-3 cells by the presence of MEHP. Biochimie 2018;149:1–8. [DOI] [PubMed] [Google Scholar]
- 35. Johnson KJ, McDowell EN, Viereck MP, Xia JQ.. Species-specific dibutyl phthalate fetal testis endocrine disruption correlates with inhibition of SREBP2-dependent gene expression pathways. Toxicol Sci 2011;120:460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang W, Shen XY, Zhang WW, Chen H, Xu WP, Wei W.. The effects of di 2-ethyl hexyl phthalate (DEHP) on cellular lipid accumulation in HepG2 cells and its potential mechanisms in the molecular level. Toxicol Mech Methods 2017;27:245–52. [DOI] [PubMed] [Google Scholar]
- 37. Horton JD, Goldstein Jl, Brown MS.. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K. et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res 2002;43:1220–35. [PubMed] [Google Scholar]
- 39. Rome S, Lecomte V, Meugnier E, Rieusset J, Debard C, Euthine V, Vidal H, Lefai E.. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiol Genomics 2008;34:327–37. [DOI] [PubMed] [Google Scholar]
- 40. Maranghi F, Lorenzetti S, Tassinari R, Moracci G, Tassinari V, Marcoccia D, Di Virgilio A, Eusepi A, Romeo A, Magrelli A. et al. In utero exposure to di-(2-ethylhexyl) phthalate affects liver morphology and metabolism in post-natal CD-1 mice. Reprod Toxicol 2010;29:427–32. [DOI] [PubMed] [Google Scholar]
- 41. Boberg J, Metzdorff S, Wortziger R, Axelstad M, Brokken L, Vinggaard AM, Dalgaard M, Nellemann C.. Impact of diisobutyl phthalate and other PPAR agonists on steroidogenesis and plasma insulin and leptin levels in fetal rats. Toxicology 2008;250:75–81. [DOI] [PubMed] [Google Scholar]
- 42. Brown MS, Goldstein Jl.. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997;89:331–40. [DOI] [PubMed] [Google Scholar]
- 43. Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res 2001;40:439–52. [DOI] [PubMed] [Google Scholar]
- 44. Ruiz R, Jideonwo V, Ahn M, Surendran S, Tagliabracci VS, Hou YY, Gamble A, Kerner J, Irimia-Dominguez JM, Puchowicz MA. et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem 2014;289:5510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gevry N, Schoonjans K, Guay F, Murphy BD.. Cholesterol supply and SREBPs modulate transcription of the Niemann–Pick C-1 gene in steroidogenic tissues. J Lipid Res 2008;49:1024–33. [DOI] [PubMed] [Google Scholar]
- 46. Eacker SM, Agrawal N, Qian K, Dichek Hl, Gong EY, Lee K, Braun RE.. Hormonal regulation of testicular steroid and cholesterol homeostasis. Mol Endocrinol 2008;22:623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martinez-Arguelles DB, Culty M, Zirkin BR, Papadopoulos V.. In utero exposure to di-(2-ethylhexyl) phthalate decreases mineralocorticoid receptor expression in the adult testis. Endocrinology 2009;150:5575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abdel-Maksoud FM, Leasor KR, Butzen K, Braden TD, Akingbemi BT.. Prenatal exposures of male rats to the environmental chemicals bisphenol A and di(2-ethylhexyl) phthalate impact the sexual differentiation process. Endocrinology 2015;156:4672–83. [DOI] [PubMed] [Google Scholar]
- 49. Wu S, Zhu J, Li Y, Lin T, Gan L, Yuan X, Xiong J, Liu X, Xu M, Zhao D. et al. Dynamic epigenetic changes involved in testicular toxicity induced by di-2-(ethylhexyl) phthalate in mice. Basic Clin Pharmacol Toxicol 2010;106:118–23. [DOI] [PubMed] [Google Scholar]
- 50. Wu SD, Zhu J, Li YS, Lin T, Gan LQ, Yuan XG, Xu MD, Wei GH.. Dynamic effect of di-2-(ethylhexyl) phthalate on testicular toxicity: epigenetic changes and their impact on gene expression. Int J Toxicol 2010;29:193–200. [DOI] [PubMed] [Google Scholar]
- 51. Moody L, Kougias D, Jung PM, Digan I, Hong A, Gorski A, Chen H, Juraska J, Pan YX.. Perinatal phthalate and high-fat diet exposure induce sex-specific changes in adipocyte size and DNA methylation. J Nutr Biochem 2019;65:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kostka G, Urbanek-Olejnik K, Wiadrowska B.. Di-butyl phthalate-induced hypomethylation of the c-myc gene in rat liver. Toxicol Ind Health 2010;26:407–16. [DOI] [PubMed] [Google Scholar]
- 53. Ge R, Tao L, Kramer PM, Cunningham Ml, Pereira MA.. Effect of peroxisome proliferators on the methylation and protein level of the c-myc protooncogene in B6C3F1 mice liver. J Biochem Mol Toxicol 2002;16:41–7. [DOI] [PubMed] [Google Scholar]
- 54. Pogribny IP, Tryndyak VP, Boureiko A, Melnyk S, Bagnyukova TV, Montgomery B, Rusyn I.. Mechanisms of peroxisome proliferator-induced DNA hypomethylation in rat liver. Mutat Res 2008;644:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Irizarry RA, Wu H, Feinberg AP.. A species-generalized probabilistic model-based definition of CpG islands. Mamm Genome 2009;20:674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S. et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 2009;41:1350–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Besnard A, Galan-Rodriguez B, Vanhoutte P, Caboche J.. Elk-1 a transcription factor with multiple facets in the brain. Front Neurosci 2011;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dimova DK, Dyson NJ.. The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005;24:2810–26. [DOI] [PubMed] [Google Scholar]
- 59. Helin K, Wu Cl, Fattaey AR, Lees JA, Dynlacht BD, Ngwu C, Harlow E.. Heterodimerization of the transcription factors E2F-1 and DP-1 leads to cooperative trans-activation. Genes Dev 1993;7:1850–61. [DOI] [PubMed] [Google Scholar]
- 60. Massague J, Seoane J, Wotton D.. Smad transcription factors. Genes Dev 2005;19:2783–810. [DOI] [PubMed] [Google Scholar]
- 61. Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci 2000;25:496–502. [DOI] [PubMed] [Google Scholar]
- 62. Pike JW, Meyer MB, Lee SM, Onal M, Benkusky NA.. The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights. J Clin Invest 2017;127:1146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ramsay RG, Gonda TJ.. MYB function in normal and cancer cells. Nat Rev Cancer 2008;8:523–34. [DOI] [PubMed] [Google Scholar]
- 64. Tantin D. Oct transcription factors in development and stem cells: insights and mechanisms. Development 2013;140:2857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gronostajski RM. Roles of the NFI/CTF gene family in transcription and development. Gene 2000;249:31–45. [DOI] [PubMed] [Google Scholar]
- 66. Hipp N, Symington H, Pastoret C, Caron G, Monvoisin C, Tarte K, Fest T, Delaloy C.. IL-2 imprints human naive B cell fate towards plasma cell through ERK/ELK1-mediated BACH2 repression. Nat Commun 2017;8:1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Allen RD 3rd, Kim HK, Sarafova SD, Siu G.. Negative regulation of CD4 gene expression by a HES-1-c-Myb complex. Mol Cell Biol 2001;21:3071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pastorcic M, Das HK.. Ets transcription factors ER81 and Elk1 regulate the transcription of the human presenilin 1 gene promoter. Brain Res Mol Brain Res 2003;113:57–66. [DOI] [PubMed] [Google Scholar]
- 69.Phthalates NRCUCotHRo (ed). Phthalates and Cumulative Risk Assessment: The Tasks Ahead. Washington (DC): National Academies Press, 2008. [PubMed] [Google Scholar]
- 70. Zhou D, Wang H, Cui H, Chen H, Pan YX.. Early-life exposure to high-fat diet may predispose rats to gender-specific hepatic fat accumulation by programming Pepck expression. J Nutr Biochem 2015;26:433–40. [DOI] [PubMed] [Google Scholar]
- 71. Herb BR, Wolschin F, Hansen KD, Aryee MJ, Langmead B, Irizarry R, Amdam GV, Feinberg AP.. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat Neurosci 2012;15:1371–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ. et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA 2012;109:10522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Holmes EE, Jung M, Meller S, Leisse A, Sailer V, Zech J, Mengdehl M, Garbe LA, Uhl B, Kristiansen G. et al. Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One 2014;9:e93933.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kint S, De Spiegelaere W, De Kesel J, Vandekerckhove L, Van Criekinge W.. Evaluation of bisulfite kits for DNA methylation profiling in terms of DNA fragmentation and DNA recovery using digital PCR. PLoS One 2018;13:e0199091.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM, Messeguer X.. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 2003;31:3651–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM.. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002;18:333–4. [DOI] [PubMed] [Google Scholar]
- 77. Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G. et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018;46:D260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.